 Maria Isabel Lovo-Martins1,2
Maria Isabel Lovo-Martins1,2 Aparecida Donizette Malvezi2
Aparecida Donizette Malvezi2 Nágela Ghabdan Zanluqui1
Nágela Ghabdan Zanluqui1 Bruno Fernando Cruz Lucchetti2
Bruno Fernando Cruz Lucchetti2 Vera Lúcia Hideko Tatakihara2
Vera Lúcia Hideko Tatakihara2 Patricia Alves Mörking1
Patricia Alves Mörking1 Admilton Gonçalves de Oliveira3
Admilton Gonçalves de Oliveira3 Samuel Goldenberg1
Samuel Goldenberg1 Pryscilla Fanini Wowk1,4*
Pryscilla Fanini Wowk1,4* Phileno Pinge-Filho2*
Phileno Pinge-Filho2*
- 1Instituto Carlos Chagas, Fiocruz - Paraná, Curitiba, Brazil
- 2Laboratório de Imunopatologia Experimental, Departamento de Ciências Patológicas, Centro de Ciências Biológicas, Universidade Estadual de Londrina, Londrina, Brazil
- 3Laboratório de Microscopia Eletrônica e Microanálises, Central de Laboratórios de Pesquisa Multiusuários, Universidade Estadual de Londrina, Londrina, Brazil
- 4Laboratório de Virologia Molecular, Instituto Carlos Chagas, Fiocruz - Paraná, Curitiba, Brazil
During the onset of Trypanosoma cruzi infection, an effective immune response is necessary to control parasite replication and ensure host survival. Macrophages have a central role in innate immunity, acting as an important trypanocidal cell and triggering the adaptive immune response through antigen presentation and cytokine production. However, T. cruzi displays immune evasion mechanisms that allow infection and replication in macrophages, favoring its chronic persistence. One potential mechanism is the release of T. cruzi strain Y extracellular vesicle (EV Y), which participate in intracellular communication by carrying functional molecules that signal host cells and can modulate the immune response. The present work aimed to evaluate immune modulation by EV Y in C57BL/6 mice, a prototype resistant to infection by T. cruzi strain Y, and the effects of direct EV Y stimulation of macrophages in vitro. EV Y inoculation in mice prior to T. cruzi infection resulted in increased parasitemia, elevated cardiac parasitism, decreased plasma nitric oxide (NO), reduced NO production by spleen cells, and modulation of cytokine production, with a reduction in TNF-α in plasma and decreased production of TNF-α and IL-6 by spleen cells from infected animals. In vitro assays using bone marrow-derived macrophages showed that stimulation with EV Y prior to infection by T. cruzi increased the parasite internalization rate and release of infective trypomastigotes by these cells. In this same scenario, EV Y induced lipid body formation and prostaglandin E2 (PGE2) production by macrophages even in the absence of T. cruzi. In infected macrophages, EV Y decreased production of PGE2 and cytokines TNF-α and IL-6 24 h after infection. These results suggest that EV Y modulates the host response in favor of the parasite and indicates a role for lipid bodies and PGE2 in immune modulation exerted by EVs.
Introduction
The protozoan Trypanosoma cruzi, the etiologic agent of Chagas disease, was discovered by Carlos Chagas more than 100 years ago when its life cycle, vector, and pathogenesis in humans were described (1). However, the elucidation of the mechanisms involved in the parasite–host interaction and modulation of the immune response remain incompletely understood. According to the World Health Organization (2017), seven to eight million people are infected with T. cruzi worldwide, and over than 10,000 deaths are recorded annually, mostly in Latin America, an endemic area for Chagas disease (2). Recently, new cases have been reported in North America, Asia, and Europe, mainly due to the migration of infected individuals from endemic areas and through non-vectorial transmissions, such as blood transfusion, organ transplants, and congenital transmissions (3).
In vector-borne transmissions, metacyclic trypomastigotes are released in the triatomine excreta when insects feed on mammalian blood. These metacyclic trypomastigotes invade host cells where they transform into replicative amastigotes. After intense replication, amastigotes differentiate into trypomastigotes, an event that culminates with the disruption of the infected cell membrane and release of a substantial number of blood trypomastigote forms that spread the pathogen to other body sites, such as heart tissues (4, 5). During early infection, the innate immune system initiates a host defense that plays an important role in controlling parasite replication and spread in host tissue, with the involvement of cytokines IFN-γ (6), TNF-α (7), and IL-12 (8) and immune cells such as macrophages, natural killers, and dendritic cells (9, 10). Later in the infection, adaptive immunity develops, potentiating innate immunity. A strong and persistent polarized T helper 1 response is established against intracellular parasites (11), and an anti-T. cruzi CD8 immune response is focused on parasitic immunodominant epitopes. These T-cell responses are important for controlling parasitemia, tissue parasitism, and polyclonal B-cell activation with antibody production (12). However, due to T. cruzi immune evasion mechanisms, this intense immune response fails to clear parasitic infection, leading to its persistence and development of the chronic phase of Chagas disease (13).
An important immune evasion mechanism in infectious diseases is the cell–cell modulatory action promoted by the release of heterogeneous extracellular vesicles (EVs). The following main classes of EVs are categorized based on their intracellular origin: the exosomes (50–150 nm diameter) formed inside multivesicular bodies and released upon fusion of these endosomal compartments with the plasma membrane and plasma membrane-derived EVs (50–1000 nm diameter) that are formed by direct budding and constriction from the plasma membrane. These vesicles can transport proteins from the membrane of the original cell, as well as intracellular proteins and nucleic acids, including small RNAs, which can modulate the target cell (14). Hence, EVs have the potential to be used for immunotherapy, immunization, and diagnosis. In fact, especally in T. cruzi, the following different mechanisms for vesicle release have been described: larger vesicles that bud from the plasma membrane and smaller vesicles that bud within the flagellar pocket are released through exocytosis of multivesicular bodies (15, 16).
Recently, the role of EVs that are secreted by different parasites during infection and disease progression have been the focus of several investigations. In some cases, EVs displayed a protective role (17–22). However, in other cases, injection of EVs resulted in downregulation of the immune response followed by an increase in parasitemia (23). Furthermore, when EVs from Trypanosoma brucei were engulfed by mammalian erythrocytes, cell clearance led to anemia (24).
Although it has been known since 1991 that shedding T. cruzi EVs contain several molecules that interfere with the immune response (25), it was only in 2009 that Trocoli-Torrecilhas et al. showed that inoculation of Balb/c mice, a classical Chagas disease susceptibility model, with EVs from T. cruzi strain Y 7 days before infection led to early mortality and severe heart damage (26). T. cruzi EVs carry proteins related to metabolism, signaling, survival, and parasite virulence (15, 27). Moreover, small RNA and proteins involved in the small RNA pathway were detected inside EVs (28). Compared with the parasite intercellular compartment, this small RNA has a distinctive profile (29) and varies between parasite stages used to obtain EVs (30). Indeed, T. cruzi EVs promote gene expression changes in host cells, mainly modifying host cell cytoskeleton, extracellular matrix, and immune responses pathways, which are essential events in the T. cruzi-host cell interplay (31). Moreover, interaction between T. cruzi EVs and lineage cells in vitro suggests that the release of EVs from T. cruzi-infected host cells is important for the modulation of host responses against the parasite (32, 33). These phenomena are strain specific, meaning that EVs shed by host cells infected by one strain do not have effects on the host response to another strain (34).
In this work, we aimed to investigate the role of EVs from T. cruzi strain Y in the innate immune compartment, peritoneal macrophages, and spleen cells from C57BL/6 mice during T. cruzi acute infection through the quantification of nitric oxide (NO), cytokines, and macrophage immunophenotyping. In addition, we developed in vitro experiments using macrophages to assess the influence of EVs released by T. cruzi in the interaction between T. cruzi and host cells. Understanding the mechanisms by which EVs from the parasite act on host cells could eventually lead to the design of new strategies to control Chagas disease.
Materials and Methods
Animals
C57BL/6 mice (male, aged 6–8 weeks) were obtained from the Instituto Carlos Chagas, Curitiba/Fiocruz-PR, Brazil, and maintained under standard conditions in the animal house of the Departamento de Ciências Patológicas, Centro de Ciências Biológicas, Universidade Estadual de Londrina. A commercial rodent diet (Nuvilab-CR1, Quimtia-Nuvital, Colombo, Brazil) and sterilized water were available ad libitum.
This study was carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the Brazilian National Council of Animal Experimentation. The protocol was approved by the Committee on the Ethics of Animal Experiments at Londrina State University (CEEA, process number 11/2015- 7045.2015.06). The use of Swiss mice for the maintenance of the parasite strain was also approved for the Committee on the Ethics of Animal Experiments at Londrina State University (CEEA, process number 28.841.2016.41).
Parasite and Cells
Trypanosoma cruzi strain Y, belonging to the T. cruzi I (TcI) lineages (35), was maintained by weekly intraperitoneal inoculations of blood trypomastigote forms into Swiss mice. Vero E6 cells (ATCC#1008) were maintained in Roswell Park Memorial Institute 1640 (RPMI-1640) medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 10 mM HEPES, 100 UI/mL penicillin, and 100 µg/mL streptomycin (all reagents from Gibco, Grand Island, NY, USA) at 37°C and under a 5% CO2 atmosphere.
Additionally, blood trypomastigotes obtained from infected Swiss mice 10 days post-infection (dpi) were used for in vitro infection of Vero E6 cell monolayers. Trypomastigote forms derived from the cell-culture supernatant were used for parasite expansion in Vero E6 and infection of bone marrow-derived macrophages (BMDMs) and mice. Trypomastigote cultures were also used for EV purification and for T. cruzi-antigen (TcAg) preparation.
T. cruzi EVs (EV Y)
Extracellular vesicles purified by tissue culture-derived trypomastigotes from T. cruzi strain Y were obtained as briefly described. Trypomastigotes collected from the supernatants of infected Vero E6 cells were washed three times in RPMI medium (2,600 × g, 10 min) and were incubated for 2 h at 37°C in RPMI (1 × 108/mL) without FBS for spontaneous EV release. Trypomastigotes were removed by centrifugation (2,600 × g 10 min), and EV-containing supernatants were filtered through a 0.45-µm sterile membrane. EV purification was performed with total exosome isolation from cell-culture media reagent (Invitrogen, Carlsbad, CA, EUA) according to the manufacturer’s instructions. The EV Y pellet was suspended in phosphate-buffered saline (PBS).
The size, distribution, and concentration of EV Y were measured in a NanoSight LM10 instrument (Malvern Instruments Ltd, Malvern, UK). The data obtained were analyzed using Nanoparticle Tracking Analysis (NTA) software (version 3.1) with default settings, according to the manufacturer’s protocol. EV preparation was sampled twice to generate two independent dilutions and reduce the number of particles in the field of view below 200 detected tracks per image. From each sample, five videos (30 s each) were analyzed. All samples were measured using the same detection threshold (Figure S1 in Supplementary Material) (36).
Experimental Design of In Vivo Experiments
C57BL/6 male mice were injected intraperitoneally with EV Y obtained from 106 T. cruzi trypomastigotes. After 7 days, the animals were infected intraperitoneally with 5 × 103 trypomastigote forms. Mice that were inoculated with EV Y or PBS but not infected and non-EV Y-treated mice infected with T. cruzi were used as controls. Parasitemia was monitored by counting the number of trypomastigotes in 5 µL of fresh blood collected from the tail vein as previously described (37). At day 12 post-infection (19 days after PBS or EV Y inoculation), the animals were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) and sacrificed by cervical dislocation. The blood, heart, and spleen were collected. The blood was centrifuged at 1,300 × g for 10 min at room temperature, and the plasma was aliquoted and frozen for nitrite and cytokine measurement. The heart was fixed in 10% buffered formalin for histological analysis. Spleen cells were cultured with medium or total antigen of T. cruzi (TcAg) as previously described (38), and the supernatants were stored for the quantification of nitrite, cytokines, and prostaglandin E2 (PGE2) (Cayman, Ann Arbor, MI, USA). The 12th day after infection was chosen during this phase, NO production is elevated, the cells of the acquired immune response are already activated, and the host response begins to control parasitemia (39–41).
Histological Analysis of the Heart
Heart halves were fixed in buffered formalin, dehydrated, and embedded in paraffin by a routine technique. Then, 5-µm-thick sections were stained with hematoxylin-eosin and analyzed by light microscopy. Cardiac parasitism was evaluated by counting the number of amastigote nests visualized at 1,000× magnification in six sections of the heart per animal, as previously described (42). The results are expressed as the mean of the number of amastigote nests per section. The data for each group are the mean ± SEM of five animals per group in two independents experiments.
Peritoneal Lavage and Macrophages
To verify the in vivo effects of EV Y at the site of inoculation, we extracted peritoneal lavage and macrophages from mice in the four groups described below. Uninfected mouse samples were collected 7 days after PBS or EV Y (from 106 T. cruzi trypomastigotes) inoculation. Infected groups were inoculated with PBS or EV Y, and after 7 days, they were infected with 5 × 103 T. cruzi. The peritoneal lavage and macrophages were collected at 7 dpi. After anesthesia and cervical dislocation, 2 mL of cold PBS was injected into the mouse peritoneum, the peritoneum was massaged, and the fluid was collected. Peritoneal lavage fluid was centrifuged and frozen for nitrite quantification. Macrophage immunophenotyping was performed by flow cytometry.
Peritoneal Macrophage Immunophenotyping
Cells were stained with a panel of immunophenotyping antibodies for 30 min at 4°C (anti-CD11b-FITC, anti-CD45-PE, anti-CD86-PECy5, anti-MHC-I-PE, anti-MHC-II-PECy5, and anti-F4/80-PE, all antibodies from BioLegend, Inc. CA, USA). Data were collected using an Accuri C5 flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and analyzed by FlowJo software (Tree Star, Ashland, OR, USA). Compensation and isotype controls were also included. Gating strategies in SSC-A/SSC-H were used to exclude the doublets, and gating in forward and side scattering (FSC/SSC) allows the separation of macrophages from other small cells. Only CD11b+ cells (macrophages) were analyzed for the frequencies of other surface markers.
Nitrite Quantification
The concentration of nitrite in spleen and macrophage cell-culture supernatants was directly determined by the Griess assay (43), which estimates total NO concentrations via nitrite analysis. Plasma and peritoneal lavage nitrite levels were evaluated by the cadmium Griess assay, as previously described (44).
Cytokine Measurement by Flow Cytometry
Cytokine concentrations in plasma and spleen-cell culture supernatants were quantified by a Cytometric Bead Array (CBA)–Mouse Inflammation Kit, according to the manufacturer’s instructions (BD Biosciences, Franklin Lakes, NJ, USA). TNF-α, IL-6, IL-12p70, IFN-γ, MCP-1, and IL-10 flow cytometry measurements were performed on an Accuri C5 flow cytometer, and a total of 1,800 events were acquired for each sample. FCAP Array software was used for data analysis.
Culture of Bone Marrow-Derived Macrophages
The differentiation of BMDMs from C57BL/6 mice was performed as previously described (45) using L929-cell-conditioned medium as a source of granulocyte/macrophage colony-stimulating factor (46). Fresh bone marrow cells were collected from two femurs and tibias in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Grand Island, NY, USA) and depleted of erythrocytes with ammonium chloride. Cells were seeded in six-well tissue culture plates (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 2–3 × 105 cells/mL and incubated at 37°C under a 5% CO2 atmosphere in DMEM supplemented with 25% FBS and 30% L929 cell-conditioned medium. Four days after the cells were seeded, 3 mL of fresh supplemented DMEM was added per well, and the cells were incubated for an additional 3 days. To harvest BMDMs, we discarded the supernatants and detached adherent cells. The cells were resuspended in supplemented DMEM that was used in all in vitro assays (10% FBS, 2 mM l-glutamine, 10 mM HEPES, 100 UI/mL penicillin, and 100 µg/mL streptomycin). The BMDM concentration was determined, and the cells were seeded in tissue culture plates for 4–12 h to ensure complete adherence.
Experimental Design With Bone Marrow-Derived Macrophages
BMDM were seeded at a density of 2 × 105 cells/well in 24-well plates with circular coverslips for a trypomastigote internalization assay and lipid body assay (47); in 48-well plates for a trypomastigote release assay; and in 96-wells plates for NO, PGE2, and cytokine quantification in the supernatants. After cells were adhered to the plates, they were incubated for 24 h at 37°C under a 5% CO2 atmosphere with EV Y obtained from 107 T. cruzi trypomastigotes for parasite internalization, lipid body, and release assays. Next, the cultures were washed with PBS at 37°C to remove available EV Y, and trypomastigote forms from the T. cruzi Y strain were added to allow parasite–macrophage interaction as follows: 1 × 106 parasites/well during 14 h of infection for the internalization and supernatant assays, 1 × 106 parasites/well for 24 h for the lipid body assay, and 2 × 104 parasites/well during 14 h of infection for the release assay. Non-internalized trypomastigotes were removed by three washes with PBS at 37°C. For the NO assay in supernatants, lipopolysaccharide (LPS from Escherichia coli 026:B6) was used as a positive control (Sigma, St. Louis, Missouri, EUA).
To investigate T. cruzi internalization, after 14 h of infection, we fixed macrophages with 100% methanol and stained them with Giemsa (Merck, Kenilworth, NJ, EUA). The coverslips were then removed from the plate and mounted on glass slides with Permount mounting medium (Fischer Chemical, Pittsburgh, PA, USA). Quantification of the number of amastigotes per macrophage was carried out using light microscopy where a total of 1,500–2,500 cells were randomly counted in each group. The internalization index was calculated by multiplying the percentage of infected cells by the mean number of parasites per infected cell (47, 48). The internalization of the control group, without EV Y, was considered 100%, and all internalization indices were normalized.
To determine the number of macrophage-released parasites, we collected the supernatants from 48-well plates daily and individually and subjected them to centrifugation (2,600 × g, 5 min, RT). Trypomastigotes were counted in a Neubauer chamber. The results are expressed as total number of T. cruzi by sample (41, 49).
Osmium Tetroxide Staining for Lipid Bodies
To evaluate if EV Y induces the formation of lipid bodies in macrophages, we stained infected and uninfected cells with osmium tetroxide as previously described (50), with some modifications. Macrophages were fixed with formaldehyde solution (formalin) 3.7%. Cells were then washed with distilled water and stained with osmium tetroxide 1% in 0.1 M cacodylate buffer solution for 30 min. Then, the cells were washed three times with distilled water and incubated with thiocarbohydrazide 0.25% for 3 min, a step that promotes reduction of osmium and highly enhances lipid labeling. The thiocarbohydrazide was removed, and osmium tetroxide 1% in 0.1 M cacodylate buffer was reintroduced for 5 min. Finally, the cells were washed three times with distilled water and mounted on glass slides with Permount mounting medium. The cells were analyzed by light microscopy, and the number of lipid bodies per macrophage was counted in at least 1,000 macrophages in each group. In this staining process, osmium tetroxide binds to unsaturated lipids and is reduced to elemental osmium, an easily visible and permanent black marker.
PGE2 Quantification
Prostaglandin E2 quantification in spleen and macrophage cell-culture supernatants was performed by a Prostaglandin E2 Express ELISA Kit from Cayman Chemical Co. (Ann Arbor, MI, USA). The test was performed according the instructions of the manufacturer, and the samples from infected mice or infected cells were diluted before analysis. The sensitivity of this assay was 15.6–2,000 pg/mL.
Statistical Analysis
For column analysis, we used Student’s t-test to compare two groups and one-way analysis of variance (ANOVA) with Tukey’s post-test for more than three groups. To analyze grouped data, we used two-way ANOVA with Tukey’s or Sidak’s post-test. The values are presented as the mean ± SEM. The results were considered significant when P < 0.05. For statistical analysis, we used the program Prism (version 6.0, GraphPad Software Inc., San Diego, CA, USA).
Results
NTA of EVs Isolated From T. cruzi Strain Y
Extracellular vesicles spontaneously shed by tissue culture-derived trypomastigote forms from strain Y were analyzed by NTA and exhibited a profile compatible with that of previously described EVs of T. cruzi (15, 27, 51). In a representative analysis, EV Y had a mean diameter size of 136.33 nm with a SD of 86.3 nm and a mode of 94 (Figure S1 in Supplementary Material). Considering the range of EV Y sizes obtained, these suspensions are likely a mix of exosomes and plasma membrane-derived EVs. In this work, we will refer to those heterogeneous samples as EVs.
T. cruzi EVs Increase Parasitemia and Cardiac Parasitism in C57BL/6 Mice
Inoculation with PBS or EV Y prior to infection (Figure 1A) did not alter the parasitic load in blood in the beginning of the infection, specifically on 3 and 5 dpi. However, at 7 dpi, EV Y-inoculated mice showed 2.5 times more parasites in blood than did control PBS mice (Figure 1B, P < 0.0001). From 9 dpi, parasitemia constantly decreased, and by 17 dpi, the parasites in the blood appeared to be controlled in both PBS and EV Y-inoculated mice. Cardiac parasitism was also evaluated in PBS or EV Y-inoculated mice at 12 dpi. There were as many as two times more amastigote nests in the hearts of mice that had received EV Y prior to infection than in those of mice that had received only PBS prior to infection (Figure 1C, P < 0.05).
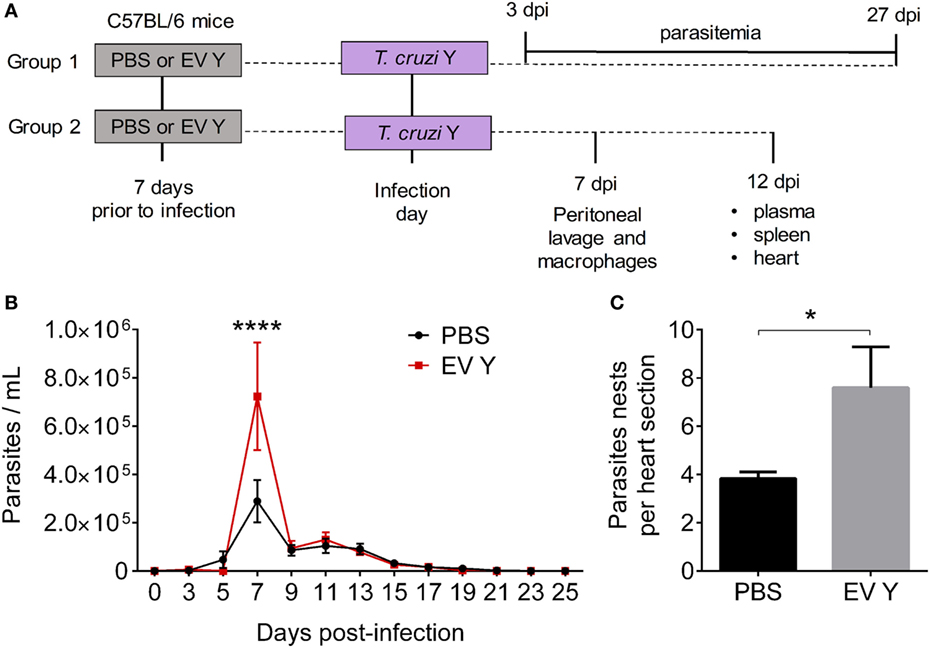
Figure 1. Previous inoculation of Trypanosoma cruzi extracellular vesicles (EVs) increases parasitemia and cardiac parasitism in C57BL/6 mice. C57BL/6 mice were inoculated with EVs derived from T. cruzi Y (EV Y) (i.p.). One week later, mice were infected with T. cruzi by the same route. (A) Experimental design for in vivo assays [days post-infection (dpi)]. (B) Parasitemia was quantified on alternating days as trypomastigotes per milliliter of blood. (C) Cardiac tissues were examined by hematoxylin and eosin staining on day 12 after T. cruzi infection. Parasitism was evaluated by counting the number of amastigote nests visualized at 1,000× magnification in six sections of the heart per animal. The results were expressed as the mean of the number of amastigote nests per section. The results are expressed as the mean ± standard error from of five mice per group in an experiment representative of two similar experiments. *P < 0.05; ****P < 0.0001 significantly different. Two-way ANOVA with (B) Sidak’s post-test and (C) Student’s t-test– GraphPad Prism.
Inoculation of T. cruzi EVs Prior to Infection Decreases the Levels of Plasma NO and TNF-α and Production of NO and Cytokines by Spleen Cells From Infected Mice
Compared with control PBS inoculation in uninfected mice, EV Y inoculation in uninfected mice did not change NO in plasma (Figure 2A). As expected, compared with PBS in the absence of infection or EV Y inoculation, infection triggered a two-fold increase in circulating NO at 12 dpi (Figure 2A, P < 0.01). Although inoculation with EV Y had no effects on uninfected mice, inoculation with EV Y prior to infection downregulated the secretion of NO in mice infected at 12 dpi to half the NO levels in control mice (Figure 2A, P < 0.05).
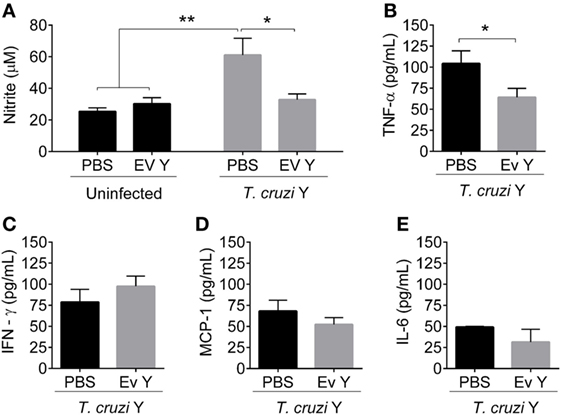
Figure 2. Inoculation of Trypanosoma cruzi extracellular vesicles (EVs) prior to infection decreases the plasmatic levels of nitric oxide and TNF-α in infected mice. C57BL/6 mice were inoculated with EVs derived from T. cruzi Y (EV Y) or phosphate-buffered saline (PBS, i.p.). One week later, the mice were infected with T. cruzi by the same route. Uninfected and PBS-inoculated mice were used as controls. Plasma was collected on day 12 after T. cruzi infection. (A) Nitrite in infected and uninfected mice was quantified by the cadmium Griess assay. Cytokines (B) TNF-α, (C) IFN-γ, (D) MCP-1, and (E) IL-6 were quantified by the Cytometric Bead Array. The results are expressed as the mean ± standard error from 5 to 9 mice per group in an experiment representative of two similar experiments. *P < 0.05; **P < 0.01 significantly different. Two-way ANOVA with (A) Sidak’s post-test and (B–E) Student’s t-test—GraphPad Prism.
Cytokine plasma levels (TNF-α, IFN-γ, MCP-1, IL-12p70, IL-10, and IL-6) in uninfected mice inoculated with PBS or EV Y were lower than the limit of detection of the assay. In infected mice, the levels of IL-12p70 and IL-10 were also below the detection threshold. However, despite the low levels of cytokines in the plasma of uninfected mice, T. cruzi infection resulted in the induction of high levels of cytokines TNF-α, IFN-γ, MCP-1, and IL-6 in plasma (Figures 2B–E). In addition, previous inoculation with EV Y in mice reduced the TNF-α level in plasma by ~50% compared with the level in PBS-inoculated infected mice (Figure 2B, P < 0.05). However, EV Y inoculation prior to infection did not alter the levels of IFN-γ, MCP-1, or IL-6 in the plasma of infected mice at 12 dpi compared with those in the plasma of PBS-inoculated infected mice (Figures 2C–E).
To assess the responsiveness of the immune cells from mice in the different experimental groups, we harvested spleen cells 12 days after T. cruzi inoculation (19 days after EV Y or PBS inoculation). Spleen cells from uninfected mice subjected to PBS or EV Y inoculation produced low and similar levels of NO when stimulated with TcAg (Figure 3A). Uninfected mouse spleen cells stimulated with TcAg resulted in increased levels of cytokines TNF-α, IFN-γ, MCP-1, IL-10, and IL-6 (Figures 3B–F). Spleen cells from uninfected mice inoculated with EV Y that were stimulated in vitro with TcAg showed at least two times lower levels of TNF-α, IFN-γ, IL-10, and IL-6 than did spleen cells from uninfected PBS-inoculated mice that were stimulated in vitro with TcAg (Figures 3B,C,E,F; P < 0.001, P < 0.01, P < 0.05, and P < 0.01, respectively).
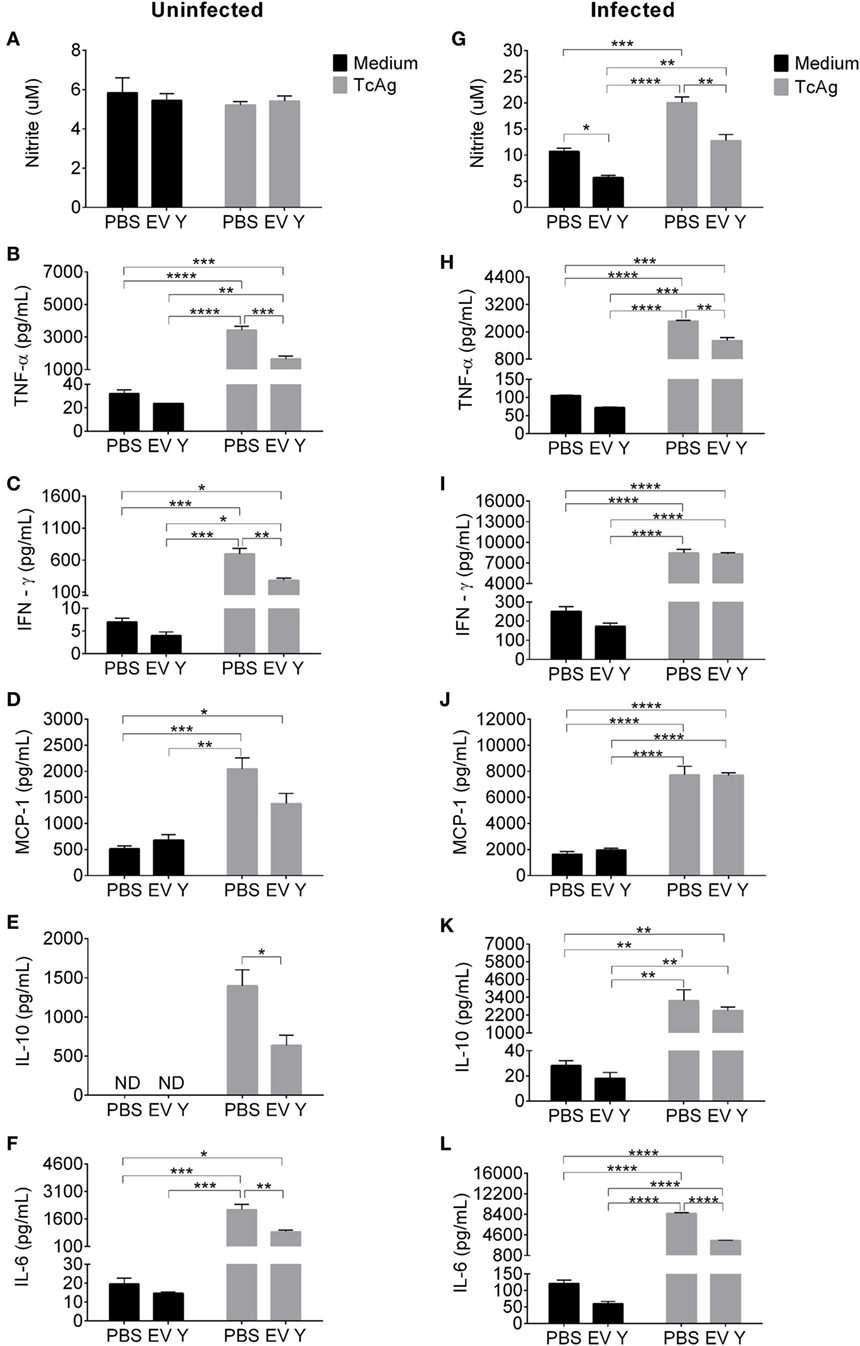
Figure 3. Inoculation of Trypanosoma cruzi extracellular vesicles (EVs) prior to infection decreases production of nitric oxide and cytokines by spleen cells from infected mice. C57BL/6 mice were inoculated with EVs derived from T. cruzi Y (EV Y) (i.p.). One week later, the mice were infected with T. cruzi (Y strain) by the same route. Infected, uninfected, and PBS-inoculated mice were used as controls. Spleens were harvested 12 days after T. cruzi infection (19 days after EV Y or PBS inoculation). The cells were cultured with medium or T. cruzi antigen (TcAg) for 48 h. (A,G) Nitrite in the supernatants of cultured spleen cells from uninfected mice (left column) and cultured spleen cells from infected mice (right column) were quantified by the Griess assay. Cytokines (B,H) TNF-α, (C,I) IFN-γ, (D,J) MCP-1, (E,K) IL-10, and (F,L) IL-6 in the supernatants of cultured spleen cells from uninfected and infected mice, respectively, were quantified by the Cytometric Bead Array. The results are expressed as the mean ± standard error from four mice per group in an experiment representative of two similar experiments. *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001 significantly different (two-way ANOVA with Tukey’s post-test)—GraphPad Prism.
As expected, compared with spleen cells from uninfected mice, spleen cells from T. cruzi-infected mice spontaneously produced more NO and cytokines when cultured with medium and even more when stimulated in vitro with TcAg (Figure 3). The spontaneous and TcAg-stimulated NO production by spleen cells isolated from infected mice inoculated with EV Y before infection was significantly lower than that by spleen cells from PBS-inoculated mice (Figure 3G, P < 0.05 and P < 0.01). In the same way, compared with spleen cells from PBS-inoculated mice that were stimulated in vitro with TcAg, spleen cells isolated from infected mice inoculated with EV Y before infection produced approximately half the levels of pro-inflammatory cytokines TNF-α and IL-6 when stimulated in vitro with TcAg (Figures 3H,L, P < 0.01 and P < 0.0001, respectively). However, EV Y inoculation in vivo was not always associated with a decrease in ex vivo production of cytokines. Even in the TcAg-stimulated spleen cells, inoculation with EV Y prior to infection did not alter the production of the pro-inflammatory cytokines IFN-γ (Figure 3I) and MCP-1 (Figures 3D,J). Notably, EV Y inoculation did not alter the production of the regulatory cytokine IL-10 (Figure 3K).
Previous Inoculation of T. cruzi Extracellular Vesicles Decreases Production of Nitrite in the Peritoneal Lavage and Alters the Expression of Surface Activation Molecules in Macrophages Harvested From T. cruzi-Infected Mouse Peritoneum
The inoculation of PBS or EV Y in vivo and infection with T. cruzi were always performed by the intraperitoneal route. Hence, we decided to analyze the peritoneal environment through the quantification of NO in the peritoneal lavage and through phenotypical analyses of the macrophages present in the peritoneum of uninfected mice at 7 days after PBS or EV Y inoculation and in that of infected mice at 7 dpi (14 days after EV Y or PBS inoculation). In the uninfected groups, the NO levels were the same between PBS- or EV Y-inoculated mice (Figure 4). Compared with uninfected mice, PBS-inoculated mice showed a two-fold increase in NO production in the peritoneal lavage after T. cruzi infection. These increased levels of NO were not observed in mice that had received EV Y prior to infection (Figure 4).
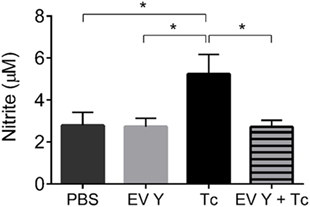
Figure 4. Previous inoculation of Trypanosoma cruzi extracellular vesicles (EVs) decreases production of nitric oxide in the peritoneal lavage of T. cruzi-infected mice. C57BL/6 mice were inoculated with EVs derived from T. cruzi Y (EV Y) (i.p.). One week later, the mice were infected with T. cruzi (Tc) by the same route or were inoculated with EV Y alone. Uninfected and PBS- or EV Y-inoculated mice were used as controls. On day 7 after T. cruzi infection and on day 7 after EV Y inoculation in uninfected animals, the peritoneum was washed, and nitrite was quantified in these fluids by the cadmium Griess assay. The results are expressed as the mean ± standard error from 4 to 9 mice per group in an experiment representative of two similar experiments. *P < 0.05 significantly different (one-way ANOVA with Tukey’s post-test)—GraphPad Prism.
The flow cytometry analysis of peritoneal macrophages is represented in Figure 5A. For this analysis, doublets were excluded, and peritoneal macrophages were identified according to size and complexity. From this gate, only CD11b+ cells were analyzed. Although most cells in the selected gate showed surface expression of the markers CD11b+ and CD45+, confirming the selection of the macrophage population (Figure 5B), the median fluorescence intensity (MIF) of CD11b after infection decreased by 50% in the PBS- and EV-inoculated groups (Figure 5C, P < 0.01). The percentage of cells expressing the co-stimulatory receptor CD86 was not significantly different between the groups (Figure 5D), but the cells from PBS-inoculated and infected mice showed more CD86 in macrophages than did cells from uninfected mice. Surprisingly, compared with the macrophages from uninfected groups, the macrophages from the group that had received EV Y before infection did not show increased CD86 (Figure 5E).
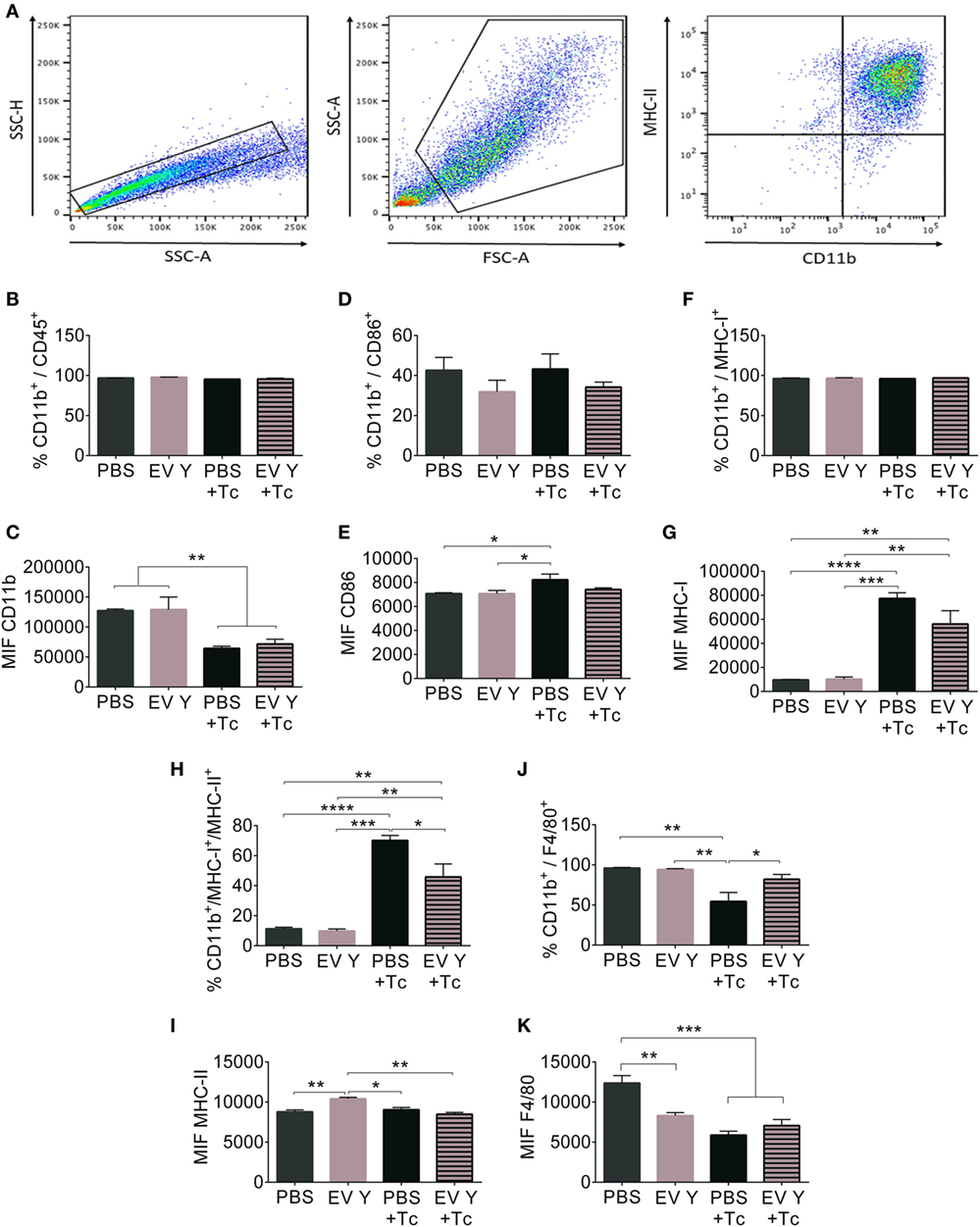
Figure 5. Previous inoculation of Trypanosoma cruzi extracellular vesicles (EVs) alters the expression and frequency of surface activation molecules in macrophages harvested from infected-mouse peritoneum. C57BL/6 mice were inoculated with extracellular vesicles derived from T. cruzi Y (EV Y) (i.p.). One week later, the mice were infected with T. cruzi by the same route or were inoculated with EV Y only. Infected, uninfected, and PBS-inoculated mice were used as controls. On day 7 after T. cruzi infection and on day 7 after EV Y inoculation in uninfected animals, peritoneum cells were harvested and analyzed by flow cytometry. (A) Gate strategy definition, (B,C) and the cells positive for the molecule CD11b were immunophenotyped for the frequency (%) and expression levels (median intensity of fluorescence (MIF)) of surface molecules (D,E) CD86, (F,G) MHC-I, (H,I) MHC-I/MHC-II, and (J,K) F4/80. The results are expressed as the mean ± standard error from 4 to 7 mice per group in an experiment representative of two similar experiments. *P < 0.05; **P < 0.01 significantly different (one-way ANOVA with Tukey’s post-test)—Graph Pad Prism.
As expected, all cells exhibited the major histocompatibility complex class I (MHC-I) protein (Figure 5F), and infection increased MHC-I in macrophages by approximately sevenfold (Figure 5G, P < 0.0001).
The percentage of macrophages positive for MHC-II molecules was similar between the uninfected groups (PBS- and EV Y-inoculated). The infection triggered an increase in macrophages presenting MHC-II in mice inoculated with PBS before infection (P < 0.0001). Despite the increase in MHC-II in macrophages from EV Y-inoculated infected mice, this increase was 34% lower than that observed in macrophages from the PBS-inoculated and infected mice (Figure 5H, P < 0.05). When the levels of MHC-II were analyzed by MIF, macrophages from mice inoculated with EV Y and from mice that were uninfected showed higher MHC-II expression than did all the other mice, even infected mice (Figure 5I).
Concerning the analysis of F4/80 protein, the percentage of macrophages that tested positive for this protein was reduced by 50% in the PBS-inoculated and infected group. The infection did not induce a reduction in F4-80 expression in mice that had received EV Y before the infection (Figure 5J, P < 0.05). The uninfected group showed higher F4/80 expression levels than did the following three groups: EV Y-inoculated and uninfected, PBS-inoculated and infected, and EV Y-inoculated and infected groups (Figure 5K, P < 0.001).
Macrophage Stimulation With T. cruzi EV Y Increases T. cruzi Infection and Trypomastigote Release From Infected Cells but Decreases Nitrite Production
In the in vitro assays, BMDMs were incubated with EV Y for 24 h, and after removal of EV Y, the cells were infected with trypomastigotes (Figure 6A). This previous contact of the macrophages with EV Y promoted a significant increase (82%) in the number of internalized amastigotes, as assessed by the internalization index (Figure 6B, P < 0.01). The mean number of amastigotes in 250 macrophages showed that macrophages previously stimulated with EV Y were more infected and presented a higher number of internalized amastigotes (Figures 6C,D).
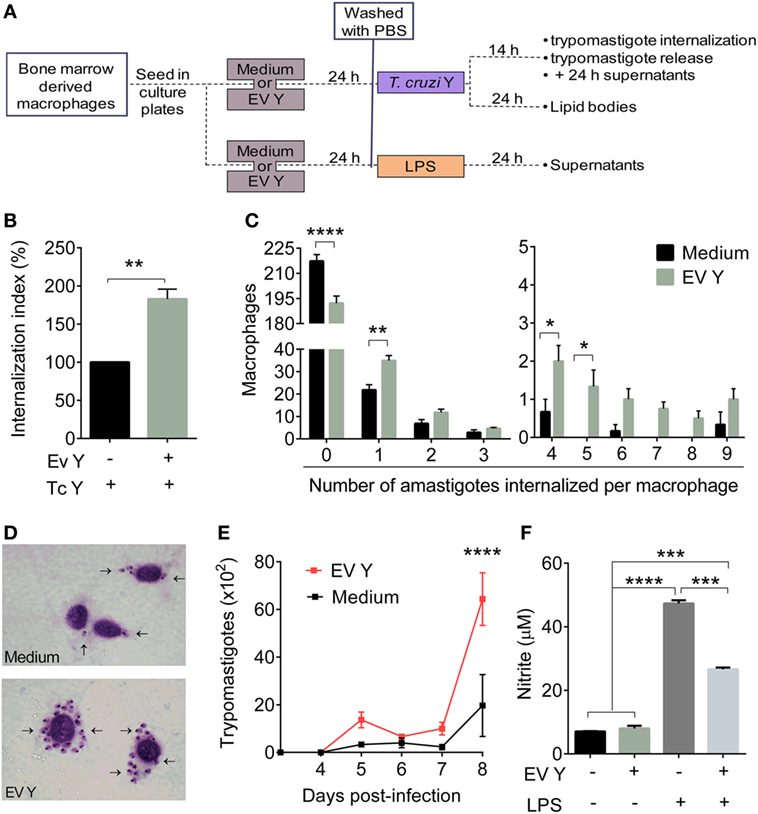
Figure 6. Macrophage in vitro stimulation with Trypanosoma cruzi extracellular vesicles (EVs) increases T. cruzi infection and trypomastigote release from infected cells but decreases nitric oxide release. Bone marrow-derived macrophages were incubated for 24 h with EVs derived from T. cruzi Y (EV Y) (i.p.). The cells were washed and infected with T. cruzi trypomastigote forms (0–1 parasites per macrophage for the trypomastigote release assay and 5 parasites per macrophage in the remaining assays) for 14 h or stimulated with 0.1 µg/mL of LPS for 24 h. After the clearance of non-internalized parasites, the cells were stained with Giemsa and analyzed by light microscopy. (A) Experimental design for macrophage in vitro assays (B) The internalization index was calculated considering the number of amastigotes internalized per macrophage. The internalization frequency of the control group, the absence of EV Y (black bar), was considered 100%. The gray bar represents macrophages stimulated with EV Y before T. cruzi infection. (C) The number of internalized amastigotes by macrophage in 250 counted cells. (D) Representative pictures of macrophages stained with Giemsa at 1000× magnification of infected cells in both groups (control and EV Y). Arrows indicate the internalized amastigotes in macrophages. (E) Daily number of trypomastigotes free in the supernatants. (F) Nitrites in the supernatants of macrophages cultured with LPS were quantified by the Griess assay. The results are expressed as the mean ± standard error from each group in triplicate analysis in an experiment representative of two similar experiments. *P < 0.05; **P < 0.01; ***P < 0.001; **** P < 0.0001 significantly different. (A) Student’s t-test, (B,D) two-way ANOVA with Sidak’s post-test and (E) one-way ANOVA with Tukey’s post-test—GraphPad Prism.
To verify the infection efficiency rate, we measured the release of trypomastigotes daily in the supernatant of infected macrophage cultures. The macrophages that had contact with EV Y before infection released three times more trypomastigotes than did the macrophages cultured only with medium, especially on the eighth day after the infection (Figure 6E, P < 0.0001).
To evaluate the modulation of NO production by EV Y in vitro, as BMDM were not activated by in vivo events such as chemotaxis gradients or cytokines, we stimulated BMDMs with LPS. LPS is a classical activator of macrophages by TLR4 stimulation; in response to LPS, macrophages produce NO, among other things (positive control) (45). Macrophages were incubated with EV Y for 24 h, and after removal of EV Y, cells were stimulated with LPS for another 24 h. Incubation with EV Y alone did not induce NO production by macrophages. By contrast, LPS induced high levels of NO release by macrophages (P < 0.0001). However, compared with macrophages stimulated with LPS alone, macrophages incubated with EV Y and then stimulated with LPS produced half of the level of NO (Figure 6F, P < 0.001).
EV Y Stimulation Induces Macrophage Lipid Body Formation and PGE2 Production and Reduces Pro-Inflammatory Cytokine Production
To verify the influence of EV Y in lipid body formation in macrophages, we incubated BMDMs with EV Y for 24 h, and after removal of EV Y, the cells were infected with trypomastigotes for another 24 h. Non-stimulated or EV Y-stimulated cells were analyzed. Osmium tetroxide staining was used to detect lipid bodies, which resemble black marks in the cytoplasm (Figure 7B). The infection increased the formation of lipid bodies in macrophages by fourfold, and previous incubation with EV Y did not alter these lipid body levels. Interestingly, incubation of macrophages with EV Y alone was sufficient to induce lipid bodies in macrophages (Figure 7A, P < 0.01).
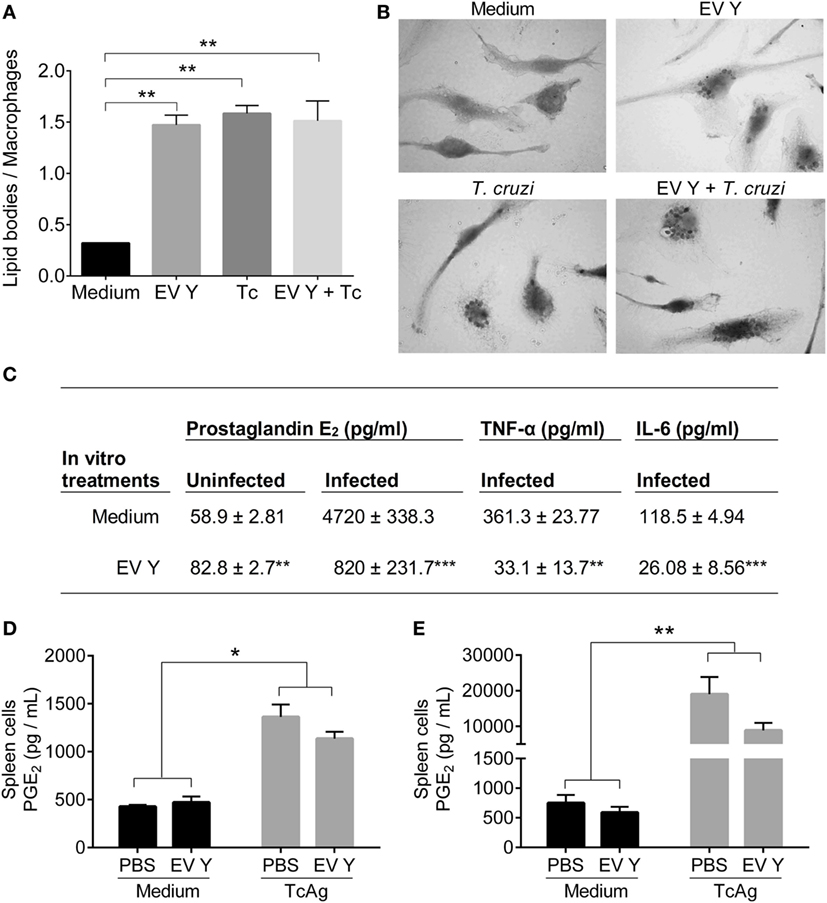
Figure 7. Trypanosoma cruzi extracellular vesicles (EVs) induce lipid body formation and prostaglandin E2 (PGE2) production by macrophages (in vitro) but not by spleen cells (ex vivo). Bone marrow-derived macrophages were incubated for 24 h with EVs derived from T. cruzi Y (EV Y) (i.p.). The cells were washed and infected with trypomastigote forms of T. cruzi (5 parasites per macrophage). After 24 h, non-internalized parasites were removed. (A) The cells were stained with osmium tetroxide and analyzed by light microscopy. The results are expressed as the mean ± standard error from each group in 250 cells counted in triplicate analysis in an experiment representative of two similar experiments. (B) Representative pictures of macrophage staining with osmium tetroxide at 1,000× magnification of uninfected cells (without and with EV Y-upper panel) and infected cells in both groups (control and EV Y- bottom panel). The lipid bodies are visible as black markers inside the macrophages. (C) PGE2 was quantified by ELISA, and cytokines (TNF-α and IL-6) were quantified by CBA in the supernatant culture of macrophages (uninfected and infected cells) after 24 h of infection. The results are expressed from two experiments. C57BL/6 mice were inoculated with EV Y (i.p.) and infected with T. cruzi by the same route 1 week later. Infected, uninfected, and PBS-inoculated mice were used as controls. Spleens were harvested 12 days after T. cruzi infection. The cells were cultured with medium or T. cruzi antigen (TcAg) for 8 h. (D,E) PGE2 was quantified by ELISA in the supernatants of cultured spleen cells from uninfected and infected mice. The results are expressed as the mean ± standard error from four mice per group in one experiment. *P < 0.05; **P < 0.01 significantly different (one-way ANOVA with Tukey’s post-test)—GraphPad Prism.
Additionally, we assessed PGE2 production by macrophages exposed to EV Y. In addition to lipid body formation, EV Y-stimulated macrophages showed higher PGE2 production than did non-stimulated macrophages (Figure 7C, P < 0.01). Previous contact with EV Y markedly reduced PGE2 production in uninfected cells compared with that in infected cells cultured with medium (P < 0.001). EV Y also decreased TNF-α (P < 0.01) and IL-6 (P < 0.001) production in infected macrophages (Figure 7C).
To assess the in vivo effect of EV Y on PGE2 production, we harvested spleen cells from mice in the different experimental groups 12 days after T. cruzi inoculation (19 days after EV Y or PBS inoculation) and cultured them in vitro with medium or stimulated them with TcAg for 8 h. Spleen cells from uninfected and infected mice stimulated with TcAg produced more PGE2 than did non-stimulated cells (Figures 7D,E, P < 0.05). In contrast to macrophages exposed to EV Y in vitro, macrophages and spleen cells inoculated with EV Y in vivo did not exhibit altered PGE2 production in these animals at this time point in either uninfected or infected mice (Figures 7D,E).
Discussion
The pathogenesis of Chagas disease is complex and involves many interactive pathways between hosts and parasites (52). In this context, only in the last decade has the role of EVs released by T. cruzi been recognized; however, the mechanisms by which EV Y exert their pathological effects are incompletely understood (53). Elucidating the underlying mechanism of the pathogenic effects of EVs shed by T. cruzi is pertinent to better understand the parasite–host relationship.
In 2009, a study reported that inoculation with EVs shed by T. cruzi prior to infection in susceptible BALB/c mice increased cardiac parasitism and accelerated mortality rates, without a significant increase in blood parasitemia (26). Herein, we explored the role of EV Y in experimental acute murine infection using resistant C57BL/6 mice and the Y strain of T. cruzi. Animal inoculation with EV Y before infection with trypomastigote forms led to a transient but substantial increase in the blood parasitic load at the beginning of the infection (7 dpi). A few days later, at 12 dpi, when the blood parasitic load was similar between the control and the EV Y-inoculated groups, amastigote parasitism in cardiac tissue was higher in mice that had received EV Y prior to infection. However, inoculation with EV Y before infection did not lead to mortality among the animals.
The role of EV Y in intercellular communication between host cells and T. cruzi has been considered in recent years (54). Bayer-Santos et al. showed that EVs released by T. cruzi are endocytosed by cells and reach the cytoplasm where they can eventually release their content (15). In another study, Neves et al. demonstrated that EVs shed by T. cruzi Y strain act in macrophages by increasing the adherence of the parasite, while EVs released by T. cruzi CL-Brener strain enhance macrophage infection by this strain through acid phosphatase activities (55). Garcia-Silva et al. showed that EV cargo promotes the life-cycle transition of epimastigotes into trypomastigotes and can be transferred between parasites and susceptible mammalian cells, increasing their infection susceptibility (28). The effects of EVs shed by parasites in infections are not exclusive to T. cruzi. In fact, in murine infections with Leishmania major and Leishmania donovani, preinoculation with EVs from these parasites resulted in increased edema and parasitism in the hind legs of mice (23). In vitro, the communication between parasites and macrophages through parasite EVs was also described in Leishmania by Silverman et al., who showed that parasite EVs induced production of IL-8 but not TNF-α by macrophages, which led to immunosuppression in the host and prepared cells for Leishmania infection (56).
During the acute phase of infection, the recognition of T. cruzi by Toll-like receptors on macrophages (57) triggers production of TNF-α and IL-12, which in turn activates other cells, such as natural killers and lymphocytes, to produce IFN-γ (58). Hence, IFN-γ activates macrophages and controls intracellular parasite growth through the production of NO by the enzyme inducible nitric oxide synthase (iNOS) and reactive oxygen molecules (6, 59, 60). NO has a trypanocidal effect (39) as it reacts with superoxide (O2−) to produce the very toxic molecule peroxynitrite (ONOO−), which leads to oxidation of thiol groups and nitrosylation of parasitic amino acids and proteins (61, 62). NO also modulates the effector immune response by regulating/enhancing inflammation during T. cruzi infection (60).
Although infection with T. cruzi triggered NO production assessed in plasma, mice that were inoculated with EV Y prior to T. cruzi challenge did not show a corresponding increase in the plasmatic levels of the trypanocidal molecule NO. In the peritoneum, the location of EV Y and T. cruzi inoculation, the levels of NO in mice that had received EV Y prior to infection were also lower than those in control mice and were similar to the levels in uninfected mice. The same phenomenon occurred in vitro when spleen cells from mice that had received EV Y in vivo before infection were stimulated with TcAg. Taken together, these data show that intraperitoneal inoculation with EV Y was able to alter the responsiveness of immune cells both locally and systemically. Inoculation of EV Y without subsequent infection did not stimulate NO production in plasma, peritoneal lavage or spleen-cell supernatants. These results suggest that EV Y stimulation had no direct trypanocidal activity in host macrophages, but it modulates these cells, creating a favorable environment for the parasite with lower NO production, which probably contributes to the higher parasitemia observed in mice treated with EV Y before infection.
The lower production of NO in infected mice induced by previous contact with EV Y partially corroborates the results of Torrecilhas et al. (26). They described the low activity of iNOS in the heart tissue of infected BALB/c mice primed with T. cruzi vesicles, but there was no significant difference in NO levels in the supernatants of spleen-cell cultures between vesicle-treated and control animals. In a chronic study developed by Nogueira et al., spleen cells from infected mice at 180 dpi were cultured in vitro with vesicles from different T. cruzi strains and responded with increased production of NO (51). However, a major difference between these studies should be mentioned and has to be taken into consideration; in the acute infection model shown here and by Torrecilhas et al., EV Y was administered in vivo before infection (26), whereas in the chronic model, the vesicles were added in vitro to the spleen cells of chronically infected animals (51).
Although inoculation was always performed in the peritoneal cavity, our study provided further evidence for the systemic effect of EV Y in infected mice; modulation of the innate immune response was demonstrated by the low plasmatic levels of TNF-α and by low production of TNF-α and IL-6 by spleen cells stimulated in vitro with TcAg. During the first weeks of infection, the pro-inflammatory cytokines TNF-α and IL-6 are required for activation of inflammatory cells and for a subsequent efficient Th1 immune response against the parasite (7, 40, 63). Low production of these cytokines induced by previous contact with EV Y probably favored the infection. Additionally, inoculation with EV Y did not change production of the regulatory cytokine IL-10 by spleen cells from infected mice stimulated in vitro with TcAg. The high concentration of IL-10 during the acute phase is associated with failed control of infection (64), and as shown here, EV Y altered the balance between the pro-inflammatory/regulatory cytokines toward an immunoregulatory profile. Consistent with these results, Torrecilhas et al. showed increased expression of IL-10 and IL-4 mRNA in heart tissue from BALB/c mice treated with T. cruzi vesicles before infection and showed low production of TNF-α by macrophages stimulated in vitro with T. cruzi vesicles (26).
EV Y inoculation without infection did not stimulate the production of NO, but spleen cells from uninfected mice inoculated with EV Y and stimulated in vitro with TcAg produced lower levels of TNF-α, IFN-γ, IL-10, and IL-6 than did spleens cells from control mice. Although these cells never had contact with the parasite, they responded to the TcAg stimulus by producing cytokines, albeit at a lower level than the infected animals. Even in these non-primed cells, EV Y exercised immunomodulatory effects by decreasing the production of cytokines after stimulus.
The phenotypic analysis of peritoneal macrophages showed a high percentage of cells positive for CD11b and CD45, confirming that the peritoneal population analyzed was composed of macrophages. CD11b belongs to the integrin β2 family and plays an important role in the migration of macrophages (65), while CD45, a protein tyrosine phosphatase expressed on all cells of hematopoietic origin, is associated with the adhesion process (66). The reduction in the amount of these superficial molecules observed in both the EV Y and control groups may be related to the internalization mechanisms of these molecules in activated macrophages (67).
Macrophages present antigens to lymphocytes through peptides associated with MHC-I (endogenous antigens) and MHC-II (exogeneous antigens) molecules (68). Additionally, co-stimulatory molecules, such as CD86 (or B-7), are necessary for efficient activation of lymphocytes by macrophages (69). EV Y inoculation before infection did not alter the percentage and expression of MHC-I in peritoneal macrophages. However, compared with PBS inoculation before infection, EV Y inoculation before infection resulted in a lower percentage of macrophages that were positive for MHC-II. Extrapolating these data, the results indicate that previous contact with EV Y might interfere with the presentation of phagocytized antigens by macrophages to lymphocytes. Reinforcing this interpretation, peritoneal macrophages from mice that received EV Y before infection showed less CD86 than did macrophages from mice that received PBS before infection. Interestingly, although EV Y inoculation without infection did not change the percentage of MHC-II positive cells, the expression or the amount of MHC-II in these cells was slightly higher than that in all the other cells, as assessed by the MIF.
The F4/80 molecule, frequently used as a marker for mouse macrophages, has been characterized as a member of the epidermal growth factor (EGF)-transmembrane 7 (TM7) family, and there is evidence for its role in the induction of immunological tolerance (70). The induction of tolerance instead of an inflammatory environment could be beneficial to the parasite. The percentage of macrophages positive for F4/80 decreased in PBS-infected mice but not in mice that had received EV Y before infection. This downregulation of F4/80 in macrophages indicates how EV Y could be modulating the immune response toward a less inflammatory environment. Surprisingly, the amount of F4/80 was lower in macrophages inoculated with EV Y without infection than in macrophages inoculated with PBS without infection.
Considering the in vivo immunomodulatory effects exerted by EV Y described here, we investigated the specific role of EV Y using in vitro assays with BMDMs. Macrophages that had contact with EV Y before infection exhibited enhanced internalization of T. cruzi trypomastigotes. In the initial steps of infection, in addition to the adherence and invasion process, T. cruzi needs to evade the trypanocidal response of macrophages to replicate and continue its life cycle (13). In addition to a higher internalization rate, EV Y enhanced the release of parasites by macrophages. Moreover, corroborating our in vivo findings, the effect of EV Y in decreasing macrophage responsivity was confirmed by the reduction in NO production in macrophage cultures activated with LPS, a classic TLR4 agonist and inducer of NO production (45).
In 2003, Melo et al. demonstrated for the first time that T. cruzi infection triggers the formation of lipid bodies in macrophages (71), and this event is directly related to PGE2 synthesis by macrophages (71, 72). They also showed that the enzyme cyclooxygenase-2 (COX-2) is co-localized with lipid bodies in infected macrophages, further strengthening these structures as substrates that are necessary for PGE2 synthesis (72). As expected, in our assays, infection with T. cruzi induced lipid body formation in macrophages. However, the contact of macrophages with EV Y without infection also induced lipid bodies in those cells but did not cause additional lipid body formation in infected macrophages. In fact, D’Avila et al. demonstrated that uninfected macrophages from the infected group also showed higher lipid body formation than did macrophages from the control group; the authors described this observation as bystander amplification (72). These results provide new insights to explain this phenomenon where EVs shed by T. cruzi induce lipid body formation in uninfected macrophages.
Additionally, EV Y alone induced production of PGE2 by macrophages. PGE2 production by activated macrophages occurs at the beginning of T. cruzi infection by the COX enzyme and plays a crucial role during the development of the infection (73–75). In macrophages, high concentrations of PGE2 diminished the production of pro-inflammatory cytokines and reduced antigen presentation and production of free radicals in these cells (76, 77). In this way, PGE2 is associated with the immunosuppression observed in infected mice with reduced lymphocyte proliferation and TNF-α production (73, 78). Moreover, uptake of apoptotic cells by macrophages during T. cruzi infection potentiates release of PGE2 and TGF-β by these cells, consequently decreasing production of NO, allowing intracellular parasite survival and growth (79). Furthermore, pharmacological inhibition of COX-2 using aspirin in peritoneal macrophages of BALB/c mice diminished T. cruzi internalization with consequent increases in IL-1β, NO and lipoxin production (80). Hence, increased production of PGE2 in macrophages that had contact with EV Y supports reduced production of pro-inflammatory cytokines (TNF-α) and NO that occurred in plasma and spleen-cell supernatants (TNF-α and IL-6) from mice inoculated with EV Y before infection. However, the release of PGE2 by infected macrophages after 24 h was significantly lower in cells that had contact with EV Y prior to infection than in cells without contact prior to infection. Although the EV Y induced lower levels of PGE2 produced by infected macrophages, the pro-inflammatory status of these cells was already downregulated, as shown by the abrupt reduction in release of the cytokines TNF-α and IL-6. Therefore, the immune modulation exerted by PGE2 that was induced by EV Y seems to be important specifically in the beginning of infection. In support of this hypothesis, in vitro production of PGE2 by spleen cells harvested from mice at 12 dpi did not change between animals inoculated with PBS or EV Y prior to infection, while the pro-inflammatory cytokines were downregulated in the same cells from animals inoculated with EV Y. Moreover, Moraes et al. showed that during the first 48 h of T. cruzi infection in H9c2 cells, COX-2 expression and activity are modulated by the parasite, leading to the control of the pro-inflammatory environment in infected cells (81). Considering that EVs shed by T. cruzi alter the genic expression of host cells (28), we hypothesize that EV Y could be modulating the expression and activity of COX-2.
In conclusion, our results indicate that EV Y exerts effects on host cells before infection, making the environment more favorable to T. cruzi in the first moments of infection. We showed that EVs released by T. cruzi modulate the immune response in vivo and demonstrated that this modulation occurs in vitro by direct effects on macrophages. These effects induce formation of lipid bodies and production of PGE2 and create a more favorable environment for parasite infection, with a reduction in inflammatory cytokines and the trypanocidal molecule NO. These results support the role of T. cruzi EVs in the complex pathogenesis of the acute phase of Chagas disease and provide new insights for a better understanding of the parasite–host relationship.
Ethics Statement
This study was carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the Brazilian National Council of Animal Experimentation. The protocol was approved by the Committee on the Ethics of Animal Experiments at Londrina State University (CEEA, process number 11/2015- 7045.2015.06). The use of Swiss mice for the maintenance of the parasite strain was also approved for the Committee on the Ethics of Animal Experiments at Londrina State University (CEEA, process number 28.841.2016.41).
Author Contributions
MM, SG, PW, and PF contributed to conception and design of the study. PM standardized and performed extracellular microvesicles isolation. MM, AM, NZ, BL and VT performed animal and cell culture experiments. AO contributed to establishment of lipid body staining and analysis. MM, PW, and PF wrote the first draft of the manuscript. All authors contributed to manuscript revision. All authors read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer HC declared a shared affiliation, though no other collaboration, with several of the authors, ML, PM, SG, PW, to the handling Editor.
Acknowledgments
The authors acknowledge their financial support and Dr. Patricia Torres Bozza Viola from the Instituto Oswaldo Cruz/Fiocruz -RJ for help with lipid body analysis. The authors also acknowledge Thiago Fernandes from Laboratório de Microscopia Eletrônica e Microanálises, Central de Laboratórios de Pesquisa Multiusuários, Universidade Estadual de Londrina for help with osmium tetroxide staining.
Funding
This work was supported by grants from CNPq-Universal (457014/2014-18), CNPq/MS/SCTIE/DECIT 31/2014 - Pesquisas sobre Doença de Chagas (466992/2014-9), and Fundação Araucária, PRONEX (255/2013). The Animal Facility from Instituto Carlos Chagas/Fiocruz-PR. Maria Isabel Lovo-Martins’ scholarship is supported by CNPq. PFW (460/2013) was a Fundação Araucária fellow, and SG (306709/2016-3) and PPF (307787/2015-0) are CNPq fellows.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.00896/full#supplementary-material.
References
1. Chagas C. Nova tripanozomiaze humana: estudos sobre a morfolojia e o ciclo evolutivo do Schizotrypanum cruzi n. gen., n. sp., ajente etiolojico de nova entidade morbida do homem. Memórias do Instituto Oswaldo Cruz (1909) 1:159–218. doi:10.1590/S0074-02761909000200008
2. World Health Organization. Chagas Disease (American Trypanosomiasis). (2017). Available from: http://www.who.int/mediacentre/factsheets/fs340/en/ (updated March 2017; Accessed: March 31, 2018).
3. Andrade DV, Gollob KJ, Dutra WO. Acute Chagas disease: new global challenges for an old neglected disease. PLoS Negl Trop Dis (2014) 8(7):e3010. doi:10.1371/journal.pntd.0003010
4. Rassi A Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet (2010) 375(9723):1388–402. doi:10.1016/S0140-6736(10)60061-X
5. Bern C, Martin DL, Gilman RH. Acute and congenital Chagas disease. Adv Parasitol (2011) 75:19–47. doi:10.1016/B978-0-12-385863-4.00002-2
6. Costa VM, Torres KC, Mendonca RZ, Gresser I, Gollob KJ, Abrahamsohn IA. Type I IFNs stimulate nitric oxide production and resistance to Trypanosoma cruzi infection. J Immunol (2006) 177(5):3193–200. doi:10.4049/jimmunol.177.5.3193
7. Wirth JJ, Kierszenbaum F. Recombinant tumor necrosis factor enhances macrophage destruction of Trypanosoma cruzi in the presence of bacterial endotoxin. J Immunol (1988) 141(1):286–8.
8. Silva JS, Aliberti JC, Martins GA, Souza MA, Souto JT, Padua MA. The role of IL-12 in experimental Trypanosoma cruzi infection. Braz J Med Biol Res (1998) 31(1):111–5. doi:10.1590/S0100-879X1998000100014
9. Zanluqui NG, Wowk PF, Pinge-Filho P. Macrophage polarization in Chagas disease. J Clin Cell Immunol (2015) 6:6. doi:10.4172/2155-9899.1000317
10. Guilmot A, Bosse J, Carlier Y, Truyens C. Monocytes play an IL-12-dependent crucial role in driving cord blood NK cells to produce IFN-g in response to Trypanosoma cruzi. PLoS Negl Trop Dis (2013) 7(6):e2291. doi:10.1371/journal.pntd.0002291
11. Hoft DF, Schnapp AR, Eickhoff CS, Roodman ST. Involvement of CD4(+) Th1 cells in systemic immunity protective against primary and secondary challenges with Trypanosoma cruzi. Infect Immun (2000) 68(1):197–204. doi:10.1128/IAI.68.1.197-204.2000
12. Cardillo F, Postol E, Nihei J, Aroeira LS, Nomizo A, Mengel J. B cells modulate T cells so as to favour T helper type 1 and CD8+ T-cell responses in the acute phase of Trypanosoma cruzi infection. Immunology (2007) 122(4):584–95. doi:10.1111/j.1365-2567.2007.02677.x
13. Cardoso MS, Reis-Cunha JL, Bartholomeu DC. Evasion of the immune response by Trypanosoma cruzi during acute infection. Front Immunol (2015) 6:659. doi:10.3389/fimmu.2015.00659
14. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol (2014) 30:255–89. doi:10.1146/annurev-cellbio-101512-122326
15. Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, Cordero EM, Marques AF, Varela-Ramirez A, et al. Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J Proteome Res (2013) 12(2):883–97. doi:10.1021/pr300947g
16. da Silveira JF, Abrahamsohn PA, Colli W. Plasma membrane vesicles isolated from epimastigote forms of Trypanosoma cruzi. Biochim Biophys Acta (1979) 550(2):222–32. doi:10.1016/0005-2736(79)90209-8
17. Del Cacho E, Gallego M, Lee SH, Lillehoj HS, Quilez J, Lillehoj EP, et al. Induction of protective immunity against Eimeria tenella infection using antigen-loaded dendritic cells (DC) and DC-derived exosomes. Vaccine (2011) 29(21):3818–25. doi:10.1016/j.vaccine.2011.03.022
18. Giri PK, Schorey JS. Exosomes derived from M. Bovis BCG infected macrophages activate antigen-specific CD4+ and CD8+ T cells in vitro and in vivo. PLoS One (2008) 3(6):e2461. doi:10.1371/journal.pone.0002461
19. Beauvillain C, Ruiz S, Guiton R, Bout D, Dimier-Poisson I. A vaccine based on exosomes secreted by a dendritic cell line confers protection against T. gondii infection in syngeneic and allogeneic mice. Microbes Infect (2007) 9(14–15):1614–22. doi:10.1016/j.micinf.2007.07.002
20. Beauvillain C, Juste MO, Dion S, Pierre J, Dimier-Poisson I. Exosomes are an effective vaccine against congenital toxoplasmosis in mice. Vaccine (2009) 27(11):1750–7. doi:10.1016/j.vaccine.2009.01.022
21. Schnitzer JK, Berzel S, Fajardo-Moser M, Remer KA, Moll H. Fragments of antigen-loaded dendritic cells (DC) and DC-derived exosomes induce protective immunity against Leishmania major. Vaccine (2010) 28(36):5785–93. doi:10.1016/j.vaccine.2010.06.077
22. Cronemberger-Andrade A, Aragao-Franca L, de Araujo CF, Rocha VJ, Borges-Silva Mda C, Figueira CP, et al. Extracellular vesicles from Leishmania-infected macrophages confer an anti-infection cytokine-production profile to naive macrophages. PLoS Negl Trop Dis (2014) 8(9):e3161. doi:10.1371/journal.pntd.0003161
23. Silverman JM, Clos J, Horakova E, Wang AY, Wiesgigl M, Kelly I, et al. Leishmania exosomes modulate innate and adaptive immune responses through effects on monocytes and dendritic cells. J Immunol (2010) 185(9):5011–22. doi:10.4049/jimmunol.1000541
24. Szempruch AJ, Sykes SE, Kieft R, Dennison L, Becker AC, Gartrell A, et al. Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell (2016) 164(1–2):246–57. doi:10.1016/j.cell.2015.11.051
25. Goncalves MF, Umezawa ES, Katzin AM, de Souza W, Alves MJ, Zingales B, et al. Trypanosoma cruzi: shedding of surface antigens as membrane vesicles. Exp Parasitol (1991) 72(1):43–53. doi:10.1016/0014-4894(91)90119-H
26. Trocoli Torrecilhas AC, Tonelli RR, Pavanelli WR, da Silva JS, Schumacher RI, de Souza W, et al. Trypanosoma cruzi: parasite shed vesicles increase heart parasitism and generate an intense inflammatory response. Microbes Infect (2009) 11(1):29–39. doi:10.1016/j.micinf.2008.10.003
27. Clemente TM, Cortez C, Novaes Ada S, Yoshida N. Surface molecules released by Trypanosoma cruzi metacyclic forms downregulate host cell invasion. PLoS Negl Trop Dis (2016) 10(8):e0004883. doi:10.1371/journal.pntd.0004883
28. Garcia-Silva MR, das Neves RF, Cabrera-Cabrera F, Sanguinetti J, Medeiros LC, Robello C, et al. Extracellular vesicles shed by Trypanosoma cruzi are linked to small RNA pathways, life cycle regulation, and susceptibility to infection of mammalian cells. Parasitol Res (2014) 113(1):285–304. doi:10.1007/s00436-013-3655-1
29. Fernandez-Calero T, Garcia-Silva R, Pena A, Robello C, Persson H, Rovira C, et al. Profiling of small RNA cargo of extracellular vesicles shed by Trypanosoma cruzi reveals a specific extracellular signature. Mol Biochem Parasitol (2015) 199(1–2):19–28. doi:10.1016/j.molbiopara.2015.03.003
30. Bayer-Santos E, Lima FM, Ruiz JC, Almeida IC, da Silveira JF. Characterization of the small RNA content of Trypanosoma cruzi extracellular vesicles. Mol Biochem Parasitol (2014) 193(2):71–4. doi:10.1016/j.molbiopara.2014.02.004
31. Garcia-Silva MR, Cabrera-Cabrera F, das Neves RF, Souto-Padrón T, de Souza W, Cayota A. Gene expression changes induced by Trypanosoma cruzi shed microvesicles in mammalian host cells: relevance of tRNA-derived halves. Biomed Res Int (2014) 2014:305239. doi:10.1155/2014/305239
32. Ramirez MI, Deolindo P, de Messias-Reason IJ, Arigi EA, Choi H, Almeida IC, et al. Dynamic flux of microvesicles modulate parasite-host cell interaction of Trypanosoma cruzi in eukaryotic cells. Cell Microbiol (2017) 19(4):e12672. doi:10.1111/cmi.12672
33. Cestari I, Ansa-Addo E, Deolindo P, Inal JM, Ramirez MI. Trypanosoma cruzi immune evasion mediated by host cell-derived microvesicles. J Immunol (2012) 188(4):1942–52. doi:10.4049/jimmunol.1102053
34. Wyllie MP, Ramirez MI. Microvesicles released during the interaction between Trypanosoma cruzi TcI and TcII strains and host blood cells inhibit complement system and increase the infectivity of metacyclic forms of host cells in a strain-independent process. Pathog Dis (2017) 75(7):1–12. doi:10.1093/femspd/ftx077
35. Zingales B, Andrade SG, Briones MR, Campbell DA, Chiari E, Fernandes O, et al. A new consensus for Trypanosoma cruzi intraspecific nomenclature: second revision meeting recommends TcI to TcVI. Mem Inst Oswaldo Cruz (2009) 104(7):1051–4. doi:10.1590/S0074-02762009000700021
36. Zeringer E, Li M, Barta T, Schageman J, Pedersen KW, Neurauter A, et al. Methods for the extraction and RNA profiling of exosomes. World J Methodol (2013) 3(1):11–8. doi:10.5662/wjm.v3.i1.11
37. Brener Z. Therapeutic activity and criterion of cure on mice experimentally infected with Trypanosoma cruzi. Rev Inst Med Trop Sao Paulo (1962) 4:389–96.
38. Lovo-Martins MI, Malvezi AD, da Silva RV, Zanluqui NG, Tatakihara VLH, Camara NOS, et al. Fish oil supplementation benefits the murine host during the acute phase of a parasitic infection from Trypanosoma cruzi. Nutr Res (2017) 41:73–85. doi:10.1016/j.nutres.2017.04.007
39. Vespa GN, Cunha FQ, Silva JS. Nitric oxide is involved in control of Trypanosoma cruzi-induced parasitemia and directly kills the parasite in vitro. Infect Immun (1994) 62(11):5177–82.
40. Munoz-Fernandez MA, Fernandez MA, Fresno M. Synergism between tumor necrosis factor-alpha and interferon-gamma on macrophage activation for the killing of intracellular Trypanosoma cruzi through a nitric oxide-dependent mechanism. Eur J Immunol (1992) 22(2):301–7. doi:10.1002/eji.1830220203
41. Silva JS, Vespa GN, Cardoso MA, Aliberti JC, Cunha FQ. Tumor necrosis factor alpha mediates resistance to Trypanosoma cruzi infection in mice by inducing nitric oxide production in infected gamma interferon-activated macrophages. Infect Immun (1995) 63(12):4862–7.
42. Roggero E, Perez AR, Tamae-Kakazu M, Piazzon I, Nepomnaschy I, Besedovsky HO, et al. Endogenous glucocorticoids cause thymus atrophy but are protective during acute Trypanosoma cruzi infection. J Endocrinol (2006) 190(2):495–503. doi:10.1677/joe.1.06642
43. Navarro-Gonzalvez JA, Garcia-Benayas C, Arenas J. Semiautomated measurement of nitrate in biological fluids. Clin Chem (1998) 44(3):679–81.
44. Panis C, Mazzuco TL, Costa CZ, Victorino VJ, Tatakihara VL, Yamauchi LM, et al. Trypanosoma cruzi: effect of the absence of 5-lipoxygenase (5-LO)-derived leukotrienes on levels of cytokines, nitric oxide and iNOS expression in cardiac tissue in the acute phase of infection in mice. Exp Parasitol (2011) 127(1):58–65. doi:10.1016/j.exppara.2010.06.030
45. Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One (2010) 5(12):e15263. doi:10.1371/journal.pone.0015263
46. Englen MD, Valdez YE, Lehnert NM, Lehnert BE. Granulocyte/macrophage colony-stimulating factor is expressed and secreted in cultures of murine L929 cells. J Immunol Methods (1995) 184(2):281–3. doi:10.1016/0022-1759(95)00136-X
47. Yoshida N, Mortara RA, Araguth MF, Gonzalez JC, Russo M. Metacyclic neutralizing effect of monoclonal antibody 10D8 directed to the 35- and 50-kilodalton surface glycoconjugates of Trypanosoma cruzi. Infect Immun (1989) 57(6):1663–7.
48. Barrias ES, Reignault LC, De Souza W, Carvalho TM. Dynasore, a dynamin inhibitor, inhibits Trypanosoma cruzi entry into peritoneal macrophages. PLoS One (2010) 5(1):e7764. doi:10.1371/journal.pone.0007764
49. Aliberti JC, Machado FS, Souto JT, Campanelli AP, Teixeira MM, Gazzinelli RT, et al. beta-Chemokines enhance parasite uptake and promote nitric oxide-dependent microbiostatic activity in murine inflammatory macrophages infected with Trypanosoma cruzi. Infect Immun (1999) 67(9):4819–26.
50. Melo RC, D’Avila H, Bozza PT, Weller PF. Imaging lipid bodies within leukocytes with different light microscopy techniques. Methods Mol Biol (2011) 689:149–61. doi:10.1007/978-1-60761-950-5_9
51. Nogueira PM, Ribeiro K, Silveira AC, Campos JH, Martins-Filho OA, Bela SR, et al. Vesicles from different Trypanosoma cruzi strains trigger differential innate and chronic immune responses. J Extracell Vesicles (2015) 4:28734. doi:10.3402/jev.v4.28734
52. Machado FS, Dutra WO, Esper L, Gollob KJ, Teixeira MM, Factor SM, et al. Current understanding of immunity to Trypanosoma cruzi infection and pathogenesis of Chagas disease. Semin Immunopathol (2012) 34(6):753–70. doi:10.1007/s00281-012-0351-7
53. Coakley G, Maizels RM, Buck AH. Exosomes and other extracellular vesicles: the new communicators in parasite infections. Trends Parasitol (2015) 31(10):477–89. doi:10.1016/j.pt.2015.06.009
54. Schorey JS, Cheng Y, Singh PP, Smith VL. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep (2015) 16(1):24–43. doi:10.15252/embr.201439363
55. Neves RF, Fernandes AC, Meyer-Fernandes JR, Souto-Padron T. Trypanosoma cruzi-secreted vesicles have acid and alkaline phosphatase activities capable of increasing parasite adhesion and infection. Parasitol Res (2014) 113(8):2961–72. doi:10.1007/s00436-014-3958-x
56. Silverman JM, Clos J, de’Oliveira CC, Shirvani O, Fang Y, Wang C, et al. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J Cell Sci (2010) 123(Pt 6):842–52. doi:10.1242/jcs.056465
57. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol (2006) 177(6):3515–9. doi:10.4049/jimmunol.177.6.3515
58. Dutra WO, Menezes CA, Magalhaes LM, Gollob KJ. Immunoregulatory networks in human Chagas disease. Parasite Immunol (2014) 36(8):377–87. doi:10.1111/pim.12107
59. Camargo MM, Andrade AC, Almeida IC, Travassos LR, Gazzinelli RT. Glycoconjugates isolated from Trypanosoma cruzi but not from Leishmania species membranes trigger nitric oxide synthesis as well as microbicidal activityin IFN-gamma-primed macrophages. J Immunol (1997) 159(12):6131–9.
60. Gutierrez FR, Mineo TW, Pavanelli WR, Guedes PM, Silva JS. The effects of nitric oxide on the immune system during Trypanosoma cruzi infection. Mem Inst Oswaldo Cruz (2009) 104(Suppl 1):236–45. doi:10.1590/S0074-02762009000900030
61. Radi R, Beckman JS, Bush KM, Freeman BA. Peroxynitrite-induced membrane lipid peroxidation: the cytotoxic potential of superoxide and nitric oxide. Arch Biochem Biophys (1991) 288(2):481–7. doi:10.1016/0003-9861(91)90224-7
62. Denicola A, Rubbo H, Rodriguez D, Radi R. Peroxynitrite-mediated cytotoxicity to Trypanosoma cruzi. Arch Biochem Biophys (1993) 304(1):279–86. doi:10.1006/abbi.1993.1350
63. Cunha-Neto E, Nogueira LG, Teixeira PC, Ramasawmy R, Drigo SA, Goldberg AC, et al. Immunological and non-immunological effects of cytokines and chemokines in the pathogenesis of chronic Chagas disease cardiomyopathy. Mem Inst Oswaldo Cruz (2009) 104(Suppl 1):252–8. doi:10.1590/S0074-02762009000900032
64. Reed SG, Brownell CE, Russo DM, Silva JS, Grabstein KH, Morrissey PJ. IL-10 mediates susceptibility to Trypanosoma cruzi infection. J Immunol (1994) 153(7):3135–40.
65. Kim GD, Lee SE, Yang H, Park HR, Son GW, Park CS, et al. beta2 integrins (CD11/18) are essential for the chemosensory adhesion and migration of polymorphonuclear leukocytes on bacterial cellulose. J Biomed Mater Res A (2015) 103(5):1809–17. doi:10.1002/jbm.a.35316
66. St-Pierre J, Ostergaard HL. A role for the protein tyrosine phosphatase CD45 in macrophage adhesion through the regulation of paxillin degradation. PLoS One (2013) 8(7):e71531. doi:10.1371/journal.pone.0071531
67. Reuter A, Panozza SE, Macri C, Dumont C, Li J, Liu H, et al. Criteria for dendritic cell receptor selection for efficient antibody-targeted vaccination. J Immunol (2015) 194(6):2696–705. doi:10.4049/jimmunol.1402535
68. van Kasteren SI, Overkleeft H, Ovaa H, Neefjes J. Chemical biology of antigen presentation by MHC molecules. Curr Opin Immunol (2014) 26:21–31. doi:10.1016/j.coi.2013.10.005
69. Sansom DM, Manzotti CN, Zheng Y. What’s the difference between CD80 and CD86? Trends Immunol (2003) 24(6):314–9. doi:10.1016/S1471-4906(03)00111-X
70. van den Berg TK, Kraal G. A function for the macrophage F4/80 molecule in tolerance induction. Trends Immunol (2005) 26(10):506–9. doi:10.1016/j.it.2005.07.008
71. Melo RC, D’Avila H, Fabrino DL, Almeida PE, Bozza PT. Macrophage lipid body induction by Chagas disease in vivo: putative intracellular domains for eicosanoid formation during infection. Tissue Cell (2003) 35(1):59–67. doi:10.1016/S0040-8166(02)00105-2
72. D’Avila H, Freire-de-Lima CG, Roque NR, Teixeira L, Barja-Fidalgo C, Silva AR, et al. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E(2) generation and increased parasite growth. J Infect Dis (2011) 204(6):951–61. doi:10.1093/infdis/jir432
73. Pinge-Filho P, Tadokoro CE, Abrahamsohn IA. Prostaglandins mediate suppression of lymphocyte proliferation and cytokine synthesis in acute Trypanosoma cruzi infection. Cell Immunol (1999) 193(1):90–8. doi:10.1006/cimm.1999.1463
74. Borges MM, Kloetzel JK, Andrade HF Jr, Tadokoro CE, Pinge-Filho P, Abrahamsohn I. Prostaglandin and nitric oxide regulate TNF-alpha production during Trypanosoma cruzi infection. Immunol Lett (1998) 63(1):1–8. doi:10.1016/S0165-2478(98)00034-0
75. Ashton AW, Mukherjee S, Nagajyothi FN, Huang H, Braunstein VL, Desruisseaux MS, et al. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J Exp Med (2007) 204(4):929–40. doi:10.1084/jem.20062432
76. Rangel Moreno J, Estrada Garcia I, De La Luz Garcia Hernandez M, Aguilar Leon D, Marquez R, Hernandez Pando R. The role of prostaglandin E2 in the immunopathogenesis of experimental pulmonary tuberculosis. Immunology (2002) 106(2):257–66. doi:10.1046/j.1365-2567.2002.01403.x
77. Alvarez Y, Valera I, Municio C, Hugo E, Padron F, Blanco L, et al. Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm (2010) 2010:1–14. doi:10.1155/2010/201929
78. Michelin MA, Silva JS, Cunha FQ. Inducible cyclooxygenase released prostaglandin mediates immunosuppression in acute phase of experimental Trypanosoma cruzi infection. Exp Parasitol (2005) 111(2):71–9. doi:10.1016/j.exppara.2005.05.001
79. Freire-de-Lima CG, Nascimento DO, Soares MB, Bozza PT, Castro-Faria-Neto HC, de Mello FG, et al. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature (2000) 403(6766):199–203. doi:10.1038/35003208
80. Malvezi AD, da Silva RV, Panis C, Yamauchi LM, Lovo-Martins MI, Zanluqui NG, et al. Aspirin modulates innate inflammatory response and inhibits the entry of Trypanosoma cruzi in mouse peritoneal macrophages. Mediators Inflamm (2014) 2014:580919. doi:10.1155/2014/580919
Keywords: extracellular vesicles, lipid bodies, macrophages, prostaglandin E2, Trypanosoma cruzi
Citation: Lovo-Martins MI, Malvezi AD, Zanluqui NG, Lucchetti BFC, Tatakihara VLH, Mörking PA, Oliveira AG, Goldenberg S, Wowk PF and Pinge-Filho P (2018) Extracellular Vesicles Shed By Trypanosoma cruzi Potentiate Infection and Elicit Lipid Body Formation and PGE2 Production in Murine Macrophages. Front. Immunol. 9:896. doi: 10.3389/fimmu.2018.00896
Received: 01 February 2018; Accepted: 11 April 2018;
Published: 27 April 2018
Edited by:
Debora Decote-Ricardo, Universidade Federal Rural do Rio de Janeiro, BrazilReviewed by:
Hugo Caire Castro-Faria-Neto, Fundação Oswaldo Cruz (Fiocruz), BrazilDanielle Oliveira Nascimento, Universidade Federal do Rio de Janeiro, Brazil
Laura Noelia Cariddi, National University of Río Cuarto, Argentina
Copyright: © 2018 Lovo-Martins, Malvezi, Zanluqui, Lucchetti, Tatakihara, Mörking, Oliveira, Goldenberg, Wowk and Pinge-Filho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pryscilla Fanini Wowk, cHJ5c2NpbGxhLndvd2tAZmlvY3J1ei5icg==;
Phileno Pinge-Filho, cGluZ2VmaWxob0B1ZWwuYnI=