 Annelisa M. Cornel
Annelisa M. Cornel Niek P. van Til
Niek P. van Til Jaap Jan Boelens
Jaap Jan Boelens Stefan Nierkens
Stefan Nierkens- 1Laboratory of Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 2Pediatric Blood and Marrow Transplantation Program, University Medical Center Utrecht, Utrecht, Netherlands
- 3Blood and Marrow Transplantation Program, Princess Maxima Center for Pediatric Oncology, Utrecht, Netherlands
Dendritic cell (DC) vaccination has been investigated as a potential strategy to target hematologic malignancies, while generating sustained immunological responses to control potential future relapse. Nonetheless, few clinical trials have shown robust long-term efficacy. It has been suggested that a combination of surmountable shortcomings, such as selection of utilized DC subsets, DC loading and maturation strategies, as well as tumor-induced immunosuppression may be targeted to maximize anti-tumor responses of DC vaccines. Generation of DC from CD34+ hematopoietic stem and progenitor cells (HSPCs) may provide potential in patients undergoing allogeneic HSPC transplantations for hematologic malignancies. CD34+ HSPC from the graft can be genetically modified to optimize antigen presentation and to provide sufficient T cell stimulatory signals. We here describe beneficial (gene)-modifications that can be implemented in various processes in T cell activation by DC, among which major histocompatibility complex (MHC) class I and MHC class II presentation, DC maturation and migration, cross-presentation, co-stimulation, and immunosuppression to improve anti-tumor responses.
Introduction
Although the overall survival rates of patients with hematologic malignancies have significantly increased in the past decades, the 5-year survival of certain acute leukemias, such as acute myeloid leukemia (AML) is still unsatisfactory due to high relapse risk (1–4). Currently, the only curative treatment consists of intense chemotherapy followed by hematopoietic cell transplantation (HCT), but only about 30% of candidates eligible for HCT transplantation have a human leukocyte antigen (HLA)-identical sibling as a donor for matched transplantation. Alternatively, bone marrow from unrelated volunteer donors could be used; however, this is limited by strict HLA-matching criteria, because of higher risks of graft-versus-host disease (GVHD), and donor availability.
Umbilical cord blood (UCB) transplantation has advantages because of its prompt availability from UCB banks, the possibility of HLA-mismatched transplantations, a lower risk of acute and chronic GVHD, and a potential higher graft-versus-leukemia effect (5–8).
Individualized dosing and timing of chemo and/or serotherapy improves overall survival of transplanted patients with hematologic malignancies after cord blood transplantation (9, 10). Cord blood T cells have shown the ability to rapidly reconstitute the immune system (9), and can mediate enhanced anti-tumor effects when compared with adult peripheral T cells (11). In addition, cord blood CD8+ T cells have shown to exhibit stronger proliferation potential and function after antigen-specific stimulation (12). The relatively low survival rate of patients with hematologic malignancies underlines the relevance to investigate novel potential effective therapies in the context of UCB transplantation to treat AML or other hematologic malignancies.
Tumor-associated antigen (TAA)-specific immunotherapy to prime the TAA-specific T cells against the leukemia to consequently induce remission has been thoroughly investigated. Four decades of research revealed the central role of dendritic cells (DCs) as a link between innate and adaptive immunity, and thereby its essential role in the control of both immune tolerance and immunity (13). The antigen presentation machinery of DCs is exploited in cellular vaccination strategies to initiate an endogenous anti-tumor response (14). The rationale for this approach is the generation of TAA-specific cytotoxic T lymphocyte (CTL) responses to specifically eradicate tumor cells and to generate immunological memory to control potential future tumor relapse (15). However, DC vaccine trials have only sporadically shown clinical responses. Insufficient DC maturation, suboptimal antigen presentation, co-stimulation, migration, or impaired initiation of anti-TAA T cell responses could be inherent to the cultured DC subset, but may also be influenced by the inhibition of immune responses by the tumor microenvironment (14, 16). Hence, efficacy of DC vaccination strategies can be improved by state-of-the-art genetic modification tools, such as messenger RNA, adenoviral and lentiviral vectors, and gene-editing techniques to enhance processes in DC activation of T cells (15, 17) and consequently boost immune responses. In this review, we will address modification of phenotypes and function of DCs, including cord blood CD34-derived DCs, to optimize the anti-tumor response to protect for relapses after HCT.
DC Subsets Eligible for Modification
Although thoroughly investigated, there is still no consensus about the most optimal DC subset to use to induce optimal TAA-specific T cell responses (18). Circulating peripheral blood DCs are difficult to isolate, hence monocyte-derived DCs (moDCs), generated from peripheral blood mononuclear cells are the most commonly used. These cells are generated from monocytes by use of granulocyte macrophage colony stimulating factor and interleukin (IL)-4 (18). Although moDC-derived vaccines are reported to be safe, clinical responses have only sporadically been observed (15, 17).
Research investigating different DC subsets pointed to differential subsets (such as conventional and plasmacytoid DCs) and functionalities (19), which could be of importance to induce favorable immune responses. The advantage of using primary DCs is that they can be promptly isolated from blood, avoiding long differentiation incubation periods before administration to the patient, thereby making this strategy suitable for standardization for multicentre trials (20). However, the differentiated status of these cells is also a drawback, as this limits expansion of the cell population. As a result, large numbers of primary DC may be required to provide effective therapeutic dosing.
Another commonly used approach is to produce DCs from CD34+ hematopoietic stem and progenitor cells (HSPCs) (21, 22), which have an extensive proliferation capacity to generate antigen presenting cells (APCs) with a primary DC phenotype (23) and the capacity to induce robust anti-tumor T cell responses. These cells are distinct from moDCs (24–27), and more resemble conventional DC or Langerhans resembling cells (28) that induce stronger anti-tumor T cell responses compared with moDCs (29). In the setting of cord blood transplantation after chemotherapy in hematologic malignancies, CD34+ HSPCs can be extracted from 20% of the remaining unit that is not transplanted, and developed into an effective DC vaccine, that can be modified at different stages of the manufacturing process, which will be discussed below.
Vaccination with UCB CD34-derived DCs has been performed in clinical trials to treat patients with melanoma and showed TAA-specific responses in some patients (23, 30). The ex vivo culturing phase to generate CD34-derived DCs provides a unique opportunity to enhance efficacy through genetic modification. Principally, the expansion phase of the protocol could be extended to 2 weeks and this does this not affect DC maturation (26). This indicates that this two-step protocol allows opportunities to modify the CD34-derived DCs at the early stage as well as during the later stages of the protocol, as compared with DCs generated from other precursor subsets.
Modulating TAA-Loading and Major Histocompatibility Complex (MHC)-I Presentation to Enhance DC Efficiency
Tumor-associated antigens are ideally over expressed on malignant cells and are simultaneously not expressed on healthy tissues or contain mutations leading to neo-antigens recognizable to T cells. Hence, a commonly used TAA is the oncoprotein Wilms’ tumor-1 (WT1), which has been ranked the number one cancer vaccine target antigen (31). WT1 is a zinc finger transcription factor with a well-established oncogenic role in WT1 overexpressing malignancies (32). WT1 overexpression is observed in the majority of acute leukemias (~90% of pediatric AML cases), as well as various solid tumors (33), making WT1 an obvious vaccine target. Despite its physiological expression in hematopoietic tissue–limited expression in the urogenital–and central nervous system (34), it has been shown that tumor overexpression of WT1 can be targeted without considerable safety concerns (35, 36). Several recent early-phase anti-WT1 DC vaccine clinical trials in multiple cancer types reported a correlation between anti-WT1 CTL responses and clinical response (35, 37, 38), showing its potential as a therapeutic strategy.
The most commonly used methods to present antigen are delivery of peptide pools or mRNA to express the tumor antigen-target, which result in the ability to transiently load DCs with antigen. An advantage to deliver mRNA is that it prevents HLA-restrictions and invasive tumor tissue isolation from patients. Alternatively, full-length WT1 mRNA can also be combined with a WT1 peptide pool to enhance its potential (14, 39). Two main modification strategies have been reported to potentially optimize TAA-loading and MHC-I presentation of WT1 epitopes: increasing translational efficiency or increasing proteasome targeting of the TAA. Codon-optimization of nucleotide sequences is commonly used to enhance expression of a transgene to increase the amount of transgene product, which could be a limiting factor in vaccinations strategies. Algorithms include selection of more commonly used codons to improve translation, but can also include features addressing transcription, mRNA processing and stability as well as protein folding. For the delivery of mRNA, transcription can be excluded as a relevant parameter for improvement, but all others may be useful. It was reported that codon-optimization of the human papillomavirus (HPV) E7 oncoprotein sequence resulted in much higher protein translation and induced CD8+ T cell responses to cryptic epitopes not harbored by wildtype E7 (40). Codon-optimization could, therefore, confer additional advantages then using native mRNA sequences.
Benteyn et al. attempted to optimize translational efficiency of full-length WT1 mRNA (41), but there was no significant advantage of the codon-optimization detected. However, transgene expression was optimized using the pST1 RNA transcription plasmid to generate in vitro synthesized mRNA with enhanced translational properties (42). This modification resulted in doubling of the interferon-γ (IFN-γ) responses in a T cell clone. Another feature employed to improve antigen presentation in both MHC-I and MHC-II was the inclusion of endosomal or lysosomal targeting sequences fused to the antigen sequence (43, 44). In particular, the fusion of the C-terminus of LAMP/DC-LAMP to the WT1 mRNA enhanced the IFN-γ also in a T cell clone (41) by increasing both MHC-I presentation and cross-presentation of WT1 peptides. These modifications only require adaptation of the WT1 mRNA sequence, which makes it relatively easy and efficient to implement in a DC vaccine.
Hosoi et al. attempted to optimize proteasome targeting to increase protein degradation and enhance presentation of full-length TAA by triggering co-translational polyubiquitination (45). This triggering of co-translational ubiquitination of the TAA resulted in more efficient priming and expansion of TAA-specific CTLs (45).
To further improve DC vaccination multi-epitope delivery may be beneficial for enhanced CTL activation, e.g., WT1 for AML treatment can be combined with proteinase 3, preferentially expressed antigen in melanoma, telomerase reverse transcriptase, or FLT3-internal tandem duplication (46) for maximal responses. In a multi-epitope vaccine combining multiple myeloma special antigen-1 and Dickkopf-1 to treat multiple myeloma enhanced responses were observed (47).
Viral vectors can also be used to deliver antigen. DCs are highly amenable to lentiviral vector transduction (48). A study using mouse DCs comparing lentiviral vectors that stably integrate into the host genome and provide constant transgene antigen expression to mRNA electroporation showed that lentiviral vector delivery enhanced IFN-γ responses to MAGE-A3 epitopes (49). In the context of UCB-derived DCs, lentiviral vectors could potentially be very useful, since <5 × 106 CD34+ progenitors can be used for the initial transduction and form the basis for expansion of large number of matured DCs (>500 × 106). Another more recent approach uses lentiviral protein transfer vectors for targeting transfer directly into APCs and inducing cytotoxic T cell responses, which could also be used for ex vivo delivery (48).
More research is necessary to confirm that the above mentioned modifications could be generally applied to other TAAs or whether this enhances efficacy of CD34-derived DC vaccines.
Modulating DC Maturation to Improve DC Efficiency
Although consensus is reached that DC vaccines should contain mature rather than immature DCs, there is no consensus about how to polarize and mature DCs to cause optimal anti-tumor responses (50). In 1997, Jonuleit et al. showed that incubation of immature DCs with a cocktail of IL-1β, TNFα, IL-6, and PGE2, similar to the GMP-grade available CYTOMIX, resulted in induction of fully matured DCs that seemed to be optimal for generation of IFN-γ producing CD4/CD8+ T cells (51), but very limited efficacy was observed. It is questionable whether to include PGE2 as it decreases the expression of IL-12p70 (50), a factor important in induction of tumor-specific Th1 T cells and CTLs facilitating tumor rejection in mouse models (52).
It is reported that DC maturation cocktails containing IFN-γ instead of PGE2 [the α-type-1-polarized DC cocktail (αDC1)] increases IL-12p70 levels in vitro and in vivo boosting TAA-specific CTL levels 40-fold in vitro in melanoma (53, 54). Superiority of αDC1-induced maturation was also observed in chronic lymphocytic leukemia assays in vitro (55). Similarly, addition of IFN-γ to the CYTOMIX maturation cocktail can increase IL-12p70 production upon CD40 stimulation in WT1 expressing DCs (26).
Another strategy to mature DCs would be to introduce maturation agents with gene therapy. A major advantage of this approach is that DCs can be used within a few hours after delivery of maturation stimuli for vaccination, whereas culturing in maturation agents requires a 24-h incubation period (56). This incubation period in vitro potentially leads to DC exhaustion and dampening of the immune response, as shown by Bonehill et al. (56). Single introduction of constitutionally active toll-like receptor 4 (caTLR4) (56, 57) and CD40L (56–58) in immature DCs has shown to induce potent DC maturation, including IL-12p70 production, and both stimuli also act synergistically to superior DC maturation. Melan-A TAA-primed DCs co-electroporated with caTLR4, CD40L, and CD70 mRNA showed an even >200-fold increase in Melan-A specific CTL responses when compared with CYTOMIX matured DCs (56). To date, direct comparisons of this strategy with αDC1-induced maturation of DCs are lacking.
The combination of these three proteins is known as the TriMix strategy, which was developed at the Free University of Brussels, introducing the danger signal caTRL4, the co-stimulatory protein CD40L both to stimulate maturation, and a co-stimulatory protein involved in early T cell activation (CD70) (41, 56, 57, 59, 60). An interesting factor in this strategy is that the DCs mature after electroporation of these factors, eliminating the need of DC incubation with maturation cocktails. A phase-II clinical trial in advanced melanoma showed that combining TriMix-matured moDCs presenting melanoma-associated antigens with ipilumab, an antagonistic CTLA4 antibody, resulted in a 6-month disease control rate of 51%, with an overall tumor response rate of 38% (59). This strategy nicely shows the potential of combining multiple modifications to improve tumor-immunity of DC vaccination.
In the TriMix DCs, maturation of DCs is maximized to improve activation, and polarization of T cells to increase tumor-immunity. However, for an optimal result, it is widely suggested that immunosuppression should be counteracted as well. This is partly established in the TriMix trial by combining the TriMix-matured DCs with ipilimumab, as it inhibits the co-inhibitory effect of the T cell membrane protein CTLA4 on CD80/CD86/CD28 co-stimulation (59).
Modulating DC Migration to Enhance DC Efficiency
There is no consensus about the most efficient administration route of DC vaccines to migrate to the draining lymph nodes (16). Administration of 111-indium labeled moDCs into patients revealed that less than 5% of the intradermally injected mature moDCs reach the draining lymph nodes (61). A major player in DC migration to the lymph nodes is the C-C motif chemokine receptor 7 (CCR7) (62). Migration to the lymph node is stimulated upon interaction with its ligand, the chemokine C-C motif ligand (CCL21) (63). Adenoviral transduction of DCs with CCR7 (64) and CCL21 (65, 66) showed an ~5.5-fold increase in DC lymph node accumulation, and enhanced tumor rejection and T cell priming in mice in vivo, respectively. This could not only increase the effectiveness of the vaccine, but may also reduce the required dose, hence, the efforts and costs associated with vaccine preparation (64). Based on these results, a GMP-grade CCL21 gene-modified monocyte-derived DC vaccine was developed (67), subsequently used in a phase-I clinical trial with non-TAA loaded CCL21 expressing DCs, which triggered TAA-specific T cell responses and enhanced CD8+ T cell tumor infiltration in a subset of patients with non-small cell lung carcinoma (68). Interestingly, CCL21 excretion attracts naive T cells and addition of TAA peptide pools and maturation of DCs may further increase the therapeutic effect. CCL21 could also be applied by mRNA delivery for transient expression similar to adenoviral vectors.
Alternatively, strategies to reduce DC tissue retention could be applied to increase DC migration by disrupting the homing factor E-cadherin (69) (or its positive regulator TGF-β) inducing upregulation of CCR7 (70). Downregulation of E-cadherin upon pro-inflammatory signaling (via TNFα, LPS, and IL-1β) further strengthens the hypothesis of involvement of E-cadherin in DC migration (69). The use of small interfering RNAs to downregulate E-cadherin expression on DCs and its effect on migratory function and immune stimulation may be an interesting option. TGF-β is also a known immunosuppressant of DCs, which makes interference of its expression a potential strategy to improve DC vaccination (71).
MHC-II Cross-Presentation to Enhance DC Function
Major histocompatibility complex-II antigen presentation is required to establish long-term memory anti-tumor immunity through stimulation of CD8+ T cells by CD4+ T cells inducing strong clonal expansion, cytokine production, tumor cell lysis, and T cell memory (72–75). MHC-II knockout DCs were able to generate potent anti-tumor CTL responses in vivo, however, without subsequent establishment of a memory anti-tumor response. Therefore, a critical factor in the development of a successful DC vaccine is the ability to present the TAA in both MHC-I and MHC-II context (39).
Full-length TAA mRNA translates into proteins ensuring the presence of MHC-I and MHC-II TAA epitopes, without the requirement of algorithms to predict epitopes per HLA-subtype (14, 39). To further boost this response, a broad TAA peptide pool can be administered in addition to the mRNA electroporation or viral vector delivery. To improve MHC-II presentation of TAA, antigen has also been targeted to endolysosomal compartments to try to improve MHC-II antigen presentation, but this resulted in increased numbers of regulatory T cells (Tregs) and attenuation of tumor immunity (76).
Many studies exploited strategies that link small epitopes to proteins increasing their likelihood of MHC-II presentation (77), however, these epitopes are difficult to predict, are MHC-II restricted, and vary per HLA-subtype and antigen. Therefore, targeting of full-length antigens to the MHC-II pathway is more desirable. Two main MHC-II pathway targeting strategies can be distinguished. The first strategy links the TAA of interest to the cytoplasmic tail of residential endolysosomal proteins, which contains the information for transport to the endolysosomal compartment. Residential endolysosomal proteins tested for this strategy include DC-LAMP (41, 43), LAMP1 (43, 44), and LIMPII (78). The second strategy entails linking of the TAA of interest to the MHC-II associated invariant chain (Ii), a protein important in MHC-II conformational regulation, thereby targeting the TAA to the endolysosomal compartment (43, 79). All studies, irrespective of the endolysosomal protein used, concluded that the increased cross-presentation enhanced CD4+ and CD8+ T cell activation and increased anti-tumor immunity in vitro and in vivo. Direct comparison of strategies using DC-LAMP, LAMP1, and Ii showed that DC-LAMP and LAMP1 have more pronounced effects than using Ii (43). Interestingly, no clinical studies incorporated these cross-presentation tools into vaccines, even though some cited papers are over 20 years old.
Modulation of Co-Stimulation to Boost DC Function
A T lymphocyte requires three signals to become fully activated (80), of which co-stimulation is provided by interaction between co-stimulatory molecules expressed on the DC and T lymphocyte. Lack of DC maturation and subsequent co-stimulation induces tolerance against the presented antigen, making these processes of vital importance in the generation of an anti-tumor response. Several co-stimulatory interactions between DCs and T cells have been explored, including CD40/CD40L, 4-1BB/4-1BBL, OX40/OX40L, CD80/86/CD28, CD27/CD70, and GITR/GITRL.
The interaction between CD40 and CD40L, expressed on DCs and T cells respectively, is one of the most potent DC activating signals (56, 81). Modifications to this axis have, therefore, been widely studied to optimize DC vaccination (41, 56–60, 82). Introduction of CD40L into DCs provides autonomous maturation and co-stimulation of DCs (83). In this way, “licensing” of DCs through CD40L interaction with CD4+ Th1 T cells is not required for initiation of a TAA-specific CTL response, and these DCs elicited superior anti-tumor immunity and inhibition of pre-existing tumor growth via induction of a TAA-specific CD4+/CD8+ anti-tumor response in vitro (56–58, 83) and in vivo (41, 82). In addition, introduction of OX40 (84), 4-1BB (85, 86), GITRL (87), and CD70 (41, 56, 57, 59) in DCs is all reported to increase the anti-tumor effect in vitro and in vivo. All the approaches used mRNA to deliver the co-stimulatory signals.
Upon maturation, OX40L expression is induced in DCs, a ligand of the T-lymphocytic membrane protein OX40. Upregulation of OX40L is stimulated by PGE2 (88), but PGE2 also downregulates IL-12p70 (50, 52). Therefore, the observed positive effect of OX40/OX40L co-stimulation on tumor rejection (89, 90), through CD4+/CD8+ T cell proliferation, prevention of T cell death, and prevention of tolerance induction, is caused by an unknown mechanism independent of IL-12p70 upregulation (84, 88, 91). Dannull et al. showed that targeting OX40L as a downstream factor of PGE2 potentially circumvents the PGE2-mediated attenuation of DC function, while utilizing its IL-12p70 independent immunostimulatory capacity in DC vaccination (84).
Another co-stimulatory interaction, 4-1BB/4-1BBL, plays a key role in activation, proliferation, and memory development of CTLs (92). 4-1BB is exploited in second and third generation chimeric antigen receptors in CTLs to provide long-lasting activation potential. 4-1BBL mRNA introduction in HER2/neu TAA expressing DCs resulted in an increased TAA-specific CTL response in vitro (85), which was also supported by studies using agonistic anti-4-1BB antibodies in vitro and in vivo (86, 93). Similar results were observed in the context of HIV-specific T cell responses (86).
A less pronounced effect has been reported for GITR/GITRL co-stimulation, which enhances CD4+/CD8+ T cell responses, while inhibiting Treg-mediated immune suppression (87, 94). A new approach to introduce heavy and light chains of an agonistic anti-GITR antibody in DCs could stimulate this pathway (87). Combining vaccination of these anti-GITR-secreting DCs with TAA-presenting DCs resulted in an increased CTL response, and inhibition of sensitivity to Treg mediated immune suppression, thereby increasing anti-tumor immunity in vitro and in vivo. This approach may cause less systemic adverse effects, while maintaining the anti-tumor response (87).
Finally, CD27/CD70 interaction promotes clonal expansion of primed CD4+/CD8+ T cells, mostly via supporting survival of primed T cell clones (87). The constitutive expression of CD27 on T cells, by contrast to the other T-lymphocytic co-stimulatory molecules, indicates an important role during early T cell priming, making its ligand an interesting molecule to modify in DC vaccination. Keller et al. showed that constitutive expression of CD70 in steady-state immature DCs loaded with TAA can overcome peripheral resistance (95), and resulted in a robust effector and memory CTL response in vitro and in vivo, even in absence of CD4+ T cells (96).
Multiple papers reported the beneficial effects of combining autonomous DC maturation via CD40L introduction with factors enhancing T cell activation through 4-1BBL (86) and CD70, in combination with caTRL4 (41, 56, 57, 59, 60), respectively, on tumor immunity. Introduction of these co-stimulatory provide multiple opportunities to enhance tumor immunity through incorporation into DC vaccines.
Interferance with Co-Inhibitory and Immunosuppressive Pathways to Enhance DC Function
Dendritic cells should live long enough to generate a potent anti-tumor response, but have a physiological short lifespan (14). Moreover, remaining activatory DCs presenting TAA in MHC-I context are killed by activated TAA-specific CTLs, which probably also is a physiological mechanism to prevent exaggeration of immune responses (97–99). A major concern in DC vaccination is that DC injection in TAA-primed mice results in DC elimination before reaching the draining lymph node (100, 101). DC elimination by CTLs can even be used as a measure for effective cytotoxic response (100, 101). DC apoptosis is triggered physiologically, as well as by the tumor microenvironment. Inhibiting DC apoptosis can prolong the DC lifespan after siRNA-mediated silencing of the pro-apoptotic proteins BAK/BAX (97, 102), BIM (98), and PTEN (99) in vitro and in vivo, which all resulted in more efficient TAA-specific CTL responses. Disadvantages inherent to modifications of pro-apoptotic proteins are the potential oncogenicity, restricting its use to temporary silencing strategies, e.g., siRNAs.
A second strategy is to inhibit tolerogenic DC development to prevent induction of anergic T cells. Silencing of several factors has been proposed, including suppressor of cytokine signaling 1 (SOCS1), IL-10, IL-10R, and TGF-βR. SOCS1 is an inducible negative feedback inhibitor of the JAK/STAT pathway and thereby negatively regulates expression of multiple cytokines, including IFN-γ, IL-2, IL-6, IL-7, IL-12, and IL-15 (103). SOCS1 deficient DCs are reported to be extremely hyperresponsive to IL-4 and IFN-γ and cause abnormal accumulation of antigen-specific T cells (104). Vaccination with HPV16mE7 pulsed, shRNA-mediated SOCS1-silenced DCs showed significantly improved anti-tumor effects compared to non-SOCS1-silenced controls in vitro and in vivo (103).
The most well-known immunosuppressive cytokines are IL-10 and TGF-β, produced by Tregs, among others, to induce DC tolerance and anergic T cells (105). The fact that high serum levels of both IL-10 and TGF-β are correlated with poor prognosis in several types of cancer indicates an interesting role of inhibition of their expression or responsiveness to their presence (71, 106). As IL-10 can be produced by DCs (107), one way to decrease its effect is to silence IL-10 expression by DCs. However, as IL-10 is also produced by other cell sources, it is probably more effective to knockout its receptor, IL-10R (106, 108), or a combination of both (109). Both studies evidently report benefits on DC maturation and anti-tumor effects in vitro and in vivo and suggest that clinical translation of this will greatly enhance DC vaccination potency. A similar effect was observed TGF-β receptor (TGF-βR) was silenced (71, 108). Ahn et al. tested the individual as well as the combined potency of siRNA-mediated silencing of IL-10R, TGF-βR, PTEN, and BIM (108). IL-10R silencing initiated the strongest individual CTL response, followed by TGF-βR. Furthermore, a cocktail combining IL-10R and TGF-βR siRNAs generated the strongest overall CTL response in vitro and in vivo.
A third strategy aims to decrease DC-mediated T cell apoptosis through co-inhibitory signals, e.g., programmed cell death 1 (PD-1) interaction with its ligand (PD-L1), which is widely described as one of the most potent immunoinhibitory interactions (110). PD-1/PD-L1 interaction is known to inhibit T cell proliferation, survival and effector function, induces apoptosis of tumor-specific T cells, and promotes Treg differentiation as well as resistance of tumor cells to CTL attacks (111). PD-1 expressing TAA-specific T cell function is inhibited by tumor PD-L1 expression, as well as by tumor-induced PD-L1 expression of DCs (111). Advanced clinical trials with PD-1 and PD-L1 antibodies show very promising results in preventing axis signaling in non-small cell lung cancer, indicating the potential that blocking this pathway enhances anti-tumor immunity (112). Silencing of PD-L1, on its own (113) or in combination with its phagocyte-restricted relative PD-L2 (114), shows augmented ex vivo TAA-specific CTL responses, which is also confirmed in vivo (115). Moreover, combined silencing of PD-L1 and IL-10 in DC vaccination showed even stronger induction of anti-tumor responses in vitro and in vivo (106) indicating the potential of combining DC modifications in maximizing anti-tumor responses.
Next to PD-1/PD-L1, indoleamine 2,3-dioxygenase (IDO) could have similar effects on T lymphocytes. IDO can be secreted by DCs and depletes the microenvironment from tryptophan, an essential amino acid required for T cell proliferation and survival (116). Furthermore, various tryptophan metabolites are directly immunosuppressive to T cells. Moreover, IDO-expressing cells are able to differentiate naive T cells into Treg cells, thereby further suppressing anti-tumor immunity. The ability to produce IDO depends on the DC subset, and signals present in the tumor microenvironment that can contribute to the amount of IDO produced (117). IDO upregulation was clearly shown in DCs used for vaccination 24 h after maturation in melanoma patients, indicating the potential relevance of IDO silencing (116). Several studies have indicated decreased tumor sizes, reduced CD4+/CD8+ T cell apoptosis, enhanced T cell proliferation and CTL activity, and decreased Treg cell numbers upon IDO silencing, which was confirmed in vitro, in vivo as well as in patient studies (116, 118).
Nonetheless, immunosuppression of the DCs by factors like IL-10, TGF-β, PD-1, and IDO is ignored in many studies, including the TriMix trial, and may cause substantial downregulation of the anti-tumor response. Especially IL-10 and PD-1 are widely reported to be important inhibitors of immune responses, making these proteins (or their ligands) interesting targets to silence. Silencing of PD-L1 in DCs is expected to cause T cell priming and activation. In the case of solid tumors, PD-L1 is often also expressed by the tumor itself, and may locally provide inhibitory signals affecting these primed T cells. In that way, effector T lymphocyte function can still be inhibited by PD-L1 binding to the T cell membrane protein PD-1. However, the remaining tumor burden in most treated AML patients is relatively low; hence, this might turn out to be less of an issue in AML therapy. The application of DC vaccine delivery during the early stages of immune reconstitution may significantly induce priming to eliminate residual AML blasts effectively. It is expected that the generation of DCs from a UCB will take approximately 4 weeks to generate. Thereafter, the initial DC vaccine can be infused into the patient, followed by multiple DC injections to further boost anti-tumor responses of de novo generated T cells. In the future, an interesting strategy might be to add TAA-specific PD-1 knockout effector T cells to the DC vaccine as well, thereby potentially stimulating and expanding these gene-modified T cells to boost anti-tumor responses. A head-to-head comparison of silencing strategies of these proteins in CD34-derived DCs is needed to select the most promising to overcome immunosuppression. However, the use of siRNA in this application may not be as effective as techniques to permanently eliminate expression, because these cells are heavily replicating.
An overview of the numerous modifications tested on DCs is summarized in Table 1.
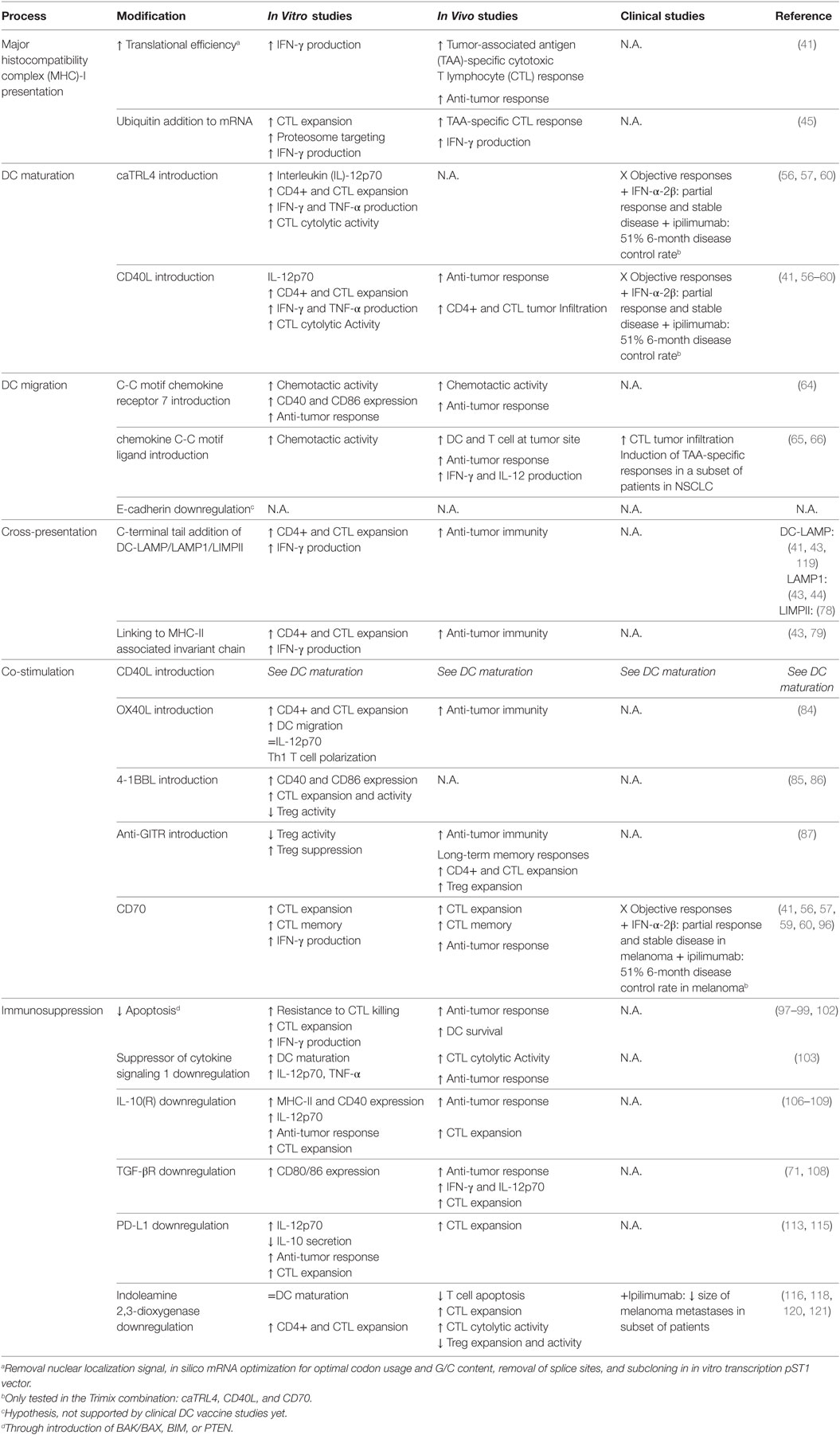
Table 1. Dendritic cell (DC) modifications to enhance anti-tumor induced immunity.
Novel Techniques to Modify CD34-Derived DCs to Potentially Improve Potency
Numerous phase-I DC vaccination-based clinical trials have confirmed the safety of using immature, mature, and TAA-expressing DC vaccines (15, 17). The use of mRNA is relatively safe, because of the temporary expression of the antigen, DC maturation signal or co-stimulatory domain. However, expression cannot be restricted to certain cell types if that is required. Integrating viral vectors may provide longer expression of molecules of interest, but has the risk to potentially cause upregulation of proto-oncogenes (122). Since DCs are generally short-lived this risk may be minimal if the genetic alterations are applied close to application into the patient. Risks of insertional oncogenesis may be increased if CD34+ progenitors are genetically altered before expansion, differentiation, and maturation, particularly because these cells are actively dividing.
The use of siRNAs in DC vaccines is promising, and has shown potential use to reduce expression of co-inhibitory signals in moDCs. Efficiency in UCB-derived DCs has not been shown yet, but may be hampered by the loss of inhibitory ability of siRNAs in cycling cells. This may require precise fine-tuning of delivery of the siRNAs to obtain effective reduction of genes of interest.
The CD34+ expansion phase of the two-step protocol (26) provides a unique environment to modify the DCs to enhance treatment efficiency. However, it is important to carefully select the factors to be removed or introduced in this phase, as this might induce differentiation or decreased proliferation of CD34+ HSPCs. Gene-editing tools to permanently eliminate expression have been used for more than a decade. Initially, zinc finger nucleases were genome sequence specific with relatively low efficiency and toxicities in hematopoietic cells. Gene-editing tools that cause insertions and deletions on a genomic level have not been applied to DCs. These techniques have been mainly used on T cells. In particular, Transcription activator-like effector nucleases mediated gene-editing and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system are used to eliminate expression of the T cell receptor or co-inhibitory signal PD-1 (123).
More recently, Gundry et al. showed efficient knockout of genes in CD34+ HSPCs (~75%) by CRISPR/Cas9 (46). They show that their strategy to electroporate CD34+ HSPCs with Cas9/sgRNA ribonucleoprotein (RNP) complexes causes efficient CRISPR/Cas9 targeting of up to four sgRNAs. No major effects of gene-editing were observed on viability and proliferation capacity. On the other hand, CRISPR/Cas9-mediated targeting of multiple targets at the same time might cause genomic translocations (124), which are potentially cytotoxic.
This strategy permits for efficient knockout of one, two, or more factors involved in immunosuppression (e.g., PD-L1, IL-10, IL-10R, TGF-β, TGF-βR, and IDO). Strikingly, Gundry et al. also reported that this method allows for efficient homology-directed repair gene-editing (125). In this way, an expression cassette containing WT1 cDNA could even be integrated at one of the gene-editing target sites, allowing for constitutive WT1 expression. Combining the TriMix strategy with gene-edited DCs could potentially be a very potent combination.
The recent advances of CRISPR/Cas9 gene-editing in CD34+ HSPCs make this strategy more efficient, commonly applicable, and technically feasible to include in DC vaccines. If the co-inhibitory genes do not affect cell-cycling or viability, gene-editing tools could be very valuable in creating more potent off-the-shelf CB-derived DC vaccines.
The major risks of CRISPR/Cas9 gene-editing are the potential off-target genome cleavage sites it can create (124). Fu et al. showed that off-target effects can be observed at sites that differ by five nucleotides from on-target sequences, indicating that this might cause efficient gene-editing of off-target sites in CRISPR/Cas9 modified cells (124). CD34+ HSPCs are highly proliferative cells and off-target cleavage might promote tumorigenesis. This risk can be reduced by using Cas9-gRNA RNP complexes rather than using mRNA or plasmids to deliver Cas9, thereby limiting their time-frame of action. Kleinstiver et al. showed that mutating four Cas9 amino acids important in DNA binding energy almost completely diminishes the off-target risk of CRISPR/Cas9, while maintaining its on-target effect (126). This indicates that switching to this mutated version of Cas9 (spCas9-HF1) will potentially further increase CRISPR/Cas9 safety.
It is important to note that, even though studies report extremely low incidences of off-target mutations with wildtype Cas9 in CD34+ HSPCs (127), more research is required to develop accurate off-target site prediction tools. Many studies report low off-target effects, but based on this in silico predicted off-target sites rather than on whole-genome sequencing. Hence, whole-genome sequencing of gene-edited cells should be performed to improve the off-target prediction algorithms.
It is also reported that multiplexing CRISPR/Cas9-mediated gene-editing targeting more than one target may result in genomic translocations (128). Poirot et al. performed CRISPR/Cas9 gene-editing in duplex and reported a translocation frequency ranging from 10−4 to 2 × 10−2 (128). After 38 days of culturing translocation frequencies remained stable or reduced, indicating that these translocations are safe and did not cause proliferative advantages. It is very important to assess the translocation frequency and the consequence of these translocations per specific gRNA sequence and the downstream effects on highly proliferative CD34+ HSPCs, and whether this may cause gene-edited related tumorigenicity is yet unknown.
When evaluating the in vivo mouse studies performed to assess DC vaccination, two main strategies could be distinguished. Most studies test the DC vaccines on a complete murine background (wildtype mice, with murine tumors and murine DCs) (87, 96, 120). Inherent to these mouse studies is that translation can be difficult due to interspecies differences. Another option is the use of humanized mouse models, e.g., NOD/SCID or more severe immune compromised NOD/SCID gamma mice that allow introduction of human DCs, TAA-specific CD8+ T cells, and human tumor cells (115). However, this also has its limitations, as these models lack the presence of interaction with human immune cells that could contribute to tumor immunity. The translation of gene-modified DCs to clinical application could be improved by the use suitable mouse models with the humanized immune systems (129).
To summarize, DC vaccination has a proven track-record of safety, but addition of genetic modifications could introduce some safety concerns that need to be addressed. The short lifespan of DCs to generate tumor immunity should improve safety of using these cells, which reduces the likelihood that DCs acquire the ability to divide uncontrollably.
An Off-The-Shelf DC Vaccination Approach
CD34-derived DC can be used as a basis to develop personalized cellular vaccines. This strategy is very promising in combination with UCB transplantation. By using the same UCB-unit the risk of adverse effects is significantly decreased by preventing mismatching. Nonetheless, the personalized nature makes this strategy laborious, relatively expensive and requires automated systems to obtain consistent high quality products. The generation of an off-the-shelf product could make this approach more cost-effective and potentially more suitable for standardization for multicentre trials.
Off-the-shelf DC vaccination products are still in their infancy, and more research and technical advances are needed to be able to generate more effective gene therapy products that have a proven quality ready for infusion into cancer patients.
Conclusion and Outlook
The sporadically observed clinical responses indicate the necessity to improve DC vaccinations. Literature suggests that intervening in early DC maturation and activation can cause a cascade-like reaction that eventually also improves downstream activatory processes. It is also widely reported that the immunosuppressive tumor microenvironment is still able to downregulate the most potently activated DCs. Hence, combining modifications of early DC activation processes, such as caTRL4, CD40L, CD70 with elimination of immunosuppressive signaling, such as IL-10R and PD-L1, may drive optimal anti-tumor T cell responses by maximizing both co-stimulatory/co-inhibitory ends of the spectrum.
Tumor-associated antigens can be delivered by optimized mRNA sequences for efficient processing and MHC-I and MHC-II presentation, which could incorporate DC-LAMP C-terminal sequences, ubiquitination or mRNA transcription from optimized transcription vectors to be electroporated in CD34+ derived DCs.
The use of CD34+ HSPCs to generate UCB-derived DCs provides an opportunity during the expansion/differentiation phase to manufacture gene-modified cellular products. Recent progress using state-of-the-art gene therapy vectors, such as self-inactivating third generation lentiviral vectors, that are used in clinical trials to treat inherited diseases and in T cell immunotherapies to treat cancer, have shown the ability to transduce hematopoietic stem cell progenitors effectively, as well as provided evidence for long-term safety. Application to DC vaccines may provide advantageous effects compared with using mRNA. Together with the recent progress in CRISPR/Cas9-mediated gene-editing efficiency of CD34+ HSPCs, this provides a unique cell pool to knockout immunosuppressive factors. The small number of CD34+ HSPCs may aid to reduce the viral vector batches and gene-editing tools required before expansion, differentiation, and maturation. It is important to investigate any negative effects on these phases during DC development.
There is a clear need for consistent comparative studies to compare DC subsets, maturation strategies, and modifications. Although many modifications have been tested in laboratory/pre-clinical studies and resulted in improved efficiency in vitro and in vivo models, very few of these modifications have translated into clinical applications. The use of state-of-the-art gene therapy vectors and gene-editing tools may create opportunities for next generation therapies with high efficacy for treating hematologic cancers and solid tumors.
Author Contributions
AC and NT wrote the manuscript. JB and SN made substantial and intellectual contributions to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Dutch Cancer Society (project number 10474).
References
1. Klusmann JH, Reinhardt D, Zimmermann M, Kremens B, Vormoor J, Dworzak M, et al. The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica (2012) 97:21–9. doi:10.3324/haematol.2011.051714
2. Singh N, Loren AW. Overview of hematopoietic cell transplantation for the treatment of hematologic malignancies. Clin Chest Med (2017) 38:575–93. doi:10.1016/j.ccm.2017.07.001
3. Zhang X, Rastogi P, Shah B, Zhang L. B lymphoblastic leukemia/lymphoma: new insights into genetics, molecular aberrations, subclassification and targeted therapy. Oncotarget (2017) 8:66728–41. doi:10.18632/oncotarget.19271
4. Yafour N, Beckerich F, Bulabois CE, Chevallier P, Daguindau E, Dumesnil C, et al. How to prevent relapse after allogeneic hematopoietic stem cell transplantation in patients with acute leukemia and myelodysplastic syndrome. Curr Res Transl Med (2017) 65:65–9. doi:10.1016/j.retram.2017.06.001
5. Gluckman E, Rocha V; EBMT Paediatric, Acute Leukemia Working Parties, Eurocord. Indications and results of cord blood transplant in children with leukemia. Bone Marrow Transplant (2008) 41(Suppl 2):S80–2. doi:10.1038/bmt.2008.60
6. Rocha V, Wagner JE Jr, Sobocinski KA, Klein JP, Zhang MJ, Horowitz MM, et al. Graft-versus-host disease in children who have received a cord-blood or bone marrow transplant from an HLA-identical sibling. Eurocord and international bone marrow transplant registry working committee on alternative donor and stem cell sources. N Engl J Med (2000) 342:1846–54. doi:10.1056/NEJM200006223422501
7. Verneris MR, Brunstein CG, Barker J, MacMillan ML, DeFor T, McKenna DH, et al. Relapse risk after umbilical cord blood transplantation: enhanced graft-versus-leukemia effect in recipients of 2 units. Blood (2009) 114:4293–9. doi:10.1182/blood-2009-05-220525
8. Brunstein CG, Gutman JA, Weisdorf DJ, Woolfrey AE, Defor TE, Gooley TA, et al. Allogeneic hematopoietic cell transplantation for hematologic malignancy: relative risks and benefits of double umbilical cord blood. Blood (2010) 116:4693–9. doi:10.1182/blood-2010-05-285304
9. Admiraal R, Lindemans CA, van Kesteren C, Bierings MB, Versluijs AB, Nierkens S, et al. Excellent T-cell reconstitution and survival depend on low ATG exposure after pediatric cord blood transplantation. Blood (2016) 128:2734–41. doi:10.1182/blood-2016-06-721936
10. Admiraal R, van Kesteren C, Jol-van der Zijde CM, Lankester AC, Bierings MB, Egberts TC, et al. Association between anti-thymocyte globulin exposure and CD4+ immune reconstitution in paediatric haemopoietic cell transplantation: a multicentre, retrospective pharmacodynamic cohort analysis. Lancet Haematol (2015) 2:e194–203. doi:10.1016/S2352-3026(15)00045-9
11. Hiwarkar P, Qasim W, Ricciardelli I, Gilmour K, Quezada S, Saudemont A, et al. Cord blood T cells mediate enhanced antitumor effects compared with adult peripheral blood T cells. Blood (2015) 126:2882–91. doi:10.1182/blood-2015-06-654780
12. Kwoczek J, Riese SB, Tischer S, Bak S, Lahrberg J, Oelke M, et al. Cord blood-derived T cells allow the generation of a more naive tumor-reactive cytotoxic T-cell phenotype. Transfusion (2018) 58:88–99. doi:10.1111/trf.14365
13. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi:10.1038/32588
14. Abraham RS, Mitchell DA. Gene-modified dendritic cell vaccines for cancer. Cytotherapy (2016) 18:1446–55. doi:10.1016/j.jcyt.2016.09.009
15. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12:265–77. doi:10.1038/nrc3258
16. Plantinga M, de Haar C, Nierkens S, Boelens JJ. Dendritic cell therapy in an allogeneic-hematopoietic cell transplantation setting: an effective strategy toward better disease control? Front Immunol (2014) 5:218. doi:10.3389/fimmu.2014.00218
17. Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, et al. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology (2017) 6:e1328341. doi:10.1080/2162402X.2017.1328341
18. Turnis ME, Rooney CM. Enhancement of dendritic cells as vaccines for cancer. Immunotherapy (2010) 2:847–62. doi:10.2217/imt.10.56
19. Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res (2013) 73:1063–75. doi:10.1158/0008-5472.CAN-12-2583
20. Wimmers F, Schreibelt G, Skold AE, Figdor CG, De Vries IJ. Paradigm shift in dendritic cell-based immunotherapy: from in vitro generated monocyte-derived DCs to naturally circulating DC subsets. Front Immunol (2014) 5:165. doi:10.3389/fimmu.2014.00165
21. Caux C, Massacrier C, Vanbervliet B, Dubois B, Durand I, Cella M, et al. CD34+ hematopoietic progenitors from human cord blood differentiate along two independent dendritic cell pathways in response to granulocyte-macrophage colony-stimulating factor plus tumor necrosis factor alpha: II. Functional analysis. Blood (1997) 90:1458–70.
22. Kumar J, Kale V, Limaye L. Umbilical cord blood-derived CD11c(+) dendritic cells could serve as an alternative allogeneic source of dendritic cells for cancer immunotherapy. Stem Cell Res Ther (2015) 6:184. doi:10.1186/s13287-015-0160-8
23. Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res (2001) 61:6451–8.
24. Mortarini R, Anichini A, Di Nicola M, Siena S, Bregni M, Belli F, et al. Autologous dendritic cells derived from CD34+ progenitors and from monocytes are not functionally equivalent antigen-presenting cells in the induction of melan-A/Mart-1(27–35)-specific CTLs from peripheral blood lymphocytes of melanoma patients with low frequency of CTL precursors. Cancer Res (1997) 57:5534–41.
25. Ferlazzo G, Klein J, Paliard X, Wei WZ, Galy A. Dendritic cells generated from CD34+ progenitor cells with flt3 ligand, c-kit ligand, GM-CSF, IL-4, and TNF-alpha are functional antigen-presenting cells resembling mature monocyte-derived dendritic cells. J Immunother (2000) 23:48–58. doi:10.1097/00002371-200001000-00007
26. de Haar C, Plantinga M, Blokland NJ, van Til NP, Flinsenberg TW, Van Tendeloo VF, et al. Generation of a cord blood-derived Wilms tumor 1 dendritic cell vaccine for AML patients treated with allogeneic cord blood transplantation. Oncoimmunology (2015) 4:e1023973. doi:10.1080/2162402X.2015.1023973
27. Thordardottir S, Schaap N, Louer E, Kester MG, Falkenburg JH, Jansen J, et al. Hematopoietic stem cell-derived myeloid and plasmacytoid DC-based vaccines are highly potent inducers of tumor-reactive T cell and NK cell responses ex vivo. Oncoimmunology (2017) 6:e1285991. doi:10.1080/2162402X.2017.1285991
28. Hutten T, Thordardottir S, Hobo W, Hubel J, van der Waart AB, Cany J, et al. Ex vivo generation of interstitial and Langerhans cell-like dendritic cell subset-based vaccines for hematological malignancies. J Immunother (2014) 37:267–77. doi:10.1097/CJI.0000000000000035
29. Ratzinger G, Baggers J, de Cos MA, Yuan J, Dao T, Reagan JL, et al. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol (2004) 173:2780–91.
30. Fay JW, Palucka AK, Paczesny S, Dhodapkar M, Johnston DA, Burkeholder S, et al. Long-term outcomes in patients with metastatic melanoma vaccinated with melanoma peptide-pulsed CD34(+) progenitor-derived dendritic cells. Cancer Immunol Immunother (2006) 55:1209–18. doi:10.1007/s00262-005-0106-6
31. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res (2009) 15:5323–37. doi:10.1158/1078-0432.CCR-09-0737
32. Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia (2007) 21:868–76. doi:10.1038/sj.leu.2404770
33. Menssen HD, Renkl HJ, Rodeck U, Maurer J, Notter M, Schwartz S, et al. Presence of Wilms’ tumor gene (WT1) transcripts and the WT1 nuclear protein in the majority of human acute leukemias. Leukemia (1995) 9:1060–7.
34. Sugiyama H. WT1 (Wilms’ tumor gene 1): biology and cancer immunotherapy. Jpn J Clin Oncol (2010) 40:377–87. doi:10.1093/jjco/hyp194
35. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic cell-based immunotherapy targeting Wilms’ tumor 1 in patients with recurrent malignant glioma. J Neurosurg (2015) 123:989–97. doi:10.3171/2015.1.JNS141554
36. Shimodaira S, Sano K, Hirabayashi K, Koya T, Higuchi Y, Mizuno Y, et al. Dendritic cell-based adjuvant vaccination targeting Wilms’ tumor 1 in patients with advanced colorectal cancer. Vaccines (Basel) (2015) 3:1004–18. doi:10.3390/vaccines3041004
37. Anguille S, Van Tendeloo VF, Berneman ZN. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia (2012) 26:2186–96. doi:10.1038/leu.2012.145
38. Coosemans A, Vanderstraeten A, Tuyaerts S, Verschuere T, Moerman P, Berneman ZN, et al. Wilms’ tumor gene 1 (WT1) – loaded dendritic cell immunotherapy in patients with uterine tumors: a phase I/II clinical trial. Anticancer Res (2013) 33:5495–500.
39. Nierkens S, Janssen EM. Harnessing dendritic cells for tumor antigen presentation. Cancers (Basel) (2011) 3:2195–213. doi:10.3390/cancers3022195
40. Lorenz FK, Wilde S, Voigt K, Kieback E, Mosetter B, Schendel DJ, et al. Codon optimization of the human papillomavirus E7 oncogene induces a CD8+ T cell response to a cryptic epitope not harbored by wild-type E7. PLoS One (2015) 10:e0121633. doi:10.1371/journal.pone.0121633
41. Benteyn D, Anguille S, Van Lint S, Heirman C, Van Nuffel AM, Corthals J, et al. Design of an optimized Wilms’ tumor 1 (WT1) mRNA construct for enhanced WT1 expression and improved immunogenicity in vitro and in vivo. Mol Ther Nucleic Acids (2013) 2:e134. doi:10.1038/mtna.2013.54
42. Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood (2006) 108:4009–17. doi:10.1182/blood-2006-04-015024
43. Bonehill A, Heirman C, Tuyaerts S, Michiels A, Breckpot K, Brasseur F, et al. Messenger RNA-electroporated dendritic cells presenting MAGE-A3 simultaneously in HLA class I and class II molecules. J Immunol (2004) 172:6649–57. doi:10.4049/jimmunol.172.11.6649
44. Lin KY, Guarnieri FG, Staveley-O’Carroll KF, Levitsky HI, August JT, Pardoll DM, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res (1996) 56:21–6.
45. Hosoi A, Takeda Y, Sakuta K, Ueha S, Kurachi M, Kimura K, et al. Dendritic cell vaccine with mRNA targeted to the proteasome by polyubiquitination. Biochem Biophys Res Commun (2008) 371:242–6. doi:10.1016/j.bbrc.2008.04.034
46. Subklewe M, Geiger C, Lichtenegger FS, Javorovic M, Kvalheim G, Schendel DJ, et al. New generation dendritic cell vaccine for immunotherapy of acute myeloid leukemia. Cancer Immunol Immunother (2014) 63:1093–103. doi:10.1007/s00262-014-1600-5
47. Lu C, Meng S, Jin Y, Zhang W, Li Z, Wang F, et al. A novel multi-epitope vaccine from MMSA-1 and DKK1 for multiple myeloma immunotherapy. Br J Haematol (2017) 178:413–26. doi:10.1111/bjh.14686
48. Uhlig KM, Schulke S, Scheuplein VA, Malczyk AH, Reusch J, Kugelmann S, et al. Lentiviral protein transfer vectors are an efficient vaccine platform and induce a strong antigen-specific cytotoxic T cell response. J Virol (2015) 89:9044–60. doi:10.1128/JVI.00844-15
49. Dullaers M, Breckpot K, Van Meirvenne S, Bonehill A, Tuyaerts S, Michiels A, et al. Side-by-side comparison of lentivirally transduced and mRNA-electroporated dendritic cells: implications for cancer immunotherapy protocols. Mol Ther (2004) 10:768–79. doi:10.1016/j.ymthe.2004.07.017
50. Kalinski P, Okada H. Polarized dendritic cells as cancer vaccines: directing effector-type T cells to tumors. Semin Immunol (2010) 22:173–82. doi:10.1016/j.smim.2010.03.002
51. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol (1997) 27:3135–42. doi:10.1002/eji.1830271209
52. Furumoto K, Mori A, Yamasaki S, Inoue N, Yang W, Nakau M, et al. Interleukin-12-gene transduction makes DCs from tumor-bearing mice an effective inducer of tumor-specific immunity in a peritoneal dissemination model. Immunol Lett (2002) 83:13–20. doi:10.1016/S0165-2478(02)00071-8
53. Vieira PL, de Jong EC, Wierenga EA, Kapsenberg ML, Kalinski P. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol (2000) 164:4507–12. doi:10.4049/jimmunol.164.9.4507
54. Mailliard RB, Wankowicz-Kalinska A, Cai Q, Wesa A, Hilkens CM, Kapsenberg ML, et al. Alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized CTL-inducing activity. Cancer Res (2004) 64:5934–7. doi:10.1158/0008-5472.CAN-04-1261
55. Lee JJ, Foon KA, Mailliard RB, Muthuswamy R, Kalinski P. Type 1-polarized dendritic cells loaded with autologous tumor are a potent immunogen against chronic lymphocytic leukemia. J Leukoc Biol (2008) 84:319–25. doi:10.1189/jlb.1107737
56. Bonehill A, Tuyaerts S, Van Nuffel AM, Heirman C, Bos TJ, Fostier K, et al. Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther (2008) 16:1170–80. doi:10.1038/mt.2008.77
57. Bonehill A, Van Nuffel AM, Corthals J, Tuyaerts S, Heirman C, Francois V, et al. Single-step antigen loading and activation of dendritic cells by mRNA electroporation for the purpose of therapeutic vaccination in melanoma patients. Clin Cancer Res (2009) 15:3366–75. doi:10.1158/1078-0432.CCR-08-2982
58. Feder-Mengus C, Schultz-Thater E, Oertli D, Marti WR, Heberer M, Spagnoli GC, et al. Nonreplicating recombinant vaccinia virus expressing CD40 ligand enhances APC capacity to stimulate specific CD4+ and CD8+ T cell responses. Hum Gene Ther (2005) 16:348–60. doi:10.1089/hum.2005.16.348
59. Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, et al. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J Clin Oncol (2016) 34:1330–8. doi:10.1200/JCO.2015.63.4121
60. Wilgenhof S, Van Nuffel AM, Corthals J, Heirman C, Tuyaerts S, Benteyn D, et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J Immunother (2011) 34:448–56. doi:10.1097/CJI.0b013e31821dcb31
61. De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res (2003) 63:12–7.
62. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell (1999) 99:23–33. doi:10.1016/S0092-8674(00)80059-8
63. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med (1999) 189:451–60. doi:10.1084/jem.189.3.451
64. Okada N, Mori N, Koretomo R, Okada Y, Nakayama T, Yoshie O, et al. Augmentation of the migratory ability of DC-based vaccine into regional lymph nodes by efficient CCR7 gene transduction. Gene Ther (2005) 12:129–39. doi:10.1038/sj.gt.3302358
65. Terando A, Roessler B, Mule JJ. Chemokine gene modification of human dendritic cell-based tumor vaccines using a recombinant adenoviral vector. Cancer Gene Ther (2004) 11:165–73. doi:10.1038/sj.cgt.7700671
66. Yang SC, Hillinger S, Riedl K, Zhang L, Zhu L, Huang M, et al. Intratumoral administration of dendritic cells overexpressing CCL21 generates systemic antitumor responses and confers tumor immunity. Clin Cancer Res (2004) 10:2891–901. doi:10.1158/1078-0432.CCR-03-0380
67. Baratelli F, Takedatsu H, Hazra S, Peebles K, Luo J, Kurimoto PS, et al. Pre-clinical characterization of GMP grade CCL21-gene modified dendritic cells for application in a phase I trial in non-small cell lung cancer. J Transl Med (2008) 6:38. doi:10.1186/1479-5876-6-38
68. Lee JM, Lee MH, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I trial of intratumoral injection of CCL21 gene-modified dendritic cells in lung cancer elicits tumor-specific immune responses and CD8(+) T-cell infiltration. Clin Cancer Res (2017) 23:4556–68. doi:10.1158/1078-0432.CCR-16-2821
69. Alvarez D, Vollmann EH, von Andrian UH. Mechanisms and consequences of dendritic cell migration. Immunity (2008) 29:325–42. doi:10.1016/j.immuni.2008.08.006
70. Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity (2007) 27:610–24. doi:10.1016/j.immuni.2007.08.015
71. Wang FL, Qin WJ, Wen WH, Tian F, Song B, Zhang Q, et al. TGF-beta insensitive dendritic cells: an efficient vaccine for murine prostate cancer. Cancer Immunol Immunother (2007) 56:1785–93. doi:10.1007/s00262-007-0322-3
72. Kalams SA, Walker BD. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J Exp Med (1998) 188:2199–204. doi:10.1084/jem.188.12.2199
73. Schnell S, Young JW, Houghton AN, Sadelain M. Retrovirally transduced mouse dendritic cells require CD4+ T cell help to elicit antitumor immunity: implications for the clinical use of dendritic cells. J Immunol (2000) 164:1243–50. doi:10.4049/jimmunol.164.3.1243
74. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science (2003) 300:337–9. doi:10.1126/science.1082305
75. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature (2003) 421:852–6. doi:10.1038/nature01441
76. Maksimow M, Miiluniemi M, Marttila-Ichihara F, Jalkanen S, Hanninen A. Antigen targeting to endosomal pathway in dendritic cell vaccination activates regulatory T cells and attenuates tumor immunity. Blood (2006) 108:1298–305. doi:10.1182/blood-2005-11-008615
77. Bonehill A, Heirman C, Thielemans K. Genetic approaches for the induction of a CD4+ T cell response in cancer immunotherapy. J Gene Med (2005) 7:686–95. doi:10.1002/jgm.713
78. Rodriguez F, Harkins S, Redwine JM, de Pereda JM, Whitton JL. CD4(+) T cells induced by a DNA vaccine: immunological consequences of epitope-specific lysosomal targeting. J Virol (2001) 75:10421–30. doi:10.1128/JVI.75.21.10421-10430.2001
79. Holst PJ, Sorensen MR, Mandrup Jensen CM, Orskov C, Thomsen AR, Christensen JP. MHC class II-associated invariant chain linkage of antigen dramatically improves cell-mediated immunity induced by adenovirus vaccines. J Immunol (2008) 180:3339–46. doi:10.4049/jimmunol.180.5.3339
80. Corthay A. A three-cell model for activation of naive T helper cells. Scand J Immunol (2006) 64:93–6. doi:10.1111/j.1365-3083.2006.01782.x
81. Caux C, Massacrier C, Vanbervliet B, Dubois B, Van Kooten C, Durand I, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med (1994) 180:1263–72. doi:10.1084/jem.180.4.1263
82. Kikuchi T, Moore MA, Crystal RG. Dendritic cells modified to express CD40 ligand elicit therapeutic immunity against preexisting murine tumors. Blood (2000) 96:91–9.
83. Koya RC, Kasahara N, Favaro PM, Lau R, Ta HQ, Weber JS, et al. Potent maturation of monocyte-derived dendritic cells after CD40L lentiviral gene delivery. J Immunother (2003) 26:451–60. doi:10.1097/00002371-200309000-00008
84. Dannull J, Nair S, Su Z, Boczkowski D, DeBeck C, Yang B, et al. Enhancing the immunostimulatory function of dendritic cells by transfection with mRNA encoding OX40 ligand. Blood (2005) 105:3206–13. doi:10.1182/blood-2004-10-3944
85. Grunebach F, Kayser K, Weck MM, Muller MR, Appel S, Brossart P. Cotransfection of dendritic cells with RNA coding for HER-2/neu and 4-1BBL increases the induction of tumor antigen specific cytotoxic T lymphocytes. Cancer Gene Ther (2005) 12:749–56. doi:10.1038/sj.cgt.7700842
86. De Keersmaecker B, Heirman C, Corthals J, Empsen C, van Grunsven LA, Allard SD, et al. The combination of 4-1BBL and CD40L strongly enhances the capacity of dendritic cells to stimulate HIV-specific T cell responses. J Leukoc Biol (2011) 89:989–99. doi:10.1189/jlb.0810466
87. Boczkowski D, Lee J, Pruitt S, Nair S. Dendritic cells engineered to secrete anti-GITR antibodies are effective adjuvants to dendritic cell-based immunotherapy. Cancer Gene Ther (2009) 16:900–11. doi:10.1038/cgt.2009.39
88. Krause P, Bruckner M, Uermosi C, Singer E, Groettrup M, Legler DF. Prostaglandin E(2) enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4-1BBL on dendritic cells. Blood (2009) 113:2451–60. doi:10.1182/blood-2008-05-157123
89. Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol (2010) 37:524–32. doi:10.1053/j.seminoncol.2010.09.013
90. Aspeslagh S, Postel-Vinay S, Rusakiewicz S, Soria JC, Zitvogel L, Marabelle A. Rationale for anti-OX40 cancer immunotherapy. Eur J Cancer (2016) 52:50–66. doi:10.1016/j.ejca.2015.08.021
91. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med (2008) 205:825–39. doi:10.1084/jem.20071341
92. Wang C, Lin GH, McPherson AJ, Watts TH. Immune regulation by 4-1BB and 4-1BBL: complexities and challenges. Immunol Rev (2009) 229:192–215. doi:10.1111/j.1600-065X.2009.00765.x
93. Lee H, Park HJ, Sohn HJ, Kim JM, Kim SJ. Combinatorial therapy for liver metastatic colon cancer: dendritic cell vaccine and low-dose agonistic anti-4-1BB antibody co-stimulatory signal. J Surg Res (2011) 169:e43–50. doi:10.1016/j.jss.2011.03.067
94. Burckhart T, Thiel M, Nishikawa H, Wuest T, Muller D, Zippelius A, et al. Tumor-specific crosslinking of GITR as costimulation for immunotherapy. J Immunother (2010) 33:925–34. doi:10.1097/CJI.0b013e3181f3cc87
95. Keller AM, Schildknecht A, Xiao Y, van den Broek M, Borst J. Expression of costimulatory ligand CD70 on steady-state dendritic cells breaks CD8+ T cell tolerance and permits effective immunity. Immunity (2008) 29:934–46. doi:10.1016/j.immuni.2008.10.009
96. Keller AM, Xiao Y, Peperzak V, Naik SH, Borst J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood (2009) 113:5167–75. doi:10.1182/blood-2008-03-148007
97. Kang TH, Lee JH, Noh KH, Han HD, Shin BC, Choi EY, et al. Enhancing dendritic cell vaccine potency by combining a BAK/BAX siRNA-mediated antiapoptotic strategy to prolong dendritic cell life with an intracellular strategy to target antigen to lysosomal compartments. Int J Cancer (2007) 120:1696–703. doi:10.1002/ijc.22377
98. Kim JH, Kang TH, Noh KH, Bae HC, Kim SH, Yoo YD, et al. Enhancement of dendritic cell-based vaccine potency by anti-apoptotic siRNAs targeting key pro-apoptotic proteins in cytotoxic CD8(+) T cell-mediated cell death. Immunol Lett (2009) 122:58–67. doi:10.1016/j.imlet.2008.12.006
99. Kim JH, Kang TH, Noh KH, Kim SH, Lee YH, Kim KW, et al. Enhancement of DC vaccine potency by activating the PI3K/AKT pathway with a small interfering RNA targeting PTEN. Immunol Lett (2010) 134:47–54. doi:10.1016/j.imlet.2010.08.008
100. Yang J, Huck SP, McHugh RS, Hermans IF, Ronchese F. Perforin-dependent elimination of dendritic cells regulates the expansion of antigen-specific CD8+ T cells in vivo. Proc Natl Acad Sci U S A (2006) 103:147–52. doi:10.1073/pnas.0509054103
101. Pedersen AE, Ronchese F. CTLA-4 blockade during dendritic cell based booster vaccination influences dendritic cell survival and CTL expansion. J Immune Based Ther Vaccines (2007) 5:9. doi:10.1186/1476-8518-5-9
102. Peng S, Kim TW, Lee JH, Yang M, He L, Hung CF, et al. Vaccination with dendritic cells transfected with BAK and BAX siRNA enhances antigen-specific immune responses by prolonging dendritic cell life. Hum Gene Ther (2005) 16:584–93. doi:10.1089/hum.2005.16.584
103. Zhu Y, Zheng Y, Mei L, Liu M, Li S, Xiao H, et al. Enhanced immunotherapeutic effect of modified HPV16 E7-pulsed dendritic cell vaccine by an adeno-shRNA-SOCS1 virus. Int J Oncol (2013) 43:1151–9. doi:10.3892/ijo.2013.2027
104. Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol (2003) 4:1169–76. doi:10.1038/ni1012
105. Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology (2006) 117:433–42. doi:10.1111/j.1365-2567.2006.02321.x
106. Llopiz D, Ruiz M, Infante S, Villanueva L, Silva L, Hervas-Stubbs S, et al. IL-10 expression defines an immunosuppressive dendritic cell population induced by antitumor therapeutic vaccination. Oncotarget (2017) 8:2659–71. doi:10.18632/oncotarget.13736
107. Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol (2010) 10:170–81. doi:10.1038/nri2711
108. Ahn YH, Hong SO, Kim JH, Noh KH, Song KH, Lee YH, et al. The siRNA cocktail targeting interleukin 10 receptor and transforming growth factor-beta receptor on dendritic cells potentiates tumour antigen-specific CD8(+) T cell immunity. Clin Exp Immunol (2015) 181:164–78. doi:10.1111/cei.12620
109. Kim JH, Kang TH, Noh KH, Bae HC, Ahn YH, Lee YH, et al. Blocking the immunosuppressive axis with small interfering RNA targeting interleukin (IL)-10 receptor enhances dendritic cell-based vaccine potency. Clin Exp Immunol (2011) 165:180–9. doi:10.1111/j.1365-2249.2011.04410.x
110. Zitvogel L, Kroemer G. Targeting PD-1/PD-L1 interactions for cancer immunotherapy. Oncoimmunology (2012) 1:1223–5. doi:10.4161/onci.21335
111. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9:562–7. doi:10.1038/nm863
112. Sgambato A, Casaluce F, Sacco PC, Palazzolo G, Maione P, Rossi A, et al. Anti PD-1 and PDL-1 immunotherapy in the treatment of advanced non-small cell lung cancer (NSCLC): a review on toxicity profile and its management. Curr Drug Saf (2016) 11:62–8. doi:10.2174/1574886311207040289
113. Wang S, Wang Y, Liu J, Shao S, Li X, Gao J, et al. Silencing B7-H1 enhances the anti-tumor effect of bladder cancer antigen-loaded dendritic cell vaccine in vitro. Onco Targets Ther (2014) 7:1389–96. doi:10.2147/OTT.S65367
114. Hobo W, Novobrantseva TI, Fredrix H, Wong J, Milstein S, Epstein-Barash H, et al. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol Immunother (2013) 62:285–97. doi:10.1007/s00262-012-1334-1
115. van der Waart AB, Fredrix H, van der Voort R, Schaap N, Hobo W, Dolstra H. siRNA silencing of PD-1 ligands on dendritic cell vaccines boosts the expansion of minor histocompatibility antigen-specific CD8(+) T cells in NOD/SCID/IL2Rg(null) mice. Cancer Immunol Immunother (2015) 64:645–54. doi:10.1007/s00262-015-1668-6
116. Wobser M, Voigt H, Houben R, Eggert AO, Freiwald M, Kaemmerer U, et al. Dendritic cell based antitumor vaccination: impact of functional indoleamine 2,3-dioxygenase expression. Cancer Immunol Immunother (2007) 56:1017–24. doi:10.1007/s00262-006-0256-1
117. Harden JL, Egilmez NK. Indoleamine 2,3-dioxygenase and dendritic cell tolerogenicity. Immunol Invest (2012) 41:738–64. doi:10.3109/08820139.2012.676122
118. Sioud M, Mobergslien A, Saeboe-Larssen S. Immunosuppressive factor blockade in dendritic cells via siRNAs results in objective clinical responses. Methods Mol Biol (2015) 1218:269–76. doi:10.1007/978-1-4939-1538-5_16
119. Kang TH, Lee JH, Bae HC, Noh KH, Kim JH, Song CK, et al. Enhancement of dendritic cell-based vaccine potency by targeting antigen to endosomal/lysosomal compartments. Immunol Lett (2006) 106:126–34. doi:10.1016/j.imlet.2006.05.004
120. Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer (2013) 132:967–77. doi:10.1002/ijc.27710
121. Sioud M, Saeboe-Larssen S, Hetland TE, Kaern J, Mobergslien A, Kvalheim G. Silencing of indoleamine 2,3-dioxygenase enhances dendritic cell immunogenicity and antitumour immunity in cancer patients. Int J Oncol (2013) 43:280–8. doi:10.3892/ijo.2013.1922
122. Biasco L, Rothe M, Buning H, Schambach A. Analyzing the genotoxicity of retroviral vectors in hematopoietic cell gene therapy. Mol Ther Methods Clin Dev (2018) 8:21–30. doi:10.1016/j.omtm.2017.10.002
123. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep (2017) 7:737. doi:10.1038/s41598-017-00462-8
124. Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol (2013) 31:822–6. doi:10.1038/nbt.2623
125. Gundry MC, Brunetti L, Lin A, Mayle AE, Kitano A, Wagner D, et al. Highly efficient genome editing of murine and human hematopoietic progenitor cells by CRISPR/Cas9. Cell Rep (2016) 17:1453–61. doi:10.1016/j.celrep.2016.09.092
126. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124:1277–87. doi:10.1182/blood-2014-01-545020
127. Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell (2014) 15:643–52. doi:10.1016/j.stem.2014.10.004
128. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res (2015) 75:3853–64. doi:10.1158/0008-5472.CAN-14-3321
Keywords: dendritic cell, vaccination, genetic modification, hematopoietic cells, hematopoietic cell transplantation, cord blood
Citation: Cornel AM, van Til NP, Boelens JJ and Nierkens S (2018) Strategies to Genetically Modulate Dendritic Cells to Potentiate Anti-Tumor Responses in Hematologic Malignancies. Front. Immunol. 9:982. doi: 10.3389/fimmu.2018.00982
Received: 14 February 2018; Accepted: 20 April 2018;
Published: 18 May 2018
Edited by:
Piergiuseppe De Berardinis, Istituto di biochimica delle proteine (IBP), ItalyReviewed by:
Paola Italiani, Consiglio Nazionale Delle Ricerche (CNR), ItalySandra Stephanie Diebold, King’s College London, United Kingdom
Copyright: © 2018 Cornel, van Til, Boelens and Nierkens. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Nierkens, cy5uaWVya2Vuc0B1bWN1dHJlY2h0Lm5s
†These authors have contributed equally to this work.