 Karolina Slowicka
Karolina Slowicka Geert van Loo
Geert van Loo- 1Unit of Cellular and Molecular Pathophysiology, VIB Center for Inflammation Research, Ghent, Belgium
- 2Department of Biomedical Molecular Biology, Ghent University, Ghent, Belgium
Optineurin (OPTN) was identified 20 years ago in a yeast-two-hybrid screen with a viral protein known to inhibit the cytolytic effects of tumor necrosis factor. Since then, OPTN has been identified as a ubiquitin-binding protein involved in many signaling pathways and cellular processes, and mutations in the OPTN gene have been associated with glaucoma, Paget’s disease of bone and neurodegenerative pathologies. Its role in autophagy, however, has attracted most attention in recent years and may explain (some of) the mechanisms behind the disease-associated mutations of OPTN. In this brief review, we focus on the role of OPTN in inflammation and immunity and describe how this may translate to its involvement in human disease.
Introduction
Optineurin (OPTN) was first identified as a binding partner of an adenoviral E3 14.7 kDa protein and named “FIP-2” (for E3-14.7K-interacting protein) but after renamed to “optineurin” (for optic neuropathy inducing) since mutations in the OPTN gene had been identified in patients with primary open-angle glaucoma (1, 2). Later on, OPTN mutations were also identified in other human pathologies including Paget disease of bone, amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (3–5), explaining the growing interest of the scientific community for this gene.
Optineurin has been characterized as a multifunctional protein regulating multiple cellular processes such as vesicular trafficking, cell division, inflammatory and antiviral signaling, anti-bacterial responses, and autophagy. OPTN can bind multiple partners; hence, disease-causing mutations may alter these interactions disturbing normal signaling (Figure 1). However, many questions remain, and more evidence is needed to clarify its multiple functions and contribution to disease.
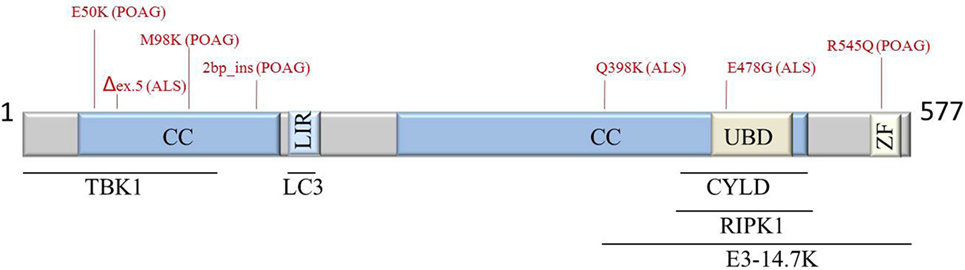
Figure 1. Schematic representation of OPTN protein structure, interaction partners, and most common POAG- and ALS-associated mutations (indicated in red). Abbreviations: ALS, amyotrophic lateral sclerosis; CC, coiled coil; CYLD, cylindromatosis; E3-14.7K, early region 3 14.7 kDa protein; LIR, LC3-interacting region; POAG, primary open-angle glaucoma; RIPK1, receptor-interacting protein kinase 1; TBK1, TANK-binding kinase 1; UBD; ubiquitin-binding domain; ZF, zinc finger; OPTN, optineurin.
OPTN and Nuclear Factor-κB (NF-κB) Signaling
Inflammatory signaling pathways, and especially NF-κB signaling, are heavily controlled by ubiquitination, a posttranslational modification of proteins. Polyubiquitin chain formation through lysine 48 (K48) of ubiquitin directs proteasomal degradation of the modified protein. By contrast, K63 or linear (M1) ubiquitination normally do not lead to degradation of the protein but mediate the binding of other proteins that contain specific ubiquitin-binding domains (UBDs), driving downstream signaling (6). OPTN’s sequence shows striking homology to NF-κB essential modulator (NEMO), the core regulatory element of the inhibitor of NF-κB kinase (IKK) complex essential for NF-κB activation (7). Both OPTN and NEMO have a similar UBD which facilitates binding to M1 and K63-linked ubiquitin chains, but not to the K48-linked ones (8). Despite their sequence homology, OPTN cannot substitute NEMO in the IKK complex (9). Initially, in vitro studies identified OPTN as a negative regulator of NF-κB signaling in response to tumor necrosis factor (TNF) by competing with NEMO for binding to ubiquitinated receptor-interacting protein kinase 1 (RIPK1), dampening downstream inflammatory signaling (7, 10) (Figure 2A). Recent structural studies showed that linear ubiquitin binding by the OPTN UBD is critical for NF-κB suppression (11). OPTN was also shown to interact with cylindromatosis (CYLD), a deubiquitinating enzyme that cleaves linear and K63-linked ubiquitin chains from proteins such as NEMO and RIPK1, to block downstream NF-κB signaling (12). OPTN has also been identified as a binding partner of interleukin-1 (IL-1) receptor-associated kinase 1 (IRAK-1), where it suppresses NF-κB activation in response to IL-1β and toll-like receptor stimulation by preventing the poly-ubiquitination of TRAF6 (13) (Figure 2B). A mutant version of OPTN that fails to recruit CYLD to inhibit NF-κB activation in response to TNF is also unable to inhibit IRAK-1-induced NF-κB signaling (12, 13). Finally, OPTN was recently shown to suppress T cell receptor-induced NF-κB activation and TNF production, in a manner dependent on ubiquitin-binding (14).
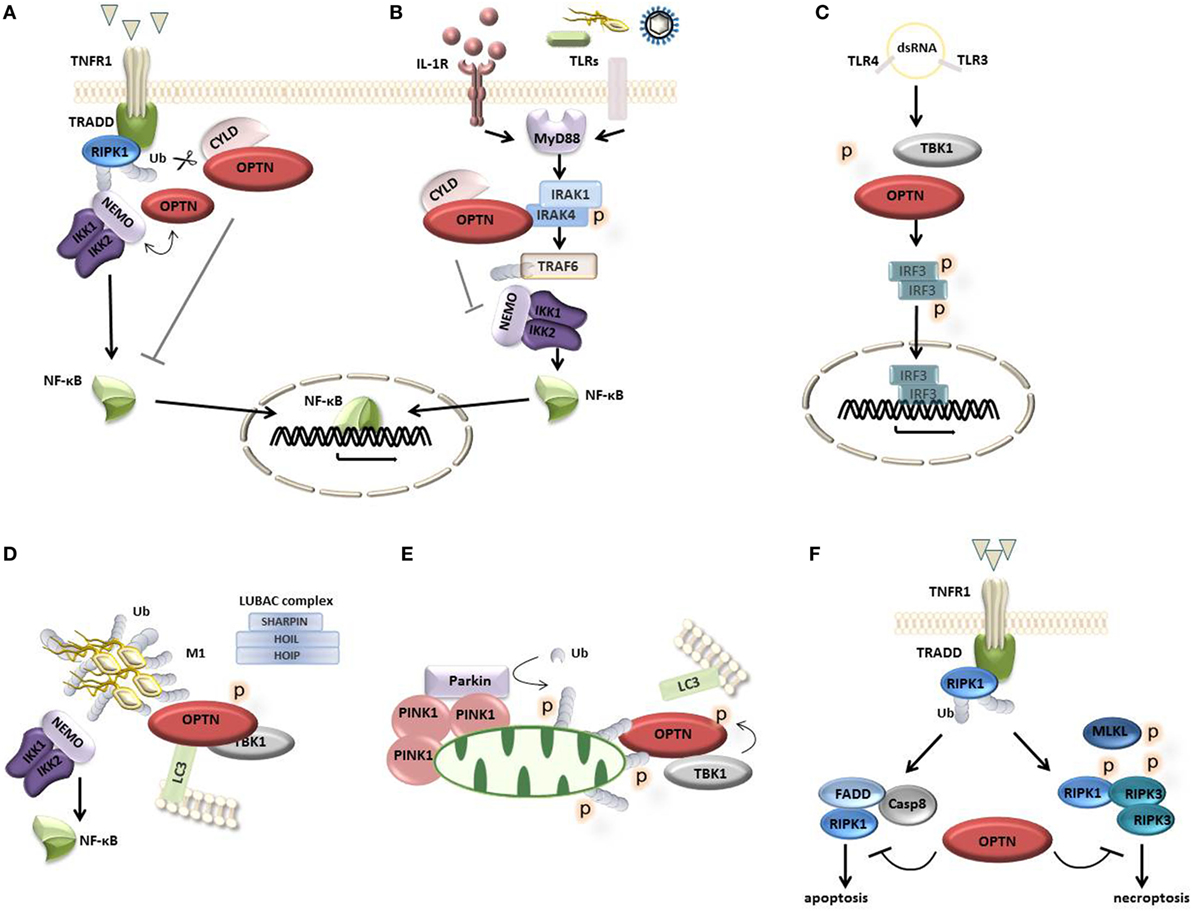
Figure 2. Optineurin (OPTN) in cellular signaling. (A) Upon activation of tumor necrosis factor (TNF) receptor 1, OPTN is recruited to the receptor complex where it competes with nuclear factor-κB (NF-κB) essential modulator (NEMO) for ubiquitinated RIPK1. OPTN can also recruit the deubiquitinating enzyme cylindromatosis (CYLD), which cleaves off ubiquitin chains from proteins such as NEMO and RIPK1, blocking downstream NF-κB signaling. (B) Upon activation of IL-1 and TLR receptors with their respective ligands, the adaptor protein MyD88 is recruited to the receptor complex where it engages the kinase interleukin-1 (IL-1) receptor-associated kinase 1 (IRAK-1) and the E3 ligase TRAF6, inducing TRAF6 auto-ubiquitination and downstream NF-κB signaling. OPTN can bind IRAK-1 through its ubiquitin-binding domain preventing TRAF6 ubiquitination and suppressing downstream NF-κB signaling. This process may involve the recruitment of CYLD. (C) Bacterial LPS and viral double-stranded RNA (dsRNA) induce the activation of TANK-binding kinase 1 (TBK1), which binds and phosphorylates OPTN, leading to the phosphorylation, dimerization, and nuclear translocation of IFN regulatory factor 3 (IRF3), mediating the transcription of interferon type I response genes. (D) OPTN acts as an autophagy receptor that associates with ubiquitin-coated cytosolic bacteria and recruits TBK1 to phosphorylate OPTN, enhancing its LC3 binding affinity, directing autophagosomal degradation. The LUBAC complex (consisting of Sharpin, HOIL-1, and HOIP) mediates the linear ubiquitination at the bacterial surface inducing the recruitment of NEMO and OPTN to activate NF-κB signaling and autophagy. (E) Damaged mitochondria are removed through mitophagy, a process involving the kinase PINK1 and the E3 ubiquitin ligase Parkin, which conjugates ubiquitin to mitochondrial outer membrane proteins, recruiting OPTN. This process involves TBK1-dependent OPTN phosphorylation promoting the autophagosomal degradation of mitochondria. (F) OPTN can bind caspase 8, limiting complex II formation and apoptosis. Alternatively, OPTN can regulate the turnover of ubiquitinated RIPK1 protecting from necroptosis.
In contrast to these in vitro studies, in vivo studies do not confirm a role for OPTN in NF-κB signaling. OPTN[D477N] knock-in mice expressing a point mutation abolishing its polyubiquitin-binding activity, as well as mice lacking either the entire C-terminal UBD or the N-terminal TBK1-binding domain, show normal NF-κB responses (8, 15, 16). Also, OPTN deficiency does not affect TNF nor TLR-induced NF-κB activation, arguing against a role for OPTN in the regulation of NF-κB signaling in vivo (17).
OPTN and Interferon (IFN) Signaling
The production of type I IFNs is an essential initial step in the host defense against infections (18). Bacterial LPS and viral double-stranded RNA (dsRNA) induce immune responses through the activation of TBK1, the phosphorylation of IFN regulatory factor 3 (IRF3), and the production of type I IFNs. OPTN can bind to TBK1, but not to the related kinase IKKε (8, 19). Bone marrow derived macrophages (BMDMs) from OPTN-deficient mice (17) or from OPTN mutant mice lacking the UBD or the TBK1-interacting region (15, 16) show diminished phosphorylation of TBK1 and IRF3 and as a result secrete lower levels of IFN-β in response to LPS or the dsRNA mimetic poly(I;C) (Figure 2C). Also BMDMs from ubiquitin-binding-defective OPTN[D477N] mutant mice show reduced TBK1 activity, IRF3 phosphorylation and production of IFN-β in response to LPS or poly(I;C) (8). Recent work from Bakshi and colleagues indeed shows that the interaction between ubiquitin chains and OPTN is required for robust phosphorylation and activation of the OPTN-TBK1 complex, triggering IRF3 activation and IFN-β production (20).
OPTN and Autophagy
With the identification of OPTN as an autophagy receptor, a new era of research on OPTN started. Autophagy is a lysosomal degradation pathway important for the removal of protein aggregates, intracellular bacteria and damaged cellular organelles (21). In 2011, the group of Ivan Dikic demonstrated that OPTN can bind to the autophagy protein LC3 via an LC3-interacting (LIR) motif, and with ubiquitin via its UBD. Upon infection of cells with Salmonella, OPTN associates with ubiquitin-coated bacteria and recruits TBK1 that phosphorylates OPTN, enhancing its LC3 binding affinity, through which it promotes the autophagic clearance of bacteria (22, 23). Also in vivo, OPTN was shown to control Salmonella infection (17, 22, 23). Upon infection, invading Salmonella bacteria become decorated with ubiquitin chains that serve as a platform to trigger various signaling cascades (24). This coat of ubiquitin around the bacteria is not uniform but contains distinct patterns of both linear and K63-linked chains eliciting different downstream signaling pathways (24, 25). Linear ubiquitination at the bacterial surface induces the recruitment of OPTN and NEMO, activating selective autophagy and inducing IKK activation and NF-κB-dependent inflammatory signaling, respectively, restricting bacterial proliferation (24, 25). The origin of M1-linked polyubiquitin in the ubiquitin coat on the bacterial surface is most probably caused by the localized recruitment of the M1-specific E3 ubiquitin ligase complex LUBAC (25) (Figure 2D). Besides OPTN, also other autophagy receptors, such as p62 and NDP52, are recruited to autophagosomal membranes through their LIR motifs. However, the recruitment of p62 and NDP52 occurs independently of LUBAC (25), demonstrating that, although there are multiple autophagy receptors, their functions are not completely redundant. Instead, they depend on their specific interacting partners and the downstream signaling cascades they activate.
Besides its role in the autophagy-mediated elimination of intracellular pathogens (xenophagy), OPTN also controls the selective autophagy of damaged mitochondria which also become conjugated with ubiquitins. This process of mitophagy depends on the kinase PINK1 and the E3 ligase Parkin, which, upon activation, ubiquitinates mitochondrial outer membrane proteins to recruit autophagy receptors. Mitophagy also involves TBK1 activation, leading to its translocation to mitochondria where it phosphorylates OPTN thereby expanding the binding capacity of OPTN to diverse ubiquitin chains, directing ubiquitin-loaded mitochondria into autophagosomes (Figure 2E) (23, 26, 27).
OPTN and Disease-Associated Mutations
Considerable interest in OPTN came from the identification of mutations in the OPTN gene in patients with degenerative diseases such as glaucoma, ALS, and FTD. Although many hypo-theses explaining the consequences of these mutations have been suggested, clear mechanisms of pathogenesis caused by OPTN mutations are still not clear.
Several OPTN mutations have been identified in patients with ALS and FTD (4, 28–31). Also mutations in TBK1 have been linked with these diseases (5, 32, 33), suggesting a common pathway defect in these pathologies. The ALS-associated TBK1 E696K mutation specifically abolishes its binding to OPTN and disrupts OPTN/TBK1 complex formation (23, 34), this in contrast to the glaucoma-associated OPTN E50K mutation, shown to cause death of retinal ganglion cells in vitro and in transgenic mice (35), which enhances the interaction between OPTN and TBK1, affecting the oligomeric state of OPTN (34). However, many ALS-associated OPTN mutations map to the C-terminal part of the protein, and not to the N-terminal TBK1-binding region, making it unlikely that these mutations will affect the interaction between OPTN and TBK1. These mutations may, however, disturb the ubiquitin-binding function of OPTN and may hint to a defect in the process of selective autophagy (34) or to a defect in OPTN’s ability to suppress NF-κB activation and apoptosis via linear ubiquitin binding (11). A recent study suggests that OPTN protects from neurodegeneration and ALS by suppressing RIPK1-dependent signaling and necroptosis, a form of regulated necrotic cell death (Figure 2F) (36). Indeed, OPTN-deficient mice develop progressive demyelination and axonal degeneration, reminiscent of ALS, due to CNS cell necroptosis and neuroinflammation, a phenotype which could be rescued by inhibiting RIPK1 kinase activity preventing necroptosis in OPTN-deficient mice (36). OPTN has also been shown to protect motor neurons from TNF-induced apoptosis, through association with caspase 8 at its dead effector domains to prevent the recruitment of FADD and downstream caspase activation (11). In agreement, cleaved caspase 3 was detected in motor neurons from OPTN-associated ALS patients, suggesting enhanced apoptosis in the absence of OPTN (11). Besides these signs of motor neuron apoptosis, intracytoplasmic inclusions in brain samples from patients with OPTN mutations often stain positive for linear ubiquitin and activated NF-κB (11).
Genetic variants of OPTN leading to lower OPTN expression have also been identified in patients with Paget’s bone disease (3, 37). OPTN was shown to act as a negative regulator of osteoclast differentiation, and mice with a loss-of-function mutation in the UBD of OPTN have increased osteoclast activity and bone turnover (38).
Finally, OPTN has been associated with Crohn’s disease (CD), and diminished expression of OPTN was observed in approximately 10% of CD patients (39, 40). Patient-derived macrophages show decreased pro-inflammatory cytokine secretion, suggesting an effect on inflammatory responses and bacterial clearance, as is observed in CD (39, 40). In agreement, loss of OPTN in mice was shown to impair cytokine production and neutrophil recruitment in a bacteria-dependent model of colitis (40, 41). However, contrary to the bacteria-driven colitis, OPTN knockout mice respond normally to the model of dextran sodium sulfate-induced colitis (17, 41). This difference in response between both colitis models might suggest a specific role for OPTN in protection from bacterial infection and infection-associated IBD. Recently, OPTN has been implicated in CD through its interaction with the endoplasmic reticulum (ER) stress sensor IRE1α, via which it was suggested to participate in the removal of ER membranes in conditions of prolonged ER stress (42). Defective autophagy and ER stress in intestinal epithelial cells induce IRE1α aggregation, triggering intestinal inflammation. However, these IRE1α aggregates can be recruited to autophagosomes via OPTN, followed by IRE1α degradation (42). Hence, OPTN-dependent selective autophagy (ERphagy) may act as a mechanism to protect from prolonged ER stress and intestinal inflammation. Alternatively, since OPTN has previously been shown to localize to protein aggregates (43), OPTN could recognize the misfolded proteins and mediate their clearance independently of IRE1α.
Concluding Remarks
Optineurin has been implicated in many signaling pathways and cellular processes. Overall, three major protective mechanisms can be considered: regulation of selective autophagy, regulation of inflammatory signaling, and protection from cell death. Mutations in OPTN, interfering with these protective activities, may eventually lead to disease. However, despite our knowledge on the role of OPTN in these cellular processes, we still know very little concerning the molecular mechanisms behind the disease-associated OPTN mutations. Most of these mutations have only been studied upon overexpression in cell culture models and have never been validated in vivo. Since OPTN knockout mice nor OPTN mutant mice defective in ubiquitin binding (15–17) develop spontaneous disease, OPTN mutations most probably induce a gain-of-function, rather than a mere loss of its normal function. Hence, the development of OPTN knock-in models expressing specific disease-associated mutations will be crucially important to clarify the importance of these mutations for disease development and will help to better understand the biological functions of OPTN.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
KS is a predoctoral fellow with the Institute for the Promotion of Innovation by Science and Technology (IWT) and is supported by a “Kom op tegen Kanker” (Stand up to Cancer) grant from the Flemish cancer society. Research in the GvL lab is supported by research grants from the FWO, the “Geneeskundige Stichting Koningin Elisabeth” (GSKE), the CBC Banque Prize, the Charcot Foundation, the “Belgian Foundation against Cancer,” and the “Concerted Research Actions” (GOA) of the Ghent University.
References
1. Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol (1998) 18:1601–10. doi:10.1128/MCB.18.3.1601
2. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science (2002) 295:1077–9. doi:10.1126/science.1066901
3. Albagha OME, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet (2010) 42:520–4. doi:10.1038/ng.562
4. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature (2010) 465:223–6. doi:10.1038/nature08971
5. Pottier C, Bieniek KF, Finch NC, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol (2015) 130:77–92. doi:10.1007/s00401-015-1436-x
6. Iwai K, Fujita H, Sasaki Y. Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat Rev Mol Cell Biol (2014) 15:503–8. doi:10.1038/nrm3836
7. Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFα-induced NF-κB activation by competing with NEMO for ubiquitinated RIP. Curr Biol (2007) 17:1438–43. doi:10.1016/j.cub.2007.07.041
8. Gleason CE, Ordureau A, Gourlay R, Arthur JSC, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J Biol Chem (2011) 286:35663–74. doi:10.1074/jbc.M111.267567
9. Schwamborn K, Weil R, Courtois G, Whiteside ST, Israël A. Phorbol esters and cytokines regulate the expression of the NEMO-related protein, a molecule involved in a NF-kappa B-independent pathway. J Biol Chem (2000) 275:22780–9. doi:10.1074/jbc.M001500200
10. Sudhakar C, Nagabhushana A, Jain N, Swarup G. NF-kappaB mediates tumor necrosis factor alpha-induced expression of optineurin, a negative regulator of NF-kappaB. PLoS One (2009) 4:e5114. doi:10.1371/journal.pone.0005114
11. Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun (2016) 7:12547. doi:10.1038/ncomms12547
12. Nagabhushana A, Bansal M, Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFα-induced NF-κB activation. PLoS One (2011) 6:e17477. doi:10.1371/journal.pone.0017477
13. Tanishima M, Takashima S, Honda A, Yasuda D, Tanikawa T, Ishii S, et al. Optineurin controls the IRAK1-TRAF6 axis identification of optineurin as an interleukin-1 receptor-associated kinase 1-binding protein and its role in regulation of MyD88-dependent signaling. J Biol Chem (2017) 292:17250–7. doi:10.1074/jbc.M117.813899
14. Montecalvo A, Watkins SC, Orange J, Kane LP. Inducible turnover of optineurin regulates T cell activation. Mol Immunol (2017) 85:9–17. doi:10.1016/j.molimm.2017.01.027
15. Munitic I, Giardino Torchia ML, Meena NP, Zhu G, Li CC, Ashwell JD. Optineurin insufficiency impairs IRF3 but not NF-κB activation in immune cells. J Immunol (2013) 191:6231–40. doi:10.4049/jimmunol.1301696
16. Meena NP, Zhu G, Mittelstadt PR, Giardino Torchia ML, Pourcelot M, Arnoult D, et al. The TBK1 binding domain of optineurin promotes type I interferon responses. FEBS Lett (2016) 590:1–11. doi:10.1002/1873-3468.12176
17. Slowicka K, Vereecke L, McGuire C, Sze M, Maelfait J, Kolpe A, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-κB signaling. Eur J Immunol (2016) 46:971–80. doi:10.1002/eji.201545863
18. Hiscott J. Convergence of the NF-κB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev (2007) 18:483–90. doi:10.1016/j.cytogfr.2007.06.002
19. Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett (2008) 582:997–1002. doi:10.1016/j.febslet.2008.02.047
20. Bakshi S, Taylor J, Strickson S, McCartney T, Cohen P. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon β. Biochem J (2017) 474:1163–74. doi:10.1042/BCJ20160992
21. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature (2011) 469:323–35. doi:10.1038/nature09782
22. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333:228–33. doi:10.1126/science.1205405
23. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113:4039–44. doi:10.1073/pnas.1523926113
24. van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hötte K, Pampaloni F, et al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-κB and restricts bacterial proliferation. Nat Microbiol (2017) 2:17066. doi:10.1038/nmicrobiol.2017.66
25. Noad J, Von Der Malsburg A, Pathe C, Michel MA, Komander D, Randow F. LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat Microbiol (2017) 17063:1–10. doi:10.1038/nmicrobiol.2017.63
26. Heo J-MM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60:7–20. doi:10.1016/j.molcel.2015.08.016
27. Harper JW, Ordureau A, Heo J-M. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol (2018) 19:93–108. doi:10.1038/nrm.2017.129
28. Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ, et al. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology (2015) 36:125–34. doi:10.1111/neup.12240
29. Tümer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, LuCamp L, et al. A novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging (2012) 33:208. doi:10.1016/j.neurobiolaging.2011.07.001
30. Millecamps S, Boillée S, Chabrol E, Camu W, Cazeneuve C, Salachas F, et al. Screening of OPTN in French familial amyotrophic lateral sclerosis. Neurobiol Aging (2011) 32:557. doi:10.1016/j.neurobiolaging.2010.11.005
31. Solski JA, Williams KL, Yang S, Nicholson GA, Blair IP. Mutation analysis of the optineurin gene in familial amyotrophic lateral sclerosis. Neurobiol Aging (2012) 33:210. doi:10.1016/j.neurobiolaging.2011.09.023
32. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci (2015) 18:631–6. doi:10.1038/nn.4000
33. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347:1436–41. doi:10.1126/science.aaa3650
34. Li F, Xie X, Wang Y, Liu J, Cheng X, Guo Y, et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun (2016) 7:12708. doi:10.1038/ncomms12708
35. Chi ZL, Akahori M, Obazawa M, Minami M, Noda T, Nakaya N, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet (2010) 19:2606–15. doi:10.1093/hmg/ddq146
36. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (2016) 353:603–8. doi:10.1126/science.aaf6803
37. Chung PYJ, Beyens G, Boonen S, Papapoulos S, Geusens P, Karperien M, et al. The majority of the genetic risk for Paget’s disease of bone is explained by genetic variants close to the CSF1, OPTN, TM7SF4, and TNFRSF11A genes. Hum Genet (2010) 128:615–26. doi:10.1007/s00439-010-0888-2
38. Obaid R, Wani SE, Azfer A, Hurd T, Jones R, Cohen P, et al. Optineurin negatively regulates osteoclast differentiation by modulating NF-κB and interferon signaling: implications for Paget’s disease. Cell Rep (2015) 13:1096–102. doi:10.1016/j.celrep.2015.09.071
39. Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJB, Sewell GW, et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J Exp Med (2009) 206:1883–97. doi:10.1084/jem.20091233090209c
40. Smith AM, Sewell GW, Levine AP, Chew TS, Dunne J, O’Shea NR, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology (2015) 144:45–55. doi:10.1111/imm.12338
41. Chew TS, O’Shea NR, Sewell GW, Oehlers SH, Mulvey CM, Crosier PS, et al. Optineurin deficiency contributes to impaired cytokine secretion and neutrophil recruitment in bacteria driven colitis. Dis Model Mech (2015) 8:817–29. doi:10.1242/dmm.020362
42. Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, et al. Defective ATG16L1-mediated removal of IRE1 α drives Crohn’s disease-like ileitis. J Exp Med (2017) 214(2):401–22. doi:10.1084/jem.20160791
Keywords: optineurin, inflammation, immunity, mitophagy/autophagy, nuclear factor-κB, ubiquitin, TANK-binding kinase 1
Citation: Slowicka K and van Loo G (2018) Optineurin Functions for Optimal Immunity. Front. Immunol. 9:769. doi: 10.3389/fimmu.2018.00769
Received: 07 February 2018; Accepted: 27 March 2018;
Published: 10 April 2018
Edited by:
Andrew Mark Smith, University College London, United KingdomReviewed by:
Oliver Florey, Babraham Institute (BBSRC), United KingdomCaroline Jefferies, Cedars-Sinai Medical Center, United States
Copyright: © 2018 Slowicka and van Loo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Geert van Loo, Z2VlcnQudmFubG9vQGlyYy52aWItdWdlbnQuYmU=