 Emma Weeding
Emma Weeding Amr H. Sawalha
Amr H. Sawalha- 1Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
- 2Division of Rheumatology, Department of Internal Medicine, University of Michigan, Ann Arbor, MI, United States
- 3Center for Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, United States
Differential deoxyribonucleic acid (DNA) methylation has emerged as a critical feature of systemic lupus erythematosus (SLE). Genome-wide DNA methylation studies have revealed methylation patterns characteristic of SLE—in particular, robust hypomethylation of interferon-regulated genes is a prominent finding in all cells of the immune system studied to date. These patterns reliably distinguish individuals with SLE from healthy controls and from individuals with other autoimmune diseases. For example, hypomethylation within IFI44L is both highly sensitive and highly specific for SLE, superior to currently available biomarkers. Furthermore, methylation status of other genetic loci has been associated with clinically relevant features of SLE including disease severity and organ-specific manifestations. Finally, DNA methylation studies have provided important insights into the pathophysiology of SLE. Most recently, there is a growing body of evidence that the transcription factor enhancer of zeste homolog 2 (EZH2) plays an important role in triggering SLE disease activity via epigenetic mechanisms, and that EZH2 blockade may be a future treatment option in SLE. In this short review, we discuss the DNA methylation patterns associated with SLE, their relationship to clinically significant features of SLE, and their implications in the development of novel diagnostic and therapeutic approaches to this complex disease.
Introduction
Systemic lupus erythematosus (SLE) is a highly heterogeneous autoimmune disease that can affect virtually any organ system in the body, resulting in protean clinical and serological manifestations which range from mild to life-threatening. The disease is broadly characterized by the production of antinuclear autoantibodies, resulting in the formation and deposition of immune complexes, which in turn leads to inflammation and damage of affected tissue. As with many other autoimmune diseases, the pathogenesis of SLE is complex and incompletely understood. A genetic component to the disease has long been presumed—first-degree relatives of individuals with SLE have up to a 30-fold higher risk of developing the disease as compared to the general population (1), which clearly suggests some degree of heritability. Indeed, extensive investigation has revealed dozens of genetic risk loci for SLE. Yet, these loci account for less than 20% of disease susceptibility (2). In the vast majority of patients, any one genetic polymorphism in isolation does not confer clinical disease. Rather, SLE arises due to some combination of genetic risk factors and various environmental factors, such as exposure to infections, chemicals, radiation, sex hormones, or other alterations to an individual’s immunologic substrate.
The study of epigenetics has emerged as an important approach in investigating the contributions of both heritable and environmental factors, as well as the interplay between them, in the pathogenesis of autoimmune disease. Epigenetic mechanisms regulate gene expression in a tissue-specific manner by controlling the accessibility of deoxyribonucleic acid (DNA) to the transcription complex without modifying the underlying nucleotide sequence. Epigenetic changes can be either inherited or induced, and can be highly dynamic over the course of a cell’s lifespan. The potentially reversible nature of epigenetic events makes them attractive candidates as biomarkers of disease activity and as targets for therapeutic strategies.
Deoxyribonucleic acid methylation is one of the most well-studied epigenetic mechanisms in humans. Methylation most commonly occurs at the C5 position of cytosine residues in CG dinucleotides and classically results in gene silencing. Conversely, demethylation is generally associated with increased chromatin accessibility and thus active gene expression. Aberrant DNA methylation has emerged as an important epigenetic feature of SLE, and the study of these abnormal methylation patterns has revealed numerous possibilities for a deepened understanding of this complex disease.
In this short review, we discuss the putative role of differential DNA methylation in the pathogenesis and pathophysiology of SLE, with a focus on how these patterns influence clinically relevant features, such as disease severity and heterogeneity, and their implications in the development of novel diagnostic and therapeutic strategies.
DNA Methylation in the Pathophysiology of SLE
In one of the earliest studies of epigenetic patterns in SLE, Richardson et al. examined the total percentage of methylated cytosine residues in T cells isolated from participants with SLE (3). This revealed significant global DNA hypomethylation of T cells in individuals with SLE when compared to healthy age-matched controls. In a similar vein, the treatment of CD4+ T cells by DNA methylation inhibitors, such as procainamide or hydralazine, has been shown to induce hypomethylation and autoreactivity in vitro (4). Furthermore, injecting these hypomethylated T cells into syngeneic mice can induce lupus-like autoimmunity in vivo (5). While these studies provide a critical foundation in understanding the epigenetic patterns in SLE, the ultimate result of autoreactivity induced via hypomethylation is almost a forgone conclusion in that the relationship between DNA methylation inhibitors and autoimmune disease in humans has already been established in clinical practice. Indeed, treatment of humans with procainamide or hydralazine can cause a lupus variant known as drug-induced lupus erythematosus, which has similar clinical manifestations as SLE, and usually resolves within months after discontinuation of the culprit medication.
A seminal work by Javierre et al. was the first to examine genome-wide DNA methylation patterns in SLE and thereby associate epigenetic changes with specific genetic loci (6). Specifically, DNA methylation status was compared in total white blood cells obtained from monozygotic twins who were discordant in SLE status. Global hypomethylation was again observed in SLE participants when compared to their healthy twins or matched controls. Furthermore, sequence-specific demethylation was found in genes associated with several cellular processes which are likely relevant to SLE pathophysiology, including immune response, cell activation, cell proliferation, and cytokine production.
Subsequent studies have examined the DNA methylation patterns of specific cells in the immune system, particularly in T cells, given the earlier evidence of T cell hypomethylation in SLE as described above. In the first study to investigate genome-wide DNA methylation changes in CD4+ T cells, the methylation status of over 25,000 CG sites (corresponding to the promoter regions of nearly 15,000 genes) was compared in SLE participants versus healthy controls (7). A total of 341 CG sites were found to be differentially methylated in SLE, with the majority of these being hypomethylated as expected. Hypomethylated genes included MMP9 and PDGFRA, both of which are involved in the development of connective tissue, as well as CD9, which has been shown to provide potent costimulatory signals promoting the activation of T cells (8). Hypermethylated genes were primarily involved in metabolic pathways, particularly folate biosynthesis, which is essential in maintaining DNA integrity and stability. Hypermethylation was also noted in RUNX3, which encodes a transcription factor that is required for T-cell maturation.
A follow-up study surveyed DNA methylation status across over 485,000 CG sites in naïve CD4+ T cells, with the intent to identify methylation changes preceding T cell differentiation and activation, and thereby revealing early epigenetic events which potentially predispose individuals to clinical manifestations of disease (9). A total of 86 differentially methylated CG sites in 47 genes were identified in SLE participants as compared to controls. Most notably, the majority of hypomethylated genes in naïve T cells found in this study are regulated by type I interferons, including IFIT1, IFIT3, MX1, STAT1, IFI44L, USP18, TRIM22, and BST2. Additionally, gene expression analysis was performed in the same naïve CD4+ T cells. This revealed that despite being hypomethylated, none of these interferon-regulated genes were overexpressed in naïve CD4+ T cells. Conversely, most of them were significantly overexpressed in total CD4+ T cells from participants with SLE. In summary, these results suggest that naïve CD4+ T cells undergo epigenetic priming toward a rapid response to type I interferons, resulting in T cell differentiation and activation, and presumably, increased disease activity.
Subsequent studies in memory T cells, regulatory T cells, and neutrophils (including low-density granulocytes) have revealed similar patterns of global hypomethylation, particularly in interferon-regulated genes (10, 11). Most recently, DNA methylation patterns in a T cell subset, specifically CD4+CD28+KIR+CD11ahi+ T cells, were examined (12). This previously undescribed subset of T cells has been found to be present in patients with SLE, with the size of the subset correlating to disease severity (13). Differential DNA methylation analysis yet again revealed global hypomethylation in this T cell subset. Moreover, this hypomethylation, in combination with increased chromatin accessibility, resulted in increased expression of pro-inflammatory genes, such as cytokine genes, adhesion molecules, Fc-gamma receptor genes, toll-like receptor genes, human leukocyte antigen molecules, and metalloproteinases. These results further emphasize the important role that this demethylated T cell subset may play in SLE pathophysiology, and suggest that blocking these downstream pro-inflammatory effects might provide novel therapeutic avenues in SLE.
DNA Methylation as a Biomarker
Even for the experienced physician, SLE can be difficult to diagnose. Owing to the significant heterogeneity of the disease, patients can present with any variety of symptoms at disease onset, some of which may be vague or non-specific. There is no single test for SLE—the diagnosis is ultimately made through clinical judgment by interpreting a patient’s symptomatology in the context of serological, radiographic, and/or histological evidence of disease. Several laboratory markers of SLE are already used in clinical practice, including antinuclear antibodies (ANAs), anti-double stranded DNA (anti-dsDNA) antibody, and anti-Smith (anti-Sm) antibody. Interpretation of autoantibody titers has significant limitations, however. While effectively all patients with SLE test positive for at least one ANA, nearly one-quarter of the general population is also ANA positive (14). Conversely, anti-dsDNA and anti-Sm antibodies are highly specific for SLE, but are only detectable in roughly half of patients (15, 16).
Zhao et al. sought to investigate whether DNA methylation status could act as a more robust biomarker in SLE (17). Specifically, they examined the methylation status of two CG sites located within the IFI44L promoter (an interferon-regulated gene found to be hypomethylated in SLE) in DNA from peripheral blood obtained from participants with SLE. They found that a given threshold of hypomethylation at either CG site had a sensitivity and specificity of greater than 90% for SLE versus healthy controls. As discussed above, this is superior to currently available biomarkers such as ANAs or anti-dsDNA antibody. Differential methylation at these CG sites also distinguished SLE from rheumatoid arthritis and primary Sjögren’s syndrome, two autoimmune diseases which can have clinical overlap with SLE. Although IFI44L is also hypomethylated in naive CD4+ T cells from individuals with primary Sjögren’s syndrome as compared to healthy controls (18), the degree of hypomethylation was found to be significantly higher among those with SLE per the work of Zhao and colleagues. Further validation is required to determine the role of IFI44L methylation status in distinguishing these two autoimmune diseases from one another. Nevertheless, the results of this study provide strong evidence that an assay for DNA methylation status in whole blood could be a powerful tool in the diagnosis of SLE. To this point, a summary of the DNA methylation patterns associated with various clinical features of SLE is provided in Table 1. These patterns are discussed in more detail below.

Table 1. Summary of differential deoxyribonucleic acid (DNA) methylation patterns in naïve CD4+ T cells associated with systemic lupus erythematosus (SLE), disease severity, and organ-specific manifestations.
Systemic lupus erythematosus can be reviewed as a relapsing-remitting disease, with the majority of patients experiencing intermittent flares of disease activity alternating with relative quiescence. Given the evidence of epigenetic T cell priming toward a robust interferon-mediated response, the question arises of whether methylation status might correlate with disease flares. To address this question, Coit et al. performed genome-wide DNA methylation analysis on naïve CD4+ T cells from participants with SLE with varying levels of disease activity as measured by SLE Disease Activity Index (SLEDAI) scores (19). They identified over 5,000 CG sites that either negatively or positively correlated with disease activity, and more broadly, discovered that higher disease activity is associated with progressive hypomethylation of genes involved in Th2, Th17, and follicular helper T cell response. Progressive hypermethylation was noted in inhibitory pathways such as the transforming growth factor beta signaling pathway. Gene expression analysis was performed and demonstrated that these epigenetic events do indeed precede gene expression. Overall, these results suggest that not only are naïve CD4+ T cells epigenetically predisposed toward an effector T cell response in SLE, but also that these epigenetic changes and putative downstream effects are associated with increased disease activity. The clinical relevance of this conclusion is that determination of DNA methylation status might provide prognostic information in predicting SLE flares, and may thus be useful in tailoring selection of medical therapy and subsequent monitoring of a patient’s response to treatment.
In current clinical practice, selection of therapy for SLE is based not only on overall disease activity but also on a given patient’s particular manifestations of disease. Understanding the methylation patterns of particular manifestations of SLE may provide additional prognostic information and help guide future development of targeted therapies. As an example, cutaneous manifestations such as malar rash or discoid rash are common in patients with SLE. Methylation patterns specific to each of these kinds of rash have been found. Specifically, Renauer et al. compared genome-wide DNA methylation profiles in naïve CD4+ T cells from participants with SLE who had a history of malar rash, discoid rash, or neither cutaneous manifestation (20). Between these three groups, they identified several hundred differentially methylated sites, the majority of which were specific to each cutaneous manifestation (or lack thereof). For those with a history of malar rash, the most extensively hypomethylated region was located in the promoter region of precursor microRNA miR-886 (VTRNA2-1). Independent studies have shown that hypomethylation of this region modulates signaling pathways which determine cell survival versus apoptosis (21). For those with a history of discoid rash, hypomethylation within RHOJ and HZF were found, both of which are also involved in determining cell survival versus apoptosis. These results correlate with older findings that the epidermis of patients with cutaneous SLE is in part characterized by an accumulation of apoptotic cells (22).
Another organ commonly affected in SLE is the kidneys. It is estimated that just over half of all patients with SLE have renal involvement, which can range in severity from mild and near-quiescent to fulminant and life-threatening. Early recognition of renal impairment is critical, as treatment is more likely to be successful when it is started as quickly as possible, and conversely, delayed diagnosis is associated with a significantly increased risk of renal failure and death (23). In an effort to determine how DNA methylation patterns might correlate with renal disease in SLE, one study examined genome-wide DNA methylation in naïve CD4+ T cells from SLE patients with and without renal involvement (24). The authors discovered 191 differentially methylated CG sites (corresponding to 121 genes) associated with the presence or absence of renal involvement. Genes which were more hypomethylated in SLE participants with renal disease included IRF7, which is a well-known genetic risk locus for SLE. Indeed the majority of hypomethylated sites were located in interferon-regulated regions, as expected. Notably, the degree of hypomethylation in these regions was significantly more robust in SLE participants with a history of renal disease, independent of overall disease activity. Genes which were more hypermethylated in SLE participants with renal disease included CD47, which has been shown to regulate T cell production of vascular endothelial growth factor (25), and CD247, which encodes the T-cell receptor zeta chain, and in turn, plays a key role in antigen receptor-mediated signaling and has been shown to be downregulated in SLE T cells (26). Finally, the authors identified a single CG site, CG10152449 in CHST12, for which hypomethylation had a sensitivity of 86% and specificity of 64% in detecting renal disease in SLE participants. No comparable biomarker currently exists in clinical practice.
DNA Methylation and Future Therapies
Arguably, the ultimate goal in the study of the epigenetics of human disease is not only to identify biological pathways which drive pathophysiology but also to use our new-found understanding of these pathways to develop novel treatment strategies. One of the most promising future treatment options revolves around a transcription regulator known as enhancer of zeste homolog 2 (EZH2). EZH2 is a histone-lysine N-methyltransferase enzyme which promotes transcriptional regulation by way of histone methylation as part of the polycomb repressive complex 2. Similar to DNA methylation, posttranslational modifications of histone proteins are epigenetic events which contribute to the pathophysiology of SLE and other autoimmune disorders by regulating gene expression (27). EZH2 trimethylates lysine 27 in histone H3, resulting in H3K27me3 and transcriptional repression. It can also recruit DNA methyltransferases DNMT1, DNMT3A, and DNMT3B (28, 29). When phosphorylated, EZH2 acts as a transcriptional activator at least in part by suppressing H3K27me3, thus disrupting gene silencing (30, 31).
In the aforementioned study by Coit et al., which examined the relationship between epigenetic changes and disease activity, methylation sites which correlated with disease activity were found to be either enriched (at hypermethylated loci) or depleted (at hypomethylated loci) in binding sites for EZH2, suggesting that it might play an important role in inducing a pro-inflammatory epigenetic shift (19). EZH2 expression in T cells is inhibited by glucose restriction via increased expression of microRNAs miR-26a and miR-101 (32). Increased glycolysis has been noted in CD4+ T cells from individuals with SLE and in mouse models of lupus, and furthermore, treatment of this abnormally enhanced glycolysis in mice resulted in a shift of immunophenotype toward that of healthy controls (33). As such, it was hypothesized that decreased levels of the above microRNAs, indicating enhanced glycolysis and subsequently increased EZH2 activity, would correlate with increased disease activity in SLE (Figure 1). This association was indeed found when comparing SLEDAI scores to levels of miR-26a expression (19).
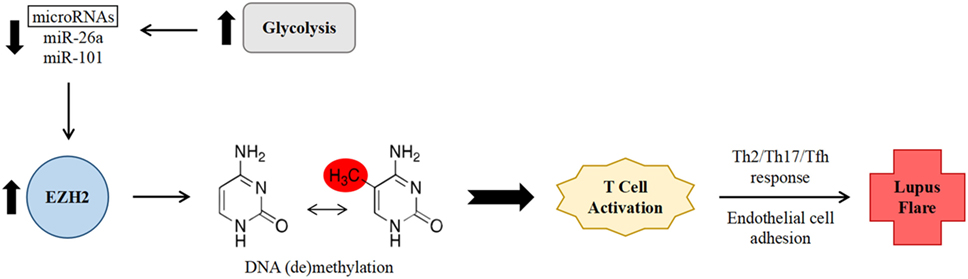
Figure 1. Proposed mechanism of increased systemic lupus erythematosus (SLE) disease activity via enhancer of zeste homolog 2 (EZH2)-mediated epigenetic remodeling within CD4+ T cells. Abnormally enhanced glycolysis in SLE results in decreased levels of the microRNAs miR-26a and miR-101. Decreased microRNA levels leads to lessened downregulation of the expression of transcription factor EZH2. EZH2 in turn promotes deoxyribonucleic acid methylation changes, leading to T cell activation, a non-Th1 effector T cell response, and increased adhesion to endothelial cells, thereby promoting SLE disease activity.
Most recently, a follow-up study examined expression levels of EZH2 in CD4+ T cells, as well as the effects on DNA methylation associated with EZH2 overexpression, in participants with SLE versus healthy controls (34). First, this study confirmed previous findings that T cell production of EZH2 is downregulated by miR-26a and miR-101. Notably, both of these microRNAs were present at reduced levels in SLE CD4+ T cells. Next, overexpression of EZH2 was induced in CD4+ T cells from healthy controls, and the resulting genome-wide DNA methylation patterns were assessed. This revealed several hundred differentially methylated CG loci, most notably in regions associated with cell adhesion and leukocyte migration. Indeed, CD4+ T cells from both the EZH2-overexpression group and the SLE group showed increased adhesion to human dermal microvascular endothelial cells. Finally, blocking EZH2 effectively reduced the capacity of these T cells to adhere to endothelial cells, providing proof of principle that EZH2 blockade may be a future therapy for SLE. Though no EZH2 inhibitor is yet widely available in clinical settings, one such agent, tazemetostat, is currently being investigated in clinical trials as a treatment for certain cancers.
Conclusion
Differential DNA methylation has emerged as a critical feature of SLE. Characterization of these methylation patterns has provided important insights into the pathophysiology of this complex disease. Furthermore, assessing an individual’s methylation status shows promise as a future clinical tool and may aid not only in the diagnosis of SLE itself but also act as a prognostic indicator to help predict disease flares and facilitate detection of organ-specific manifestations. Finally, the study of differential DNA methylation and its downstream functional effects on the immunologic environment has now revealed an encouraging potential future treatment option for SLE, namely EZH2 blockade.
Author Contributions
EW and AS drafted and critically revised the manuscript.
Conflict of Interest Statement
AS is listed as inventor on a patent application for using IFI44L methylation as a biomarker in SLE. EW declares no conflict of interest.
The reviewer HL and handling Editor declared their shared affiliation.
Funding
This work was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health grants number R01AI097134 and U19AI110502.
Abbreviations
BST2, bone marrow stromal cell antigen 2; CD, cluster of differentiation; CHST12, carbohydrate sulfotransferase 12; DNA, deoxyribonucleic acid; EZH2, enhancer of zeste homolog 2; HLA, human leukocyte antigen; HZF, ring finger protein 39; IFI44L, interferon-induced protein 44 like; IFIT, interferon-induced proteins with tetratricopeptide repeats; IRF7, interferon regulatory factor 7; MMP9, matrix metallopeptidase 9; MX1, MX dynamin like GTPase 1; PDGFRA, platelet-derived growth factor receptor A; RHOJ, ras homolog family member J; RNA, ribonucleic acid; RUNX3, Runt Related Transcription Factor 3; SLE, systemic lupus erythematosus; STAT1, signal transducer and activator of transcription 1; TGFB, transforming growth factor beta; TRIM22, tripartite motif containing 22; USP18, ubiquitin-specific peptidase 18; VEGF, vascular endothelial growth factor; VTRNA2-1, vault RNA 2-1.
References
1. Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum (2005) 52(4):1138–47. doi:10.1002/art.20999
2. Rullo OJ, Tsao BP. Recent insights into the genetic basis of systemic lupus erythematosus. Ann Rheum Dis (2013) 72(Suppl 2):ii56–61. doi:10.1136/annrheumdis-2012-202351
3. Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum (1990) 33(11):1665–73. doi:10.1002/art.1780331109
4. Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J Immunol (1988) 140(7):2197–200.
5. Richardson B, Sawalha AH, Ray D, Yung R. Murine models of lupus induced by hypomethylated T cells. Methods Mol Biol (2012) 900:169–80. doi:10.1007/978-1-60761-720-4_8
6. Javierre BM, Fernandez AF, Richter J, Al-Shahrour F, Martin-Subero JI, Rodriguez-Ubreva J, et al. Changes in the pattern of DNA methylation associate with twin discordance in systemic lupus erythematosus. Genome Res (2010) 20(2):170–9. doi:10.1101/gr.100289.109
7. Jeffries MA, Dozmorov M, Tang Y, Merrill JT, Wren JD, Sawalha AH. Genome-wide DNA methylation patterns in CD4+ T cells from patients with systemic lupus erythematosus. Epigenetics (2011) 6(5):593–601. doi:10.4161/epi.6.5.15374
8. Tai XG, Yashiro Y, Abe R, Toyooka K, Wood CR, Morris J, et al. A role for CD9 molecules in T cell activation. J Exp Med (1996) 184:753–8. doi:10.1084/jem.184.2.753
9. Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naïve CD4+ T cells from lupus patients. J Autoimmun (2013) 43:78–84. doi:10.1016/j.jaut.2013.04.003
10. Absher DM, Li X, Waite LL, Gibson A, Roberts K, Edberg J, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet (2013) 9:e1003678. doi:10.1371/journal.pgen.1003678
11. Coit P, Yalavarthi S, Ognenovski M, Zhao W, Hasni S, Wren JD, et al. Epigenome profiling reveals significant DNA demethylation of interferon signature genes in lupus neutrophils. J Autoimmun (2015) 58:59–66. doi:10.1016/j.jaut.2015.01.004
12. Gensterblum E, Renauer P, Coit P, Strickland FM, Kilian NC, Miller S, et al. CD4+CD28+KIR+CD11ahi T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients. J Autoimmun (2018) 86:19–28. doi:10.1016/j.jaut.2017.09.011
13. Strickland FM, Patel D, Khanna D, Somers E, Robida AM, Pihalja M, et al. Characterisation of an epigenetically altered CD4(+) CD28(+) Kir(+) T cell subset in autoimmune rheumatic diseases by multiparameter flow cytometry. Lupus Sci Med (2016) 3(1):e000147. doi:10.1136/lupus-2016-000147
14. Wandstrat A, Carr-Johnson F, Branch V, Gray H, Fairhurst A, Reimold A, et al. Autoantibody profiling to identify individuals at risk for systemic lupus erythematosus. J Autoimmun (2006) 27:153–60. doi:10.1016/j.jaut.2006.09.001
15. Kurien BT, Scofield RH. Autoantibody determination in the diagnosis of systemic lupus erythematosus. Scand J Immunol (2006) 64:227–35. doi:10.1111/j.1365-3083.2006.01819.x
16. Zieve GW, Khusial PR. The anti-Sm immune response in autoimmunity and cell biology. Autoimmun Rev (2003) 2:235–40. doi:10.1016/S1568-9972(03)00018-1
17. Zhao M, Zhou Y, Zhu B, Wan M, Jiang T, Tan Q, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis (2016) 75(11):1998–2006. doi:10.1136/annrheumdis-2015-208410
18. Altorok N, Coit P, Hughes T, Koelsch KA, Stone DU, Rasmussen A, et al. Genome-wide DNA methylation patterns in naive CD4+ T cells from patients with primary Sjögren’s syndrome. Arthritis Rheumatol (2014) 66(3):731–9. doi:10.1002/art.38264
19. Coit P, Dozmorov MG, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, Wren JD, et al. Epigenetic reprogramming in naive CD4+ T cells favoring T cell activation and non-Th1 effector T cell immune response as an early event in lupus flares. Arthritis Rheumatol (2016) 68(9):2200–9. doi:10.1002/art.39720
20. Renauer P, Coit P, Jeffries MA, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, et al. DNA methylation patterns in naïve CD4+ T cells identify epigenetic susceptibility loci for malar rash and discoid rash in systemic lupus erythematosus. Lupus Sci Med (2015) 2(1):e000101. doi:10.1136/lupus-2015-000101
21. Treppendahl MB, Qiu X, Søgaard A, Yang X, Nandrup-Bus C, Hother C, et al. Allelic methylation levels of the noncoding VTRNA2-1 located on chromosome 5q31.1 predict outcome in AML. Blood (2012) 119(1):206–16. doi:10.1182/blood-2011-06-362541
22. Chung JH, Kwon OS, Eun HC, Youn JI, Song YW, Kim JG, et al. Apoptosis in the pathogenesis of cutaneous lupus erythematosus. Am J Dermatopathol (1998) 20(3):233–41. doi:10.1097/00000372-199806000-00002
23. Faurschou M, Starklint H, Halberg P, Jacobsen S. Prognostic factors in lupus nephritis: diagnostic and therapeutic delay increases the risk of terminal renal failure. J Rheumatol (2006) 33(8):1563–9.
24. Coit P, Renauer P, Jeffries MA, Merrill JT, McCune WJ, Maksimowicz-McKinnon K, et al. Renal involvement in lupus is characterized by unique DNA methylation changes in naïve CD4+ T cells. J Autoimmun (2015) 61:29–35. doi:10.1016/j.jaut.2015.05.003
25. Kaur S, Chang T, Singh SP, Lim L, Mannan P, Garfield SH, et al. CD47 signaling regulates the immunosuppressive activity of VEGF in T cells. J Immunol (2014) 193:3914–24. doi:10.4049/jimmunol.1303116
26. Nambiar MP, Enyedy EJ, Fisher CU, Krishnan S, Warke VG, Gilliland WR, et al. Abnormal expression of various molecular forms and distribution of T cell receptor zeta chain in patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:163–74. doi:10.1002/1529-0131(200201)46:1<163::AID-ART10065>3.0.CO;2-J
27. Hu N, Qiu X, Luo Y, Yuan J, Li Y, Lei W, et al. Abnormal histone modification patterns in lupus CD4+ T cells. J Rheumatol (2008) 35(5):804–10.
28. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science (2002) 298:1039–43. doi:10.1126/science.1076997
29. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439:871–4. doi:10.1038/nature04431
30. Yan J, Li B, Lin B, Lee PT, Chung TH, Tan J, et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood (2016) 128:948–58. doi:10.1182/blood-2016-01-690701
31. Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science (2005) 310:306–10. doi:10.1126/science.1118947
32. Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, et al. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol (2016) 17(1):95–103. doi:10.1038/ni.3313
33. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med (2015) 7(274):274ra18. doi:10.1126/scitranslmed.aaa0835
Keywords: autoimmunity, biomarker, EZH2, IFI44L, lupus, methylation, T cells, therapeutic
Citation: Weeding E and Sawalha AH (2018) Deoxyribonucleic Acid Methylation in Systemic Lupus Erythematosus: Implications for Future Clinical Practice. Front. Immunol. 9:875. doi: 10.3389/fimmu.2018.00875
Received: 29 January 2018; Accepted: 09 April 2018;
Published: 24 April 2018
Edited by:
George C. Tsokos, Harvard Medical School, United StatesReviewed by:
Hao Li, Harvard Medical School, United StatesDipyaman Ganguly, Indian Institute of Chemical Biology (CSIR), India
Copyright: © 2018 Weeding and Sawalha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amr H. Sawalha, YXNhd2FsaGFAdW1pY2guZWR1