 Valentina Pegoretti
Valentina Pegoretti Wia Baron
Wia Baron Jon D. Laman
Jon D. Laman Ulrich L. M. Eisel
Ulrich L. M. Eisel- 1Department of Molecular Neurobiology (GELIFES), University of Groningen, Groningen, Netherlands
- 2Department of Cell Biology, University Medical Center Groningen (UMCG), University of Groningen, Groningen, Netherlands
- 3Department of Neuroscience, University Medical Center Groningen (UMCG), University of Groningen, Groningen, Netherlands
Autoimmunity develops when self-tolerance mechanisms are failing to protect healthy tissue. A sustained reaction to self is generated, which includes the generation of effector cells and molecules that destroy tissues. A way to restore this intrinsic tolerance is through immune modulation that aims at refurbishing this immunologically naïve or unresponsive state, thereby decreasing the aberrant immune reaction taking place. One major cytokine has been shown to play a pivotal role in several autoimmune diseases such as rheumatoid arthritis (RA) and multiple sclerosis (MS): tumor necrosis factor alpha (TNFα) modulates the induction and maintenance of an inflammatory process and it comes in two variants, soluble TNF (solTNF) and transmembrane bound TNF (tmTNF). tmTNF signals via TNFR1 and TNFR2, whereas solTNF signals mainly via TNFR1. TNFR1 is widely expressed and promotes mainly inflammation and apoptosis. Conversely, TNFR2 is restricted mainly to immune and endothelial cells and it is known to activate the pro-survival PI3K-Akt/PKB signaling pathway and to sustain regulatory T cells function. Anti-TNFα therapies are successfully used to treat diseases such as RA, colitis, and psoriasis. However, clinical studies with a non-selective inhibitor of TNFα in MS patients had to be halted due to exacerbation of clinical symptoms. One possible explanation for this failure is the non-selectivity of the treatment, which avoids TNFR2 stimulation and its immune and tissue protective properties. Thus, a receptor-selective modulation of TNFα signal pathways provides a novel therapeutic concept that might lead to new insights in MS pathology with major implications for its effective treatment.
Introduction
Multiple sclerosis (MS) affects approximately 2.5 million people worldwide. It is considered as an autoimmune disease characterized by white and gray matter lesions in the central nervous system (CNS) caused by autoreactive T cells that escaped from central and peripheral tolerance patrolling mechanisms. These cells travel along with activated B cells and monocytes to the CNS where they infiltrate, starting a synergistic attack against myelin (1). As demyelination is a key feature of MS pathology, several myelin proteins have been investigated as targets of these autoreactive lymphocytes. It has been shown that myelin basic protein and myelin oligodendrocyte glycoprotein are recognized by mature autoreactive T-helper cells in MS patients but also in healthy individuals (2). The identification of a major T cell autoantigen in MS is still a matter of controversy. It may be due to technical limitations in autoantibodies’ detection or epitope spreading (3). Anyway, the search of pathological anti-myelin immune responses is still open-ended.
Currently, the etiology of MS has been investigated from another angle that favors the idea that initial pathology occurs within the CNS, similarly to other neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases (4). This theory argues that degeneration of oligodendrocytes and/or myelin initiates pathology by releasing autoantigen, which in turn are responsible for the autoimmune, inflammatory response in the organism. Of importance, mitochondrial dysfunction (5), ROS production (6), misfolding of proteins (7), and release of proapoptotic signals (8) are just few of the consequences (9). Myelin-loaded microglia/macrophages are also largely involved in this pathological process (10). They are constantly producing ROS through oxidative burst giving rise to mitochondrial dysfunction and proapoptotic signals release causing oligodendrocytes death and demyelination.
Being it autoimmune or neurodegenerative, the study of the nature of this disease is yet mostly descriptive than causative. With this limited understanding, the animal models currently available seem to mimic only few and separated features of the disease, which further restrict our view of the underlying mechanisms causing MS. Even though it seems very difficult to achieve a solid and unifying explanation, great efforts have been made to develop treatments to reduce the symptomatic incidences in MS patients. The available therapeutic strategies are primarily focused on suppressing or modulating certain immune functions thereby leading to a partial and temporary recovery sometimes with major side effects (11). There is still a strong urge for an effective treatment that slows down MS disease progression or to prevent its development.
This review will focus on the potential value for MS treatment of tumor necrosis factor alpha (TNFα), a major cytokine involved in several biological functions. Furthermore, the different and dual functions of TNFα are specified by the two receptors (TNFR1 and TNFR2) that it activates. Current research highlights a great potential of selectively targeting TNF–TNFRs signaling with promising immune protective, tissue regenerative, and neuroprotective therapeutic properties.
Anti-TNF Therapies in Autoimmunity
Self-recognition is an essential biological process that gives rise to immune tolerance: a state of indifference or non-reactivity toward a substance that would normally be expected to excite an immunological response (12). Yet, when the immune system erroneously identifies a self-antigen as a danger, it initiates an inflammatory response against it. The latter mechanism is defined as autoimmunity, which encompasses tissue damage, caused by T-cell or antibody reactivity to self. Many inflammatory diseases are autoimmune diseases, including rheumatoid arthritis (RA), MS, Graves’ disease, type 1 diabetes mellitus, Crohns’ disease, and others. Nowadays, they affect 12.5% of the world’s population (13) and they can be distinguished based on their primary target organ (joints, skin—psoriasis; CNS—MS; intestine—inflammatory bowel disease (IBD); pancreas—type 1 diabetes mellitus). For many years, the standard treatment relied on diminishing autoimmune pathology with general immunosuppressive agents, anti-proliferative drugs, and corticosteroids. Halting the immune system has always major and diffuse side effects that increase the toxicity of the intervention thereby decreasing its therapeutic value. Immunosuppressant drugs are widely used by clinicians to reduce inflammatory attacks on tissues but, due to their low efficacy, disease-modifying drugs with greater specificity and lower toxicity were implemented. Monoclonal antibodies and engineered biological products have become now standard interventions for several autoimmune diseases, including MS. A long standing class of biologics used for many autoimmune diseases is TNF blockers which includes infliximab, etanercept, adalimumab, PEGylated certolizumab, and golimumab. These are the FDA-approved anti-TNF biologics for the treatment of Crohn’s disease, ulcerative colitis, RA, ankylosing spondylitis (AS), psoriatic arthritis, and plaque psoriasis (14). Even though it has been extensively studied, the potential therapeutic value of blocking TNF is limited by its partial efficacy in different diseases. Anti-TNF treatment is discontinued in 1/3 of RA patients within the first year of treatment (15). Around 10–30% of IBD patients do not respond to initial treatment while 23–46% lose response over time (16). Similarly, 27% of patients with psoriasis discontinue anti-TNF treatment after a year or lose its efficacy over time (17). So far, there is little evidence explaining the reasons and risk factors for primary or secondary non-response. Therefore, other strategies are implemented by clinicians to maintain efficacy with acceptable tolerability such as using a different TNF blocker, switching class of biologic, dose adjustments, and change in route of administration, when possible. Moreover, failure of anti-TNF therapies can also be due to development of adverse effects such as infections, malignancies, acute infusion and injection reactions, autoimmunity, and cardiovascular effects (18, 19).
In 1999, a clinical trial testing the efficacy of a TNF inhibitor, Lenercept, for MS treatment had to be halted due to exacerbations of symptoms when compared to placebo-treated MS patients (20). Likewise, there are several clinical reports of RA and AS patients treated with TNF blockers that developed CNS demyelination after treatment (21, 22).
Although partially effective in other autoimmune diseases, anti-TNF therapies in MS patients seem to worsen pathology and clinical symptoms. A possible explanation for this failure is the inability of the drug to grant access to the CNS (23). In other tissues is rather easy to penetrate and exert a local effect, the brain is a privileged site that instead restricts entry to macromolecules such as biologics. Furthermore, non-selective TNF inhibitors dampen down the active inflammatory response ongoing in certain diseases such as RA and IBD. While for these diseases the anti-inflammatory effects could be enough for (at least partial) recovery, MS treatment requires a more profound reestablishment of homeostasis that includes tissue protective and regenerative properties. Interestingly, the last two decades of research on TNFα signaling showed that the soluble form of TNF (solTNF) triggers apoptotic and proinflammatory signals to the cell via TNFR1 while the transmembrane form (tmTNF) is able to promote cell survival through TNFR2 activation (see Figure 1). The following chapter recapitulates the current studies trying to specify and optimize selectively targeting TNF–TNFRs within an MS therapeutic frame.
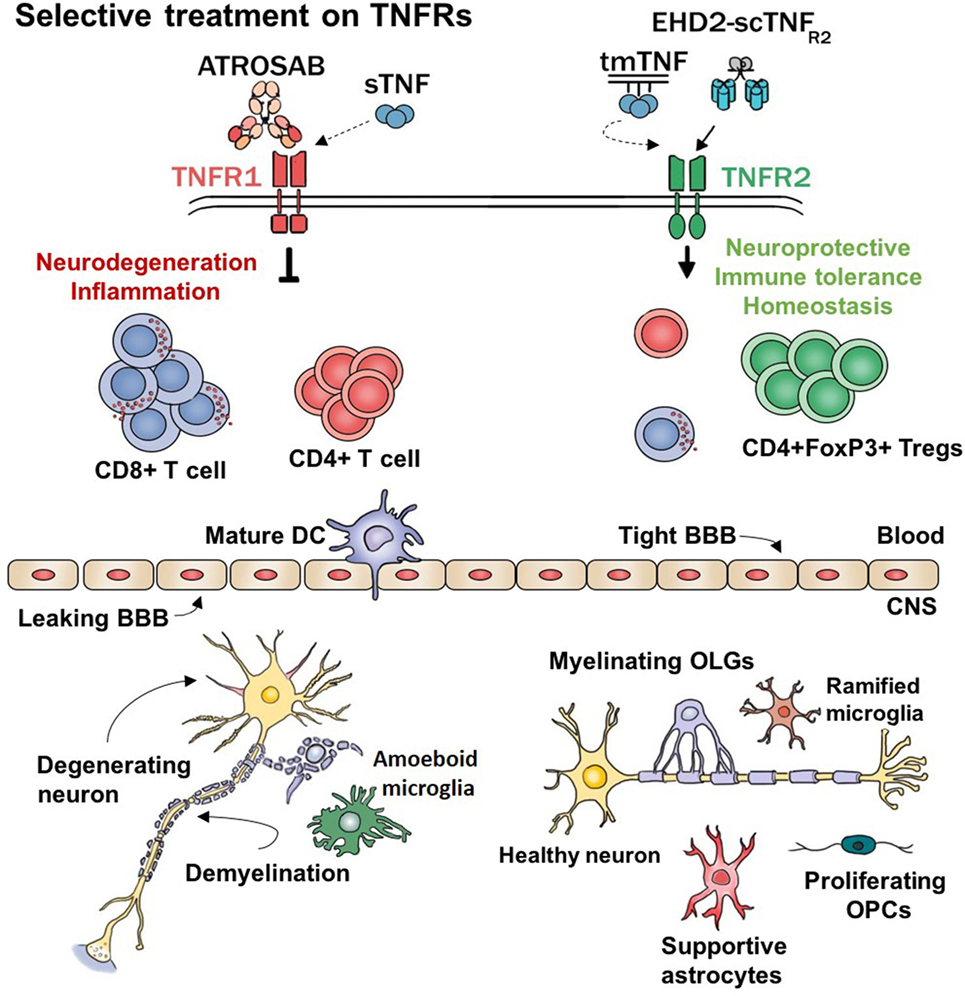
Figure 1. Hypothetical working model. TNFα and its receptors regulate major functions of several cell types. This model represents the expected effects of selectively modulating TNF–TNFRs signaling. sTNF, soluble TNF; tmTNF, transmembrane TNF; ATROSAB, TNFR1 antagonistic antibody; EHD2-scTNFR2, TNFR2 agonist; CD8+ T cell, cytotoxic T cells; CD4+ T cells, helper T cells; DC, dendritic cell; BBB, blood–brain barrier; Tregs, regulatory T cells; CNS, central nervous system; OPCs, oligodendrocyte’s precursor cells; OLGs, oligodendrocytes; TNFα, tumor necrosis factor alpha.
TNF–TNFR Signaling: Therapeutic Implications for MS
Tumor necrosis factor alpha is a pleiotropic cytokine regulating many physiological and pathological functions such as cell survival (24), apoptosis (25), inflammation (26–28), autoimmunity (29), demyelination (30), and cancer (31). TNFα is synthesized as a transmembrane protein of 26 kDa and forms a stable homo-trimeric molecule (tmTNF). Proteolytic cleavage of the protein via TNFα converting enzyme (TACE/ADAM17) produces a 17-kDa monomeric protein, a soluble homo-trimeric molecule of 51 kDa (solTNF). TNFα signaling is then generated through the interaction with two distinct transmembrane receptors, the 55-kDa TNF receptor type I (TNFR1) and the 75-kDa TNF receptor type II (TNFR2). The two TNFα variants display different affinities for the two receptors. TNFR1 is activated by both soluble and transmembrane forms with higher affinity for solTNF while activation of TNFR2 is solely due to tmTNF. Furthermore, the two receptors differ in the intracellular pathways that they trigger leading to various cellular responses (32–34). TNFR1 has been described as stimulator of effector caspase-mediated apoptosis (35, 36), while TNFR2 promotes cell survival through PI3K-Akt/PKB signaling pathway (37, 38). However, TNFR1 activation may also prevent TNF-induced apoptosis by activating the classical NF-κB pathway (39) and receptor-interacting protein 1 (RIP1) ubiquitination (40). Upon TNFR1 stimulation, the intracellular death domain (DD) recruits RIP1 and TNFR-associated death domain (TRADD). TRADD engages TNFR-associated factor 2 (TRAF2), inhibitor of apoptosis protein 1 (cIAP1) and inhibitor of apoptosis protein 2 (cIAP2) thereby leading to the formation of complex I (41). RIP1 ubiquitination and complex I activation later stimulates catalytic IκB kinase (IKK) complex, which favors the activation of NF-κB pathway (42). If this signaling fails, complex II will trigger caspase 8-mediated apoptosis upon TNFR1 ligand binding (43). Importantly, the initiation of this apoptotic process heavily relies on the levels of the inhibitory protein (cFLIP). The more NF-κB is activated by complex I, the more cFLIP will be available to inhibit caspase-mediated apoptosis (44).
In addition, it was shown that TNFRs cross talk intracellularly giving rise to TNFR1-induced cell survival and TNFR2-induced apoptosis (33). In contrast with TNFR1, TNFR2 does not contain a DD but it is still capable of inducing apoptosis upon its activation (45). A common intracellular molecule family in the TNFα signaling cascade is TRAFs, which are recruited by both TNFRs complexes. In CD30 and CD40 cells, TNFR2 stimulation might lead to TRAF2 degradation, which results in caspase 8 activation and eventually apoptosis (46). TRAF2 is also an important regulator of cell survival through TNFR1-mediated activation of C-Jun and NF-κB. In vitro studies showed that NF-κB activation leads to production of TRAF1, which blocks TNFR2-mediated degradation of TRAF2 (47).
Another distinctive feature of these two receptors is their differential expression in different tissues. While TNFR1 is ubiquitously expressed, TNFR2 can be found mainly on endothelial cells, various immune cells, and certain CNS cells (48).
All these peculiar features enable such a complex cytokine to have major, sometimes conflicting, effects depending on its form, the receptor that it triggers and the cell type on which it may act (see Figure 1). Because of this pleiotropic effect, the function of TNFα will depend on the ratio of co-expression of its receptors which will shift the balance between cellular survival and apoptosis.
The following sections will highlight the beneficial properties of targeting selectively TNFRs’ signaling pathway found in in vitro and in vivo models of MS (see Table 1). This may help to further elucidate the therapeutic value of TNFα in the treatment of MS and other autoimmune diseases.
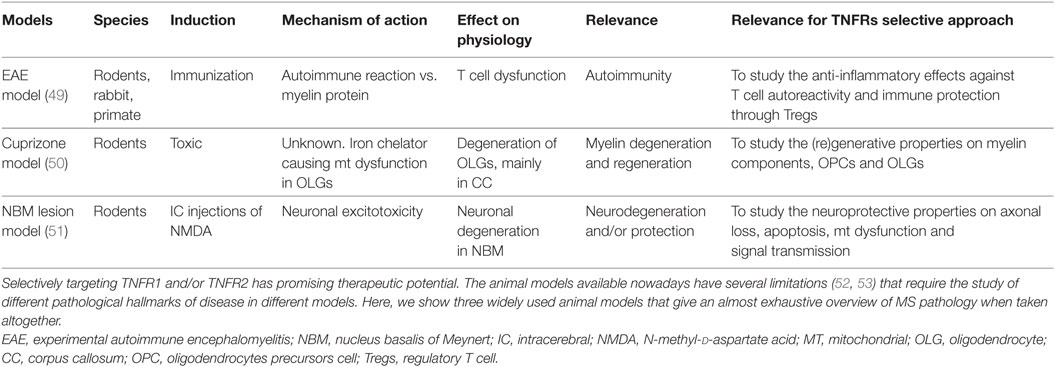
Table 1. Animal models to investigate pharmacological interventions for multiple sclerosis (MS).
Selective Targeting of TNFRs: Immune Protection Properties
Growing evidence suggest that proinflammatory factors are intertwined with a complex resolution program of inflammation after few hours an inflammatory response begins (54–56). In a coculture experiment with murine CD4+CD25+FoxP3+ regulatory T cells (Tregs) and CD4+CD25− effector T cells (Teffs), short-term exposure to TNFα promotes Teffs expansion while a more prolonged treatment favors proliferation and activation of Tregs (57). Moreover, TNFR2−/− mice show normal pool of Tregs but, when stimulated with septic challenge, they fail to expand. This suggests that TNFα might have a proinflammatory action in the early phases of inflammation through Teffs to leave then Tregs to re-establish homeostatic balance through TNFR2 signaling. Similar evidence comes from leukocytes isolated from RA patients on anti-TNF medications (58). Adalimumab, an anti-TNF antibody, but not the solTNFR2 Etanercept, promotes the interaction between monocytes and Tregs leading to expansion of FoxP3+ Tregs and suppression of Th17 cells through IL-2/STAT5 pathway. This effect is caused by adalimumab binding to tmTNF on monocytes, which is able to enhance both expression of tmTNF and its binding to TNFR2 on Tregs. Additionally, increased IL-17 production in TNFR2-deficient T cells is prevented by exogenous IL-2 showing that tmTNF–TNFR2 signaling suppresses Th17 differentiation by promoting IL-2 expression (59). Furthermore, an in vivo EAE study with TNFRs−/− mice showed a reduction of clinical symptoms, demyelination score, CD3+ T cell infiltrates, and activated microglia/macrophages in TNFR1−/− mice. On the contrary, lacking TNFR2 seems to worsen EAE disease course. In the same study, EAE was induced in normal C57BL/6 to investigate the effect of antibody-mediated TNFR1 inhibition. These results show attenuated EAE severity and delayed the onset of disease in the treated group mainly through decreased demyelination score and neuronal loss while there is only a mild reduction in immune infiltrates into the CNS (60). If silencing TNFR1 is not enough, activation of TNFR2 in mouse microglia culture promotes expression of anti-inflammatory and neuroprotective genes as granulocyte colony-stimulating factor, adrenomedullin, IL-10, and IFN-γ (61). Specifically, conditional knockout of microglial TNFR2 reveals earlier EAE onset by means of increased number of infiltrates, T cell activation, and demyelination scores. On the other hand, ablation of microglial/macrophage TNFR2 leads to EAE suppression (62). This experiment further expands our knowledge of TNFα functions on its receptors: TNFR2 has dual roles depending on its location in central or peripheral myeloid cells as much as solTNF and tmTNF have detrimental or protective properties, respectively.
Selective Targeting of TNFRs: Tissue Regeneration Properties
A crucial pathological hallmark of MS is white matter lesions caused by axonal demyelination. An effective pharmacological intervention for this disease requires tissue regenerative properties to counteract tissue damage at the lesion site. In vitro studies reveal that TNFR2 activation protects oligodendrocyte’s progenitor cells (OPCs) from oxidative stress (63). OPCs are increasingly being studied in MS research as they are shown to be essential to the remyelination process (64). The beneficial effect of TNFR2 seems to continue in later stages of development of these critical cells. In a primary coculture setup, maturation of oligodendrocytes into myelinating cells appears to be boosted through astrocyte-specific TNFR2 stimulation (65). In 2001, an important in vivo study investigated the different role of TNFRs in the cuprizone model for demyelination (see Table 1) in mice lacking either TNFα or one its receptors (66). In this study, the absence of TNFα delays the remyelination process due to reduction of proliferating OPCs and mature oligodendrocytes when compared to wild type mice. Interestingly, similar effects are found in mice lacking TNFR2, but not TNFR1, underling a substantial role of TNFR2 in promoting oligodendrocytes proliferation and regeneration. In this vein, inhibition of solTNF shows that tmTNF increases axon preservation and improves myelin compaction in an EAE mouse model for MS (67). In the same study, myelin-specific genes and increased number of OPCs are found upon tmTNF treatment. A recent EAE study with conditional knockout mice highlights that TNFR2 specifically on oligodendrocytes drives their differentiation and remyelination (68). Furthermore, treatment with XPro1595, a selective solTNF inhibitor, in a cuprizone mouse model (see Table 1) shows faster remyelination due to improved myelin phagocytosis by microglia (69).
Selective Targeting of TNFRs: Neuroprotection Properties
In the progressive stages of MS, axonal loss and neurodegeneration seem to take over, at least partially, inflammation as main pathological hallmarks (70). Several in vitro studies underline the potential neuroprotective effect of selective targeting of TNFRs. In a human dopaminergic neuronal cell line (LUHMES), TNFR2 stimulation of the PI3K-PKB/Akt pro-survival pathway rescues neurons from oxidative stress-induced cell death (71). Furthermore, similar results were found in an in vitro model of glutamate-induced excitotoxicity in primary cortical neurons. TNFR2, and not TNFR1, induces persistent PI3K PKB/Akt-mediated NF-κB activation leading to neuroprotection, which is enhanced by N-methyl-d-aspartate receptor co-stimulation (37). Using the same in vitro model, another study shows that activation of TNFR2 signaling pathway mediated lovastatin-induced neuroprotection against glutamate excitotoxicity (72). Statins are widely prescribed in clinical practice for lowering cholesterol levels. Nonetheless, a specific statin, called Simvastatin, has been shown to be effective in decreasing whole-brain atrophy in patients with secondary progressive MS in a phase II trial (73). The neuroprotective effect of TNFR2 was also found in an in vivo model using TNFR1−/− and TNFR2−/− mice. After retinal ischemia, TNFR1 deficiency leads to strong decrease in neuronal death while absence of TNFR2 leads to enhanced neurodegeneration (36). Another in vivo model using genetic ablation of solTNF shows neuroprotection against focal cerebral ischemia (74). Interestingly, a recent study reveals that the neuroprotective and anti-inflammatory effects found by antagonizing TNFR1 in the nucleus basalis lesion (NBM) model (see Table 1) is enhanced through TNFR2 signaling (75).
Future Perspectives
Due to contradicting results concerning TNFα and TNFR signaling in neurodegenerative diseases in the late 90s, major advances have been made in recent years in understanding the biology of TNF–TNFRs signaling in health and disease. This review highlighted the potential therapeutic value of this target, specifically within MS pathology. Of importance, the available MS treatments are focused on limiting the burden and occurrence of autoreactive peripheral immune cells. We can see them as drugs boosting the immune system’s resistance toward an insult against self-tissue. Obviously, this leads to a temporary effect of the treatment, which is followed by a partial decrease in symptoms, mainly in patients with relapsing-remitting MS. Moreover, most of these drugs are not able to slow down disease progression. Recently, several immunologists and evolutionary ecologists introduced the concept of disease tolerance as a defense mechanism against infectious agents (76–78). In flies (79), rodents (80), and humans (81) studies, modulating disease tolerance resulted in protection against several types of infection and restored homeostasis (82). As in different patrolling mechanisms, attack is not always the best defense mechanism: damage control is as important as pathogen control. Within autoimmunity, dysregulated disease tolerance can be seen as a failure of the immune system to control tissue damage caused by autoreactive immune cells. Interestingly, selective modulation of TNFRs triggers a variety of protective and pro-survival properties, which in turn are positively affecting the pathological milieu derived from autoreactive lymphocytes. Breaking the vicious circle of chronic inflammation and protect tissue against further damage are essential features for a therapeutic agent that aims at restoring proper immune functions and general homeostasis.
Nevertheless, some challenges need to be addressed to further elucidate the potential of this treatment target. As briefly mentioned above, activating TNFR2 in peripheral or central myeloid cells resulted in opposing therapeutic effects (62) underling the need for a pharmacological approach that minimizes peripheral immune activation. However, blood–brain barrier (BBB) permeability of these compounds might be an obstacle to overcome in order to reach all beneficial effects of this target. In the past decade, great progress has been made in developing nanoparticles (83, 84) and cell-specific drug carriers (85) through the BBB, giving a promising perspective for CNS diseases (86).
To conclude, selective modulation of TNFRs through TNFR2 activation and/or TNFR1 silencing has great therapeutic potential in terms of immune, tissue, and neuroprotective properties, especially for MS treatment.
Author Contributions
The authors contributed equally to this work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported by the Dutch MS Research Foundation (grant # 15-898 MS to ULME, WB and JDL).
References
1. Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune privileged sites: absolute barriers versus educational gates. Nat Rev Immunol (2013) 13:206–18. doi:10.1038/nri3391
2. Hellings N, Barée M, Verhoeven C, D’hooghe MB, Medaer R, Bernard CC, et al. T-cell reactivity to multiple myelin antigens in multiple sclerosis patients and healthy controls. J Neurosci Res (2001) 63:290–302. doi:10.1002/1097-4547(20010201)63:3<290::AID-JNR1023>3.0.CO;2-4
3. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol (2015) 15(9):545–58. doi:10.1038/nri3871
4. Stys PK, Zamponi GW, van Minnen J, Geurts JJ. Will the real multiple sclerosis please stand up? Nat Rev Neurosci (2012) 13(7):507–14. doi:10.1038/nrn3275
5. Witte ME, Mahad DJ, Lassmann H, van Horssen J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med (2014) 20(3):179–87. doi:10.1016/j.molmed.2013.11.007
6. LeVine SM. The role of reactive oxygen species in the pathogenesis of multiple sclerosis. Med Hypotheses (1992) 39(3):271–4. doi:10.1016/0306-9877(92)90121-R
7. David MA, Tayebi M. Detection of protein aggregates in brain and cerebrospinal fluid derived from multiple sclerosis patients. Front Neurol (2014) 5:251. doi:10.3389/fneur.2014.00251
8. Macchi B, Marino-Merlo F, Nocentini U, Pisani V, Cuzzocrea S, Grelli S, et al. Role of inflammation and apoptosis in multiple sclerosis: comparative analysis between the periphery and the central nervous system. J Neuroimmunol (2015) 287:80–7. doi:10.1016/j.jneuroim.2015.08.016
9. Friese MA, Schattling B, Fugger L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol (2014) 10(4):225–38. doi:10.1038/nrneurol.2014.37
10. Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett (2011) 585(23):3715–23. doi:10.1016/j.febslet.2011.08.004
11. Wingerchuk DM, Carter JL. Multiple sclerosis: current and emerging disease-modifying therapies and treatment strategies. Mayo Clin Proc (2014) 89(2):225–40. doi:10.1016/j.mayocp.2013.11.002
12. Burnet FM, Medawar P. Nobel Lectures, Physiology of Medicine 1942-1962. Amsterdam: Elsevier (1964).
13. Lerner A, Jeremias P, Matthias T. The world incidence and prevalence of autoimmune diseases is increasing. Int J Celiac Dis (2015) 3-4:151–5. doi:10.12691/ijcd-3-4-8
14. Lis K, Kuzawińska O, Bałkowiec-Iskra E. Tumor necrosis factor inhibitors – state of knowledge. Arch Med Sci (2014) 10(6):1175–85. doi:10.5114/aoms.2014.47827
15. Silva-Fernandez L, Hyrich K. Rheumatoid arthritis: when TNF inhibitors fail in RA—weighing up the options. Nat Rev Rheumatol (2014) 10:262–4. doi:10.1038/nrrheum.2014.34
16. Roda G, Jharap B, Neeraj N, Colombel J-F. Loss of response to anti-TNFs: definition, epidemiology, and management. Clin Transl Gastroenterol (2016) 7(1):e135. doi:10.1038/ctg.2015.63
17. Esposito M, Gisondi P, Cassano N, Ferrucci G, Del Giglio M, Loconsole F, et al. Survival rate of antitumour necrosis factor-α treatments for psoriasis in routine dermatological practice: a multicentre observational study. Br J Dermatol (2013) 169:666–72. doi:10.1111/bjd.12422
18. Connor V. Anti-TNF therapies: a comprehensive analysis of adverse effects associated with immunosuppression. Rheumatol Int (2011) 31(3):327–37. doi:10.1007/s00296-009-1292-x
19. Antoni C, Braun J. Side effects of anti-TNF therapy: current knowledge. Clin Exp Rheumatol (2002) 20(6 Suppl 28):S152–7.
20. The Lenercept Multiple Sclerosis Study Group, The University of British Columbia MS/MRI Analysis Group. TNF neutralization in MS: results of a randomized, placebo-controlled multicenter study. Neurology (1999) 53:457–65. doi:10.1212/WNL.53.3.457
21. Richez C, Blanco P, Lagueny A, Schaeverbeke T, Dehais J. Neuropathology resembling CIDP in patients receiving tumour necrosis factor-a blockers. Neurology (2005) 64:1468–70. doi:10.1212/01.WNL.0000158681.29117.8B
22. Sicotte NL, Voskuhl RR. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology (2001) 57:1885–8. doi:10.1212/WNL.57.10.1885
23. Robinson WH, Genovese MC, Moreland LW. Demyelinating and neurologic events reported in association with tumor necrosis factor α antagonism: by what mechanisms could tumor necrosis factor α antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum (2001) 44:1977–83. doi:10.1002/1529-0131(200109)44:9<1977::AID-ART345>3.0.CO;2-6
24. Van Herreweghe F, Festjens N, Declercq W, Vandenabeele P. Tumor necrosis factor-mediated cell death: to break or to burst, that’s the question. Cell Mol Life Sci (2010) 67:1567–79. doi:10.1007/s00018-010-0283-0
25. Martinez TN, Chen X, Bandyopadhyay S, Merrill AH, Tansey MG. Ceramide sphingolipid signaling mediates tumor necrosis factor (TNF)-dependent toxicity via caspase signaling in dopaminergic neurons. Mol Neurodegener (2012) 7:45. doi:10.1186/1750-1326-7-45
26. Probert L, Eugster HP, Akassoglou K, Bauer J, Frei K, Lassmann H, et al. TNFR1 signalling is critical for the development of demyelination and the limitation of T-cell responses during immune-mediated CNS disease. Brain (2000) 123(10):2005–19. doi:10.1093/brain/123.10.2005
27. Smyth MJ, Johnstone RW. Role of TNF in lymphocyte-mediated cytotoxicity. Microsc Res Tech (2000) 50:196–208. doi:10.1002/1097-0029(20000801)50:3<196::AID-JEMT3>3.0.CO;2-9
28. Kim EY, Priatel JJ, Teh SJ, Teh HS. TNF receptor type 2 (p75) functions as a costimulator for antigen-driven T cell responses in vivo. J Immunol (2006) 176:1026–35. doi:10.4049/jimmunol.176.2.1026
29. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov (2010) 9:482–93. doi:10.1038/nrd3030
30. Probert L. TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience (2015) 302:2–22. doi:10.1016/j.neuroscience.2015.06.038
31. Waters JP, Pober JS, Bradley JR. Tumour necrosis factor and cancer. J Pathol (2013) 230(3):241–8. doi:10.1002/path.4188
32. Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ (2003) 10(1):45–65. doi:10.1038/sj.cdd.4401189
33. Naudé PJW, den Boer JA, Luiten PGM, Eisel ULM. Tumor necrosis factor receptor cross-talk. FEBS J (2011) 278:888–98. doi:10.1111/j.1742-4658.2011.08017.x
34. Dong Y, Dekens DW, De Deyn PP, Naudé PJW, Eisel ULM. Targeting of tumor necrosis factor alpha receptors as a therapeutic strategy for neurodegenerative disorders. Antibodies (2015) 4:369–408. doi:10.3390/antib4040369
35. Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell (2003) 114:181–90. doi:10.1016/S0092-8674(03)00521-X
36. Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci (2002) 216:1–7. doi:10.1523/JNEUROSCI.22-07-j0001.2002
37. Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel U. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-d-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem (2004) 279:32869–81. doi:10.1074/jbc.M311766200
38. Eisel ULM, Biber K, Luiten PGM. Life and death of nerve cells: therapeutic cytokine signaling pathways. Curr Signal Transduct Ther (2006) 1:133–46. doi:10.2174/157436206777012057
39. Wajant H, Scheurich P. TNFR1-induced activation of the classical NF-κB pathway. FEBS J (2011) 278(6):862–76. doi:10.1111/j.1742-4658.2011.08015.x
40. O’Donnell MA, Ting AT. RIP1 comes back to life as a cell death regulator in TNFR1 signaling. FEBS J (2011) 278:877–87. doi:10.1111/j.1742-4658.2011.08016.x
41. Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science (1998) 281:1680–3. doi:10.1126/science.281.5383.1680
42. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell (2006) 22:245–57. doi:10.1016/j.molcel.2006.03.026
43. Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol (1998) 8:1001–8. doi:10.1016/S0960-9822(07)00420-4
44. Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol (2001) 21:5299–305. doi:10.1128/MCB.21.16.5299-5305.2001
45. Bigda J, Beletsky I, Brakebusch C, Varfolomeev Y, Engelmann H, Bigda J, et al. Dual role of the p75 tumor necrosis factor (TNF) receptor in TNF cytotoxicity. J Exp Med (1994) 180:445–60. doi:10.1084/jem.180.2.445
46. Fotin-Mleczek M, Henkler F, Samel D, Reichwein M, Hausser A, Parmryd I, et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J Cell Sci (2002) 115:2757–70.
47. Wicovsky A, Henkler F, Salzmann S, Scheurich P, Kneitz C, Wajant H. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1–TNFR2 cooperation. Oncogene (2009) 28:1769–81. doi:10.1038/onc.2009.219
48. Richter C, Messerschmidt S, Holeiter G, Tepperink J, Osswald S, Zappe A, et al. The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol Cell Biol (2012) 32:2515–29. doi:10.1128/MCB.06458-11
49. Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol (2011) 164(4):1079–106. doi:10.1111/j.1476-5381.2011.01302.x
50. Zendedel A, Beyer C, Kipp M. Cuprizone-induced demyelination as a tool to study remyelination and axonal protection. J Mol Neurosci (2013) 51(2):567–72. doi:10.1007/s12031-013-0026-4
51. Luiten PG, Douma BR, Van der Zee EA, Nyakas C. Neuroprotection against NMDA induced cell death in rat nucleus basalis by Ca2+ antagonist nimodipine, influence of aging and developmental drug treatment. Neurodegeneration (1995) 4(3):307–14. doi:10.1016/1055-8330(95)90020-9
52. Denic A, Johnson AJ, Bieber AJ, Warrington AE, Rodriguez M, Pirko I.The relevance of animal models in multiple sclerosis research. Pathophysiology (2011) 18(1):21–9. doi:10.1016/j.pathophys.2010.04.004
53. Ransohoff RM. Animal models of multiple sclerosis: the good, the bad and the bottom line. Nat Neurosci (2012) 15(8):1074–7. doi:10.1038/nn.3168
54. Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov (2004) 3(5):401–16. doi:10.1038/nrd1383
55. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol (2005) 6(12):1191–7. doi:10.1038/ni1276
56. Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol (2013) 13(1):59–66. doi:10.1038/nri3362
57. Chen X, Bäumel M, Männel DN, Howard OM, Oppenheim JJ. Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol (2007) 179(1):154–61. doi:10.4049/jimmunol.179.1.154
58. Nguyen DX, Ehrenstein MR. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF-TNF-RII binding in rheumatoid arthritis. J Exp Med (2016) 213(7):1241–53. doi:10.1084/jem.20151255
59. Miller PG, Bonn MB, McKarns SC. Transmembrane TNF-TNFR2 impairs Th17 differentiation by promoting Il2 expression. J Immunol (2015) 195(6):2633–47. doi:10.4049/jimmunol.1500286
60. Williams SK, Maier O, Fischer R, Fairless R, Hochmeister S, Stojic A, et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One (2014) 9(2):e90117. doi:10.1371/journal.pone.0090117
61. Veroni C, Gabriele L, Canini I, Castiello L, Coccia E, Remoli ME, et al. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol Cell Neurosci (2010) 45(3):234–44. doi:10.1016/j.mcn.2010.06.014
62. Gao H, Danzi MC, Choi CS, Taherian M, Dalby-Hansen C, Ellman DG, et al. Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep (2017) 18(1):198–212. doi:10.1016/j.celrep.2016.11.083
63. Maier O, Fischer R, Agresti C, Pfizenmaier K. TNF receptor 2 protects oligodendrocyte progenitor cells against oxidative stress. Biochem Biophys Res Commun (2013) 440(2):336–41. doi:10.1016/j.bbrc.2013.09.083
64. Blakemore WF, Keirstead HS. The origin of remyelinating cells in the central nervous system. J Neuroimmunol (1999) 98:69–76. doi:10.1016/S0165-5728(99)00083-1
65. Fischer R, Wajant H, Kontermann R, Pfizenmaier K, Maier O. Astrocyte-specific activation of TNFR2 promotes oligodendrocyte maturation by secretion of leukemia inhibitory factor. Glia (2014) 62:272–83. doi:10.1002/glia.22605
66. Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat Neurosci (2001) 4:1116–22. doi:10.1038/nn738
67. Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, et al. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain (2011) 134:2736–54. doi:10.1093/brain/awr199
68. Madsen PM, Motti D, Karmally S, Szymkowski DE, Lambertsen KL, Bethea JR, et al. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J Neurosci (2016) 36(18):5128–43. doi:10.1523/JNEUROSCI.0211-16.2016
69. Karamita M, Barnum C, Möbius W, Tansey MG, Szymkowski DE, Lassmann H, et al. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight (2017) 2(8):e87455. doi:10.1172/jci.insight.87455
70. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol (2012) 8(11):647–56. doi:10.1038/nrneurol.2012.168
71. Fischer R, Maier O, Siegemund M, Wajant H, Scheurich P, Pfizenmaier K. A TNF receptor 2 selective agonist rescues human neurons from oxidative stress-induced cell death. PLoS One (2011) 6:e27621. doi:10.1371/journal.pone.0027621
72. Dolga AM, Nijholt IM, Ostroveanu A, Ten Bosch Q, Luiten PG, Eisel UL. Lovastatin induces neuroprotection through tumor necrosis factor receptor 2 signaling pathways. J Alzheimers Dis (2008) 13(2):111–22. doi:10.3233/JAD-2008-13201
73. Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet (2014) 383(9936):2213–21. doi:10.1016/S0140-6736(13)62242-4
74. Madsen PM, Clausen BH, Degn M, Thyssen S, Kristensen LK, Svensson M, et al. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J Cereb Blood Flow Metab (2016) 36(9):1553–69. doi:10.1177/0271678X15610339
75. Dong Y, Fischer R, Naudé PJ, Maier O, Nyakas C, Duffey M, et al. Essential protective role of tumor necrosis factor receptor 2 in neurodegeneration. Proc Natl Acad Sci U S A (2016) 113(43):12304–9. doi:10.1073/pnas.1605195113
76. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335(6071):936–41. doi:10.1126/science.1214935
77. Read AF, Graham AL, Råberg L. Animal defenses against infectious agents: is damage control more important than pathogen control? PLoS Biol (2008) 6(12):10. doi:10.1371/journal.pbio.1000004
78. Ayres JS, Schneider DS. Two ways to survive an infection: what resistance and tolerance can teach us about treatments for infectious diseases. Nat Rev Immunol (2008) 8(11):889–95. doi:10.1038/nri2432
79. Ayres JS, Freitag N, Schneider DS. Identification of Drosophila mutants altering defense of and endurance to Listeria monocytogenes infection. Genetics (2008) 178:1807–15. doi:10.1534/genetics.107.083782
80. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci U S A (2009) 106:15837–42. doi:10.1073/pnas.0903419106
81. Gozzelino R, Andrade BB, Larsen R, Luz NF, Vanoaica L, Seixas E, et al. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe (2012) 12:693–704. doi:10.1016/j.chom.2012.10.011
82. Soares MP, Teixeira L, Moita LF. Disease tolerance and immunity in host protection against infection. Nat Rev Immunol (2017) 17(2):83–96. doi:10.1038/nri.2016.136
83. Gao W, Zhang L. Coating nanoparticles with cell membranes for targeted drug delivery. J Drug Target (2015) 23(7–8):619–26. doi:10.3109/1061186X.2015.1052074
84. Singh R, Lillard JW Jr. Nanoparticle-based targeted drug delivery. Exp Mol Pathol (2009) 86(3):215–23. doi:10.1016/j.yexmp.2008.12.004
85. Frenz T, Grabski E, Durán V, Hozsa C, Stępczyńska A, Furch M, et al. Antigen presenting cell-selective drug delivery by glycan-decorated nanocarriers. Eur J Pharm Biopharm (2015) 95(Pt A):13–7. doi:10.1016/j.ejpb.2015.02.008
Keywords: tumor necrosis factor alpha, TNFR2, TNFR1, immune tolerance, multiple sclerosis, neurodegeneration
Citation: Pegoretti V, Baron W, Laman JD and Eisel ULM (2018) Selective Modulation of TNF–TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 9:925. doi: 10.3389/fimmu.2018.00925
Received: 31 December 2017; Accepted: 13 April 2018;
Published: 30 April 2018
Edited by:
Magdalena Plebanski, RMIT University, AustraliaReviewed by:
Harley Y. Tse, Wayne State University, United StatesBruce Milne Hall, University of New South Wales, Australia
Copyright: © 2018 Pegoretti, Baron, Laman and Eisel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentina Pegoretti, di5wZWdvcmV0dGlAcnVnLm5s;
Ulrich L. M. Eisel, dS5sLm0uZWlzZWxAcnVnLm5s