 Ahmed El-Shamy
Ahmed El-Shamy Andrea D. Branch
Andrea D. Branch Thomas D. Schiano1
Thomas D. Schiano1- 1Division of Liver Disease, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Department of Pharmaceutical and Biological Sciences, California Northstate University, Elk Grove, CA, United States
- 3Division of Rheumatology, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
The complement system bridges innate and adaptive immunity against microbial infections, with viral infection being a major trigger. Activation of the classical, alternative, and lectin pathways have been reported in chronic hepatitis C virus (HCV) infection and/or cryoglobulinemia. HCV infection leads to dysregulation of complement-mediated immune responses. Clinical and experimental evidence support involvement of complement in intra- and extrahepatic manifestations of HCV infection, such as liver fibrosis and type II cryoglobulinemia. In this review, we summarize studies that have investigated the interplay between HCV and the complement system to establish chronic infection and autoimmunity, as well as the association between HCV pathogenesis and abnormal complement profiles. Several unanswered questions are highlighted which suggest additional informative lines of investigation.
Introduction
The complement system includes major host defense mechanisms that bridge innate and adaptive immunity against microbial infections. It is also a critical mediator of the clearance of immune complexes and injured cells (1–3). Dysregulation may be associated with chronic autoimmune inflammatory conditions, such as systemic lupus erythematosus, cryoglobulinemia, and rheumatoid arthritis. Viral infections may trigger dysregulation by direct effects on complement components for the purpose of immune evasion, by effects on specific receptors used for viral entrance into cells or by promoting a pathogenic antiglobulin [i.e., rheumatoid factor (RF)] response as part of chronic immune stimulation. In particular, hepatitis C virus (HCV) infection has been associated with a number of extrahepatic disorders, such as type II mixed cryoglobulinemia (MC) and B cell lymphoma that may be accompanied by complement dysregulation. The recent introduction of direct-acting antiviral (DAA) therapy for HCV allows most patients to achieve a sustained virological response (SVR)/cure. Treatment reduces liver inflammation and improves extrahepatic disease manifestations, but cryoglobulinemia and liver dysfunction may persist (4–14).
The majority of HCV-infected patients with evidence of B cell clonality have abnormal complement profiles; a low serum level of C4 is a “signature” for type II MC patients (15, 16). However, only limited information is available regarding the mechanisms by which the complement system is involved in HCV-induced intra- and extrahepatic disease. Therefore, in this review, we aim to highlight the interplay between HCV and the complement system that become apparent with chronic infection and lymphoproliferation.
Production Sites of Complement Proteins and Receptors
More than 40 complement-related proteins have been identified in the plasma and on cell surfaces, constituting more than 15% of the of plasma globulins (17, 18). The liver makes ~90% of the plasma components of classical, alternative, and lectin pathways. By contrast, whereas hepatocytes are the predominant source of C1r and C1s in blood, activated monocytes/macrophages and immature dendritic cells are the primary source of C1q, a recognition molecule for classical pathway activation that also has significant non-complement functions (19). Although the liver produces the majority of C4, multiple tissues may also produce this protein for local consumption, particularly in response to interferon-gamma (20). Several complement receptors, such as complement receptor type 1 (CR1), the complement receptor of the immunoglobulin superfamily (CRIg), the integrin complement receptors 3 (CR3) and 4 (CR4), and the complement component 5a receptor 1 (C5aR), are expressed on liver cells (hepatocytes, endothelial cells, Kupffer cells, and stellate cells), and contribute to a variety of functions, such as induction of gluconeogenesis, synthesis of acute-phase proteins, hepatocyte proliferation, and phagocytosis (21).
Fibrosis and Regeneration
Increasing evidence implicates activation of the complement system in the pathogenesis and response to acute and chronic liver injury (22). In a clinical study investigating the relationship between complement activation and development of liver fibrosis, blood samples from 50 chronically infected HCV patients were compared to 35 patients with other various liver diseases and 50 healthy controls (23). Complement system activation, as indicated by a significant decrease in total plasma complement activity (CH50) and increase in SC5b-9 [a marker for generation of the membrane-attack complex (MAC)] was associated with high necroinflammatory activity in the HCV patients and patients with other liver diseases compared to controls. The level of SC5b-9 significantly correlated with liver fibrosis stage in HCV-infected patients but not in patients with other liver injuries. In a proteomic survey of serum samples from HCV-infected patients, liver fibrosis stage was associated with a decrease in C3, C4, and Factor H-related protein-1, a regulatory C3b-binding protein (24). C4a, a cleavage product of C4 that contrasts with C3a and C5a with regard to biologic function, was reported to be negatively correlated with the stage of liver fibrosis in children with chronic HCV infection, serum levels being significantly lower in HCV children with advanced fibrosis than those with no/mild fibrosis (25); the significance of this study, however, was limited by the low numbers of HCV-infected children, and the fact that C4a may increase as a function of classical pathway activation and C4 consumption. In a murine genomic study, the gene encoding C5 was identified as a quantitative trait locus associated with the development of liver fibrosis (26). Expression of C5aR1 was significantly upregulated on hepatic stellate cells during trans-differentiation to myofibroblasts in culture. Since myofibroblasts synthesize collagens and other extracellular matrix proteins, elevated C5aR1 expression is consistent with the concept that C5 is a modifier of liver fibrogenesis. Indeed, blockade of C5aR1 reduces hepatic fibrosis in mice (26). Critical roles for C3a and C5a for liver regeneration following carbon tetrachloride injury have been shown in C3, C5, and C3aR knock-out and double knock-out mice, reversible by restoration of C3a, C5, or C5a (27). Interestingly, in 277 patients with chronic HCV infection, two C5 single-nucleotide polymorphisms (SNPs), rs17611 and rs2300929, were associated with advanced fibrosis (26). However, in a study of 1,435 HCV-infected patients and 1,003 patients with other liver diseases, there was no significant association between these C5 SNPs and fibrosis in either patient group (28).
Hepatocellular Carcinoma (HCC)
Cancer growth is determined by intrinsic properties of malignant cells as well as several modifiers, including the complement system, which may either reduce or increase progression (29). HCC is the second leading cause of organ-specific cancer-related death worldwide (30) and is the most rapidly increasing cause of cancer-related death in the United States, Europe, and Japan (31). Of note, C3a was identified by mass spectrometry and 2-dimensional gel electrophoresis (2-DE) of serum samples obtained from HCV-HCC, chronic HCV, HBV-HCC, chronic HBV, and healthy subjects as a differentially expressed biomarker protein with significantly higher levels among HCV-HCC patients compared to the other groups (32).
Treatment
Until recently, HCV treatment has centered on interferon-alpha (IFNα), a cytokine with both antiviral and immunologic effects. However, IFNα-based treatment failed to eliminate HCV in many patients and was often poorly tolerated, particularly because of its ability to induce, uncover and/or exacerbate autoimmune/inflammatory disorders (33, 34). The CC genotype associated with the rs285009 SNP of the C4 gene closely correlated with decreased level of mRNA expression and C4 protein which was more striking at baseline in HCV patients compared to healthy controls. More importantly, the presence of this SNP was significantly associated with a poor response to IFNα-based therapy as well as the development of a high degree of liver fibrosis (35). Interestingly, a significant reduction in C4 activity was also observed in relapsers after IFNα treatment compared to patients who achieved SVR (36). Polymorphism at the rs2230201 SNP of C3 was also associated with IFNα treatment outcome (37). The rs2230201 ‘C’ allele was associated with increased serum C3 levels compared to the ‘T’ allele, which conferred an advantage in attaining SVR, especially in homozygotes. Patients with serum C3 value < 53 mg/dL and non-CC genotypes may not respond to IFNα treatment (37). Recently developed DAA therapies provide an opportunity for HCV patients with autoimmune/inflammatory disorders to be cured with a low risk of side effects (38–40). In the era of DAA, how a patient’s complement profile contributes to the treatment response remains to be defined.
In summary, accumulating clinical observations support a role for the complement system in mediating liver inflammation and fibrosis in HCV infection (Table 1). However, the mechanisms underlying these observations are still unclear. Thus, further experimental and molecular studies are required to dissect how the complement system contributes to intrahepatic HCV pathogenesis, including roles in innate and adaptive immunity, regulation of apoptosis, fibrosis, and regeneration.
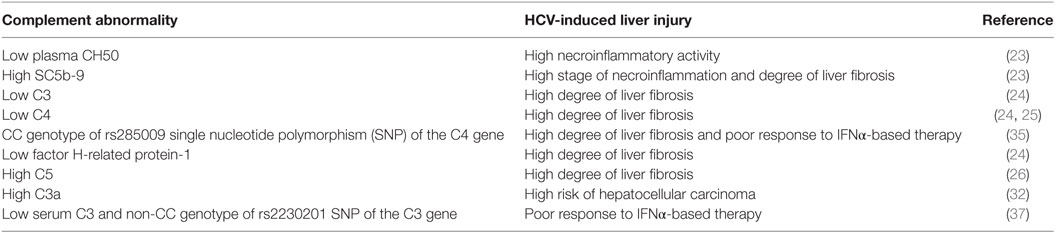
Table 1. Complement system abnormalities in HCV-induced liver injury.
HCV Strategies to Overcome Antiviral Responses of the Complement System
Hepatitis C virus lacks a DNA intermediate; thus, it is incapable of integrating into host chromosomal DNA. Despite this, unlike most RNA viruses, chronic infection is established in ~80% of cases (41) through multiple strategies to evade innate and adaptive antiviral responses (42). In part, this is accomplished directly by inhibition of complement components and/or indirectly by induction of regulators of complement activation (RCA) (Table 2). Mazumdar et al. examined the relationship between HCV infection and C3 concentrations in blood. C3 has a central role in modulating all three pathways of the complement system. In matched serum and liver biopsy samples from HCV patients, both the levels of C3 in serum and the expression of mRNA in biopsies were significantly lower compared to serum and tissue obtained from healthy donors (43). Further in vitro studies showed that HCV-NS5A protein strongly downregulated C3 promoter activity at the basal level. In addition, expression of the transcription factor C/EBP-β, which induces C3 promoter activity, was reduced in immortalized human hepatocytes and human hepatoma cells (Huh7) that were either infected with cell culture-adapted HCV or transfected with HCV-NS5A (43). Moreover, HCV inhibited C3 convertase activity, which is critical in promoting the activity of classical and lectin pathways of complement system (44). Infection of a hepatoma cell line with HCV resulted in inhibition of C2 expression and hence impairment of C3 convertase function. On the other hand, C3b deposition onto bacterial membrane was reduced by sera from HCV patients as compared to healthy controls, which further indicates impaired C3 convertase (44). C4 contributes to the eradication of several viral infections by its role as opsonin and by its central role in promoting the activity of the classical and lectin pathways (3). Notably, C4 protein levels in the serum and mRNA expression levels in liver tissue were lower in HCV patients compared to patients with unrelated liver diseases (45). In vitro studies showed that the expression levels of the two C4 isoforms (C4A and C4B) were significantly reduced in hepatocytes transfected with a full-length HCV genome. In particular, among different HCV proteins, only core and NS5A contributed to HCV’s inhibitory effect on C4 as shown by in vitro transfection experiments, using the Huh7 hepatoma cell line and plasmids containing different HCV proteins (45). Consistent with these in vitro results, the expression levels of C4 mRNA in liver tissue of HCV-core or NS5A transgenic mice were also significantly reduced. Mechanistic studies showed that HCV-core downregulated the expression of upstream stimulatory factor-1, a transcription factor critical for C4 expression, while NS5A inhibited the expression of interferon regulatory factor-1, which is required for IFN-γ-induced C4 promoter activation (45).

Table 2. HCV strategies to evade innate and adaptive immunity using complement system-related components.
Likewise, Kim et al., showed that liver biopsies from HCV patients had lower expression of C9 mRNA compared to samples from unrelated diseases or healthy controls. This indicates that HCV regulates the MAC via C9. C9 mRNA was significantly downregulated in cultured hepatocytes infected with HCV (46). In particular, HCV-core protein had a critical role in regulating C9 promoter activity. Furthermore, in a subsequent in vitro study, HCV NS2 and NS5B proteins were found to be responsible for HCV-associated inhibition of the hepatocyte protein major histocompatibility complex class I-related chains A and B (MICA/B) which functions as a key receptor ligand for NKG2D on NK cells resulting in downregulation of C3 and C4 hepatocyte synthesis (47).
A general role for lectins and pattern recognition of viral glycoproteins has been identified for HIV and HCV (48). In vitro studies showed that mannan-binding lectin (MBL) bound to the HCV E2 ectodomain and E1/E2 heterodimers through its lectin domain, as well as activate complement through MBL-associated serine protease 2 (49). Ficolin-2, a known lectin pathway activator was found to inhibit attachment of HCV envelope E1 and E2 N-glycans to their low-density lipoprotein (LDL) and scavenger B1 receptors (50), with elevated blood levels of L-ficolin or MBL being found in the serum of some patients, possibly correlating with MBL2 genetic variants and response to IFNα (51).
Host expression of RCAs, such as CD35, CD46, CD55, and CD59, serves to protect the cells from MAC lysis (52, 53). HCV has developed strategies to attenuate complement activation by regulating RCAs. Amet et al. first showed that CD59, a key member of RCA, associated with the external membrane of HCV particles obtained from infected patients and Huh7.5.1 cells and had a direct role in abrogating antibody-dependent complement-mediated lysis (54). In vitro studies by Ejaz et al. indicated that HCV selectively incorporates CD59 in its envelope, which inhibits the formation of the MAC complex (55). Also, it was found that HCV infection upregulates the expression of CD55, which accelerates the decay of C3 convertase (56). Taken together, HCV has the capability to attenuate the complement system at multiple steps to weaken the innate immune response.
Role of gC1q Receptor (gC1qR) in HCV Pathogenesis
gC1q receptor is an acidic multifunctional cellular protein ubiquitously expressed on somatic cells (57). It binds to the globular heads of C1q and modulates complement activation (58). Apart from its interaction with C1q, gC1qR binds to several host cell-surface ligands, such as vitronectin and high molecular weight kininogen (59). Interaction of these ligands with gC1qR leads to classical complement pathway activation with generation of inflammatory cytokines, cell adhesion, and activation of the intrinsic coagulation cascade leading to the production of bradykinin, increased vascular permeability, and infiltration of vascular tissue with proinflammatory cells (60). In addition to cellular proteins, gC1qR interacts with several microbial proteins, such as adenovirus core protein (61), HIV rev (62), and protein A of Staphylococcus aureus (63), suggesting its role in the pathogenesis of these infections. Interestingly, by using HCV-core protein as bait in yeast two-hybrid assay, Kittlesen et al. was the first to report the interaction between gC1qR and HCV-core (64). The interaction of HCV-core to gC1qR on T-lymphocytes resulted in inhibition of T-cell proliferation and function through impairment of ERK/MEK phosphorylation (65) and Lck/Akt activation (66). Also, engagement of gC1qR on monocyte-derived dendritic cells with HCV-core resulted in an impaired capacity to generate type 1 CD4+ T cell immunity via inhibition of TLR-induced IL-12 production (67). Therefore, HCV might utilize the direct interaction of its core protein with gC1qR on T cells as a tool to suppress cellular immunity which might imply an important role in persistent infection, an observation that might extend to minicore isoforms of this protein, which lack the RNA binding domain of the p21 core (68).
By contrast to the inhibitory influence of HCV-core protein and gC1qR interaction on T cell responses, this interaction on B cells resulted in hyper-activation and proliferation indicated by upregulation of CD69, overexpression of costimulatory and chemokine receptors, and increased production of IgM and IgG (69). This might partially explain the link between chronic HCV infection, B-cell lymphoproliferative disorders, and several autoimmune-related diseases (70–72). In support of this, the level of circulating gC1qR and gC1qR mRNA of PBMC in HCV patients with MC, one of the major B-cell disorders associated HCV infection, is significantly increased compared to HCV patients without MC or healthy controls (73). Interestingly, there was also a positive correlation between circulating gC1qR with RF activity and C1q concentrations in HCV patients with MC (73). Taken together, these observations suggest the involvement of gC1qR in the pathogenesis of HCV-induced autoimmunity.
Role of C1q in HCV-Induced MC
Mixed cryoglobulins are cold-precipitable complexes of monoclonal or polyclonal IgM RF with polyclonal or oligoclonal IgG (15). Type II MCs, which are composed of monoclonal IgMκ RF and polyclonal IgG, are heavily represented among cryoglobulins associated with chronic HCV infection, and those found in patients with primary Sjogrens syndrome, both of which may be complicated by clonal B-cell proliferations and specific types of non-Hodgkin’s lymphoma (15). HCV patients with symptomatic type II MC suffer from various extrahepatic manifestations, including vascular, renal, and neurological lesions (74), i.e., cryoglobulinemic vasculitis (CryoVas) (75).
In type III MCs, both the IgM and IgG components appear to be polyclonal; extrahepatic disease manifestations may occur, but cryoglobulin levels are lower than type II, and an association with asymptomatic disease is more frequent. In addition, intermediate types characterized by oligoclonality or mixed IgM clonality with polyclonal IgM (type IIa) have also been described (76). Type III MCs may be found in HCV infection, as well as associated with rheumatic diseases [e.g., systemic lupus erythematosus (SLE)] in which complement activation may occur (16). The significance of Type IIa and related intermediate forms with regard to the progression to clonality that may occur in cirrhosis associated with HCV, and in primary Sjogrens syndrome, remains to be fully defined (77, 78).
As noted, a low serum level of C4 is a significant “signature” of type II MC patients (15, 16). This selective depression of C4 strongly implicates classical pathway activation of the complement in cryoglobulin formation. However, the level and function of C4 may be significantly influenced by inter-individual copy-number variation of C4A or C4B genes, charge variation, or isotype deficiency of these genes (79). Incorporation of C1q into isolated cryoprecipitates was first demonstrated as an 11S peak on density gradient ultracentrifugation in patients with lupus nephritis (80). More recent studies have confirmed that cryoprecipitates from patients with HCV-associated CryoVas are enriched in C1q (81), antibodies to HCV antigens (82), and may contain HCV-core protein as indicated by results obtained using an enhanced highly sensitive chemiluminescent microparticle immunoassay (81). Based on evidence that C1q and HCV-core bind to gC1qR, gC1qR/HCV-core complexes might provide a platform for complement activation and deposition of C4D at sites of vasculopathy (83). Additional factors that might be reflected in depletion of C4/C1q and localization to cryocomplexes include the ability of C1q to bind promiscuously to >100 known ligands, including both IgG- and IgM-containing immune complexes, surface-bound C-reactive protein, and molecules exposed at the surface of apoptotic cells, with binding through charged residues on the apex of the gC1q heterotrimer (19, 84), acquired C1-inhibitor deficiency (85), regulation of activation by C4-binding protein (86, 87), and antibodies to C1q and/or potentially to other components of the C1 complex (88).
Role of RF in MC Pathogenesis
Although MC may be associated with a rheumatoid-like arthritis, it is distinct from RA in that antiglobulin activity is restricted to the IgM isotype; although it is presumed that the antigen specificity is directed primarily to determinants in the Fc portion of IgG uncovered by aggregation or complexing to antigen, there have been some suggestions of F(ab)′2 anti-hinge or anti-idiotypic specificity. Low-affinity RFs (Kd~10−5M) are natural antibodies cross reactive with other autoantigens, whereas high-affinity RFs (Kd~10−7M) have undergone affinity maturation and are hypermutated (89). The RF response is broadly represented in a number of infectious etiologies, either as a response to immune complexes formed by the infecting microbe and antibodies or as a function of direct infection and polyclonal B-cell stimulation. In Type II cryoglobulins, the contribution of IgM and IgM RF to total protein content presents a spectrum of concentrations and clonality, ranging from cryocomplexes with RF activity greatly exceeding serum levels and enhanced representation of IgM cross-reactive idiotypes (notably the Wa idiotype first described 45 years ago) (90). This allows for the observation of RF in mixed cryoglobulins in patients with apparently negative RFs, and mixed cryoglobulins that are composed almost entirely of IgM with kappa light chains. It has also been reported that HCV virion in fractionated mixed cryoglobulins is complexed with VLDL, providing a mechanism for localization of virus to sites of pathology via the LDL receptor (91), and the potential for LDL receptor genetic polymorphisms to influence disease outcome (92). RFs studied from B-cell clusters isolated by microdissection of liver biopsies from HCV-infected patients were hypermutated and overlapped in regard to variable region gene usage with blood and bone marrow (93).
Apoptotic Role of C1q in SLE
Activation of complement is a central feature of SLE, intimately related to the pathogenesis of lupus nephritis, and a marker for disease activity and relapse; deficiencies or polymorphisms of molecules central to the classical, alternative, and lectin pathways have been linked to disease susceptibility, immune-complex nephritis, and severity (94). In particular, a central role for C1q and C1q receptors, both with regard to deficiency and as molecular cell-surface sensors for innate and acquired immune responses, has been reviewed (95). The observation that SLE develops in ~90% patients genetically deficient in C1q highlighted a function of C1q as “protector” against autoimmunity that may be independent of its classical role ion complement activation (96). In SLE, C1q deficiency may result either from genetic disorders or anti-C1q autoantibodies (97). The contributions of C1q deficiency in the development of SLE can be related to abrogation of binding to molecules (phosphatidylserine, double-stranded DNA, glyceraldehyde-3-posphate dehydrogenase, annexins 2 and 5, calreticulin) expressed on the surface of dying cells (17) and resultant lack of generation of activated C1s to cleave these apoptotic autoantigens (19). In addition, all three pathways of complement can be activated on the surface of apoptotic cells without further activation of innate or adaptive immune components (17); impaired clearance of dying cells and immune complexes in absolute or functional C1q deficiency is linked to the development of self-reactive B cells with affinity toward multiple autoantigens, and effects on monocyte and dendritic cell differentiation (98, 99).
Anti-C1q autoantibodies have been reported in 30–50% of SLE patients, most commonly correlating with antibodies to double-stranded DNA, nephritis, and low levels of C3 and C4 (100, 101). While antibodies with unique specificities for the globular head and collagen tail of C1q have been identified, the impact of blocking C1q domains on biological activity remains uncertain compared to a number of other (e.g., calreticulin) known inhibitors (17). Although the C1q is a major component of HCV-induced cryoprecipitate, to the best of our knowledge there are no published studies addressing this issue. Low levels of C1q and a significant prevalence of anti-C1q autoantibodies are shared features of SLE and HCV-induced cryoglobulinemia (36, 73, 102, 103).
Interferon-alpha is well-known to have both antiviral and inflammatory effects (104). Plasmacytoid dendritic cells (pCD) are the major producers of IFNα (105). Interestingly, C1q collagen tail interacts with LAIR-1 (CD305), an inhibitory receptor for C1q (106), on pCDs and restricts the production of IFNα (107). Therefore, anti-C1q autoantibodies might contribute to HCV-induced cryoglobulinemia by blocking the interaction between the C1q tail and its inhibitory receptor, LAIR-1, on pDCs resulting in uncontrolled overproduction of IFNα, which may in turn drive the inflammation associated with progression of MC in HCV patients. Alternatively, elevated levels of IFNα produced by uncontrolled pCDs might promote differentiation of B cells into plasma cells resulting in production of pathogenic autoantibodies reported in SLE (108).
Conclusion
The complement system plays a central role in rheumatic and autoimmune diseases, several of which are associated with the presence in blood of cold-perceptible immune complexes enriched in IgM RF, specific antibody activities, putative antigens and C1q as part of a cascade capable of activating the classical pathway, leading in turn to the generation of anaphylatoxins, chemotactic factors, and inflammatory mediators. Both C1q and its globular receptor are promiscuous with regard to ligand specificity, allowing for alternative functions that include binding to specific intracellular antigens expressed on the surface of apoptotic cells, as well as to specific domains of HCV. A research agenda includes the mapping of C1q epitopes responsible for binding to diverse ligands, anti-C1q antibodies, heterotrimeric formation, and C4/C2 serine protease generation that might in turn be targets for therapy. Similar therapeutic strategies might be targeted to gC1qR binding to C1q and High Molecular Weight Kininogen in plasma, on the surface of endothelial cells as a mechanism for vasculopathy, and the regulation of danger sensors on mononuclear cells and immature dendritic cells. A second line of investigation is the delineation of factors responsible for the strikingly low C4 levels in sera of patients with Type II MC and some patients with SLE with regard to mechanisms such as copy-number variation, polymorphisms, cleavage and deposition in tissue, and specific inhibitors. With regard to HCV, a focus on liver pathology would provide an arena to identify complement-defined mechanisms of disease, including immune activation in lymphoid follicles, steatosis, fibrosis, and regeneration.
Author Contributions
AE and PG wrote the review. AB and TS revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Seaver Foundation and an Investigator-initiated grant from Gilead Sciences.
References
1. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi:10.1056/NEJM200104053441406
2. Sarma JV, Ward PA. The complement system. Cell Tissue Res (2011) 343:227–35. doi:10.1007/s00441-010-1034-0
3. Holers VM. Complement and its receptors: new insights into human disease. Annu Rev Immunol (2014) 32:433–59. doi:10.1146/annurev-immunol-032713-120154
4. D’Ambrosio R, Aghemo A, Rumi MG, Ronchi G, Donato MF, Paradis V, et al. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology (2012) 56:532–43. doi:10.1002/hep.25606
5. Chekuri S, Nickerson J, Bichoupan K, Sefcik R, Doobay K, Chang S, et al. Liver stiffness decreases rapidly in response to successful hepatitis C treatment and then plateaus. PLoS One (2016) 11:e0159413. doi:10.1371/journal.pone.0159413
6. Gragnani L, Visentini M, Fognani E, Urraro T, De Santis A, Petraccia L, et al. Prospective study of guideline-tailored therapy with direct-acting antivirals for hepatitis C virus-associated mixed cryoglobulinemia. Hepatology (2016) 64:1473–82. doi:10.1002/hep.28753
7. Mandorfer M, Kozbial K, Schwabl P, Freissmuth C, Schwarzer R, Stern R, et al. Sustained virologic response to interferon-free therapies ameliorates HCV-induced portal hypertension. J Hepatol (2016) 65:692–9. doi:10.1016/j.jhep.2016.05.027
8. Sise ME, Bloom AK, Wisocky J, Lin MV, Gustafson JL, Lundquist AL, et al. Treatment of hepatitis C virus-associated mixed cryoglobulinemia with direct-acting antiviral agents. Hepatology (2016) 63:408–17. doi:10.1002/hep.28297
9. Bonacci M, Lens S, Londono MC, Marino Z, Cid MC, Ramos-Casals M, et al. Virologic, clinical, and immune response outcomes of patients with hepatitis C virus-associated cryoglobulinemia treated with direct-acting antivirals. Clin Gastroenterol Hepatol (2017) 15:575–83.e1. doi:10.1016/j.cgh.2016.09.158
10. Comarmond C, Garrido M, Pol S, Desbois AC, Costopoulos M, Le Garff-Tavernier M, et al. Direct-acting antiviral therapy restores immune tolerance to patients with hepatitis C virus-induced cryoglobulinemia Vasculitis. Gastroenterology (2017) 152:2052–62.e2. doi:10.1053/j.gastro.2017.02.037
11. Emery JS, Kuczynski M, La D, Almarzooqi S, Kowgier M, Shah H, et al. Efficacy and safety of direct acting antivirals for the treatment of mixed cryoglobulinemia. Am J Gastroenterol (2017) 112:1298–308. doi:10.1038/ajg.2017.49
12. Lauletta G, Russi S, Pavone F, Vacca A, Dammacco F. Direct-acting antiviral agents in the therapy of hepatitis C virus-related mixed cryoglobulinaemia: a single-centre experience. Arthritis Res Ther (2017) 19:74. doi:10.1186/s13075-017-1280-6
13. Saadoun D, Pol S, Ferfar Y, Alric L, Hezode C, Si Ahmed SN, et al. Efficacy and safety of sofosbuvir plus daclatasvir for treatment of HCV-associated cryoglobulinemia vasculitis. Gastroenterology (2017) 153:49–52.e5. doi:10.1053/j.gastro.2017.03.006
14. Welsch C, Efinger M, Von Wagner M, Herrmann E, Zeuzem S, Welzel TM, et al. Ongoing liver inflammation in patients with chronic hepatitis C and sustained virological response. PLoS One (2017) 12:e0171755. doi:10.1371/journal.pone.0171755
15. Gorevic PD. Rheumatoid factor, complement, and mixed cryoglobulinemia. Clin Dev Immunol (2012) 2012:439018. doi:10.1155/2012/439018
16. Gorevic PD, Galanakis DK. Cryoglobulins, cryofibrinogenemia, and pyroglobulins. 8th ed. In: Detrick B, Schmitz J, Hamilton R, editors. Manual of Molecular and Clinical Laboratory Immunology. Washington, DC: ASM Press (2016). p. 101–11.
17. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I – molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi:10.3389/fimmu.2015.00262
18. Reddy YN, Siedlecki AM, Francis JM. Breaking down the complement system: a review and update on novel therapies. Curr Opin Nephrol Hypertens (2017) 26:123–8. doi:10.1097/MNH.0000000000000305
19. Lu J, Kishore U. C1 complex: an adaptable proteolytic module for complement and non-complement functions. Front Immunol (2017) 8:592. doi:10.3389/fimmu.2017.00592
20. Mitchell TJ, Naughton M, Norsworthy P, Davies KA, Walport MJ, Morley BJ. IFN-gamma up-regulates expression of the complement components C3 and C4 by stabilization of mRNA. J Immunol (1996) 156:4429–34.
22. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol (2016) 12:383–401. doi:10.1038/nrneph.2016.70
23. Vasel M, Rutz R, Bersch C, Feick P, Singer MV, Kirschfink M, et al. Complement activation correlates with liver necrosis and fibrosis in chronic hepatitis C. Clin Immunol (2014) 150:149–56. doi:10.1016/j.clim.2013.11.014
24. Gangadharan B, Antrobus R, Dwek RA, Zitzmann N. Novel serum biomarker candidates for liver fibrosis in hepatitis C patients. Clin Chem (2007) 53:1792–9. doi:10.1373/clinchem.2007.089144
25. Behairy BE, El-Mashad GM, Abd-Elghany RS, Ghoneim EM, Sira MM. Serum complement C4a and its relation to liver fibrosis in children with chronic hepatitis C. World J Hepatol (2013) 5:445–51. doi:10.4254/wjh.v5.i8.445
26. Hillebrandt S, Wasmuth HE, Weiskirchen R, Hellerbrand C, Keppeler H, Werth A, et al. Complement factor 5 is a quantitative trait gene that modifies liver fibrogenesis in mice and humans. Nat Genet (2005) 37:835–43. doi:10.1038/ng1599
27. Rutkowski MJ, Sughrue ME, Kane AJ, Ahn BJ, Fang S, Parsa AT. The complement cascade as a mediator of tissue growth and regeneration. Inflamm Res (2010) 59:897–905. doi:10.1007/s00011-010-0220-6
28. Halangk J, Sarrazin C, Neumann K, Puhl G, Mueller T, Teuber G, et al. Evaluation of complement factor 5 variants as genetic risk factors for the development of advanced fibrosis in chronic hepatitis C infection. J Hepatol (2008) 49:339–45. doi:10.1016/j.jhep.2008.05.021
29. Markiewski MM, Deangelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol (2008) 9:1225–35. doi:10.1038/ni.1655
30. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2015) 136:E359–86. doi:10.1002/ijc.29210
31. Hoshida Y, Fuchs BC, Bardeesy N, Baumert TF, Chung RT. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. J Hepatol (2014) 61:S79–90. doi:10.1016/j.jhep.2014.07.010
32. Lee IN, Chen CH, Sheu JC, Lee HS, Huang GT, Chen DS, et al. Identification of complement C3a as a candidate biomarker in human chronic hepatitis C and HCV-related hepatocellular carcinoma using a proteomics approach. Proteomics (2006) 6:2865–73. doi:10.1002/pmic.200500488
33. McHutchison JG, Gordon SC, Schiff ER, Shiffman ML, Lee WM, Rustgi VK, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. Hepatitis Interventional Therapy Group. N Engl J Med (1998) 339:1485–92. doi:10.1056/NEJM199811193392101
34. Liang TJ, Ghany MG. Therapy of hepatitis C – back to the future. N Engl J Med (2014) 370:2043–7. doi:10.1056/NEJMe1403619
35. Chowdhury SJ, Karra VK, Bharali R, Kar P. Role of complement component C4 in treatment response and disease progression in chronic hepatitis C patients. J Viral Hepat (2015) 22:671–4. doi:10.1111/jvh.12383
36. Dumestre-Perard C, Ponard D, Drouet C, Leroy V, Zarski JP, Dutertre N, et al. Complement C4 monitoring in the follow-up of chronic hepatitis C treatment. Clin Exp Immunol (2002) 127:131–6. doi:10.1046/j.1365-2249.2002.01729.x
37. Chowdhury SJ, Karra VK, Gumma PK, Bharali R, Kar P. rs2230201 polymorphism may dictate complement C3 levels and response to treatment in chronic hepatitis C patients. J Viral Hepat (2015) 22:184–91. doi:10.1111/jvh.12280
38. Lynch SM, Wu GY. Hepatitis C virus: a review of treatment guidelines, cost-effectiveness, and access to therapy. J Clin Transl Hepatol (2016) 4:310–9. doi:10.14218/JCTH.2016.00027
39. Zhang J, Nguyen D, Hu KQ. Chronic hepatitis C virus infection: a review of current direct-acting antiviral treatment strategies. N Am J Med Sci (Boston) (2016) 9(2):47–54.
40. Falade-Nwulia O, Suarez-Cuervo C, Nelson DR, Fried MW, Segal JB, Sulkowski MS. Oral direct-acting agent therapy for hepatitis C virus infection: a systematic review. Ann Intern Med (2017) 166:637–48. doi:10.7326/M16-2575
41. Thrift AP, El-Serag HB, Kanwal F. Global epidemiology and burden of HCV infection and HCV-related disease. Nat Rev Gastroenterol Hepatol (2017) 14:122–32. doi:10.1038/nrgastro.2016.176
42. Gokhale NS, Vazquez C, Horner SM. Hepatitis C virus. Strategies to evade antiviral responses. Future Virol (2014) 9:1061–75. doi:10.2217/fvl.14.89
43. Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, et al. Hepatitis C virus proteins inhibit C3 complement production. J Virol (2012) 86:2221–8. doi:10.1128/JVI.06577-11
44. Kim H, Meyer K, Di Bisceglie AM, Ray R. Inhibition of c3 convertase activity by hepatitis C virus as an additional lesion in the regulation of complement components. PLoS One (2014) 9:e101422. doi:10.1371/journal.pone.0101422
45. Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. Transcriptional repression of C4 complement by hepatitis C virus proteins. J Virol (2011) 85:4157–66. doi:10.1128/JVI.02449-10
46. Kim H, Meyer K, Di Bisceglie AM, Ray R. Hepatitis C virus suppresses C9 complement synthesis and impairs membrane attack complex function. J Virol (2013) 87:5858–67. doi:10.1128/JVI.00174-13
47. Kim H, Bose SK, Meyer K, Ray R. Hepatitis C virus impairs natural killer cell-mediated augmentation of complement synthesis. J Virol (2014) 88:2564–71. doi:10.1128/JVI.02988-13
48. Mason CP, Tarr AW. Human lectins and their roles in viral infections. Molecules (2015) 20:2229–71. doi:10.3390/molecules20022229
49. Brown KS, Keogh MJ, Owsianka AM, Adair R, Patel AH, Arnold JN, et al. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell (2010) 1:664–74. doi:10.1007/s13238-010-0088-9
50. Zhao Y, Ren Y, Zhang X, Zhao P, Tao W, Zhong J, et al. Ficolin-2 inhibits hepatitis C virus infection, whereas apolipoprotein E3 mediates viral immune escape. J Immunol (2014) 193:783–96. doi:10.4049/jimmunol.1302563
51. Zupin L, Polesello V, Alberi G, Moratelli G, Croce SL, Masutti F, et al. MBL2 genetic variants in HCV infection susceptibility, spontaneous viral clearance and pegylated interferon plus ribavirin treatment response. Scand J Immunol (2016) 84:61–9. doi:10.1111/sji.12444
52. Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology (2011) 411:362–73. doi:10.1016/j.virol.2010.12.045
53. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion strategies of viruses: an overview. Front Microbiol (2017) 8:1117. doi:10.3389/fmicb.2017.01117
54. Amet T, Ghabril M, Chalasani N, Byrd D, Hu N, Grantham A, et al. CD59 incorporation protects hepatitis C virus against complement-mediated destruction. Hepatology (2012) 55:354–63. doi:10.1002/hep.24686
55. Ejaz A, Steinmann E, Banki Z, Anggakusuma, Khalid S, Lengauer S, et al. Specific acquisition of functional CD59 but not CD46 or CD55 by hepatitis C virus. PLoS One (2012) 7:e45770. doi:10.1371/journal.pone.0045770
56. Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, et al. Hepatitis C virus infection upregulates CD55 expression on the hepatocyte surface and promotes association with virus particles. J Virol (2013) 87:7902–10. doi:10.1128/JVI.00917-13
57. Dembitzer FR, Kinoshita Y, Burstein D, Phelps RG, Beasley MB, Garcia R, et al. gC1qR expression in normal and pathologic human tissues: differential expression in tissues of epithelial and mesenchymal origin. J Histochem Cytochem (2012) 60:467–74. doi:10.1369/0022155412440882
58. Ghebrehiwet B, Lim BL, Kumar R, Feng X, Peerschke EI. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol Rev (2001) 180:65–77. doi:10.1034/j.1600-065X.2001.1800106.x
59. Joseph K, Ghebrehiwet B, Peerschke EI, Reid KB, Kaplan AP. Identification of the zinc-dependent endothelial cell binding protein for high molecular weight kininogen and factor XII: identity with the receptor that binds to the globular “heads” of C1q (gC1q-R). Proc Natl Acad Sci U S A (1996) 93:8552–7. doi:10.1073/pnas.93.16.8552
60. Joseph K, Ghebrehiwet B, Kaplan AP. Activation of the kinin-forming cascade on the surface of endothelial cells. Biol Chem (2001) 382:71–5. doi:10.1515/BC.2001.012
61. Matthews DA, Russell WC. Adenovirus core protein V interacts with p32 – a protein which is associated with both the mitochondria and the nucleus. J Gen Virol (1998) 79(Pt 7):1677–85. doi:10.1099/0022-1317-79-7-1677
62. Luo Y, Yu H, Peterlin BM. Cellular protein modulates effects of human immunodeficiency virus type 1 Rev. J Virol (1994) 68:3850–6.
63. Nguyen T, Ghebrehiwet B, Peerschke EI. Staphylococcus aureus protein A recognizes platelet gC1qR/p33: a novel mechanism for staphylococcal interactions with platelets. Infect Immun (2000) 68:2061–8. doi:10.1128/IAI.68.4.2061-2068.2000
64. Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest (2000) 106:1239–49. doi:10.1172/JCI10323
65. Yao ZQ, Nguyen DT, Hiotellis AI, Hahn YS. Hepatitis C virus core protein inhibits human T lymphocyte responses by a complement-dependent regulatory pathway. J Immunol (2001) 167:5264–72. doi:10.4049/jimmunol.167.9.5264
66. Yao ZQ, Eisen-Vandervelde A, Waggoner SN, Cale EM, Hahn YS. Direct binding of hepatitis C virus core to gC1qR on CD4+ and CD8+ T cells leads to impaired activation of Lck and Akt. J Virol (2004) 78:6409–19. doi:10.1128/JVI.78.12.6409-6419.2004
67. Waggoner SN, Hall CH, Hahn YS. HCV core protein interaction with gC1q receptor inhibits Th1 differentiation of CD4+ T cells via suppression of dendritic cell IL-12 production. J Leukoc Biol (2007) 82:1407–19. doi:10.1189/jlb.0507268
68. Eng FJ, El-Shamy A, Doyle EH, Klepper A, Muerhoff AS, Branch AD. Newly discovered hepatitis C virus minicores circulate in human blood. Hepatol Commun (2018) 2:21–8. doi:10.1002/hep4.1125
69. Yao ZQ, Prayther D, Trabue C, Dong ZP, Moorman J. Differential regulation of SOCS-1 signalling in B and T lymphocytes by hepatitis C virus core protein. Immunology (2008) 125:197–207. doi:10.1111/j.1365-2567.2008.02829.x
70. McMurray RW, Elbourne K. Hepatitis C virus infection and autoimmunity. Semin Arthritis Rheum (1997) 26:689–701. doi:10.1016/S0049-0172(97)80005-4
71. Strassburg CP, Vogel A, Manns MP. Autoimmunity and hepatitis C. Autoimmun Rev (2003) 2:322–31. doi:10.1016/S1568-9972(03)00036-3
72. Paroli M, Iannucci G, Accapezzato D. Hepatitis C virus infection and autoimmune diseases. Int J Gen Med (2012) 5:903–7. doi:10.2147/IJGM.S37580
73. Sansonno D, Tucci FA, Ghebrehiwet B, Lauletta G, Peerschke EI, Conteduca V, et al. Role of the receptor for the globular domain of C1q protein in the pathogenesis of hepatitis C virus-related cryoglobulin vascular damage. J Immunol (2009) 183:6013–20. doi:10.4049/jimmunol.0902038
74. Cacoub P, Gragnani L, Comarmond C, Zignego AL. Extrahepatic manifestations of chronic hepatitis C virus infection. Dig Liver Dis (2014) 46(Suppl 5):S165–73. doi:10.1016/j.dld.2014.10.005
75. Quartuccio L, Isola M, Corazza L, Maset M, Monti G, Gabrielli A, et al. Performance of the preliminary classification criteria for cryoglobulinaemic vasculitis and clinical manifestations in hepatitis C virus-unrelated cryoglobulinaemic vasculitis. Clin Exp Rheumatol (2012) 30(Suppl 70):S48–52.
76. Tissot JD, Invernizzi F, Schifferli JA, Spertini F, Schneider P. Two-dimensional electrophoretic analysis of cryoproteins: a report of 335 samples. Electrophoresis (1999) 20:606–13. doi:10.1002/(SICI)1522-2683(19990301)20:3<606::AID-ELPS606>3.0.CO;2-N
77. Sene D, Ghillani-Dalbin P, Thibault V, Guis L, Musset L, Duhaut P, et al. Longterm course of mixed cryoglobulinemia in patients infected with hepatitis C virus. J Rheumatol (2004) 31(11):2199–206.
78. De Rosa FG, Abel G, Agnello V. Observations on cryoglobulin testing: II. The association of oligoclonal mixed cryoglobulinemia with cirrhosis in patients infected with hepatitis C virus. J Rheumatol (2009) 36:1956–7. doi:10.3899/jrheum.090189
79. Menegatti E, Messina M, Oddone V, Rubini E, Sciascia S, Naretto C, et al. Immunogenetics of complement in mixed cryoglobulinaemia. Clin Exp Rheumatol (2016) 34(Suppl 97):S12–5.
80. Hanauer LB, Christian CL. Studies of cryoproteins in systemic lupus erythematosus. J Clin Invest (1967) 46:400–8. doi:10.1172/JCI105541
81. Sansonno D, Lauletta G, Nisi L, Gatti P, Pesola F, Pansini N, et al. Non-enveloped HCV core protein as constitutive antigen of cold-precipitable immune complexes in type II mixed cryoglobulinaemia. Clin Exp Immunol (2003) 133:275–82. doi:10.1046/j.1365-2249.2003.02204.x
82. Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med (1992) 327:1490–5. doi:10.1056/NEJM199211193272104
83. Dammacco F, Racanelli V, Russi S, Sansonno D. The expanding spectrum of HCV-related cryoglobulinemic vasculitis: a narrative review. Clin Exp Med (2016) 16:233–42. doi:10.1007/s10238-016-0410-9
84. Weiner SM, Prasauskas V, Lebrecht D, Weber S, Peter HH, Vaith P. Occurrence of C-reactive protein in cryoglobulins. Clin Exp Immunol (2001) 125:316–22. doi:10.1046/j.1365-2249.2001.01606.x
85. Casali P, Borzini P, Pioltelli P, Invernizzi F, Zanussi C. Acquired C1-inhibitor deficiency in essential cryoglobulinemia and macrocryoglobulinemia. Acta Haematol (1978) 59:277–84. doi:10.1159/000207773
86. Haydey RP, Patarroyo De Rojas M, Gigli I. A newly described control mechanism of complement activation in patients with mixed cryoglobulinemia (cryoglobulins and complement). J Invest Dermatol (1980) 74:328–32. doi:10.1111/1523-1747.ep12543575
87. Zadura AF, Theander E, Blom AM, Trouw LA. Complement inhibitor C4b-binding protein in primary Sjogren’s syndrome and its association with other disease markers. Scand J Immunol (2009) 69:374–80. doi:10.1111/j.1365-3083.2009.02229.x
88. Lintner KE, Wu YL, Yang Y, Spencer CH, Hauptmann G, Hebert LA, et al. Early components of the complement classical activation pathway in human systemic autoimmune diseases. Front Immunol (2016) 7:36. doi:10.3389/fimmu.2016.00036
89. Posnett DN, Edinger J. When do microbes stimulate rheumatoid factor? J Exp Med (1997) 185:1721–3. doi:10.1084/jem.185.10.1721
90. De Rosa FG, Agnello V. Observations on cryoglobulin testing: I. The association of cryoglobulins containing rheumatoid factors with manifestation of cryoglobulinemic vasculitis. J Rheumatol (2009) 36:1953–5. doi:10.3899/jrheum.081035
91. Agnello V. The Kunkel legacy and hepatitis C virus infection. Clin Immunol (2016) 172:78–82. doi:10.1016/j.clim.2016.07.023
92. Caruz A, Neukam K, Rivero-Juarez A, Herrero R, Real LM, Camacho A, et al. Association of low-density lipoprotein receptor genotypes with hepatitis C viral load. Genes Immun (2014) 15:16–24. doi:10.1038/gene.2013.56
93. Dammacco F, Sansonno D, Piccoli C, Racanelli V, D’amore FP, Lauletta G. The lymphoid system in hepatitis C virus infection: autoimmunity, mixed cryoglobulinemia, and overt B-cell malignancy. Semin Liver Dis (2000) 20:143–57. doi:10.1055/s-2000-9613
94. Bryan AR, Wu EY. Complement deficiencies in systemic lupus erythematosus. Curr Allergy Asthma Rep (2014) 14:448. doi:10.1007/s11882-014-0448-2
95. Ghebrehiwet B, Peerschke EI. Role of C1q and C1q receptors in the pathogenesis of systemic lupus erythematosus. Curr Dir Autoimmun (2004) 7:87–97. doi:10.1159/000075688
96. Macedo AC, Isaac L. Systemic lupus erythematosus and deficiencies of early components of the complement classical pathway. Front Immunol (2016) 7:55. doi:10.3389/fimmu.2016.00055
97. Son M, Diamond B, Santiago-Schwarz F. Fundamental role of C1q in autoimmunity and inflammation. Immunol Res (2015) 63:101–6. doi:10.1007/s12026-015-8705-6
98. Botto M, Walport MJ. C1q, autoimmunity and apoptosis. Immunobiology (2002) 205:395–406. doi:10.1078/0171-2985-00141
99. Ghebrehiwet B, Hosszu KK, Valentino A, Ji Y, Peerschke EI. Monocyte expressed macromolecular C1 and C1q receptors as molecular sensors of danger: implications in SLE. Front Immunol (2014) 5:278. doi:10.3389/fimmu.2014.00278
100. Seelen MA, Trouw LA, Daha MR. Diagnostic and prognostic significance of anti-C1q antibodies in systemic lupus erythematosus. Curr Opin Nephrol Hypertens (2003) 12:619–24. doi:10.1097/00041552-200311000-00008
101. Marto N, Bertolaccini ML, Calabuig E, Hughes GR, Khamashta MA. Anti-C1q antibodies in nephritis: correlation between titres and renal disease activity and positive predictive value in systemic lupus erythematosus. Ann Rheum Dis (2005) 64:444–8. doi:10.1136/ard.2004.024943
102. Saadoun D, Sadallah S, Trendelenburg M, Limal N, Sene D, Piette JC, et al. Anti-C1q antibodies in hepatitis C virus infection. Clin Exp Immunol (2006) 145:308–12. doi:10.1111/j.1365-2249.2006.03153.x
103. Fadda SH, Bassyouni IH, Hamdy A, Foad NA, Wali IE. Anti-C1q in chronic hepatitis C virus genotype IV infection: association with autoimmune rheumatologic manifestations. Immunol Invest (2015) 44:45–55. doi:10.3109/08820139.2014.932378
104. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi:10.1038/nri3581
105. Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, et al. Viral infection and toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol (2004) 34:796–805. doi:10.1002/eji.200324610
106. Meyaard L. The inhibitory collagen receptor LAIR-1 (CD305). J Leukoc Biol (2008) 83:799–803. doi:10.1189/jlb.0907609
107. Son M, Santiago-Schwarz F, Al-Abed Y, Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U S A (2012) 109:E3160–7. doi:10.1073/pnas.1212753109
Keywords: liver, hepatitis C virus, complement, C1q, gC1qR, mixed cryoglobulinemia
Citation: El-Shamy A, Branch AD, Schiano TD and Gorevic PD (2018) The Complement System and C1q in Chronic Hepatitis C Virus Infection and Mixed Cryoglobulinemia. Front. Immunol. 9:1001. doi: 10.3389/fimmu.2018.01001
Received: 24 January 2018; Accepted: 23 April 2018;
Published: 29 May 2018
Edited by:
Uday Kishore, Brunel University London, United KingdomReviewed by:
Laura Gragnani, Università degli Studi di Firenze, ItalyAlexander William Tarr, University of Nottingham, United Kingdom
Copyright: © 2018 El-Shamy, Branch, Schiano and Gorevic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed El-Shamy, YWhtZWQuZWxzaGFteUBjbnN1LmVkdQ==;
Peter D. Gorevic, cGV0ZXIuZ29yZXZpY0Btb3VudHNpbmFpLm9yZw==