 Christian Koenecke
Christian Koenecke Andreas Krueger
Andreas Krueger- 1Clinic for Hematology, Hemostasis, Oncology and Stem Cell Transplantation, Hannover Medical School, Hannover, Germany
- 2Institute of Immunology, Hannover Medical School, Hannover, Germany
- 3Institute for Molecular Medicine, Goethe-University Frankfurt, Frankfurt am Main, Germany
Acute graft-versus-host disease (GvHD) is still a major cause of treatment-related mortality after allogeneic stem cell transplantation. Allo-antigen recognition of donor T cells after transplantation account for the onset and persistence of this disease. MicroRNAs (miRNAs) are molecular regulators involved in numerous processes during T-cell development, homeostasis, and activation. Thus, miRNAs also contribute to pathological T-cell function during GvHD. Given their capacity of fine-tuning T-cell function, miRNAs have emerged as promising therapeutic targets to curtail acute GvHD, but simultaneously maintain T-cell-mediated graft-versus-tumor effects. Here, we review the role of key miRNAs contributing to the pathophysiology of GvHD. We focus on those miRNAs acting in T cells and for which a role in GvHD has been established in preclinical models. Finally, we provide an outlook for clinical application of this new therapeutic target for GvHD prevention and treatment.
Introduction
Acute graft-versus-host disease (GvHD) is still a major complication after allogeneic hematopoietic stem cell or bone marrow transplantation (alloSCT). Allogeneic donor T cells are the main inducers of this too often lethal disease. Donor T cells recognize and respond to allo-antigen, use chemokine receptors and integrins to home to epithelial organs, such as the liver, the intestine, or the skin and orchestrate a significant immune response, which frequently results in severe organ damage. Unspecific prophylactic and therapeutic immunosuppressive treatment is the current clinical practice to improve patients’ outcome. However, the repertory of therapeutic and preventive options is limited and, therefore, new targets to control acute GvHD are urgently needed (1).
Since T cells orchestrate this disease, they are first choice to be targeted. Only naïve donor CD4 and CD8 T cells respond to allo-antigen via the T-cell receptor (TCR) in alloSCT, whereas central and/or memory T cells are not able to induce acute GvHD (2). Depending on several signals from the microenvironment, naïve T cells respond to allo-antigens in different ways. TCR signals are usually provided by antigen-presenting cells (APCs) via MHC class I or II molecules to CD8 and CD4 T cells, respectively. Recipient and donor APCs have differential impact on GvHD-induction by donor T cells (3–9). Furthermore, additional signals via cytokines are provided by the inflamed microenvironment and lead to onset and/or acceleration of this immune response (10). Whereas the plasticity of donor CD8 T cells seem to be limited, CD4 T cells develop into different subtypes during activation. T helper (Th) subtypes, such as Th1, Th2, Th17, and regulatory T cells (Treg) have distinct functions in the course of GvHD. The main drivers of acute GvHD, at least in rodents, are Th1 and Th17 cells (11–14). The cytokine release of such subtypes ultimately leads to tissue damage, which defines the clinical outcome of the disease. However, Th2 responses with cytokines such as IL-4, IL-5, IL-9, and IL-13 contribute to acute GvHD as well (15–17). It is believed that the impact on the pathophysiology of such cytokines depends on the timing and location of cytokines released by CD4 subsets. This is especially true for the Th1 cytokine IFN-γ, which is involved in inflammatory processes but can also facilitate immunosuppressive effects (18, 19). Further Th1 type cytokines TNFα and IL-2 have been tested for the prevention and treatment of GvHD not only in experimental models but also in patients with heterogeneous results (20). Th17 cells produce cytokines such as IL-17A, IL-17F, and IL-22 under the influence of IL-23 (21). A role for Th17 and associated cytokines such as IL-17A and IL-22 during acute GvHD has been shown, however, with controversial results. In one study, IL-17A deficiency leads to disease reduction (22), whereas another study shows that absence of Th17 cells exacerbates acute GvHD (23). IL-22 has been shown to be protective during GvHD by protection of recipients’ intestinal stem cells (24). A critical role in the pathophysiology of acute GvHD is attributed to Treg cells (25–27). It has been demonstrated in preclinical animal models that thymic-derived CD4+CD25+ natural Treg cells prevent the development of severe acute GvHD while preserving graft-versus-tumor (GvT) effects (28). Clinical studies are currently underway to test the therapeutic potential of natural Treg cells as a cellular therapy (29). However, the role of induced Treg cells in the context of GvHD is less clear (30), and it is controversially discussed whether such cells are suitable for therapeutic usage. Other CD4 T cell subsets, such as T follicular helper (Tfh) cells seem to have a role in chronic GvHD, but not acute GvHD (31). Furthermore, there is some evidence that also NK cells, natural killer T cell and invariant natural killer (iNK) T cells contribute to acute GvHD pathophysiology (25).
MicroRNAs (miRNAs) Controlling T-Cell Development and Function
MicroRNAs act as post-transcriptional regulators predominantly by facilitating mRNA degradation or inhibiting translation. For most miRNAs, multiple, even hundreds, of target mRNAs have been predicted in silico, and many of those have been validated experimentally at least in some cell types (32). Conversely, mRNAs frequently contain binding sites for multiple miRNAs. Thus, miRNAs are likely to play diverse roles in controlling gene expression in different contexts dependent on both miRNA and mRNA concentration and their binding affinity. Of note, many mRNAs display a limited degree of repression through the action of an individual miRNA (33, 34). Accordingly, a major function of miRNAs might lie in controlling noise in protein expression of lowly expressed genes (35, 36) or in generating expression switches (37). In addition, it has been suggested that individual miRNAs co-target multiple mRNAs within the same pathway, thus being able to functionally control biological processes despite low levels of repression of individual mRNAs (38). In turn, some mRNAs appear to be key targets of a wide variety of miRNAs. One example constitutes the negative regulator of PI3K signaling, Pten. In addition, a pseudogene homologous to Pten has been described to act as negative regulator of miRNA-mediated regulation of Pten via competition for miRNA binding (39–41). Although the hypothesis that miRNA function is regulated via the abundance of corresponding miRNA-binding sites in competing mRNAs is compelling, quantitative analysis of miRNA copies and abundance of miRNA response elements suggested that individual competing RNAs are unlikely to significantly contribute to target derepression (42–45). Recently, Heissmeyer and colleagues demonstrated that the RNA binding Protein Roquin blocks miRNA-mediated regulation by occupying a binding site for miR-17–92 in the 3′ untranslated region (UTR) of Pten mRNA, thus adding another level of complexity to the system (46).
Despite the described complexity in miRNA–mRNA interdependence, functionally relevant regulatory one miRNA—one mRNA relationships have been demonstrated using targeted deletion of defined miRNA-binding sites in individual genes. For instance, some, but not all, functions of miR-155 in the immune system could be ascribed to its repression of Socs-1 (47). On the other hand, targeted deletion of a miR-142-binding site in Cdkn1b did not phenocopy aberrant proliferation of thymocytes observed in miR-142-deficient mice (48).
Unsurprisingly, miRNAs also play a fundamental role in T-cell development and differentiation. Thus, T-lineage specific ablation of genes essential for processing of the vast majority of miRNAs, such as Dicer, Drosha, or Dgcr8 results in aberrant T-cell development and function. Loss of virtually all miRNAs early during T-cell development results in increased levels of apoptosis of thymocytes and, concomitantly, reduced thymocyte numbers (49, 50). Deletion of Dicer at later developmental stages showed that miRNAs are critical for maintenance of peripheral T cells, most notably, the CD8 T cell compartment (51). Within the CD4 T cell compartment, loss of virtually all miRNAs resulted in a bias toward a Th1 differentiation program and against differentiation toward the Th2 and Th17 lineages (51, 52). In addition, generation of Tfh cells is dependent on miRNA (53). Furthermore, miRNAs are critical for intrathymic development as well as peripheral induction of Treg cells (54). Treg-cell specific deletion of all miRNAs resulted in fatal autoimmunity due to defective peripheral Treg-cell homeostasis (55–57).
In both T-cell development and differentiation, individual miRNAs have been extensively characterized [reviewed in Ref. (58–60)]. Here, we focus mainly on describing physiological roles of miRNAs that have been directly linked to GvHD in preclinical models or may affect critical T-cell function in the context of GvHD (Figure 1; Table 1). By comparing consequences of deletion of individual miRNAs versus deletion of the total pool of miRNAs, Dooley and colleagues identified those individual miRNAs as dominant, deletion of which helps to explain the consequences of loss of the complete miRNAome (59). In contrast, loss of individual miRNAs that does not mirror complete miRNA deficiency rather indicates functions in line with the role of miRNAs to fine-tune biological processes.
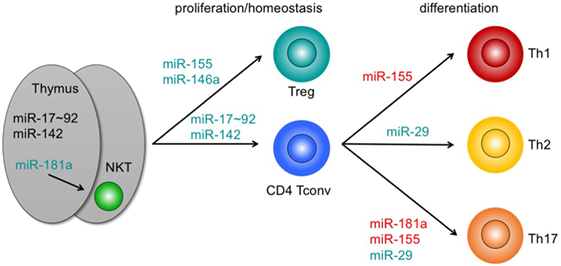
Figure 1. Pathophysiologically relevant microRNAs (miRNAs) in graft-versus-host disease with a functional role in T-cell development, homeostasis, and differentiation. miRNAs adjacent to arrows indicate those miRNAs that positively contribute to intrathymic development (left), proliferation and/or homeostasis (center), or differentiation of T cell subsets. miRNAs highlighted in green mirror exert major functions. Loss of these miRNAs partially mirrors depletion of all miRNAs in mice. miRNAs highlighted in red indicate miRNAs with non-dominant functions. For details, see text. Abbreviations: NKT, natural killer T cell; Treg, regulatory T cell; Tconv, conventional T cell; Th, T helper cell.
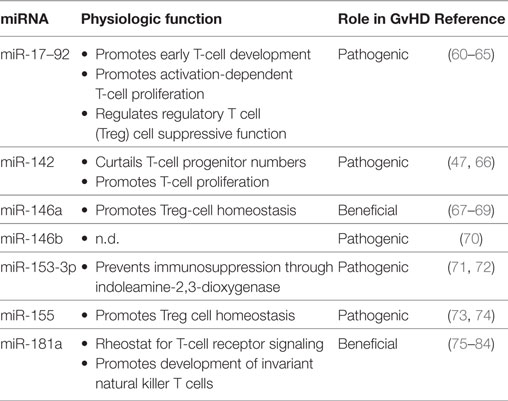
Table 1. MicroRNAs (miRNAs) acting in T cells, for which a role in graft-versus-host disease (GvHD) has been established in preclinical models.
A cluster of six related miRNAs, miR-17–92, confers competitive fitness to the earliest T-cell progenitors in the thymus and pre-thymic progenitors by regulating IL-7 receptor levels and IL-7 signaling (85). Conversely, miR-142 curtails numbers of early T-lineage progenitors, although loss of this miRNA inhibits proliferation and ultimately results in peripheral lymphopenia (48). Together, miR-17–92 and miR-142 help to explain the majority of T-lineage developmental defects observed upon early depletion of all miRNAs. Members of the miR-181 family are among the most prominently expressed miRNA species in thymocytes preceding and/or undergoing selection (75, 86). miR-181a targets a group of phosphatases, all of which act as negative regulators of TCR signaling (76). Accordingly, miR-181a sensitizes cells toward TCR signaling and may serve as a rheostat during positive selection (76, 77). Conversely, loss of miR-181a-1 and miR-181b-1 dampens TCR signaling in double-positive thymocytes (78, 79). Nevertheless, the effect of loss of miR-181a/b-1 on selection of conventional T cells (Tconv) is comparatively mild. In an HY-TCR transgenic model, loss of miR-181a/b-1 results in an increase in positively selected TCR-transgenic T cells (79). In contrast, miR-181a/b-1 is critical for development of agonist-selected iNK T cells (78, 80, 81). Although control of agonist-selected populations by miR-181 can be plausibly explained by regulation of TCR signal strength and elevated TCR expression was able to completely restore iNKT cell development, alternative mechanisms of actions, such as via regulation of Pten, cannot be fully ruled out (66, 78, 80).
Once outside the thymus miRNAs are critical for peripheral maintenance and proliferation of T cells. Thus, T cells deficient in miR-142 fail to expand after egress from the thymus and do not efficiently proliferate upon TCR triggering (48, 61). Similarly, T cells lacking miR-17–92 proliferate inefficiently after stimulation in vitro and after infection, a defect that can be partially restored by re-introduction of miR-17 and miR-92a (62, 63, 82). Despite its low levels of expression when compared to thymocytes, miR-181a has been proposed to regulate responsiveness in peripheral CD4 T cells, especially in humans. Notably, expression of miR-181a progressively declines with age, resulting high sensitivity toward TCR signals in cord blood T cells and impaired sensitivity in naive T cells from aged individuals (83, 87).
Loss of miR-29 results in a bias toward Th1 differentiation similar to that observed in pan-miRNA-deficient T cells via derepression of key transcription factors T-bet and Eomes as well as IFN-γ (62, 67). restoration of miR-29 expression in these mice can partially overcome this defect (62). miR-146a and miR-155 constitute critical miRNAs for function and homeostasis of Treg cells. Loss of miR-146a derepressed transcription factor Stat1 and, consequently, resulted in dysregulated expression of IFN-γ and accompanying immunopathology (73). The Foxp3-dependent miR-155 promotes Treg-cell homeostasis by controlling responsiveness to IL-2 signals via repression of Socs-1 (64). Combined these data place Socs-1 in a central miRNA-controlled hub of Treg cell function, because both effector signals, such as Stat1/IFN-γ as well as homeostatic signals via Stat5/IL-2 are regulated by Socs-1. Of note, disruption of the miR-155/Socs-1 axis by introducing mutations into miR-155-binding sites in the Socs1 3′UTR perturbed Treg-cell homeostasis under competitive conditions, but not at steady state, highlighting the complexity of miRNA-dependent gene regulation (47). Interestingly, although miR-17–92 contributes to regulating suppressive function via regulating production of IL-10, this cluster does not control proliferation or survival of Treg cells (88). Specific induction of miR-181a in differentiating Th17 cells can selectively sensitize these cells toward TCR-mediated signals, thus contributing to controlled memory T cell formation (89).
miRNA Function in T Cells in the Context of Acute GvHD
For a number of miRNAs, a role in the pathophysiology of acute GvHD has been described. miR-155 is important for Treg cell homeostasis and was also linked to differentiation of Tconv in autoimmune responses such as in experimental autoimmune encephalomyelitis (74, 90). miR-155 expression is upregulated in donor T cells in an experimental GvHD model and also in intestinal tissue of GvHD patients. Furthermore, donor T cells lacking miR-155 induce reduced GvHD, whereas miR-155 overexpression accelerates the course of the disease in experimental alloSCT. Consistently, blocking miR-155 by locked-nucleic-acid-modified (LNA−) anti-miR-155 oligonucelotides diminishes GvHD. In addition to alterations in T cell differentiation, impaired homing to GvHD target organs due to decreased expression of homing receptors CCR5, CXCR4, and S1P1 on effector T cells, has been proposed as the underlying molecular mechanism of blocking miR-155 (65).
Since miR-142 and miR-17–92 constitute important mediators of T cell proliferation, their contribution to GvHD pathophysiology was not unexpected. Donor T cells deficient for miR-142 show impaired cell cycle progression and simultaneously reduced proliferation, which in turn leads to reduced GvHD severity and mortality in multiple experimental alloSCT models. Mir-142 deficiency in T cells leads to upregulation of atypical E2F transcription factors E2f7 and E2f8, which are both known targets of this miRNA. Thus, targeting the interaction between miR-142 and E2F transcription factors, might serve as a potential therapeutic approach (61). T cells deficient in miR-17–92 proliferate inefficiently after stimulation in vitro and after infection (63, 82). In line with these data, miR-17–92-deficient donor T cells were unable to induce lethal acute GvHD, which could be mostly ascribed to miR-17 and miR-19b (68). Interestingly, in this model, the GvT effect was preserved, which might be due to the fact that proliferation, survival, and cytotoxic functions of CD8 T cells are at least partially maintained in the context of GvHD, when miR-17–92 is targeted. These data highlight the capacity even of broadly acting miRNAs to fine tune T cell function in a context-dependent manner.
The miR-146 family includes two members, miR-146a and miR-146b, which are both involved in acute GvHD pathophysiology. miR-146a overexpression in donor T cells reduces GvHD and in turn lack of this miRNA leads to increased GvHD severity with high levels of TNFα. In line with these experimental data, the occurrence of the SNP rs2910164 in alloSCT donors, which reduces miR-146a expression, is associated with higher incidence of severe GvHD in a patient cohort (69). One target of miR-146a in T cells is the TNF-receptor-associated factor 6 (TRAF6) (70). TRAF6 regulates transcription of TNFα via NF-κB and high levels of miR-146a reduces TNFα transcription in donor T cells and thereby reduces GvHD. TRAF6 is also a target of miR-146b and antagomir-mediated knockdown of miR-146b leads to enhanced TRAF6 expression. Increased TRAF6, in turn, activates NF-κB and leads to enhanced survival, proliferation, and suppressive activity of natural Treg cells. Hence, miR-146b antagomir-treated human Treg cells decrease mortality in a xenogenic GvHD-model and might, therefore, improve the adoptive Treg cell therapy (91). The opposing functions of the closely related miR-146a and miR-146b shed light on the complexity of miRNA-based intervention to treat GvHD.
Indoleamine-2,3-dioxygenase (IDO) is a critical enzyme in providing essential amino acids for T cell proliferation and exerts immunosuppressive effects. Increased levels of IDO levels prevent acute GvHD (72). IDO is directly targeted by miR-153-3p and miR-153-3p. Antagomir for miR-153-3p and miR-153-3p led to higher IDO expression in experimental GvHD and delayed the course of disease (84).
miR-181a acts as a rheostat for T cell sensitivity to antigen by downregulation of several phosphatases downstream of the TCR (76). Primary T cells overexpressing miR-181a failed to induce experimental GvHD, whereas, conversely, donor T cells lacking miR-181a/b-1 accelerated acute GvHD in the same model (92). Overexpression of miR-181a resulted in decreased T cell survival, most likely because of reduced expression of anti-apoptotic BCL-2 protein expression. Repression of BCL-2 protein in this context likely results from a combination of it being a direct target of miR-181a as well as a consequence of apoptosis regulation dependent on TCR signal strength. This study points toward BCL-2 inhibition as a novel therapeutic strategy in prevention of allo-reactivity and highlights miRNAs as titrable therapeutic targets in order to prevent GvHD while preserving GvT effects.
Future Directions
Understanding the role of miRNAs in physiological T cell function and in the context of disease has the potential to open up new avenues for therapy. Most of this knowledge is derived from the analysis of rodent models and is only beginning to be complemented by studies in human primary T cells (93). To a large extent, miRNAs and their mRNA target sites are evolutionary conserved. However, some notable exceptions have been described. Thus, miR-125b controls human T cell differentiation via a number of mRNA targets lacking corresponding miR-125b binding sites in mouse, including the genes encoding IFN-γ and IL-10RA (93). In terms of therapeutic delivery, alloSCT has the distinct advantage that cells might already be manipulated prior to transplantation ex vivo. Recently, methods for efficient CRISPR/Cas9-mediated genome engineering of primary human T cells have been reported (94). In addition, highly potent chemically modified miRNA analogs or antagonists are beginning to emerge that can be directly delivered in vivo (95, 96), some of which have already been employed experimentally in rodent models of GvHD (65, 69). In order to prevent off-target effects in case of systemic delivery such miRNAs can be further modified to generate compounds that can be selectively activated through light in typical GvHD target organs, such as skin and gut (97).
Our knowledge of miRNA action is far from complete and a number of miRNAs with a role in T cell function, for instance, miR-148a, miR-182, and miR-326, may ultimately also play a role in GvHD (98–100). Similarly, a number of miRNAs have been identified as putative biomarkers for GvHD, such as miR-194, but a functional role in pathogenesis remains to be established (101). Finally, miRNAs also play a role in non-T cells during GvHD. For instance, miR-100 has been reported to limit neovascularization in the intestine and miR-155, which functions as a key regulator of Treg cells as well as Tconv, negatively impacts upon dendritic cell function during GvHD (102, 103).
Conclusion
MicroRNAs play critical role in many biological processes including T-cell development and differentiation. Given that GvHD is a predominantly T-cell driven disease, miRNAs have emerged as attractive therapeutic targets for ex vivo and potentially even in vivo modulation. A better understanding of miRNAs controlling physiologic T-cell function has resulted in efficient translation of manipulation of miRNAs in preclinical models of GvHD. However, despite the generally broad interspecies conservation of miRNAs, translation into clinical treatment of GvHD remains a challenge.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Deutsche Forschungsgemeinschaft (www.dfg.de): SFB738/A8 and SFB900/B8 to CK and SFB902/B15 and KR2320/5-1 to AK.
References
1. Zeiser R, Blazar BR. Acute graft-versus-host disease – biologic process, prevention, and therapy. N Engl J Med (2017) 377(22):2167–79. doi:10.1056/NEJMra1609337
2. Zheng H, Matte-Martone C, Li H, Anderson BE, Venketesan S, Sheng Tan H, et al. Effector memory CD4+ T cells mediate graft-versus-leukemia without inducing graft-versus-host disease. Blood (2008) 111(4):2476–84. doi:10.1182/blood-2007-08-109678
3. Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, et al. Donor APCs are required for maximal GVHD but not for GVL. Nat Med (2004) 10(9):987–92. doi:10.1038/nm1089
4. Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science (1999) 285(5426):412–5. doi:10.1126/science.285.5426.412
5. Anderson BE, McNiff JM, Jain D, Blazar BR, Shlomchik WD, Shlomchik MJ. Distinct roles for donor- and host-derived antigen-presenting cells and costimulatory molecules in murine chronic graft-versus-host disease: requirements depend on target organ. Blood (2005) 105(5):2227–34. doi:10.1182/blood-2004-08-3032
6. Duffner UA, Maeda Y, Cooke KR, Reddy P, Ordemann R, Liu C, et al. Host dendritic cells alone are sufficient to initiate acute graft-versus-host disease. J Immunol (2004) 172(12):7393–8. doi:10.4049/jimmunol.172.12.7393
7. Markey KA, Banovic T, Kuns RD, Olver SD, Don AL, Raffelt NC, et al. Conventional dendritic cells are the critical donor APC presenting alloantigen after experimental bone marrow transplantation. Blood (2009) 113(22):5644–9. doi:10.1182/blood-2008-12-191833
8. Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, et al. Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med (2002) 8(6):575–81. doi:10.1038/nm0902-1039a
9. Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, et al. Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med (2011) 18(1):135–42. doi:10.1038/nm.2597
10. Chakraverty R, Cote D, Buchli J, Cotter P, Hsu R, Zhao G, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med (2006) 203(8):2021–31. doi:10.1084/jem.20060376
11. Imanguli MM, Swaim WD, League SC, Gress RE, Pavletic SZ, Hakim FT. Increased T-bet+ cytotoxic effectors and type I interferon-mediated processes in chronic graft-versus-host disease of the oral mucosa. Blood (2009) 113(15):3620–30. doi:10.1182/blood-2008-07-168351
12. Murphy WJ, Welniak LA, Taub DD, Wiltrout RH, Taylor PA, Vallera DA, et al. Differential effects of the absence of interferon-gamma and IL-4 in acute graft-versus-host disease after allogeneic bone marrow transplantation in mice. J Clin Invest (1998) 102(9):1742–8. doi:10.1172/JCI3906
13. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 mediate acute graft-versus-host disease, each with distinct end-organ targets. J Clin Invest (2000) 105(9):1289–98. doi:10.1172/JCI7894
14. Ratajczak P, Janin A, Peffault de Latour R, Leboeuf C, Desveaux A, Keyvanfar K, et al. Th17/Treg ratio in human graft-versus-host disease. Blood (2010) 116(7):1165–71. doi:10.1182/blood-2009-12-255810
15. Fowler DH, Gress RE. Th2 and Tc2 cells in the regulation of GVHD, GVL, and graft rejection: considerations for the allogeneic transplantation therapy of leukemia and lymphoma. Leuk Lymphoma (2000) 38(3–4):221–34. doi:10.3109/10428190009087014
16. Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood (2011) 117(11):3220–9. doi:10.1182/blood-2010-08-303008
17. Tawara I, Maeda Y, Sun Y, Lowler KP, Liu C, Toubai T, et al. Combined Th2 cytokine deficiency in donor T cells aggravates experimental acute graft-vs-host disease. Exp Hematol (2008) 36(8):988–96. doi:10.1016/j.exphem.2008.02.010
18. Lu Y, Waller EK. Dichotomous role of interferon-gamma in allogeneic bone marrow transplant. Biol Blood Marrow Transplant (2009) 15(11):1347–53. doi:10.1016/j.bbmt.2009.07.015
19. Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, et al. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol (2012) 189(6):2890–6. doi:10.4049/jimmunol.1200413
20. Alousi AM, Weisdorf DJ, Logan BR, Bolanos-Meade J, Carter S, Difronzo N, et al. Etanercept, mycophenolate, denileukin, or pentostatin plus corticosteroids for acute graft-versus-host disease: a randomized phase 2 trial from the Blood and Marrow Transplant Clinical Trials Network. Blood (2009) 114(3):511–7. doi:10.1182/blood-2009-03-212290
21. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol (2014) 14(9):585–600. doi:10.1038/nri3707
22. Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, et al. IL-17 contributes to CD4-mediated graft-versus-host disease. Blood (2009) 113(4):945–52. doi:10.1182/blood-2008-08-172155
23. Yi T, Zhao D, Lin CL, Zhang C, Chen Y, Todorov I, et al. Absence of donor Th17 leads to augmented Th1 differentiation and exacerbated acute graft-versus-host disease. Blood (2008) 112(5):2101–10. doi:10.1182/blood-2007-12-126987
24. Hanash AM, Kappel LW, Yim NL, Nejat RA, Goldberg GL, Smith OM, et al. Abrogation of donor T-cell IL-21 signaling leads to tissue-specific modulation of immunity and separation of GVHD from GVL. Blood (2011) 118(2):446–55. doi:10.1182/blood-2010-07-294785
25. Zeiser R, Socie G, Blazar BR. Pathogenesis of acute graft-versus-host disease: from intestinal microbiota alterations to donor T cell activation. Br J Haematol (2016) 175(2):191–207. doi:10.1111/bjh.14295
26. Prinz I, Koenecke C. Therapeutic potential of induced and natural FoxP3(+) regulatory T cells for the treatment of graft-versus-host disease. Arch Immunol Ther Exp (Warsz) (2012) 60(3):183–90. doi:10.1007/s00005-012-0172-3
27. Hippen KL, Loschi M, Nicholls J, MacDonald KPA, Blazar BR. Effects of microRNA on regulatory T cells and implications for adoptive cellular therapy to ameliorate graft-versus-host disease. Front Immunol (2018) 9:57. doi:10.3389/fimmu.2018.00057
28. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med (2003) 9(9):1144–50. doi:10.1038/nm915
29. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51. doi:10.1182/blood-2015-06-653667
30. Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, et al. Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur J Immunol (2009) 39(11):3091–6. doi:10.1002/eji.200939432
31. Flynn R, Du J, Veenstra RG, Reichenbach DK, Panoskaltsis-Mortari A, Taylor PA, et al. Increased T follicular helper cells and germinal center B cells are required for cGVHD and bronchiolitis obliterans. Blood (2014) 123(25):3988–98. doi:10.1182/blood-2014-03-562231
32. Chou CH, Shrestha S, Yang CD, Chang NW, Lin YL, Liao KW, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res (2018) 46(D1):D296–302. doi:10.1093/nar/gkx1067
33. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature (2008) 455(7209):58–63. doi:10.1038/nature07228
34. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature (2010) 466(7308):835–40. doi:10.1038/nature09267
35. Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Bluthgen N, Marks DS, et al. Gene expression. MicroRNA control of protein expression noise. Science (2015) 348(6230):128–32. doi:10.1126/science.aaa1738
36. Herranz H, Cohen SM. MicroRNAs and gene regulatory networks: managing the impact of noise in biological systems. Genes Dev (2010) 24(13):1339–44. doi:10.1101/gad.1937010
37. Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet (2011) 43(9):854–9. doi:10.1038/ng.905
38. Tsang JS, Ebert MS, van Oudenaarden A. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Mol Cell (2010) 38(1):140–53. doi:10.1016/j.molcel.2010.03.007
39. Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature (2010) 465(7301):1033–8. doi:10.1038/nature09144
40. Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell (2011) 147(2):344–57. doi:10.1016/j.cell.2011.09.029
41. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell (2011) 146(3):353–8. doi:10.1016/j.cell.2011.07.014
42. Seitz H. Redefining microRNA targets. Curr Biol (2009) 19(10):870–3. doi:10.1016/j.cub.2009.03.059
43. Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell (2012) 149(3):515–24. doi:10.1016/j.cell.2012.04.005
44. Denzler R, McGeary SE, Title AC, Agarwal V, Bartel DP, Stoffel M. Impact of microRNA levels, target-site complementarity, and cooperativity on competing endogenous RNA-regulated gene expression. Mol Cell (2016) 64(3):565–79. doi:10.1016/j.molcel.2016.09.027
45. Jens M, Rajewsky N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat Rev Genet (2015) 16(2):113–26. doi:10.1038/nrg3853
46. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T, et al. Roquin suppresses the PI3K-mTOR signaling pathway to inhibit T helper cell differentiation and conversion of Treg to Tfr cells. Immunity (2017) 47(6):1067–82 e12. doi:10.1016/j.immuni.2017.11.008
47. Lu LF, Gasteiger G, Yu IS, Chaudhry A, Hsin JP, Lu Y, et al. A single miRNA-mRNA interaction affects the immune response in a context- and cell-type-specific manner. Immunity (2015) 43(1):52–64. doi:10.1016/j.immuni.2015.04.022
48. Mildner A, Chapnik E, Varol D, Aychek T, Lampl N, Rivkin N, et al. MicroRNA-142 controls thymocyte proliferation. Eur J Immunol (2017) 47(7):1142–52. doi:10.1002/eji.201746987
49. Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, et al. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med (2005) 201(9):1367–73. doi:10.1084/jem.20050572
50. Chong MM, Zhang G, Cheloufi S, Neubert TA, Hannon GJ, Littman DR. Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev (2010) 24(17):1951–60. doi:10.1101/gad.1953310
51. Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med (2005) 202(2):261–9. doi:10.1084/jem.20050678
52. Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity (2016) 44(6):1284–98. doi:10.1016/j.immuni.2016.05.015
53. Baumjohann D, Kageyama R, Clingan JM, Morar MM, Patel S, de Kouchkovsky D, et al. The microRNA cluster miR-17 approximately 92 promotes TFH cell differentiation and represses subset-inappropriate gene expression. Nat Immunol (2013) 14(8):840–8. doi:10.1038/ni.2642
54. Cobb BS, Hertweck A, Smith J, O’Connor E, Graf D, Cook T, et al. A role for Dicer in immune regulation. J Exp Med (2006) 203(11):2519–27. doi:10.1084/jem.20061692
55. Liston A, Lu LF, O’Carroll D, Tarakhovsky A, Rudensky AY. Dicer-dependent microRNA pathway safeguards regulatory T cell function. J Exp Med (2008) 205(9):1993–2004. doi:10.1084/jem.20081062
56. Chong MM, Rasmussen JP, Rudensky AY, Littman DR. The RNAseIII enzyme Drosha is critical in T cells for preventing lethal inflammatory disease. J Exp Med (2008) 205(9):2005–17. doi:10.1084/jem.20081219
57. Zhou X, Jeker LT, Fife BT, Zhu S, Anderson MS, McManus MT, et al. Selective miRNA disruption in T reg cells leads to uncontrolled autoimmunity. J Exp Med (2008) 205(9):1983–91. doi:10.1084/jem.20080707
58. Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat Rev Immunol (2013) 13(9):666–78. doi:10.1038/nri3494
59. Dooley J, Linterman MA, Liston A. MicroRNA regulation of T-cell development. Immunol Rev (2013) 253(1):53–64. doi:10.1111/imr.12049
60. Monticelli S. MicroRNAs in T helper cell differentiation and plasticity. Semin Immunol (2013) 25(4):291–8. doi:10.1016/j.smim.2013.10.015
61. Sun Y, Oravecz-Wilson K, Mathewson N, Wang Y, McEachin R, Liu C, et al. Mature T cell responses are controlled by microRNA-142. J Clin Invest (2015) 125(7):2825–40. doi:10.1172/JCI78753
62. Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, et al. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity (2011) 35(2):169–81. doi:10.1016/j.immuni.2011.07.009
63. Wu T, Wieland A, Lee J, Hale JS, Han JH, Xu X, et al. Cutting edge: miR-17-92 is required for both CD4 Th1 and T follicular helper cell responses during viral infection. J Immunol (2015) 195(6):2515–9. doi:10.4049/jimmunol.1500317
64. Lu LF, Thai TH, Calado DP, Chaudhry A, Kubo M, Tanaka K, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity (2009) 30(1):80–91. doi:10.1016/j.immuni.2008.11.010
65. Ranganathan P, Heaphy CE, Costinean S, Stauffer N, Na C, Hamadani M, et al. Regulation of acute graft-versus-host disease by microRNA-155. Blood (2012) 119(20):4786–97. doi:10.1182/blood-2011-10-387522
66. Blume J, Zur Lage S, Witzlau K, Georgiev H, Weiss S, Lyszkiewicz M, et al. Overexpression of Valpha14Jalpha18 TCR promotes development of iNKT cells in the absence of miR-181a/b-1. Immunol Cell Biol (2016) 94(8):741–6. doi:10.1038/icb.2016.40
67. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol (2011) 12(9):861–9. doi:10.1038/ni.2073
68. Wu Y, Heinrichs J, Bastian D, Fu J, Nguyen H, Schutt S, et al. MicroRNA-17-92 controls T-cell responses in graft-versus-host disease and leukemia relapse in mice. Blood (2015) 126(11):1314–23. doi:10.1182/blood-2015-02-627356
69. Stickel N, Prinz G, Pfeifer D, Hasselblatt P, Schmitt-Graeff A, Follo M, et al. miR-146a regulates the TRAF6/TNF-axis in donor T cells during GVHD. Blood (2014) 124(16):2586–95. doi:10.1182/blood-2014-04-569046
70. Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a controls the resolution of T cell responses in mice. J Exp Med (2012) 209(9):1655–70. doi:10.1084/jem.20112218
71. Zhang Y, Liu W, Chen Y, Liu J, Wu K, Su L, et al. A cellular microRNA facilitates regulatory T lymphocyte development by targeting the FOXP3 promoter TATA-box motif. J Immunol (2018) 200(3):1053–63. doi:10.4049/jimmunol.1700196
72. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the tryptophan catabolic pathway, indoleamine 2,3-dioxygenase (IDO), for suppression of graft-versus-host disease (GVHD) lethality. Blood (2009) 114(24):5062–70. doi:10.1182/blood-2009-06-227587
73. Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell (2010) 142(6):914–29. doi:10.1016/j.cell.2010.08.012
74. O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity (2010) 33(4):607–19. doi:10.1016/j.immuni.2010.09.009
75. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity (2010) 32(6):828–39. doi:10.1016/j.immuni.2010.05.009
76. Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, Liu G, et al. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell (2007) 129(1):147–61. doi:10.1016/j.cell.2007.03.008
77. Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat Immunol (2009) 10(11):1162–9. doi:10.1038/ni.1797
78. Zietara N, Lyszkiewicz M, Witzlau K, Naumann R, Hurwitz R, Langemeier J, et al. Critical role for miR-181a/b-1 in agonist selection of invariant natural killer T cells. Proc Natl Acad Sci U S A (2013) 110(18):7407–12. doi:10.1073/pnas.1221984110
79. Schaffert SA, Loh C, Wang S, Arnold CP, Axtell RC, Newell EW, et al. mir-181a-1/b-1 modulates tolerance through opposing activities in selection and peripheral T cell function. J Immunol (2015) 195(4):1470–9. doi:10.4049/jimmunol.1401587
80. Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limon P, Kaech SM, et al. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity (2013) 38(5):984–97. doi:10.1016/j.immuni.2013.02.021
81. Sandrock I, Zietara N, Lyszkiewicz M, Oberdorfer L, Witzlau K, Krueger A, et al. MicroRNA-181a/b-1 is not required for innate gammadelta NKT effector cell development. PLoS One (2015) 10(12):e0145010. doi:10.1371/journal.pone.0145010
82. Wu T, Wieland A, Araki K, Davis CW, Ye L, Hale JS, et al. Temporal expression of microRNA cluster miR-17-92 regulates effector and memory CD8+ T-cell differentiation. Proc Natl Acad Sci U S A (2012) 109(25):9965–70. doi:10.1073/pnas.1207327109
83. Palin AC, Ramachandran V, Acharya S, Lewis DB. Human neonatal naive CD4+ T cells have enhanced activation-dependent signaling regulated by the microRNA miR-181a. J Immunol (2013) 190(6):2682–91. doi:10.4049/jimmunol.1202534
84. Zhao XS, Wang YN, Lv M, Kong Y, Luo HX, Ye XY, et al. miR-153-3p, a new bio-target, is involved in the pathogenesis of acute graft-versus-host disease via inhibition of indoleamine-2,3-dioxygenase. Oncotarget (2016) 7(30):48321–34. doi:10.18632/oncotarget.10220
85. Regelin M, Blume J, Pommerencke J, Vakilzadeh R, Witzlau K, Lyszkiewicz M, et al. Responsiveness of developing T cells to IL-7 signals is sustained by miR-17 approximately 92. J Immunol (2015) 195(10):4832–40. doi:10.4049/jimmunol.1402248
86. Kirigin FF, Lindstedt K, Sellars M, Ciofani M, Low SL, Jones L, et al. Dynamic microRNA gene transcription and processing during T cell development. J Immunol (2012) 188(7):3257–67. doi:10.4049/jimmunol.1103175
87. Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med (2012) 18(10):1518–24. doi:10.1038/nm.2963
88. de Kouchkovsky D, Esensten JH, Rosenthal WL, Morar MM, Bluestone JA, Jeker LT. microRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J Immunol (2013) 191(4):1594–605. doi:10.4049/jimmunol.1203567
89. Mele F, Basso C, Leoni C, Aschenbrenner D, Becattini S, Latorre D, et al. ERK phosphorylation and miR-181a expression modulate activation of human memory TH17 cells. Nat Commun (2015) 6:6431. doi:10.1038/ncomms7431
90. Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol (2011) 187(5):2213–21. doi:10.4049/jimmunol.1003952
91. Lu Y, Hippen KL, Lemire AL, Gu J, Wang W, Ni X, et al. miR-146b antagomir-treated human Tregs acquire increased GVHD inhibitory potency. Blood (2016) 128(10):1424–35. doi:10.1182/blood-2016-05-714535
92. Lee CW, Wohlan K, Dallmann I, Forster R, Ganser A, Krueger A, et al. miR-181a expression in donor T cells modulates graft-versus-host disease after allogeneic bone marrow transplantation. J Immunol (2016) 196(9):3927–34. doi:10.4049/jimmunol.1502152
93. Rossi RL, Rossetti G, Wenandy L, Curti S, Ripamonti A, Bonnal RJ, et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat Immunol (2011) 12(8):796–803. doi:10.1038/ni.2057
94. Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med (2018) 215(3):985–97. doi:10.1084/jem.20171626
95. Maschmeyer P, Petkau G, Siracusa F, Zimmermann J, Zugel F, Kuhl AA, et al. Selective targeting of pro-inflammatory Th1 cells by microRNA-148a-specific antagomirs in vivo. J Autoimmun (2017) 89:41–52. doi:10.1016/j.jaut.2017.11.005
96. Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, et al. Silencing of microRNAs in vivo with ’antagomirs’. Nature (2005) 438(7068):685–9. doi:10.1038/nature04303
97. Lucas T, Schafer F, Muller P, Eming SA, Heckel A, Dimmeler S. Light-inducible antimiR-92a as a therapeutic strategy to promote skin repair in healing-impaired diabetic mice. Nat Commun (2017) 8:15162. doi:10.1038/ncomms15162
98. Haftmann C, Stittrich AB, Zimmermann J, Fang Z, Hradilkova K, Bardua M, et al. miR-148a is upregulated by Twist1 and T-bet and promotes Th1-cell survival by regulating the proapoptotic gene Bim. Eur J Immunol (2015) 45(4):1192–205. doi:10.1002/eji.201444633
99. Stittrich AB, Haftmann C, Sgouroudis E, Kuhl AA, Hegazy AN, Panse I, et al. The microRNA miR-182 is induced by IL-2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol (2010) 11(11):1057–62. doi:10.1038/ni.1945
100. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol (2009) 10(12):1252–9. doi:10.1038/ni.1798
101. Gimondi S, Dugo M, Vendramin A, Bermema A, Biancon G, Cavane A, et al. Circulating miRNA panel for prediction of acute graft-versus-host disease in lymphoma patients undergoing matched unrelated hematopoietic stem cell transplantation. Exp Hematol (2016) 44(7):624–34 e1. doi:10.1016/j.exphem.2016.03.005
102. Leonhardt F, Grundmann S, Behe M, Bluhm F, Dumont RA, Braun F, et al. Inflammatory neovascularization during graft-versus-host disease is regulated by alphav integrin and miR-100. Blood (2013) 121(17):3307–18. doi:10.1182/blood-2012-07-442665
Keywords: T cell, microRNA, graft-versus-host disease, graft-versus-tumor effect, differentiation
Citation: Koenecke C and Krueger A (2018) MicroRNA in T-Cell Development and T-Cell Mediated Acute Graft-Versus-Host Disease. Front. Immunol. 9:992. doi: 10.3389/fimmu.2018.00992
Received: 27 February 2018; Accepted: 20 April 2018;
Published: 07 May 2018
Edited by:
Robert Zeiser, Albert Ludwigs Universität Freiburg, GermanyReviewed by:
Benedetto Bruno, Università degli Studi di Torino, ItalyRaphael Carapito, Université de Strasbourg, France
Copyright: © 2018 Koenecke and Krueger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christian Koenecke, a29lbmVja2UuY2hyaXN0aWFuQG1oLWhhbm5vdmVyLmRl;
Andreas Krueger, YW5kcmVhcy5rcnVlZ2VyQGtndS5kZQ==