 Aleksandra Leligdowicz
Aleksandra Leligdowicz Melissa Richard-Greenblatt
Melissa Richard-Greenblatt Julie Wright
Julie Wright Valerie M. Crowley
Valerie M. Crowley Kevin C. Kain
Kevin C. Kain- Sandra Rotman Centre for Global Health, University Health Network and University of Toronto, Toronto, ON, Canada
Sepsis, a dysregulated host response to infection that causes life-threatening organ dysfunction, is a highly heterogeneous syndrome with no specific treatment. Although sepsis can be caused by a wide variety of pathogenic organisms, endothelial dysfunction leading to vascular leak is a common mechanism of injury that contributes to the morbidity and mortality associated with the syndrome. Perturbations to the angiopoietin (Ang)/Tie2 axis cause endothelial cell activation and contribute to the pathogenesis of sepsis. In this review, we summarize how the Ang/Tie2 pathway is implicated in sepsis and describe its prognostic as well as therapeutic utility in life-threatening infections.
Introduction
Sepsis is a state of life-threatening organ dysfunction caused by a dysregulated host response to infection (1). Despite being a leading cause of global morbidity and mortality, sepsis has no known specific therapies (2). The current critical illness classification defines organ dysfunction by an increase in the Sequential [sepsis-related] Organ Failure Assessment (SOFA) score (3). However, sepsis is a heterogeneous syndrome that is not completely characterized using non-specific clinical variables. The use of generic classification models for complex, critically ill patients may impede appropriate triage and management and limit the application of personalized treatment strategies (4). A more comprehensive characterization of sepsis pathophysiology may reveal new opportunities for precision medicine-based therapies with either novel or repurposed agents that target specific pathways contributing to disease (4, 5).
Microvascular dysfunction is the endpoint of many life-threatening infections (6), and a well-established relationship exists between endothelial injury and sepsis (7–9). Determining the severity of microbial infections is challenging early in the course of disease when clinical scoring systems have limited prognostic utility. Many classic markers of end-organ compromise (such as serum lactate, bilirubin, and creatinine clearance) are not informative until significant clinical deterioration has occurred. However, the timely recognition of sepsis is critical as early aggressive management can considerably reduce morbidity and mortality (10). Early prognostic indicators of critical illness severity are needed to improve early recognition, appropriate triage, management, and outcomes, as well as to enable rational health resource allocation. This review examines the role of the angiopoietin (Ang)/Tie2 axis in sepsis and summarizes its potential applications in the early recognition of sepsis and as a therapeutic target to improve clinical outcomes.
The Ang/Tie Axis in Sepsis
Vascular function and permeability are regulated by endothelial-specific receptor tyrosine kinases and their ligands, including the vascular endothelial growth factor (VEGF)-VEGF-receptors (VEGFRs), and the Ang-Tie receptors. The Tie1 and Tie2 receptors constitute the Tie receptor family and are almost exclusively expressed in the endothelium (11, 12). Tie2 functions as a receptor for the Ang family of proteins (Ang1, Ang2, and Ang4), while Tie1 is an orphan receptor that can be activated by Angs via its interaction with Tie2 (13). Binding of Angs to Tie2 in the stable vasculature promotes the formation of a Tie1/Tie2 heterodimer in a β1 integrin-dependent manner, resulting in Tie2 trafficking to cell–cell junctions (14, 15).
During vascular quiescence, mesenchymal cells secrete Ang1, a strong Tie2 agonist, to support endothelial survival and vascular stability (16). Under these conditions, oligomerized Ang1 promotes the trans-association of Tie2 at cell–cell contacts and can also anchor Tie2 to the extracellular matrix (ECM) through binding fibronectin, collagen, and vitronectin with high affinity (17). In addition to forming adhesive structures between cell–cell and cell–substratum contacts, Tie2 activation by Ang1 induces a number of downstream signaling cascades as shown in Figure 1. Notably, the serine kinase, Akt, is activated and results in the phosphorylation of the Forkhead box protein O1 (FOXO1) transcription factor, leading to the nuclear exclusion of FOXO1 and decreased expression of its target genes (18–20). The inhibition of Foxo1 transcriptional activity in endothelial cells (ECs) induces expression of genes involved in vessel stability and the repression of genes involved in vascular destabilization, including Ang2. Consequently, during quiescence, Ang2 is constitutively expressed at low levels and co-localizes with von Willebrand factor (vWF) within the Weibel Palade bodies (WPBs) of ECs (21).
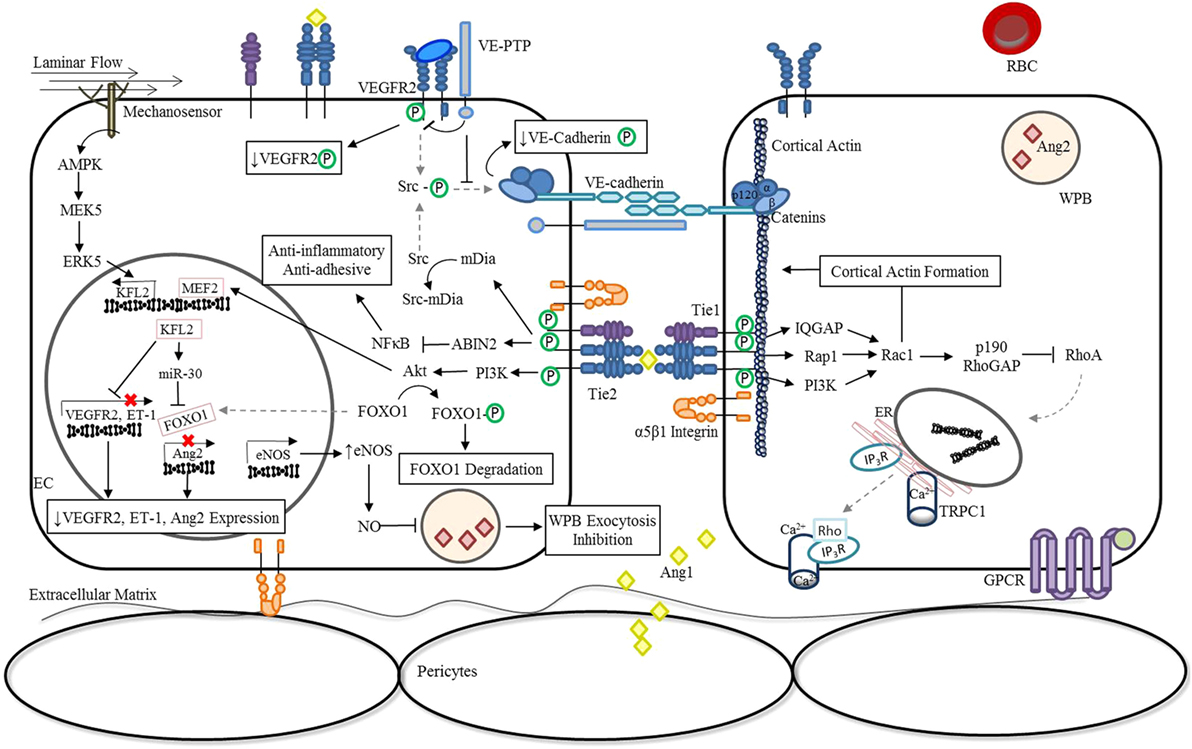
Figure 1. Ang/Tie pathway regulation of vascular stability. In the absence of inflammation or infection (stable vasculature), Ang1 is secreted by mesenchymal cells (22, 23) and low levels of autocrine Ang2 are constitutively secreted (24). Binding of Angs to Tie2 promotes the interaction of Tie1 and Tie2 in a β1 integrin-dependent manner and regulates Ang-induced Tie2 trafficking to cell–cell junctions (14, 15). Under these conditions, endothelial Tie1 enhances the activity of Ang1 and is essential for Ang2 agonistic activity (14, 15). Oligomerized Ang1 also induces the translocation of Tie2 to cell–cell contacts and induces an Ang1-bridged Tie2 trans-association (17). The resulting phosphorylation of Tie2 leads to the activation a number of downstream signaling pathways that are involved in vessel stability and endothelial barrier function. Activation of the downstream serine kinase, AKT, leads to the phosphorylation and nuclear exclusion of FOXO1, and repression of its target genes which include Ang2 (18–20). Ang1 inhibition of NF-κB reporter gene activity via activation of ABIN2 dampens the expression of adhesion molecules and pro-inflammatory cytokines (25, 26), preventing further activation of the endothelium through localized inflammatory mediators. In parallel, Ang/Tie2 signaling stimulates the transcriptional activity of MEF2 through the PI3K/AKT pathway to induce the expression of a second transcription factor KFL2 to ultimately counteract VEGF-mediated vascular permeability (↑eNOS expression; ↓VEGFR2 and ET-1 expression) (27). The increase in NO generated by eNOS combined with the negative regulation of Ang2 expression during quiescence significantly reduces luminal concentrations of Ang2 (28). In addition, KFL2 induces miR-30 expression, further blocking the transcription of Ang2 (29). The phosphorylation of Src, which generally culminates in the phosphodependent internalization of VE-cadherin, is also inhibited by Ang1/Tie2. Signaling through Ang1 leads to the activation of mDia, resulting in the sequestration of Src, and prevention of subsequent phosphorylation by VEGFR2 (30). At cell–cell junctions, Ang1/Tie2 also blocks VEGF signaling by promoting the interaction of VEGFR2 with VE-PTP (31). Lastly, activation of Tie2 can lead to the activation of the GTPase, Rac1, via IQGAP (32), Rap1 (33), or PI3K/Akt (34)-dependent pathways to stabilize the cortical actin cytoskeleton and maintain adherens and tight junctions between cells (35). In the presence of LPS, activation of the RhoA-specific GTPase activating protein, p190RhoGAP, by Rac1, is essential for shifting the balance away from RhoA rearrangement of the actin cytoskeleton and preventing vascular permeability (32, 35). Abbreviations: ABIN2, A20-binding inhibitor of nuclear factor-κB-2; Ang1, angiopoietin-1; Ang2, angiopoietin-2; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; ER, endoplasmic reticulum; ET-1, endothelin-1; FOXO1, forkhead box protein O1; GPCR, G-protein-coupled receptor; IP3R, inositol triphosphate receptor; IQGAP, IQ motif containing GTPase activating protein; KLF2, Krüppel-like factor-2; LPS, lipopolysaccharide; mDia, mammalian diaphanous; MEF2, myocyte enhancer factor-2; miR-30, microRNA-30-5p; NF-κB, nuclear factor-κB; NO, nitric oxide; PI3K, phosphoinositide triphosphate kinase; Rac1, RAS-related C3 botulinum toxin substrate 1; Rap1, Ras-related protein 1; RBC, red blood cell; RhoA, Ras homolog gene family, member A; Src, proto-oncogene tyrosine-protein kinase; TRPC1, transient receptor potential channel-1; VE-cadherin, vascular endothelial-cadherin; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; VE-PTP, vascular endothelial protein tyrosine phosphatase; WPB, Weibel-Palade Body; P, phosphorylation.
Upon stimulation of ECs by inflammatory cytokines or VEGF, Ang2 expression and secretion from WPB are increased, creating an autocrine regulatory mechanism of Tie2 signaling (36, 37). However, in contrast to Ang1, the action of Ang2 on Tie2 signaling has an additional level of complexity that is dependent on the microenvironment of ECs (38–41). While the Tie1/Tie2 heterodimeric complex enables both Ang1 and Ang2 to function as Tie2 agonists (14, 15), in the presence of an infection or inflammation ECs shed the Tie1 ectodomain, and Ang2 binding results in Tie2 antagonism (14). Similarly, Tie1 shedding decreases Ang1 agonistic activity (reduced Tie2 phosphorylation), demonstrating that Tie1 is required for the full activation of Tie2 (14, 15). Taken together, infection increases Ang2 expression and its release from WPBs, tipping the luminal Ang balance in favor of Ang2. Consequently, the increase in Ang2/Tie2 binding, particularly under conditions of enhanced Tie1 shedding, blocks Tie2 activation and contributes to the destabilization of the endothelium.
In addition, binding of Ang1 to Tie2 can also stimulate the association of vascular endothelial (VE)-protein tyrosine phosphatase (PTP) with the Tie receptor complex (42). Under conditions of hypoxia, such as that resulting from infection-induced reduction in laminar flow, VE-PTP expression is up-regulated (43), and a negative feedback loop is triggered to limit Tie2 activation (42, 44). As outlined in Figure 2, there are a number of mechanisms employed by ECs to modulate Ang/Tie2 signaling during infection-induced endothelial activation. Findings from our group (45–53) and many others (54–60) have shown that disruption of any of these components related to the Ang/Tie axis may result in endothelial dysregulation and microvascular leak, regardless of the microbial etiology.
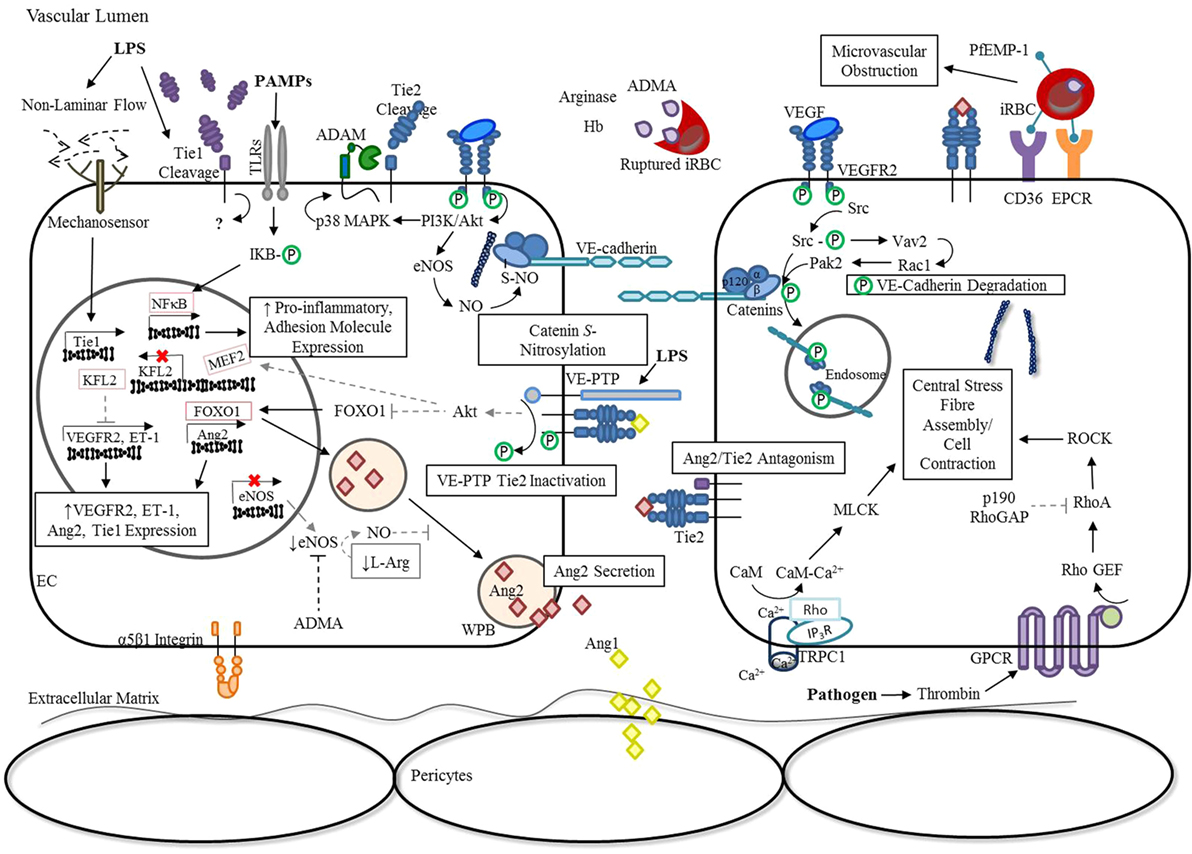
Figure 2. Endothelial activation: dysregulation of Ang/Tie signaling during severe infection. In response to inflammation, ECs release Ang2 from WPB into the vascular lumen, tipping Ang2:Ang1 ratios in favor of Ang2 (21). The release of ADMA, arginase, and hemoglobin from ruptured Plasmodium falciparum infected erythrocytes additionally decrease the availability of NO, enhancing WPB exocytosis (61, 62). Simultaneously, the presence of infection or TNF-α leads to Tie1 inactivation by ectodomain cleavage, thereby reducing the agnostic activity of Ang1 and promoting the antagonistic action of Ang2 (14, 15). Under conditions of hypoxia, such as an infection-induced reduction in laminar flow, VE-PTP expression is up-regulated (43), and a negative feedback loop is triggered that limits Tie2 activation by enhancing its association with VE-PTP (42). Interestingly, the transcription of Tie1 is also up-regulated in response to reduced laminar flow, despite ectodomain cleavage, suggesting an unknown role for the intracellular tyrosine kinase domain in signaling under these conditions (63). The resulting inactivation of Tie2 during infection and/or inflammation promotes FOXO1 transcriptional activity, thereby increasing Ang2 expression (15, 18, 20). The reduction in KFL2 expression associated with Tie2 inactivation stimulates VEGF-induced monocyte adhesion and vascular permeability (27, 64, 65). Furthermore, increased VEGFR2-signaling during infection leads to the activation of downstream pathways, such as PI3K/Akt and Src. Consequently, p38 MAPK-dependent activation of the protease, ADAM-15, induces Tie2 shedding and prevents its downstream signaling (66). In parallel, VEGFR2 activation of eNOS and Src further disrupt adherens junction complexes through S-nitrosylation of β-catenin (67) and phosphorylation of VE-cadherin (68, 69), respectively. Pathogens can indirectly lead to the activation of RhoA through GPCRs (70), for example, thrombin binding of protease-activated receptors (71, 72), to promote the formation of actin stress fibers that increase centripetal tension throughout the cytoskeleton (73). Furthermore, limited Tie2 activation during infection prevents p190RhoGAP inhibition of RhoA (35), and subsequent coupling of IP3R and TRPC1 to form a Ca2+ channel in the plasma membrane (74). This rise in intracellular Ca2+ leads to CaM-dependent activation of MLCK to further support EC contraction (35, 75). Lastly, TLR activation by PAMPs triggers the downstream activation of NF-κB to induce the expression of pro-inflammatory cytokines and adhesion molecules (76). This generates a positive feedback mechanism for endothelial destabilization, until infection and the associated inflammatory response are resolved (77). Abbreviations: ADAM-15, disintegrin and metalloproteinase domain-containing protein 15; ADMA, asymmetric dimethylarginine; Ang1, angiopoietin-1; Ang2, angiopoietin-2; Ca2+, calcium; CaM, calmodulin; CD36, cluster differentiation 36; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; EPCR, endothelial protein C receptor ET-1, endothelin-1; FOXO1, forkhead box protein O1; GPCR, G-protein-coupled receptor; iRBC, Plasmodium-infected red blood cell; IP3R, inositol triphosphate receptor; IQGAP, IQ motif containing GTPase activating protein; KLF2, Krüppel-like factor-2; LPS, lipopolysaccharide; mDia, mammalian diaphanous; MEF2, myocyte enhancer factor-2; miR-30, microRNA-30-5p; MLCK, myosin light-chain kinase; NF-κB, nuclear factor-κB; NO, nitric oxide; p190RhoGAP, p190Rho GTPase-activating protein; p38 MAPK, p38 mitogen-activated protein kinase; PAMPs, pathogen-associated molecular patterns; PI3K, phosphoinositide triphosphate kinase; Rac1, RAS-related C3 botulinum toxin substrate 1; Rap1, Ras-related protein 1; RhoA, Ras homolog gene family, member A; RhoGEF: Rho guanine nucleotide exchange factor; S-NO, S-nitrosylation; Src, proto-oncogene tyrosine-protein kinase; TRPC1, transient receptor potential channel-1; VE-cadherin, vascular endothelial-cadherin; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; VE-PTP, vascular endothelial protein tyrosine phosphatase; WPB, Weibel-Palade Body; P, phosphorylation.
Although beyond the scope of this review it should be noted that both coagulation and complement activation contribute to the course and outcome of sepsis. A connection between the Ang/Tie2 pathway and coagulation in sepsis was revealed in a proteomic analysis of septic patients with disseminated coagulation (DIC). Findings from this study demonstrated that changes in Tie2 signaling was an initiating event in septic DIC and, at least in a mouse model, restoring Tie2 activation was sufficient to mitigate thrombosis (78). Additionally, the anticoagulant, activated protein C (APC) has been shown to bind and activate Tie2, leading to improved endothelial barrier integrity (79). Still, little remains known about the interplay between complement activation and Ang/Tie2 pathways in sepsis; this is an area that requires further investigation.
The Ang/Tie Pathway in Severe Bacterial Infections
The incidence of severe sepsis in the United States is estimated at 3 cases per 1,000 population (80, 81). Mortality due to sepsis is high at approximately 20–55% (80–85), estimates that are relatively stable over nearly the past decade (86). Despite a high burden of disease and improved application of management strategies (87), there is a lack of effective treatments specific for sepsis (5, 88).
Gram-negative and Gram-positive infections occur with similar frequencies in hospitalized patients (89), and both can trigger sepsis mediated in part by the production of exotoxins or the release of bacterial cell wall components into the systemic circulation (32). These microbial products modulate many host response pathways, including in the Ang/Tie axis. The suppression of Ang1/Tie2 signaling and the associated microvascular leak are common features of many severe infections including bacterial sepsis (54). Reduced Tie2 activation during sepsis is the result of several perturbations to the pathway, including decreased Tie2 and Ang1 expression (24, 54, 57, 90–92), generation of soluble Tie receptors (14, 15, 40, 41, 66, 93, 94), and the antagonistic activity of Ang2 (14, 15).
Preclinical models of sepsis that have been used to explore the mechanisms and consequences of Tie2 signaling have shown that Tie2 expression and phosphorylation are greatly reduced during systemic infection. Inhibition of Tie2 activation leads to the nuclear localization of FOXO1 and transcription of its target genes, resulting in increased microvascular permeability (14, 15, 57, 91, 92). Tie2 mRNA levels have been shown to decline in response to decreases in endothelial shear stress associated with severe infection in a nuclear factor (NF)-κB dependent manner (92, 95). In contrast, the downregulation of Tie2 protein on the EC surface is not mediated by NF-κB but rather by the proteolytic cleavage of the Tie2 extracellular domain (92). In vitro studies have demonstrated both constitutive and VEGF-stimulated Tie2 receptor cleavage result in the release of a 75-kDa soluble Tie2 (sTie2) protein (66, 94, 96). The cleavage of the Tie2 ectodomain prevents Ang/Tie2 signaling and the circulating sTie2 which is generated may then function as a ligand trap, binding, and further inhibiting Ang activity. Indeed, the intravenous administration of adenoviral vectors expressing sTie2 blocked Ang/Tie2 signaling in Mycoplasma pulmonis-infected mice (97). Notably, the presence of sTie2 has been documented in vivo (93, 98, 99) and its levels are significantly increased in septic versus non-septic Intensive Care Units (ICU) patients (100). However, recent evidence incorporating mathematical and in vitro experimental modeling suggests that the molar ratio of sTie2:Ang1 levels found in patients with severe sepsis would have little influence on Ang/Tie2 activation in vivo (101).
In addition to reducing Tie2 expression, bacterial infections may also alter the functional activation state of this receptor, contributing to a leaky microvascular phenotype. Murine models of sepsis demonstrate a significant decline in the phosphorylated Tie2:total Tie2 ratio following LPS administration (91). This may be due, at least partly, to the influence of Tie1 on Tie2 activation. As already discussed, under baseline conditions, Ang1 or Ang2 stimulation of Tie2 promotes its interaction with Tie1 to form a heteromeric complex that is translocated to areas of cell–cell contacts (14, 17, 102). Oligomerized Ang1 bridges Tie2 at cell junctions resulting in the formation of trans-associations with Tie2 that preferentially activate Akt and its downstream signaling pathways, maintaining vascular quiescence (17). However in the mouse model, both LPS challenge and M. pulmonis infection induce cleavage of the Tie1 ectodomain responsible for its interaction with Tie2, thereby reducing Tie2 activation (14, 15). During endotoxemia, Tie1 cleavage also promotes antagonistic Ang2 activity resulting in the suppression of Tie2 signaling. This restores FOXO1 activity and establishes a positive feedback loop whereby FOXO1-driven Ang2 expression promotes microvascular leak during infection (14, 15).
Interestingly in murine models, Gram-negative bacteria may increase Ang2 expression by a different mechanism than that of Gram-positive bacteria. LPS has been shown to increase Ang2 expression via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 signaling through the inhibition of NFκ-B kinase subunit β/NF-κB and mitogen-activated protein kinase (MAPK)/activator protein 1 pathways (103), as well as the histone/protein deacetylase, Sirtuins 3 (24). Although mice treated with Gram-positive cell wall components (peptidoglycan and lipoteichoic acid) also exhibit elevated levels of Ang2 (24, 104), a comparison of bacterial etiologies among septic ICU patients revealed that Ang2/Ang1 ratios were significantly higher among patients with Gram-negative than those with Gram-positive infections (105). Recent evidence suggests that increased circulating Ang2 in Gram-positive infections is not the result of de novo biosynthesis, but rather the stimulated secretion of intracellular storage pools in response to the binding of cell wall components to TLR2 (24). While mechanistic differences may account for the more robust Ang2 increase observed in Gram-negative bacterial infections, these findings require in vivo confirmation.
During sepsis, an increase in Ang1 or decrease in Ang2 levels can enhance survival in murine bacterial sepsis models (56, 90, 106, 107). The overexpression of Ang1 using an adenoviral construct expressing human Ang1 (rh-Ang1) was found to attenuate LPS-induced expression of endothelial adhesion molecules in mouse lungs and kidneys, resulting in decreased leukocyte infiltration into interstitial spaces and minimizing hemodynamic instability (106, 107). In these mice, endothelial nitric oxide synthase (eNOS) expression was preserved and inducible nitric oxide (NO) synthase activity was decreased, contributing to reduced microvascular permeability in major organs (106, 107). The corresponding anti-inflammatory and anti-permeability effects of overexpressing Ang1 ultimately resulted in reduced organ injury as well as enhanced survival in endotoxemic mice compared to their Fc-controls. Despite the observed protective effect, little remains known as to how elevated levels of Ang1 impact the innate immune response. However, a recent study demonstrated that the improved survival conferred by the administration of rh-Ang1 in a mouse model of cerebral malaria (CM) was independent of its direct effects on parasitemia as both mice receiving rh-Ang1 and the Fc control had comparable parasite burdens (45). These findings suggest that increasing Ang1 levels during severe infection does not impair the host’s ability to resolve infection.
The protective effect observed with Ang1 has also been described in a cecal ligation and perforation (CLP) model of mice with one functional Ang2 allele (Ang2±), suggesting Ang2 contributes to multi-organ dysfunction and death in sepsis (56). In contrast, compared to their wild-type littermates, Ang2−/− knockout mice developed acute kidney injury following LPS exposure (90). Interestingly, the complete loss of Ang2 was previously observed to result in developmental abnormalities in mouse vasculature (108) likely rendering Ang2−/− mice susceptible to LPS-induced kidney injury. Nevertheless, these pre-clinical studies suggest that increasing circulating Ang1 and reducing Ang2 are associated with improved endothelial function during bacterial sepsis.
Bacterial pathogens are an important cause of sepsis, at least in part, due to their ability to induce systemic microvascular dysfunction through their interactions with the Ang/Tie system. A detailed understanding of mechanisms that regulate this pathway, and the ways in which bacteria modulate Tie2 activity, may suggest intervention strategies to maintain endothelial quiescence and microvascular integrity during severe bacterial infections. Preclinical studies of Tie2 directed therapies are reviewed in a later section.
Endothelial Dysfunction in Malaria Infection
Malaria is an acute infectious disease caused by parasitic protozoa of the genus Plasmodium. Severe malaria is a sepsis syndrome that was responsible for an estimated 429,000 deaths in 2016 (109). It is typically caused by Plasmodium falciparum and is a complex multisystem disease with cerebral involvement (CM) being the most severe manifestation. Despite treatment with intravenous artesunate, CM has a reported case fatality rate of 30% in adults and 18% in children, with approximately one-third of survivors left with long-term neurological and neurocognitive deficits (110–114). While the pathophysiology of severe malaria is incompletely understood, it is characterized by marked inflammation, oxidative stress, and endothelial dysfunction and microvascular leak associated with disruption of the Ang/Tie2 axis.
The interaction between parasitized red blood cells (RBCs) and the host endothelium is central to the pathobiology of malaria infection and the development of severe disease. P. falciparum-infected RBCs (iRBCs) express the P. falciparum erythrocyte membrane protein 1 (PfEMP-1) on their cell surface, which mediates binding of the iRBCs to host EC receptors, including intracellular adhesion molecule-1 (ICAM-1), cluster of differentiation 36 (CD36), endothelial protein C receptor (EPCR), and gC1qR (115–118). This cytoadhesion leads to organ-specific sequestration of iRBCs, resulting in microvascular obstruction, impaired perfusion, hypoxia and metabolic derangements (119), all of which can contribute to endothelial activation (37, 63, 120, 121).
Endothelial stability is mediated in large part by two distinct but inter-related pathways, the Ang/Tie2 axis and the NO biosynthetic pathway (discussed below). Similar to bacterial sepsis, Ang1 has been shown to play a protective role in the severe malaria models (45), while several human studies have demonstrated a positive correlation between increased circulating Ang2 levels and poor clinical outcomes in pediatric and adult malaria infections (49, 52, 61, 122–124). Increased levels of Ang2 have also been implicated in the pathogenesis of placental malaria and its associated adverse birth outcomes, including stillbirth and fetal growth restriction (125–127).
The murine model of experimental CM (ECM) recapitulates several features of human severe and CM (128–132), including Ang dysregulation (45, 133). Deletion of the Ang1 locus increased susceptibility to ECM, while restoring circulating Ang1 levels maintained blood–brain barrier (BBB) integrity and enhanced survival, demonstrating that Ang1 is required to stabilize the microvasculature and improve outcome (45). The pathophysiology of human CM is debated, but likely results from both parasite and host determinants (134–138). Similar to ECM, recent human MRI data has provided evidence that BBB dysfunction and microvascular leak may contribute to BBB breakdown and cerebral edema in both adult and pediatric patients with CM (139, 140). In both pre-clinical and clinical studies, higher levels of Ang1 are associated with better outcomes, whereas higher levels of Ang2 correlate with disease severity and mortality. Thus, interventions that enhance Ang1 and/or Tie2 expression and activation may be beneficial in reducing malaria-associated adverse outcomes.
Nitric oxide is produced by a family of NO synthases (NOS) that use l-arginine as a substrate and the cofactor tetrahydrobiopterin. The rupture of iRBCs releases free hemoglobin, arginase, and contributes to the generation of asymmetric dimethylarginine (ADMA) in the circulation (Figure 2). These compounds collectively limit NO bioavailability via several mechanisms. Hemoglobin does so by reacting with NO, converting it to a biologically inactive nitrate. Proteolysis of erythrocyte proteins releases ADMA (an endogenous inhibitor of NOS) as well as arginase and converts arginine to ornithine, limiting the pool of arginine available for NO production (62, 141). Reduced levels of bioavailable NO lower the threshold for cytokine-induced EC activation and exocytosis of vWF and Ang2 from WPBs (21). Therefore, malaria infection fosters microenvironment conditions that facilitate the release of Ang2 and promote the switch from a quiescent to an activated endothelial phenotype. Interventions that improve NO bioavailability may be promising candidates for the treatment of severe malaria [discussed in Ref. (142–144)].
Less is known about the role of the Tie2 component of the pathway in malaria infection but reduced expression of Tie2 is observed in the lungs of malaria-infected mice (54) and increased levels of circulating sTie2 are observed in human severe malaria (48). These observations support the hypothesis that therapeutics that increase Tie2 expression may restore endothelial quiescence and reduce the risk of ALI and acute respiratory distress syndrome (ARDS) in human malaria infection (145). Ang/Tie2 targeted therapeutics in the treatment of malaria are discussed later in this review.
Bench to Bedside: The Ang/Tie Pathway Components as Biomarkers of Life-Threatening Infections
Septic shock is the quintessential state of systemic endothelial dysfunction and microvascular leak. Over the last two decades, multiple studies of adult and pediatric populations have consistently shown that sepsis is marked by decreased levels of circulating Ang1 and increased levels of Ang2 [reviewed in Ref. (146–148)] and that the Ang2/Ang1 ratio can risk-stratify patients with critical illness. Notably, increased levels of Ang2 or a higher Ang2/Ang1 ratio predict mortality in septic patients (50, 53, 56, 100, 149–163). Among critically ill patients admitted to ICUs, plasma levels of Ang2 or the Ang2/Ang1 ratio increased across the spectrum of patients with sepsis and septic shock independent of the infecting pathogen (53, 56, 60, 150, 153, 155, 156, 158, 160, 161, 163–168). Ang2 levels correlated with surrogates of disease severity, including markers of tissue hypoperfusion, such as serum lactate (56, 60, 151, 155, 161), kidney injury (151, 154, 161), hepatic dysfunction (151), coagulopathy (151, 152), and markers of systemic inflammation (56, 60, 167). Ang2 levels were also associated with other clinical correlates of disease severity including Acute Physiology and Chronic Health Evaluation II (APACHE II) scores (56, 155, 161, 167), ICU length of stay (60), bacteremia (159), positive fluid balance (151, 162), need for corticosteroid support (151), and measures of organ failure (50, 53, 56, 60, 151–155, 160, 161, 164, 167, 169). Importantly, Ang2 levels measured early in the course of sepsis, including within 24 h of symptoms onset (158, 163, 166, 168), presentation to an Emergency Department (56, 150), and admission to ICU (50, 53, 60, 155, 157, 159, 160, 162, 164, 165), were associated with disease severity and predicted hospital mortality.
The circulating mediators of Tie2 signaling are particularly valuable in predicting outcomes of lung injury, likely owing to the high level of Tie2 expression in pulmonary vascular endothelium (147). In the lungs, Ang2 contributes to microvascular leak leading to pulmonary edema, ALI, and ARDS (55, 163, 168–173) [reviewed in Ref. (174)]. Elevated levels of circulating Ang2 not only correlate with disease severity, but also predict the degree of pulmonary microvascular leak (100, 163), duration of mechanical ventilation (163, 175), the partial pressure arterial oxygen to fraction of inspired oxygen (PaO2/FiO2) ratio (161–163, 168), and mortality (163, 169, 172, 173, 176, 177). Ang2 levels were a strong predictor of death in infection-mediated ARDS (149), an association that holds regardless of the inflammatory trigger.
Ang1 and Ang2 proteins have been evaluated as potential biomarkers of malaria disease severity and mortality, with Ang2 levels predicting not only in-hospital but also post-discharge mortality in children with severe malaria (46). Increased circulating levels of circulating Ang2 and decreased levels of Ang1 are associated with both severe P. falciparum (48, 49, 52, 122–124, 141, 178–183) and severe Plasmodium vivax infections (184–186) [reviewed in Ref. (187)]. Furthermore, the degree of Ang derangement correlates with the severity and outcome of P. falciparum infection (48, 49, 61, 122, 141, 181–184, 188), including anemia, jaundice, hypoglycemia (180), kidney injury (178, 180, 182), respiratory distress (181), CM (48, 52, 122, 178, 180, 189), and death (48, 49, 52, 61, 141, 180, 182, 189–191). Incorporating Ang2 concentrations into clinical scoring tools significantly improved the prediction accuracy of the models for mortality (48). Moreover, circulating Ang2 levels were informative in monitoring response to therapy and were predictive of short and long-term mortality (61, 122, 192).
Therapeutic Interventions Targeting the Ang/Tie Axis
The observation that the Ang/Tie2 pathway contributes to disease pathobiology and that circulating ligands of Tie2, Ang1, and Ang2 can risk-stratify critically ill patients suggests that this pathway is a therapeutic target to prevent microvascular leak associated with sepsis. Off target effects of corticosteroids and HMG-CoA reductase inhibitors, both of which can reduce the severity of critical illness (193–198), have been shown to modulate Ang1 and Ang2 levels (199, 200) [reviewed in Ref. (36, 201, 202)]. Ang/Tie2-directed anti-angiogenic pharmacotherapies are in preclinical and clinical trials for the treatment of several malignancies and neovascular eye diseases [reviewed in Ref. (203)]; however, the development of adjunctive therapies for the management of sepsis and other critical illnesses associated with microvascular dysfunction have lagged behind. To date, pre-clinical studies with interventions that have stabilized the Tie2 receptor provide evidence that targeting this pathway may enable precision medicine approaches to improve outcomes of severe infections in humans [Table 1; reviewed in Ref. (146, 148)].
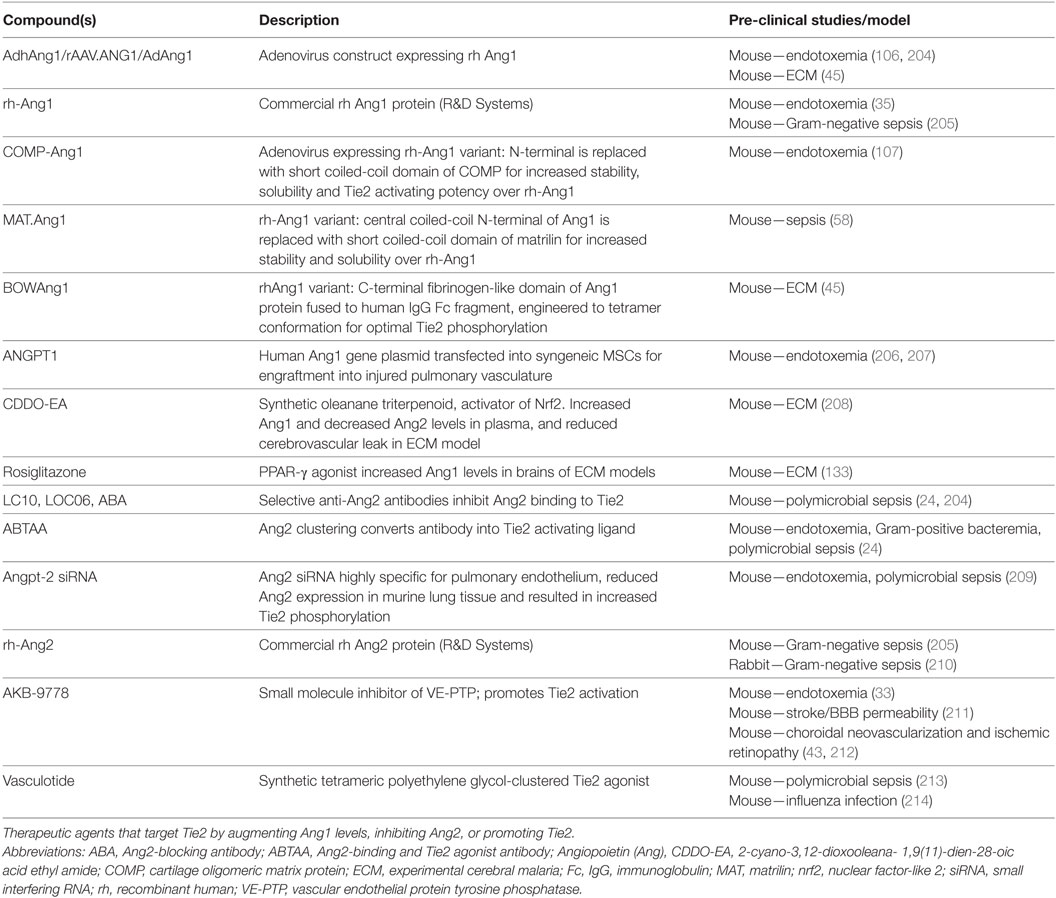
Table 1. Ang/Tie2-targeted therapies in pre-clinical studies of sepsis.
Studies using murine models of sepsis have demonstrated that therapeutic compounds that augment Ang1 expression can attenuate many of the adverse outcomes associated with endotoxemia. Increased Ang1, driven by adenovirus-mediated rh gene delivery prior to an LPS challenge, preserved eNOS activity in lung tissue and reduced lung injury, prevented up-regulation of cellular adhesion molecules, improved hemodynamics, and reduced mortality (106, 204). A more potent and stable Tie2 phosphorylating molecule, cartilage oligomatrix protein (COMP)-Ang1 (215) similarly prevented adhesion molecule expression and conferred renal protection in the sepsis model (107). Although these findings establish the protective benefit of sustaining Tie2 phosphorylation through Ang1 treatment in sepsis, the use of an adenoviral delivery vector is problematic for translation to human therapy. Subsequent studies demonstrated that rh Ang1 (rh-Ang1) delivered systemically to mice undergoing cecal ligation and perforation (CLP) stabilized endothelial barrier function, preventing pulmonary capillary leak, and decreased leukocyte infiltration into both lungs and kidneys by suppressing ICAM-1 expression (35). Despite the short half-life of rh-Ang1 (216), treatment was able to avert multi-organ dysfunction and increased survival following CLP (59). Matrilin-1-Ang1 (MAT.Ang1) was developed as a stable Ang1 variant amenable to direct intravenous administration; it too stabilized the endothelium in the setting of LPS-induced endotoxemia (58).
Ang1 therapy has also been shown to mitigate the adverse sequelae of severe malaria infection. Using BowAng1, a rh Ang1 protein capable of phosphorylating Tie2 (217), it was demonstrated that its addition to artesunate therapy preserved the integrity of the BBB and improved survival in a murine model of CM, even when administered during the late stage of infection (45). Other therapeutic agents that increase Ang1 expression also prevent BBB leak and improve survival in ECM. Mice treated with the ethyl amide of a synthetic oleanane triterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acid (CDDO-EA), had increased Ang1, reduced Ang2 and Ang2:Ang1 ratio, and this was associated with improved BBB integrity (208). Similarly, treatment of malaria infected mice with the PPAR-γ agonist rosiglitazone in combination with artesunate at the onset of neurological symptoms achieved higher plasma and brain levels of Ang1 and a lower Ang2:Ang1 ratio compared to mice treated with artesunate alone. Furthermore, these mice had enhanced BBB integrity, improved survival and better cognitive and motor outcomes than mice treated with anti-malarials alone (133). Rosiglitazone has entered human clinical trials. To date, a randomized clinical trial of rosiglitazone in young adults with uncomplicated malaria showed reduced levels of pro-inflammatory mediators, a lower Ang2:Ang1 ratio, and higher levels of brain-derived neurotrophic factor, a protein involved in neuronal survival and proliferation (133, 218). A phase IIa study has shown rosiglitazone to be safe and well tolerated in pediatric patients with uncomplicated malaria and is currently being tested in a phase IIb trial in children with severe malaria (219).
Other novel modes of augmenting Ang1 expression are currently in preclinical trials. Preliminary work utilizing cell-based therapy has demonstrated that mesenchymal stem cells (MSCs) transfected with Ang1 are able to engraft the pulmonary endothelium damaged during sepsis, preserve pulmonary endothelial integrity, and ameliorate ALI/ARDS (206, 207).
In contrast to the constitutive expression of Ang1, Ang2 is relea-sed in response to infectious triggers with considerable dynamic range (56, 168), making this molecule an appealing target for pharmacologic inhibition in sepsis. In preclinical trials, lung-targeted small interfering RNA (siRNA) against Ang2 delivered both pre- and post-sepsis induction reduced pulmonary inflammatory cytokine levels, ICAM-1 expression, neutrophil organ infiltration, and overall disease severity while improving survival (209). Functional inhibition of Ang2/Tie2 binding using anti-Ang2 antibodies decreased rates of hemodynamic shock and mortality in murine sepsis and ARDS models (24, 204).
Furthermore, the novel Ang2-binding and Tie2-activating antibody (ABTAA) utilizes a mechanism through which Ang2 clustering converts the antibody-antigen cluster into a Tie2 activating ligand, thus allowing simultaneous Ang2 inhibition and Tie2 activation (24). When compared to the conventional anti-Ang2 antibody, ABTAA conferred increased protection against microvascular dysfunction, end-organ damage, and mortality in CLP, endotoxemia, and Staphylococcus aureus models of sepsis (24). In combination with broad-spectrum antibiotics (imipenem/cilastatin), ABTAA improved survival to 70%, compared to 20% survival in animals treated with antibiotics alone in the CLP model (24). Seemingly paradoxically, studies in murine and rabbit models of pyelonephritis and sepsis found that rh-Ang2 administration prolonged survival in Gram-negative sepsis (205, 210). Notably, these studies did not assess Tie2 phosphorylation status, leaving the mechanism of Ang2-mediated survival in these models unclear.
In light of these findings, further analyses have been performed examining the role of simultaneous Ang2 inhibition and Tie2 activation in vascular protection during sepsis (24). When treated with the antibody ABTAA, mice with high-grade CLP had significantly improved survival rates (40%) compared to the conventional Ang2-blocking antibody (ABA; 13%). These findings were further extended to two other sepsis models used in this study: endotoxemia (rate of survival increase: 63% ABTAA vs 33% ABA) and S. aureus bacteremia (rate of survival increase: 55% ABTAA vs. 9% ABA). In these models, it was observed that ABTAA ameliorated endotoxemic and CLP-induced sepsis by preserving endothelial glycocalyx and microvascular integrity of major organs (24). Taken together, these studies underline the importance of Tie2 activation in ameliorating the progression of sepsis and demonstrate that solely blocking Ang2 is insufficient for preserving endothelial integrity during severe bacterial sepsis.
In addition to targeting the Tie2 receptor through Ang1 and Ang2, several other agents have been used to maintain Tie2 phosphorylation in animal models of sepsis. For example, pharmacologic inhibition of the Tie2 phosphatase, VE-PTP, with AKB-9778 stabilized the pulmonary endothelium following LPS administration in mice (33), offering another potential mechanism to modulate the activity of Tie2 in sepsis. This compound has already been used in human trials to treat diabetic macular edema and ocular neovascularization (43, 212). Alternatively, Vasculotide, a synthetic polyethylene glycol-clustered Tie2 agonist, has been shown to sustain Tie2 activation in vivo. Its administration both pre- and post-CLP reduced end-organ dysfunction and mortality in the murine abdominal sepsis model (155). Vasculotide administration also preserved pulmonary endothelial barrier function and survival following murine infection with several strains of influenza. Importantly, the protective effect was realized even with therapy delayed up to 72 h after infection, conditions similar to typical septic patient presentations (214). Unlike interventions that manipulate Ang1 or Ang2 expression, Vasculotide is highly specific for the Tie2 receptor and does not displace Ang1 or Ang2. As such, Vasculotide may avoid interfering with off-target effects of Ang1 during the dynamic host sepsis response.
Although the pathophysiology Tie2-mediated vascular dysfunction in sepsis remains incompletely understood, the effects of these therapies in preclinical sepsis models warrant further investigation to develop human Tie2-directed therapies.
Conclusion
The Ang/Tie2 axis plays an essential role in maintaining endothelial barrier stability and its disruption during systemic infection contributes to the pathologic cascade that culminates in end-organ failure and death. In addition to its mechanistic role in the pathobiology of sepsis, components of the Ang/Tie2 system can function as prognostic biomarkers of disease severity and outcomes, and potentially serve as important therapeutic targets in the management of sepsis.
The dysregulation of Ang/Tie2 signaling is “pathogen agnostic” and appears to represent a final common pathway in many different types of microbial infections, including bacterial and parasitic processes described in this review. As such, therapeutic interventions to restore Tie2 activity may be useful in the early management of serious infections where there is a high degree of diagnostic uncertainty. Furthermore, use of Ang/Tie2 adjunctive therapy in sepsis may confer protection against the collateral systemic damage that results in significant morbidity and mortality. The pathophysiology Tie2-mediated microvascular dysfunction in sepsis remains incompletely understood, but the findings from preclinical sepsis models warrant further investigation with the aim of developing human Tie2-directed therapies to improve outcomes of life-threatening infections.
Author Contributions
AL, MR-G, JW, VC, and KK conceived the ideas for preparing this review. AL, MR-G, JW, VC, and KK contributed to the writing, editing, and approval of the final review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by a Collaborative Research Agreement Grant from Intellectual Ventures/Global Good (KK), the Canadian Institutes of Health Research (CIHR) grants MOP-13721, MOP-115160, MOP-136813, a CIHR Foundation grant FDN-148439 (KK.), the Canada Research Chair Program (KK), a CIHR Banting fellowship (AL), Mitacs Elevate Fellowship (MR-G.) and donations from Kim Kertland and the Tesari Foundation. KK. is named an inventor on a patent “Biomarkers for early determination of a critical or life-threatening response to illness and/or treatment response” held by University Health Network.
Abbreviations
ABIN2, A20-binding inhibitor of nuclear factor-κB-2; ABA, Ang2-blocking antibody; ABTAA, Ang2-binding and Tie2-activating antibody; ADAM-15, disintegrin and metalloproteinase domain-containing protein 15; ADMA, asymmetric dimethyl arginine; Ang, angiopoietin; ARDS, acute respiratory distress syndrome; BBB, blood brain barrier; BDNF, brain-derived neurotrophic factor; Ca2+, calcium; CaM, calmodulin; CD36, cluster of differentiation 36; CDDO-EA, 2-cyano-3,12-dioxooleana- 1,9(11)-dien-28-oic acid ethyl amide; CM, cerebral malaria; COMP-Ang1, cartilage oligomatrix protein Ang1; EC, endothelial cell; ECM, experimental cerebral malaria; EPCR, endothelial protein C receptor; ER, endoplasmic reticulum; ET-1, endothelin-1; FOXO1, forkhead box protein O1; GPCR, G-protein-coupled receptor; PAMP, pathogen-associated molecular pattern; ICAM-1, intracellular adhesion molecule-1; ICU, intensive care unit; IP3R, inositol triphosphate receptor; IQGAP, IQ motif containing GTPase activating protein; KLF2, Krüppel-like factor-2; iRBC, infected red blood cell; LPS, lipopolysaccharide; MAT, matrilin; mDia, mammalian diaphanous; MEF2, myocyte enhancer factor-2; miR-30, micro ribonucleic acid-30-5p; MLCK, myosin light-chain kinase; NF-κB, nuclear factor-κB; NO, nitric oxide; NOS, nitric oxide synthase; eNOS, endothelial nitric oxide; NFR2, nuclear factor erythroid 2-related factor 2; p38 MAPK, p38 mitogen-activated protein kinase; p190RhoGAP, p190 Rho GTPase-activating protein; PfEMP-1, Plasmodium falciparum erythrocyte membrane protein-1; PI3K, phosphoinositide triphosphate kinase; Rac1, RAS-related C3 botulinum toxin substrate 1; rh-Ang1, recombinant Ang1; Rap1, Ras-related protein 1; RBC, red blood cell; rh, recombinant human; RhoA, Ras homolog gene family, member A; RhoGEF, Rho guanine nucleotide exchange factor; SOFA, sequential organ failure assessment; Src, proto-oncogene tyrosine-protein kinase; TRPC1, transient receptor potential channel-1; VE-cadherin, vascular endothelial-cadherin; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; VE-PTP, vascular endothelial protein tyrosine phosphatase; WPB, Weibel-Palade bodies; p38 MAPK, p38 mitogen-activated protein kinase; PPAR, peroxisome proliferator-activated receptor.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315:801–10. doi:10.1001/jama.2016.0287
2. Cohen J, Vincent JL, Adhikari NK, Machado FR, Angus DC, Calandra T, et al. Sepsis: a roadmap for future research. Lancet Infect Dis (2015) 15:581–614. doi:10.1016/S1473-3099(15)70112-X
3. Vincent JL, Moreno R, Takala J, Willatts S, De Mendonca A, Bruining H, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med (1996) 22:707–10. doi:10.1007/BF01709751
4. Calfee CS. Precision medicine: an opportunity to improve outcomes of patients with sepsis. Am J Respir Crit Care Med (2016) 194:137–9. doi:10.1164/rccm.201604-0697ED
5. Matthay MA, Liu KD. New strategies for effective therapeutics in critically ill patients. JAMA (2016) 315:747–8. doi:10.1001/jama.2016.0661
6. Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence (2013) 4:507–16. doi:10.4161/viru.24530
7. Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascon GA, et al. The endothelium in sepsis. Shock (2016) 45:259–70. doi:10.1097/SHK.0000000000000473
8. Lee WL, Liles WC. Endothelial activation, dysfunction and permeability during severe infections. Curr Opin Hematol (2011) 18:191–6. doi:10.1097/MOH.0b013e328345a3d1
9. Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med (2010) 363:689–91. doi:10.1056/NEJMcibr1007320
10. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock: 2016. Crit Care Med (2017) 45:486–552. doi:10.1097/CCM.0000000000002255
11. Partanen J, Armstrong E, Makela TP, Korhonen J, Sandberg M, Renkonen R, et al. A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains. Mol Cell Biol (1992) 12:1698–707. doi:10.1128/MCB.12.4.1698
12. Dumont DJ, Yamaguchi TP, Conlon RA, Rossant J, Breitman ML. tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene (1992) 7:1471–80.
13. Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res (2006) 98:1014–23. doi:10.1161/01.RES.0000218275.54089.12
14. Korhonen EA, Lampinen A, Giri H, Anisimov A, Kim M, Allen B, et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J Clin Invest (2016) 126:3495–510. doi:10.1172/JCI84923
15. Kim M, Allen B, Korhonen EA, Nitschke M, Yang HW, Baluk P, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest (2016) 126:3511–25. doi:10.1172/JCI84871
16. Koh GY. Orchestral actions of angiopoietin-1 in vascular regeneration. Trends Mol Med (2013) 19:31–9. doi:10.1016/j.molmed.2012.10.010
17. Fukuhara S, Sako K, Minami T, Noda K, Kim HZ, Kodama T, et al. Differential function of Tie2 at cell-cell contacts and cell-substratum contacts regulated by angiopoietin-1. Nat Cell Biol (2008) 10:513–26. doi:10.1038/ncb1714
18. Kim I, Kim HG, So JN, Kim JH, Kwak HJ, Koh GY. Angiopoietin-1 regulates endothelial cell survival through the phosphatidylinositol 3’-Kinase/Akt signal transduction pathway. Circ Res (2000) 86:24–9. doi:10.1161/01.RES.86.1.24
19. Kontos CD, Cha EH, York JD, Peters KG. The endothelial receptor tyrosine kinase Tie1 activates phosphatidylinositol 3-kinase and Akt to inhibit apoptosis. Mol Cell Biol (2002) 22:1704–13. doi:10.1128/MCB.22.6.1704-1713.2002
20. Daly C, Wong V, Burova E, Wei Y, Zabski S, Griffiths J, et al. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev (2004) 18:1060–71. doi:10.1101/gad.1189704
21. Fiedler U, Scharpfenecker M, Koidl S, Hegen A, Grunow V, Schmidt JM, et al. The Tie-2 ligand angiopoietin-2 is stored in and rapidly released upon stimulation from endothelial cell Weibel-Palade bodies. Blood (2004) 103:4150–6. doi:10.1182/blood-2003-10-3685
22. Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell (1996) 87:1171–80. doi:10.1016/S0092-8674(00)81813-9
23. Sundberg C, Kowanetz M, Brown LF, Detmar M, Dvorak HF. Stable expression of angiopoietin-1 and other markers by cultured pericytes: phenotypic similarities to a subpopulation of cells in maturing vessels during later stages of angiogenesis in vivo. Lab Invest (2002) 82:387–401. doi:10.1038/labinvest.3780433
24. Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2alpha/Notch3 pathways. Sci Rep (2016) 6:20931. doi:10.1038/srep20931
25. Hughes DP, Marron MB, Brindle NP. The antiinflammatory endothelial tyrosine kinase Tie2 interacts with a novel nuclear factor-kappaB inhibitor ABIN-2. Circ Res (2003) 92:630–6. doi:10.1161/01.RES.0000063422.38690.DC
26. Tadros A, Hughes DP, Dunmore BJ, Brindle NP. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood (2003) 102:4407–9. doi:10.1182/blood-2003-05-1602
27. Sako K, Fukuhara S, Minami T, Hamakubo T, Song H, Kodama T, et al. Angiopoietin-1 induces Kruppel-like factor 2 expression through a phosphoinositide 3-kinase/AKT-dependent activation of myocyte enhancer factor 2. J Biol Chem (2009) 284:5592–601. doi:10.1074/jbc.M806928200
28. Matsushita K, Morrell CN, Cambien B, Yang SX, Yamakuchi M, Bao C, et al. Nitric oxide regulates exocytosis by S-nitrosylation of N-ethylmaleimide-sensitive factor. Cell (2003) 115:139–50. doi:10.1016/S0092-8674(03)00803-1
29. Demolli S, Doebele C, Doddaballapur A, Lang V, Fisslthaler B, Chavakis E, et al. MicroRNA-30 mediates anti-inflammatory effects of shear stress and KLF2 via repression of angiopoietin 2. J Mol Cell Cardiol (2015) 88:111–9. doi:10.1016/j.yjmcc.2015.10.009
30. Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell (2008) 14:25–36. doi:10.1016/j.devcel.2007.10.019
31. Hayashi M, Majumdar A, Li X, Adler J, Sun Z, Vertuani S, et al. VE-PTP regulates VEGFR2 activity in stalk cells to establish endothelial cell polarity and lumen formation. Nat Commun (2013) 4:1672. doi:10.1038/ncomms2683
32. David S, Ghosh CC, Mukherjee A, Parikh SM. Angiopoietin-1 requires IQ domain GTPase-activating protein 1 to activate Rac1 and promote endothelial barrier defense. Arterioscler Thromb Vasc Biol (2011) 31:2643–52. doi:10.1161/ATVBAHA.111.233189
33. Frye M, Dierkes M, Kuppers V, Vockel M, Tomm J, Zeuschner D, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med (2015) 212:2267–87. doi:10.1084/jem.20150718
34. Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett (2003) 546:93–7. doi:10.1016/S0014-5793(03)00454-X
35. Mammoto T, Parikh SM, Mammoto A, Gallagher D, Chan B, Mostoslavsky G, et al. Angiopoietin-1 requires p190 RhoGAP to protect against vascular leakage in vivo. J Biol Chem (2007) 282:23910–8. doi:10.1074/jbc.M702169200
36. Ghosh CC, Thamm K, Berghelli AV, Schrimpf C, Maski MR, Abid T, et al. Drug repurposing screen identifies foxo1-dependent angiopoietin-2 regulation in sepsis*. Crit Care Med (2015) 43:e230–40. doi:10.1097/CCM.0000000000000993
37. Mandriota SJ, Pepper MS. Regulation of angiopoietin-2 mRNA levels in bovine microvascular endothelial cells by cytokines and hypoxia. Circ Res (1998) 83:852–9. doi:10.1161/01.RES.83.8.852
38. Maisonpierre PC, Suri C, Jones PF, Bartunkova S, Wiegand SJ, Radziejewski C, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science (1997) 277:55–60. doi:10.1126/science.277.5322.55
39. Daly C, Pasnikowski E, Burova E, Wong V, Aldrich TH, Griffiths J, et al. Angiopoietin-2 functions as an autocrine protective factor in stressed endothelial cells. Proc Natl Acad Sci U S A (2006) 103:15491–6. doi:10.1073/pnas.0607538103
40. Scharpfenecker M, Fiedler U, Reiss Y, Augustin HG. The Tie-2 ligand angiopoietin-2 destabilizes quiescent endothelium through an internal autocrine loop mechanism. J Cell Sci (2005) 118:771–80. doi:10.1242/jcs.01653
41. Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol (2009) 29:2011–22. doi:10.1128/MCB.01472-08
42. Winderlich M, Keller L, Cagna G, Broermann A, Kamenyeva O, Kiefer F, et al. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol (2009) 185:657–71. doi:10.1083/jcb.200811159
43. Shen J, Frye M, Lee BL, Reinardy JL, McClung JM, Ding K, et al. Targeting VE-PTP activates TIE2 and stabilizes the ocular vasculature. J Clin Invest (2014) 124:4564–76. doi:10.1172/JCI74527
44. Saharinen P, Eklund L, Miettinen J, Wirkkala R, Anisimov A, Winderlich M, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell-cell and cell-matrix contacts. Nat Cell Biol (2008) 10:527–37. doi:10.1038/ncb1715
45. Higgins SJ, Purcell LA, Silver KL, Tran V, Crowley V, Hawkes M, et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci Transl Med (2016) 8:358ra128. doi:10.1126/scitranslmed.aaf6812
46. Conroy AL, Hawkes M, McDonald CR, Kim H, Higgins S, Barker KR, et al. Host biomarkers are associated with response to therapy and long-term mortality in pediatric severe malaria. Open Forum Infect Dis (2016) 3:1–10. doi:10.1093/ofid/ofw134
47. Conroy AL, Gelvez M, Hawkes M, Rajwans N, Tran V, Liles WC, et al. Host biomarkers are associated with progression to dengue haemorrhagic fever: a nested case-control study. Int J Infect Dis (2015) 40:45–53. doi:10.1016/j.ijid.2015.07.027
48. Conroy AL, Glover SJ, Hawkes M, Erdman LK, Seydel KB, Taylor TE, et al. Angiopoietin-2 levels are associated with retinopathy and predict mortality in Malawian children with cerebral malaria: a retrospective case-control study*. Crit Care Med (2012) 40:952–9. doi:10.1097/CCM.0b013e3182373157
49. Erdman LK, Dhabangi A, Musoke C, Conroy AL, Hawkes M, Higgins S, et al. Combinations of host biomarkers predict mortality among Ugandan children with severe malaria: a retrospective case-control study. PLoS One (2011) 6:e17440. doi:10.1371/journal.pone.0017440
50. Ricciuto DR, dos Santos CC, Hawkes M, Toltl LJ, Conroy AL, Rajwans N, et al. Angiopoietin-1 and angiopoietin-2 as clinically informative prognostic biomarkers of morbidity and mortality in severe sepsis. Crit Care Med (2011) 39:702–10. doi:10.1097/CCM.0b013e318206d285
51. Page AV, Kotb M, McGeer A, Low DE, Kain KC, Liles WC. Systemic dysregulation of angiopoietin-1/2 in streptococcal toxic shock syndrome. Clin Infect Dis (2011) 52:e157–61. doi:10.1093/cid/cir125
52. Lovegrove FE, Tangpukdee N, Opoka RO, Lafferty EI, Rajwans N, Hawkes M, et al. Serum angiopoietin-1 and -2 levels discriminate cerebral malaria from uncomplicated malaria and predict clinical outcome in African children. PLoS One (2009) 4:e4912. doi:10.1371/journal.pone.0004912
53. Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, et al. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PLoS One (2015) 10:e0141251. doi:10.1371/journal.pone.0141251
54. Ghosh CC, David S, Zhang R, Berghelli A, Milam K, Higgins SJ, et al. Gene control of tyrosine kinase TIE2 and vascular manifestations of infections. Proc Natl Acad Sci U S A (2016) 113:2472–7. doi:10.1073/pnas.1519467113
55. Agrawal A, Matthay MA, Kangelaris KN, Stein J, Chu JC, Imp BM, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med (2013) 187:736–42. doi:10.1164/rccm.201208-1460OC
56. David S, Mukherjee A, Ghosh CC, Yano M, Khankin EV, Wenger JB, et al. Angiopoietin-2 may contribute to multiple organ dysfunction and death in sepsis*. Crit Care Med (2012) 40:3034–41. doi:10.1097/CCM.0b013e31825fdc31
57. Ghosh CC, Mukherjee A, David S, Knaus UG, Stearns-Kurosawa DJ, Kurosawa S, et al. Impaired function of the Tie-2 receptor contributes to vascular leakage and lethality in anthrax. Proc Natl Acad Sci U S A (2012) 109:10024–9. doi:10.1073/pnas.1120755109
58. Alfieri A, Watson JJ, Kammerer RA, Tasab M, Progias P, Reeves K, et al. Angiopoietin-1 variant reduces LPS-induced microvascular dysfunction in a murine model of sepsis. Crit Care (2012) 16:R182. doi:10.1186/cc11666
59. David S, Park JK, Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, et al. Acute administration of recombinant angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine (2011) 55:251–9. doi:10.1016/j.cyto.2011.04.005
60. Davis JS, Yeo TW, Piera KA, Woodberry T, Celermajer DS, Stephens DP, et al. Angiopoietin-2 is increased in sepsis and inversely associated with nitric oxide-dependent microvascular reactivity. Crit Care (2010) 14:R89. doi:10.1186/cc9020
61. Yeo TW, Lampah DA, Gitawati R, Tjitra E, Kenangalem E, Piera K, et al. Angiopoietin-2 is associated with decreased endothelial nitric oxide and poor clinical outcome in severe falciparum malaria. Proc Natl Acad Sci U S A (2008) 105:17097–102. doi:10.1073/pnas.0805782105
62. Omodeo-Sale F, Cortelezzi L, Vommaro Z, Scaccabarozzi D, Dondorp AM. Dysregulation of L-arginine metabolism and bioavailability associated to free plasma heme. Am J Physiol Cell Physiol (2010) 299:C148–54. doi:10.1152/ajpcell.00405.2009
63. Woo KV, Qu X, Babaev VR, Linton MF, Guzman RJ, Fazio S, et al. Tie1 attenuation reduces murine atherosclerosis in a dose-dependent and shear stress-specific manner. J Clin Invest (2011) 121:1624–35. doi:10.1172/JCI42040
64. Babaei S, Teichert-Kuliszewska K, Zhang Q, Jones N, Dumont DJ, Stewart DJ. Angiogenic actions of angiopoietin-1 require endothelium-derived nitric oxide. Am J Pathol (2003) 162:1927–36. doi:10.1016/S0002-9440(10)64326-X
65. Atkins GB, Jain MK. Role of Kruppel-like transcription factors in endothelial biology. Circ Res (2007) 100:1686–95. doi:10.1161/01.RES.0000267856.00713.0a
66. Findley CM, Cudmore MJ, Ahmed A, Kontos CD. VEGF induces Tie2 shedding via a phosphoinositide 3-kinase/Akt dependent pathway to modulate Tie2 signaling. Arterioscler Thromb Vasc Biol (2007) 27:2619–26. doi:10.1161/ATVBAHA.107.150482
67. Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, et al. S-nitrosylation of beta-catenin by eNOS-derived NO promotes VEGF-induced endothelial cell permeability. Mol Cell (2010) 39:468–76. doi:10.1016/j.molcel.2010.07.013
68. Esser S, Lampugnani MG, Corada M, Dejana E, Risau W. Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J Cell Sci (1998) 111(Pt 13):1853–65.
69. Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol (2006) 8:1223–34. doi:10.1038/ncb1486
70. Yu OM, Brown JH. G protein-coupled receptor and RhoA-stimulated transcriptional responses: links to inflammation, differentiation, and cell proliferation. Mol Pharmacol (2015) 88:171–80. doi:10.1124/mol.115.097857
71. van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res (2000) 87:335–40. doi:10.1161/01.RES.87.4.335
72. Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev (2006) 86:279–367. doi:10.1152/physrev.00012.2005
73. Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science (1996) 273:245–8. doi:10.1126/science.273.5272.245
74. Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, et al. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ Res (2005) 96:1282–90. doi:10.1161/01.RES.0000171894.03801.03
75. Wysolmerski RB, Lagunoff D. Involvement of myosin light-chain kinase in endothelial cell retraction. Proc Natl Acad Sci U S A (1990) 87:16–20. doi:10.1073/pnas.87.1.16
76. Kawai T, Akira S. Signaling to NF-kappaB by toll-like receptors. Trends Mol Med (2007) 13:460–9. doi:10.1016/j.molmed.2007.09.002
77. Ding J, Song D, Ye X, Liu SF. A pivotal role of endothelial-specific NF-kappaB signaling in the pathogenesis of septic shock and septic vascular dysfunction. J Immunol (2009) 183:4031–8. doi:10.4049/jimmunol.0900105
78. Higgins SJ, De Ceunynck K, Kellum JA, Chen X, Gu X, Chaudhry SA, et al. Tie2 protects the vasculature against thrombus formation in systemic inflammation. J Clin Invest (2018) 128(4):1471–84. doi:10.1172/JCI97488
79. Minhas N, Xue M, Jackson CJ. Activated protein C binds directly to Tie2: possible beneficial effects on endothelial barrier function. Cell Mol Life Sci (2017) 74:1895–906. doi:10.1007/s00018-016-2440-6
80. Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med (2001) 29:1303–10. doi:10.1097/00003246-200107000-00002
81. Kumar G, Kumar N, Taneja A, Kaleekal T, Tarima S, McGinley E, et al. Nationwide trends of severe sepsis in the 21st century (2000–2007). Chest (2011) 140:1223–31. doi:10.1378/chest.11-0352
82. Liu V, Escobar GJ, Greene JD, Soule J, Whippy A, Angus DC, et al. Hospital deaths in patients with sepsis from 2 independent cohorts. JAMA (2014) 312:90–2. doi:10.1001/jama.2014.5804
83. Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, et al. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med (2010) 38:367–74. doi:10.1097/CCM.0b013e3181cb0cdc
84. Kaukonen KM, Bailey M, Suzuki S, Pilcher D, Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000–2012. JAMA (2014) 311:1308–16. doi:10.1001/jama.2014.2637
85. Boussekey N, Cantrel J, Dorchin Debrabant L, Langlois J, Devos P, Meybeck A, et al. Epidemiology, prognosis, and evolution of management of septic shock in a French intensive care unit: a five years survey. Crit Care Res Pract (2010) 2010:436427. doi:10.1155/2010/436427
86. Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, et al. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009–2014. JAMA (2017) 318:1241–9. doi:10.1001/jama.2017.13836
87. Gupta RG, Hartigan SM, Kashiouris MG, Sessler CN, Bearman GM. Early goal-directed resuscitation of patients with septic shock: current evidence and future directions. Crit Care (2015) 19:286. doi:10.1186/s13054-015-1011-9
88. Angus DC. The search for effective therapy for sepsis: back to the drawing board? JAMA (2011) 306:2614–5. doi:10.1001/jama.2011.1853
89. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5:4–11. doi:10.4161/viru.27372
90. Kurniati NF, van Meurs M, Vom Hagen F, Jongman RM, Moser J, Zwiers PJ, et al. Pleiotropic effects of angiopoietin-2 deficiency do not protect mice against endotoxin-induced acute kidney injury. Nephrol Dial Transplant (2013) 28:567–75. doi:10.1093/ndt/gfs336
91. Mofarrahi M, Nouh T, Qureshi S, Guillot L, Mayaki D, Hussain SN. Regulation of angiopoietin expression by bacterial lipopolysaccharide. Am J Physiol Lung Cell Mol Physiol (2008) 294:L955–63. doi:10.1152/ajplung.00449.2007
92. Kurniati NF, Jongman RM, vom Hagen F, Spokes KC, Moser J, Regan ER, et al. The flow dependency of Tie2 expression in endotoxemia. Intensive Care Med (2013) 39:1262–71. doi:10.1007/s00134-013-2899-7
93. Findley CM, Mitchell RG, Duscha BD, Annex BH, Kontos CD. Plasma levels of soluble Tie2 and vascular endothelial growth factor distinguish critical limb ischemia from intermittent claudication in patients with peripheral arterial disease. J Am Coll Cardiol (2008) 52:387–93. doi:10.1016/j.jacc.2008.02.045
94. Reusch P, Barleon B, Weindel K, Martiny-Baron G, Godde A, Siemeister G, et al. Identification of a soluble form of the angiopoietin receptor TIE-2 released from endothelial cells and present in human blood. Angiogenesis (2001) 4:123–31. doi:10.1023/A:1012226627813
95. Hay DC, Beers C, Cameron V, Thomson L, Flitney FW, Hay RT. Activation of NF-kappaB nuclear transcription factor by flow in human endothelial cells. Biochim Biophys Acta (2003) 1642:33–44. doi:10.1016/S0167-4889(03)00084-3
96. Singh N, Macnamara E, Rashid S, Ambati J, Kontos CD, Higgins E, et al. Systemic soluble Tie2 expression inhibits and regresses corneal neovascularization. Biochem Biophys Res Commun (2005) 332:194–9. doi:10.1016/j.bbrc.2005.04.108
97. Fuxe J, Lashnits E, O’Brien S, Baluk P, Tabruyn SP, Kuhnert F, et al. Angiopoietin/Tie2 signaling transforms capillaries into venules primed for leukocyte trafficking in airway inflammation. Am J Pathol (2010) 176:2009–18. doi:10.2353/ajpath.2010.090976
98. Chung NA, Makin AJ, Lip GY. Measurement of the soluble angiopoietin receptor tie-2 in patients with coronary artery disease: development and application of an immunoassay. Eur J Clin Invest (2003) 33:529–35. doi:10.1046/j.1365-2362.2003.01173.x
99. Harris AL, Reusch P, Barleon B, Hang C, Dobbs N, Marme D. Soluble Tie2 and Flt1 extracellular domains in serum of patients with renal cancer and response to antiangiogenic therapy. Clin Cancer Res (2001) 7:1992–7.
100. van der Heijden M, van Nieuw Amerongen GP, van Hinsbergh VW, Groeneveld AB. The interaction of soluble Tie2 with angiopoietins and pulmonary vascular permeability in septic and nonseptic critically ill patients. Shock (2010) 33:263–8. doi:10.1097/SHK.0b013e3181b2f978
101. Alawo DOA, Tahir TA, Fischer M, Bates DG, Amirova SR, Brindle NPJ. Regulation of angiopoietin signalling by soluble Tie2 ectodomain and engineered ligand trap. Sci Rep (2017) 7:3658. doi:10.1038/s41598-017-03981-6
102. Saharinen P, Kerkela K, Ekman N, Marron M, Brindle N, Lee GM, et al. Multiple angiopoietin recombinant proteins activate the Tie1 receptor tyrosine kinase and promote its interaction with Tie2. J Cell Biol (2005) 169:239–43. doi:10.1083/jcb.200411105
103. Menden H, Welak S, Cossette S, Ramchandran R, Sampath V. Lipopolysaccharide (LPS)-mediated angiopoietin-2-dependent autocrine angiogenesis is regulated by NADPH oxidase 2 (Nox2) in human pulmonary microvascular endothelial cells. J Biol Chem (2015) 290:5449–61. doi:10.1074/jbc.M114.600692
104. Lee JY, Linge HM, Ochani K, Lin K, Miller EJ. N-ethylmaleimide sensitive factor (NSF) inhibition prevents vascular instability following gram-positive pulmonary challenge. PLoS One (2016) 11:e0157837. doi:10.1371/journal.pone.0157837
105. Hahn WO, Mikacenic C, Price BL, Harju-Baker S, Katz R, Himmelfarb J, et al. Host derived biomarkers of inflammation, apoptosis, and endothelial activation are associated with clinical outcomes in patients with bacteremia and sepsis regardless of microbial etiology. Virulence (2016) 7:387–94. doi:10.1080/21505594.2016.1144003
106. Witzenbichler B, Westermann D, Knueppel S, Schultheiss HP, Tschope C. Protective role of angiopoietin-1 in endotoxic shock. Circulation (2005) 111:97–105. doi:10.1161/01.CIR.0000151287.08202.8E
107. Kim DH, Jung YJ, Lee AS, Lee S, Kang KP, Lee TH, et al. COMP-angiopoietin-1 decreases lipopolysaccharide-induced acute kidney injury. Kidney Int (2009) 76:1180–91. doi:10.1038/ki.2009.387
108. Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell (2002) 3:411–23. doi:10.1016/S1534-5807(02)00217-4
110. Boivin MJ. Effects of early cerebral malaria on cognitive ability in Senegalese children. J Dev Behav Pediatr (2002) 23:353–64. doi:10.1097/00004703-200210000-00010
111. Boivin MJ, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cognitive impairment after cerebral malaria in children: a prospective study. Pediatrics (2007) 119:e360–6. doi:10.1542/peds.2006-2027
112. Fernando SD, Rodrigo C, Rajapakse S. The ‘hidden’ burden of malaria: cognitive impairment following infection. Malar J (2010) 9:366. doi:10.1186/1475-2875-9-366
113. John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, et al. Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics (2008) 122:e92–9. doi:10.1542/peds.2007-3709
114. Idro R, Kakooza-Mwesige A, Asea B, Ssebyala K, Bangirana P, Opoka RO, et al. Cerebral malaria is associated with long-term mental health disorders: a cross sectional survey of a long-term cohort. Malar J (2016) 15:184. doi:10.1186/s12936-016-1233-6
115. Rowe JA, Claessens A, Corrigan RA, Arman M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: molecular mechanisms and therapeutic implications. Expert Rev Mol Med (2009) 11:e16. doi:10.1017/S1462399409001082
116. Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature (2013) 498:502–5. doi:10.1038/nature12216
117. Turner E, Brewster JA, Simpson NA, Walker JJ, Fisher J. Imidazole-based erythrocyte markers of oxidative stress in preeclampsia – an NMR investigation. Reprod Sci (2009) 16:1040–51. doi:10.1177/1933719109340928
118. Biswas AK, Hafiz A, Banerjee B, Kim KS, Datta K, Chitnis CE. Plasmodium falciparum uses gC1qR/HABP1/p32 as a receptor to bind to vascular endothelium and for platelet-mediated clumping. PLoS Pathog (2007) 3:1271–80. doi:10.1371/journal.ppat.0030130
119. Medana IM, Turner GD. Human cerebral malaria and the blood-brain barrier. Int J Parasitol (2006) 36:555–68. doi:10.1016/j.ijpara.2006.02.004
120. Oh H, Takagi H, Suzuma K, Otani A, Matsumura M, Honda Y. Hypoxia and vascular endothelial growth factor selectively up-regulate angiopoietin-2 in bovine microvascular endothelial cells. J Biol Chem (1999) 274:15732–9. doi:10.1074/jbc.274.22.15732
121. Simon MP, Tournaire R, Pouyssegur J. The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol (2008) 217:809–18. doi:10.1002/jcp.21558
122. Conroy AL, Phiri H, Hawkes M, Glover S, Mallewa M, Seydel KB, et al. Endothelium-based biomarkers are associated with cerebral malaria in Malawian children: a retrospective case-control study. PLoS One (2010) 5:e15291. doi:10.1371/journal.pone.0015291
123. Jain A, Kaushik R, Kaushik RM. Malarial hepatopathy: clinical profile and association with other malarial complications. Acta Trop (2016) 159:95–105. doi:10.1016/j.actatropica.2016.03.031
124. Petersen JE, Mkumbaye SI, Vaaben AV, Manjurano A, Lyimo E, Kavishe RA, et al. Plasma Ang2 and ADAM17 levels are elevated during clinical malaria; Ang2 level correlates with severity and expression of EPCR-binding PfEMP1. Sci Rep (2016) 6:35950. doi:10.1038/srep35950
125. Ataíde R, Murillo O, Dombrowski JG, Souza RM, Lima FA, Lima GFMC, et al. Malaria in pregnancy interacts with and alters the angiogenic profiles of the placenta. PLoS Negl Trop Dis (2015) 9:e0003824. doi:10.1371/journal.pntd.0003824
126. Conroy AL, Silver KL, Zhong K, Rennie M, Ward P, Sarma JV, et al. Complement activation and the resulting placental vascular insufficiency drives fetal growth restriction associated with placental malaria. Cell Host Microbe (2013) 13:215–26. doi:10.1016/j.chom.2013.01.010
127. Silver KL, Zhong K, Leke RGF, Taylor DW, Kain KC. Dysregulation of angiopoietins is associated with placental malaria and low birth weight. PLoS One (2010) 5:e9481. doi:10.1371/journal.pone.0009481
128. Lamb TJ, Brown DE, Potocnik AJ, Langhorne J. Insights into the immunopathogenesis of malaria using mouse models. Expert Rev Mol Med (2006) 8:1–22. doi:10.1017/S1462399406010581
129. Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J, et al. Neuropathogenesis of human and murine malaria. Trends Parasitol (2010) 26:277–8. doi:10.1016/j.pt.2010.03.002
130. Hunt NH, Grau GE, Engwerda C, Barnum SR, van der Heyde H, Hansen DS, et al. Murine cerebral malaria: the whole story. Trends Parasitol (2010) 26:272–4. doi:10.1016/j.pt.2010.03.006
131. Stevenson MM, Gros P, Olivier M, Fortin A, Serghides L. Cerebral malaria: human versus mouse studies. Trends Parasitol (2010) 26:274–5. doi:10.1016/j.pt.2010.03.008
132. de Souza JB, Hafalla JC, Riley EM, Couper KN. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology (2010) 137:755–72. doi:10.1017/S0031182009991715
133. Serghides L, McDonald CR, Lu Z, Friedel M, Cui C, Ho KT, et al. PPARgamma agonists improve survival and neurocognitive outcomes in experimental cerebral malaria and induce neuroprotective pathways in human malaria. PLoS Pathog (2014) 10:e1003980. doi:10.1371/journal.ppat.1003980
134. Patnaik JK, Das BS, Mishra SK, Mohanty S, Satpathy SK, Mohanty D. Vascular clogging, mononuclear cell margination, and enhanced vascular permeability in the pathogenesis of human cerebral malaria. Am J Trop Med Hyg (1994) 51:642–7. doi:10.4269/ajtmh.1994.51.642
135. Brown H, Hien TT, Day N, Mai NT, Chuong LV, Chau TT, et al. Evidence of blood-brain barrier dysfunction in human cerebral malaria. Neuropathol Appl Neurobiol (1999) 25:331–40. doi:10.1046/j.1365-2990.1999.00188.x
136. Brown HC, Chau TT, Mai NT, Day NP, Sinh DX, White NJ, et al. Blood-brain barrier function in cerebral malaria and CNS infections in Vietnam. Neurology (2000) 55:104–11. doi:10.1212/WNL.55.1.104
137. Medana IM, Day NP, Sachanonta N, Mai NT, Dondorp AM, Pongponratn E, et al. Coma in fatal adult human malaria is not caused by cerebral oedema. Malar J (2011) 10:267. doi:10.1186/1475-2875-10-267
138. Mohanty S, Mishra SK, Patnaik R, Dutt AK, Pradhan S, Das B, et al. Brain swelling and mannitol therapy in adult cerebral malaria: a randomized trial. Clin Infect Dis (2011) 53:349–55. doi:10.1093/cid/cir405
139. Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, et al. Brain swelling and death in children with cerebral malaria. N Engl J Med (2015) 372:1126–37. doi:10.1056/NEJMoa1400116
140. Mohanty S, Benjamin LA, Majhi M, Panda P, Kampondeni S, Sahu PK, et al. Magnetic resonance imaging of cerebral malaria patients reveals distinct pathogenetic processes in different parts of the brain. mSphere (2017) 2:1–14. doi:10.1128/mSphere.00193-17
141. Yeo TW, Lampah DA, Tjitra E, Gitawati R, Darcy CJ, Jones C, et al. Increased asymmetric dimethylarginine in severe falciparum malaria: association with impaired nitric oxide bioavailability and fatal outcome. PLoS Pathog (2010) 6:e1000868. doi:10.1371/journal.ppat.1000868
142. Weinberg JB, Lopansri BK, Mwaikambo E, Granger DL. Arginine, nitric oxide, carbon monoxide, and endothelial function in severe malaria. Curr Opin Infect Dis (2008) 21:468–75. doi:10.1097/QCO.0b013e32830ef5cf
143. John CC, Kutamba E, Mugarura K, Opoka RO. Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev Anti Infect Ther (2010) 8:997–1008. doi:10.1586/eri.10.90
144. Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med (2013) 19:156–67. doi:10.1038/nm.3073
145. Taylor WRJ, Hanson J, Turner GDH, White NJ, Dondorp AM. Respiratory manifestations of malaria. Chest (2012) 142:492–505. doi:10.1378/chest.11-2655
146. Lukasz A, Kümpers P, David S. Role of angiopoietin/tie2 in critical illness: promising biomarker, disease mediator, and therapeutic target? Scientifica (Cairo) (2012) 2012:160174–8. doi:10.6064/2012/160174
147. Parikh SM. Dysregulation of the angiopoietin-Tie-2 axis in sepsis and ARDS. Virulence (2013) 4:517–24. doi:10.4161/viru.24906
148. van der Heijden M, van Nieuw Amerongen GP, Chedamni S, van Hinsbergh VWM, Johan Groeneveld AB. The angiopoietin-Tie2 system as a therapeutic target in sepsis and acute lung injury. Expert Opin Ther Targets (2009) 13:39–53. doi:10.1517/14728220802626256
149. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA. Plasma angiopoietin-2 in clinical acute lung injury. Crit Care Med (2012) 40:1731–7. doi:10.1097/CCM.0b013e3182451c87
150. Fang Y, Li C, Shao R, Yu H, Zhang Q, Zhao L. Prognostic significance of the angiopoietin-2/angiopoietin-1 and angiopoietin-1/Tie-2 ratios for early sepsis in an emergency department. Crit Care (2015) 19:879. doi:10.1186/s13054-015-1075-6
151. Fisher J, Douglas JJ, Linder A, Boyd JH, Walley KR, Russell JA. Elevated plasma angiopoietin-2 levels are associated with fluid overload, organ dysfunction, and mortality in human septic shock. Crit Care Med (2016) 44:2018–27. doi:10.1097/CCM.0000000000001853
152. Jesmin S, Wada T, Gando S, Sultana SS, Zaedi S. The dynamics of angiogenic factors and their soluble receptors in relation to organ dysfunction in disseminated intravascular coagulation associated with sepsis. Inflammation (2013) 36:186–96. doi:10.1007/s10753-012-9534-6
153. Kranidioti H, Orfanos SE, Vaki I, Kotanidou A, Raftogiannis M, Dimopoulou I, et al. Angiopoietin-2 is increased in septic shock: evidence for the existence of a circulating factor stimulating its release from human monocytes. Immunol Lett (2009) 125:65–71. doi:10.1016/j.imlet.2009.06.006
154. Kümpers P, Hafer C, David S, Hecker H, Lukasz A, Fliser D, et al. Angiopoietin-2 in patients requiring renal replacement therapy in the ICU: relation to acute kidney injury, multiple organ dysfunction syndrome and outcome. Intensive Care Med (2010) 36:462–70. doi:10.1007/s00134-009-1726-7
155. Kümpers P, Lukasz A, David S, Horn R, Hafer C, Faulhaber-Walter R, et al. Excess circulating angiopoietin-2 is a strong predictor of mortality in critically ill medical patients. Crit Care (2008) 12:R147. doi:10.1186/cc7130
156. Kümpers P, van Meurs M, David S, Molema G, Bijzet J, Lukasz A, et al. Time course of angiopoietin-2 release during experimental human endotoxemia and sepsis. Crit Care (2009) 13:R64. doi:10.1186/cc7866
157. Lin S-M, Chung F-T, Kuo C-H, Chou P-C, Wang T-Y, Chang P-J, et al. Circulating angiopopietin-1 correlates with the clinical course of multiple organ dysfunction syndrome and mortality in patients with severe sepsis. Medicine (2015) 94:e878. doi:10.1097/MD.0000000000000878
158. Luz Fiusa MM, Costa-Lima C, de Souza GR, Vigorito AC, Penteado Aranha FJ, Lorand-Metze I, et al. A high angiopoietin-2/angiopoietin-1 ratio is associated with a high risk of septic shock in patients with febrile neutropenia. Crit Care (2013) 17:R169. doi:10.1186/cc12848
159. Mankhambo LA, Banda DL, Group IS, Jeffers G, White SA, Balmer P, et al. The role of angiogenic factors in predicting clinical outcome in severe bacterial infection in Malawian children. Crit Care (2010) 14:R91. doi:10.1186/cc9025
160. Palud A, Parmentier-Decrucq E, Pastre J, De Freitas Caires N, Lassalle P, Mathieu D. Evaluation of endothelial biomarkers as predictors of organ failures in septic shock patients. Cytokine (2015) 73:213–8. doi:10.1016/j.cyto.2015.02.013
161. Siner JM, Bhandari V, Engle KM, Elias JA, Siegel MD. Elevated serum angiopoietin 2 levels are associated with increased mortality in sepsis. Shock (2009) 31:348–53. doi:10.1097/SHK.0b013e318188bd06
162. van der Heijden M, Pickkers P, van Nieuw Amerongen GP, van Hinsbergh VWM, Bouw MPWJM, van der Hoeven JG, et al. Circulating angiopoietin-2 levels in the course of septic shock: relation with fluid balance, pulmonary dysfunction and mortality. Intensive Care Med (2009) 35:1567–74. doi:10.1007/s00134-009-1560-y
163. van der Heijden M, van Nieuw Amerongen GP, Koolwijk P, van Hinsbergh VWM, Groeneveld ABJ. Angiopoietin-2, permeability oedema, occurrence and severity of ALI/ARDS in septic and non-septic critically ill patients. Thorax (2008) 63:903–9. doi:10.1136/thx.2007.087387
164. Giuliano JS, Lahni PM, Harmon K, Wong HR, Doughty LA, Carcillo JA, et al. Admission angiopoietin levels in children with septic shock. Shock (2007) 28:650–4. doi:10.1097/shk.0b013e318123867b
165. Giuliano JS, Tran K, Li F-Y, Shabanova V, Tala JA, Bhandari V. The temporal kinetics of circulating angiopoietin levels in children with sepsis. Pediatr Crit Care Med (2014) 15:e1–8. doi:10.1097/PCC.0b013e3182a553bb
166. Lymperopoulou K, Velissaris D, Kotsaki A, Antypa E, Georgiadou S, Tsaganos T, et al. Angiopoietin-2 associations with the underlying infection and sepsis severity. Cytokine (2015) 73:163–8. doi:10.1016/j.cyto.2015.01.022
167. Orfanos SE, Kotanidou A, Glynos C, Athanasiou C, Tsigkos S, Dimopoulou I, et al. Angiopoietin-2 is increased in severe sepsis: correlation with inflammatory mediators. Crit Care Med (2007) 35:199–206. doi:10.1097/01.CCM.0000251640.77679.D7
168. Parikh SM, Mammoto T, Schultz A, Yuan H-T, Christiani D, Karumanchi SA, et al. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med (2006) 3:e46. doi:10.1371/journal.pmed.0030046
169. Yehya N, Thomas NJ, Meyer NJ, Christie JD, Berg RA, Margulies SS. Circulating markers of endothelial and alveolar epithelial dysfunction are associated with mortality in pediatric acute respiratory distress syndrome. Intensive Care Med (2016) 42:1137–45. doi:10.1007/s00134-016-4352-1
170. Bhandari V, Choo-Wing R, Lee CG, Zhu Z, Nedrelow JH, Chupp GL, et al. Hyperoxia causes angiopoietin 2-mediated acute lung injury and necrotic cell death. Nat Med (2006) 12:1286–93. doi:10.1038/nm1494
171. Fremont RD, Koyama T, Calfee CS, Wu W, Dossett LA, Bossert FR, et al. Acute lung injury in patients with traumatic injuries: utility of a panel of biomarkers for diagnosis and pathogenesis. J Trauma (2010) 68:1121–7. doi:10.1097/TA.0b013e3181c40728
172. Gallagher DC, Parikh SM, Sukhatme VP, Balonov K, Talmor D, Miller A. Circulating angiopoietin 2 correlates with mortality in a surgical population with ALI/ARDS. Crit Care Med (2006) 34:A28. doi:10.1097/00003246-200612002-00103
173. Wada T, Jesmin S, Gando S, Yanagida Y, Mizugaki A, Sultana S, et al. The role of angiogenic factors and their soluble receptors in acute lung injury (ALI)/acute respiratory distress syndrome (ARDS) associated with critical illness. J Inflamm (2013) 10:6. doi:10.1186/1476-9255-10-6
174. Terpstra ML, Aman J, van Nieuw Amerongen GP, Groeneveld ABJ. Plasma biomarkers for acute respiratory distress syndrome: a systematic review and meta-analysis*. Crit Care Med (2014) 42:691–700. doi:10.1097/01.ccm.0000435669.60811.24
175. Uchida T, Ito H, Yamamoto H, Ohno N, Asahara M, Yamada Y, et al. Elevated levels of angiopoietin-2 as a biomarker for respiratory failure after cardiac surgery. J Cardiothorac Vasc Anesth (2014) 28:1293–301. doi:10.1053/j.jvca.2014.03.004
176. Ong T, McClintock DE, Kallet RH, Ware LB, Matthay MA, Liu KD. Ratio of angiopoietin-2 to angiopoietin-1 as a predictor of mortality in acute lung injury patients. Crit Care Med (2010) 38:1845–51. doi:10.1097/CCM.0b013e3181eaa5bf
177. Zinter MS, Spicer A, Orwoll BO, Alkhouli M, Dvorak CC, Calfee CS, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiol Lung Cell Mol Physiol (2016) 310:L224–31. doi:10.1152/ajplung.00336.2015
178. Conroy AL, Lafferty EI, Lovegrove FE, Krudsood S, Tangpukdee N, Liles WC, et al. Whole blood angiopoietin-1 and -2 levels discriminate cerebral and severe (non-cerebral) malaria from uncomplicated malaria. Malar J (2009) 8:295. doi:10.1186/1475-2875-8-295
179. MacMullin G, Mackenzie R, Lau R, Khang J, Zhang H, Rajwans N, et al. Host immune response in returning travellers infected with malaria. Malar J (2012) 11:148. doi:10.1186/1475-2875-11-148
180. Prapansilp P, Medana I, Mai NT, Day NP, Phu N, Yeo TW, et al. A clinicopathological correlation of the expression of the angiopoietin-Tie-2 receptor pathway in the brain of adults with Plasmodium falciparum malaria. Malar J (2013) 12:50. doi:10.1186/1475-2875-12-50
181. Abdi AI, Fegan G, Muthui M, Kiragu E, Musyoki JN, Opiyo M, et al. Plasmodium falciparum antigenic variation: relationships between widespread endothelial activation, parasite PfEMP1 expression and severe malaria. BMC Infect Dis (2014) 14:1399. doi:10.1186/1471-2334-14-170
182. Hanson J, Lee SJ, Hossain MA, Anstey NM, Charunwatthana P, Maude RJ, et al. Microvascular obstruction and endothelial activation are independently associated with the clinical manifestations of severe falciparum malaria in adults: an observational study. BMC Med (2015) 13:122. doi:10.1186/s12916-015-0365-9
183. Rubach MP, Mukemba J, Florence S, Lopansri BK, Hyland K, Volkheimer AD, et al. Impaired systemic tetrahydrobiopterin bioavailability and increased oxidized biopterins in pediatric falciparum malaria: association with disease severity. PLoS Pathog (2015) 11:e1004655. doi:10.1371/journal.ppat.1004655
184. Barber BE, William T, Grigg MJ, Parameswaran U, Piera KA, Price RN, et al. Parasite biomass-related inflammation, endothelial activation, microvascular dysfunction and disease severity in vivax malaria. PLoS Pathog (2015) 11:e1004558. doi:10.1371/journal.ppat.1004558
185. Gomes LT, Alves-Junior ER, Rodrigues-Jesus C, Nery AF, Gasquez-Martin TO, Fontes CJ. Angiopoietin-2 and angiopoietin-2/angiopoietin-1 ratio as indicators of potential severity of Plasmodium vivax malaria in patients with thrombocytopenia. PLoS One (2014) 9:e109246. doi:10.1371/journal.pone.0109246
186. Yeo TW, Lampah DA, Tjitra E, Piera K, Gitawati R, Kenangalem E, et al. Greater endothelial activation, Weibel-Palade body release and host inflammatory response to Plasmodium vivax, compared with Plasmodium falciparum: a prospective study in Papua, Indonesia. J Infect Dis (2010) 202:109–12. doi:10.1086/653211
187. de Jong GM, Slager JJ, Verbon A, van Hellemond JJ, van Genderen PJJ. Systematic review of the role of angiopoietin-1 and angiopoietin-2 in Plasmodium species infections: biomarkers or therapeutic targets? Malar J (2016) 15:581. doi:10.1186/s12936-016-1624-8
188. Weinberg JB, Yeo TW, Mukemba JP, Florence SM, Volkheimer AD, Wang H, et al. Dimethylarginines: endogenous inhibitors of nitric oxide synthesis in children with falciparum malaria. J Infect Dis (2014) 210:913–22. doi:10.1093/infdis/jiu156
189. Jain V, Lucchi NW, Wilson NO, Blackstock AJ, Nagpal AC, Joel PK, et al. Plasma levels of angiopoietin-1 and -2 predict cerebral malaria outcome in Central India. Malar J (2011) 10:383. doi:10.1186/1475-2875-10-383
190. Hawkes M, Opoka RO, Namasopo S, Miller C, Thorpe KE, Lavery JV, et al. Inhaled nitric oxide for the adjunctive therapy of severe malaria: protocol for a randomized controlled trial. Trials (2011) 12:e92. doi:10.1186/1745-6215-12-176
191. Moxon CA, Chisala NV, Wassmer SC, Taylor TE, Seydel KB, Molyneux ME, et al. Persistent endothelial activation and inflammation after Plasmodium falciparum infection in Malawian children. J Infect Dis (2013) 209:610–5. doi:10.1093/infdis/jit419
192. Brouwers J, Noviyanti R, Fijnheer R, de Groot PG, Trianty L, Mudaliana S, et al. Platelet activation determines angiopoietin-1 and VEGF levels in malaria: implications for their use as biomarkers. PLoS One (2014) 8:e64850. doi:10.1371/journal.pone.0064850
193. Björkhem-Bergman L, Bergman P, Andersson J, Lindh JD. Statin treatment and mortality in bacterial infections – a systematic review and meta-analysis. PLoS One (2010) 5:e10702. doi:10.1371/journal.pone.0010702
194. Kwong JC, Li P, Redelmeier DA. Influenza morbidity and mortality in elderly patients receiving statins: a cohort study. PLoS One (2009) 4:e8087. doi:10.1371/journal.pone.0008087
195. Leung S, Pokharel R, Gong MN. Statins and outcomes in patients with bloodstream infection: a propensity-matched analysis. Crit Care Med (2012) 40:1064–71. doi:10.1097/CCM.0b013e31823bc9e7
196. Minneci PC, Deans KJ, Eichacker PQ, Natanson C. The effects of steroids during sepsis depend on dose and severity of illness: an updated meta-analysis. Clin Microbiol Infect (2009) 15:308–18. doi:10.1111/j.1469-0691.2009.02752.x
197. Mortensen EM, Nakashima B, Cornell J, Copeland LA, Pugh MJ, Anzueto A, et al. Population-based study of statins, angiotensin II receptor blockers, and angiotensin-converting enzyme inhibitors on pneumonia-related outcomes. Clin Infect Dis (2012) 55:1466–73. doi:10.1093/cid/cis733
198. Vandermeer ML, Thomas AR, Kamimoto L, Reingold A, Gershman K, Meek J, et al. Association between use of statins and mortality among patients hospitalized with laboratory-confirmed influenza virus infections: a multistate study. J Infect Dis (2012) 205:13–9. doi:10.1093/infdis/jir695
199. Köksal BT, Ozbek OY, Bayraktar N, Yazici AC. Evaluation of angiopoietin 1 and 2, vascular endothelial growth factor, and tumor necrosis factor alpha levels in asthmatic children. Allergy Asthma Proc (2014) 35:482–8. doi:10.2500/aap.2014.35.3793
200. Lee P-H, Kim B-G, Seo H-J, Park J-S, Lee J-H, Park S-W, et al. Circulating angiopoietin-1 and -2 in patients with stable and exacerbated asthma. Ann Allergy Asthma Immunol (2016) 116:339–43. doi:10.1016/j.anai.2015.12.011
201. Fedson DS. Treating the host response to emerging virus diseases: lessons learned from sepsis, pneumonia, influenza and Ebola. Ann Transl Med (2016) 4:421–421. doi:10.21037/atm.2016.11.03
202. van Meurs M, Kümpers P, Ligtenberg JJM, Meertens JHJM, Molema G, Zijlstra JG. Bench-to-bedside review: angiopoietin signalling in critical illness – a future target? Crit Care (2009) 13:207. doi:10.1186/cc7153
203. Saharinen P, Eklund L, Alitalo K. Therapeutic targeting of the angiopoietin-TIE pathway. Nat Rev Drug Discov (2017) 16:635–61. doi:10.1038/nrd.2016.278
204. Ziegler T, Horstkotte J, Schwab C, Pfetsch V, Weinmann K, Dietzel S, et al. Angiopoietin 2 mediates microvascular and hemodynamic alterations in sepsis. J Clin Invest (2013) 123:3436–45. doi:10.1172/JCI66549
205. Tzepi IM, Giamarellos-Bourboulis EJ, Carrer DP, Tsaganos T, Claus RA, Vaki I, et al. Angiopoietin-2 enhances survival in experimental sepsis induced by multidrug-resistant Pseudomonas aeruginosa. J Pharmacol Exp Ther (2012) 343:278–87. doi:10.1124/jpet.112.195180
206. Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med (2007) 4:e269. doi:10.1371/journal.pmed.0040269
207. Xu J, Qu J, Cao L, Sai Y, Chen C, He L, et al. Mesenchymal stem cell-based angiopoietin-1 gene therapy for acute lung injury induced by lipopolysaccharide in mice. J Pathol (2008) 214:472–81. doi:10.1002/path.2302
208. Crowley VM, Ayi K, Lu Z, Liby KT, Sporn M, Kain KC. Synthetic oleanane triterpenoids enhance blood brain barrier integrity and improve survival in experimental cerebral malaria. Malar J (2017) 16:463. doi:10.1186/s12936-017-2109-0
209. Stiehl T, Thamm K, Kaufmann J, Schaeper U, Kirsch T, Haller H, et al. Lung-targeted RNA interference against angiopoietin-2 ameliorates multiple organ dysfunction and death in sepsis. Crit Care Med (2014) 42:e654–62. doi:10.1097/CCM.0000000000000524
210. Safioleas K, Giamarellos-Bourboulis EJ, Carrer DP, Pistiki A, Sabracos L, Deliveliotis C, et al. Reverse kinetics of angiopoietin-2 and endotoxins in acute pyelonephritis: implications for anti-inflammatory treatment? Cytokine (2016) 81:28–34. doi:10.1016/j.cyto.2016.01.019
211. Gurnik S, Devraj K, Macas J, Yamaji M, Starke J, Scholz A, et al. Angiopoietin-2-induced blood-brain barrier compromise and increased stroke size are rescued by VE-PTP-dependent restoration of Tie2 signaling. Acta Neuropathol (2016) 131:753–73. doi:10.1007/s00401-016-1551-3
212. Campochiaro PA, Sophie R, Tolentino M, Miller DM, Browning D, Boyer DS, et al. Treatment of diabetic macular edema with an inhibitor of vascular endothelial-protein tyrosine phosphatase that activates Tie2. Ophthalmology (2015) 122:545–54. doi:10.1016/j.ophtha.2014.09.023
213. Kumpers P, Gueler F, David S, Slyke PV, Dumont DJ, Park JK, et al. The synthetic tie2 agonist peptide vasculotide protects against vascular leakage and reduces mortality in murine abdominal sepsis. Crit Care (2011) 15:R261. doi:10.1186/cc10523
214. Sugiyama MG, Armstrong SM, Wang C, Hwang D, Leong-Poi H, Advani A, et al. The Tie2-agonist vasculotide rescues mice from influenza virus infection. Sci Rep (2015) 5:11030. doi:10.1038/srep11030
215. Cho C-H, Kammerer RA, Lee HJ, Steinmetz MO, Ryu YS, Lee SH, et al. COMP-Ang1: a designed antiopoietin-1 variant with nonleaky angiogenic activity. Proc Natl Acad Sci U S A (2004) 101:6. doi:10.1073/pnas.0307574101
216. Zhu YG, Qu JM, Zhang J, Jiang HN, Xu JF. Novel interventional approaches for ALI/ARDS: cell-based gene therapy. Mediators Inflamm (2011) 2011:560194. doi:10.1155/2011/560194
217. Davis S, Papadopoulos N, Aldrich TH, Maisonpierre PC, Huang T, Kovac L, et al. Angiopoietins have distinct modular domains essential for receptor binding, dimerization and superclustering. Nat Struct Biol (2003) 10:38–44. doi:10.1038/nsb880
218. Boggild AK, Krudsood S, Patel SN, Serghides L, Tangpukdee N, Katz K, et al. Use of peroxisome proliferator-activated receptor gamma agonists as adjunctive treatment for Plasmodium falciparum malaria: a randomized, double-blind, placebo-controlled trial. Clin Infect Dis (2009) 49:841–9. doi:10.1086/605431
Keywords: endothelial dysfunction, Tie2 receptor, angiopoietins, sepsis, malaria, critical care
Citation: Leligdowicz A, Richard-Greenblatt M, Wright J, Crowley VM and Kain KC (2018) Endothelial Activation: The Ang/Tie Axis in Sepsis. Front. Immunol. 9:838. doi: 10.3389/fimmu.2018.00838
Received: 07 December 2017; Accepted: 05 April 2018;
Published: 24 April 2018
Edited by:
Thomas Luft, Universitätsklinikum Heidelberg, GermanyReviewed by:
Adriane Feijo Evangelista, Barretos Cancer Hospital, BrazilJoerg Halter, Universität Basel, Switzerland
Copyright: © 2018 Leligdowicz, Richard-Greenblatt, Wright, Crowley and Kain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin C. Kain, a2V2aW4ua2FpbkB1aG4uY2E=
†Co-first authors.