 Reka P. Toth
Reka P. Toth Julie D. Atkin
Julie D. Atkin- 1Motor Neuron Disease Research Centre, Department of Biomedical Sciences, Macquarie University, Sydney, NSW, Australia
- 2Department of Biochemistry, La Trobe Institute for Molecular Science, Melbourne, VIC, Australia
Neurodegenerative disorders, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia, and glaucoma, affect millions of people worldwide. ALS is caused by the loss of motor neurons in the spinal cord, brainstem, and brain, and genetic mutations are responsible for 10% of all ALS cases. Glaucoma is characterized by the loss of retinal ganglion cells and is the most common cause of irreversible blindness. Interestingly, mutations in OPTN, encoding optineurin, are associated with both ALS and glaucoma. Optineurin is a highly abundant protein involved in a wide range of cellular processes, including the inflammatory response, autophagy, Golgi maintenance, and vesicular transport. In this review, we summarize the role of optineurin in cellular mechanisms implicated in neurodegenerative disorders, including neuroinflammation, autophagy, and vesicular trafficking, focusing in particular on the consequences of expression of mutations associated with ALS and glaucoma. This review, therefore showcases the impact of optineurin dysfunction in ALS and glaucoma.
Introduction
Neurodegenerative diseases are characterized by the degeneration and consequent death of neurons in specific regions of the brain and/or spinal cord. A wide range of neurodegenerative disorders manifest in humans, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease, that affect movement, speech, memory, and sensory or cognitive functions. Similarly, glaucoma is a primary optic neuropathy that has also been categorized as a neurodegenerative disorder (1). A variable proportion of cases of these diseases result from the presence of mutations in specific genes. One of the genes mutated in ALS, as well as in glaucoma, encodes optineurin, a ubiquitously expressed protein involved in neuroinflammation, Golgi maintenance, vesicular trafficking, and autophagy.
A common pathological hallmark of neurodegenerative diseases is the presence of misfolded proteins that aggregate into insoluble intra- or extracellular inclusions. Extensive research has been undertaken to determine whether these inclusions contribute to the development of disease, but it remains controversial whether the inclusions are either harmful, by directly inducing toxicity or beneficial, by sequestering misfolded, toxic proteins. Similarly, the etiology of these conditions remains unclear. Optineurin is present within the inclusions in several neurodegenerative disorders, including ALS (2), FTD (3), Alzheimer’s disease (4), and Huntington’s disease (5). However, the role of optineurin in the pathogenesis of ALS and other diseases is not well understood. In this review, we summarize the current understanding of the role of optineurin in ALS, glaucoma, and also other neurodegenerative diseases, focusing on autophagy, vesicular trafficking, and neuroinflammation.
ALS and FTD
Amyotrophic lateral sclerosis, also known as motor neuron disease or Lou Gehrig’s disease, is a mainly adult-onset, rapidly progressing neurodegenerative disorder. ALS is associated with the loss of upper motor neurons in the motor cortex and brainstem and lower motor neurons in the ventral horn of the spinal cord. The median period of survival following diagnosis is approximately 3 years (6), although 10% of patients live up to 10 years after symptom onset (7). Over the course of ALS, patients gradually lose the ability to walk, move, swallow, or speak, and eventually to breath. The worldwide incidence of ALS is 1.75 in 100,000 people and it is slightly higher in men than women (8).
Most cases of ALS (90%) arise sporadically, with no previous family history. However, approximately 10% of cases are familial (fALS), resulting from the presence of one or more genetic mutations. It was previously thought that sporadic ALS (sALS) has no genetic component; however, recent studies have revealed that genetic mutations may be the underlying cause of a significant proportion of sALS cases (9–11). However, studies of identical twins have also implicated both environmental and epigenetic factors in the development of ALS (12–16).
Due to improvements in next-generation sequencing methods, extensive advances have been made in the last 20 years in the underlying genetic causes of fALS. The first gene linked to ALS was superoxide dismutase-1 (SOD1) (17), and since then, more than 30 genes have been associated (18), either strongly or weakly, with ALS. Mutations in the Chromosome 9 open reading frame 72 (C9orf72) gene are responsible for most fALS cases (40%) in Caucasian populations (19), followed by mutations in genes encoding SOD1 (12%), TAR-DNA-binding protein of 43 kDa (TDP-43, 4%), and fused in sarcoma (FUS, 4%) (19). Rarer cases of ALS are caused by mutations in the genes encoding TANK-binding kinase-1 (TBK-1) (20), valosin containing protein (VCP) (21), ubiquilin-2 (22), cyclin-F (23), and optineurin (2), as well as several others (Table 1). Optineurin mutations have been identified in both fALS and sALS patients, and their frequency depends on ethnicity. In Asian populations, mutations in OPTN account for 4% of fALS and 0.4% of sALS cases, whereas this frequency is much lower in European populations (1.5 and 0.3%, respectively) (24). Dysfunction in several cellular mechanisms has been linked with the pathophysiology of ALS (and/or glaucoma), including excitotoxicity, deregulation of RNA metabolism, endoplasmic reticulum (ER) stress, dysfunction of mitochondria, autophagy, neuroinflammation, cytoskeletal defects, and altered axonal transport (Figure 1).
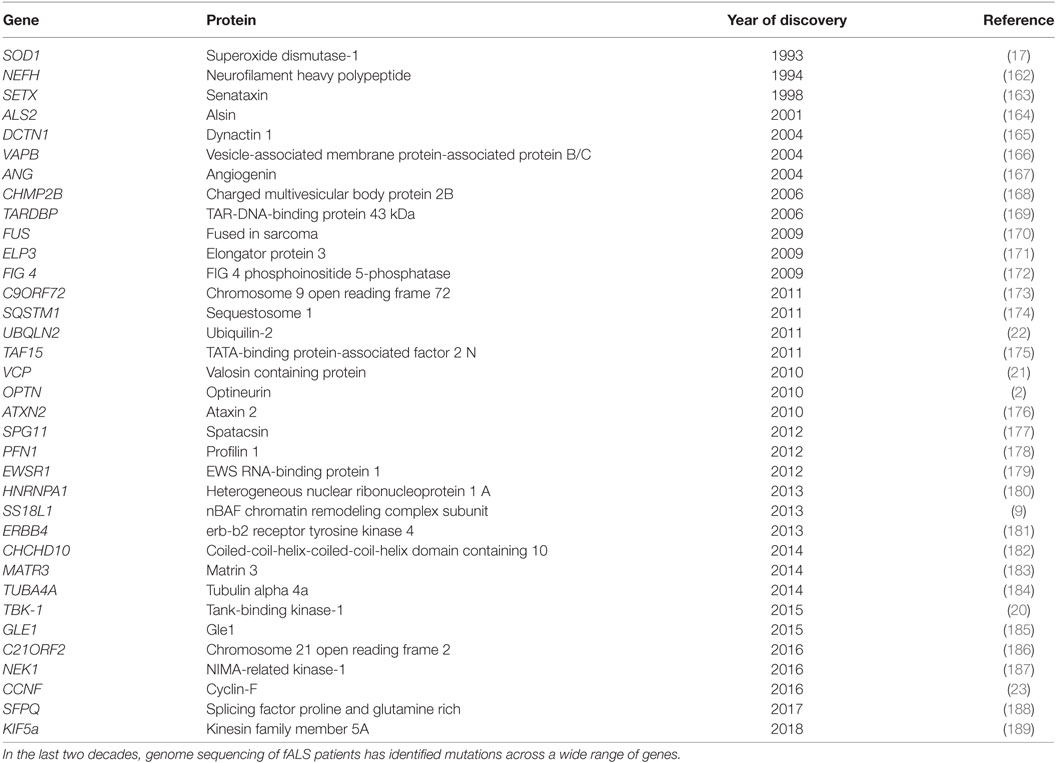
Table 1. Genes associated with familial ALS.
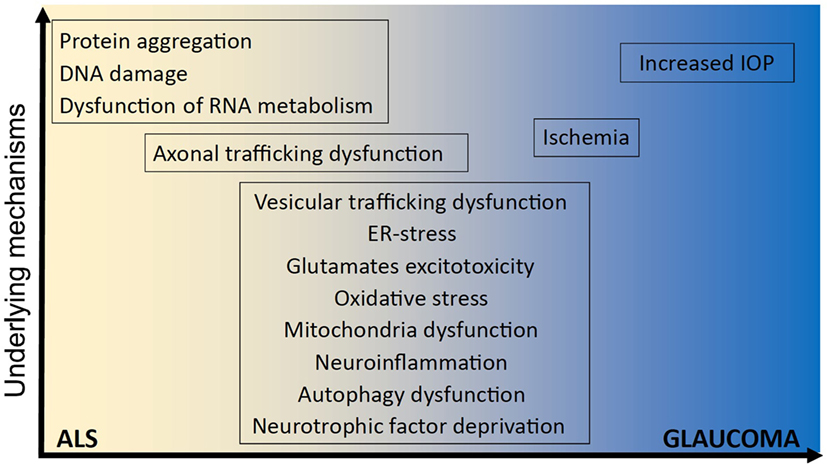
Figure 1. Underlying mechanisms implicated in ALS and glaucoma. It remains unclear how optineurin mutations cause amyotrophic lateral sclerosis (ALS) and glaucoma and whether neurodegeneration involves mechanisms that are unique to one disorder or common to both. However, several shared cellular mechanisms have been implicated in both disorders, namely vesicular trafficking, endoplasmic reticulum (ER) stress, excitotoxicity, oxidative stress, mitochondria dysfunction, neuroinflammation, autophagy dysfunction, neurotrophic factor deprivation, axonal trafficking dysfunction, and ischemia. In contrast, other cellular processes have been implicated in either ALS; dysregulation of RNA metabolism, DNA damage, and protein aggregation; or glaucoma; increased intraocular pressure (IOP), and IOP; but not both.
In recent years, it has become increasingly recognized that ALS overlaps genetically and pathologically with FTD. Moreover, it is now an accepted dogma that ALS and FTD represent two ends of the same disease spectrum (25). FTD is the second most common form of dementia after Alzheimer’s disease, with a prevalence estimated between 10 and 20 per 100,000 individuals (26, 27). FTD is a complex disorder associated with progressive atrophy of both the frontal and temporal lobes of the brain. Clinical symptoms include behavioral changes and difficulties in speech (27, 28) [for recent reviews, see Ref. (26, 29)]. Up to 50% of ALS patients will develop FTD-like symptoms (29, 30), and approximately 15% of patients will receive a definite diagnosis of FTD. Similarly, up to half of FTD patients will experience motor neuron dysfunction, and eventually 15% will be diagnosed with ALS. Genetic mutations are present in 10–20% of FTD cases and mutations in C9orf72, TARDBP, FUS, VCP, or SQSTM1, and TBK-1 are also associated with FTD as well as ALS. Although the role of OPTN mutations in FTD is controversial, they are implicated in >1% of patients (31).
Glaucoma
Glaucoma describes a group of disorders characterized by the death of retinal ganglion cells (RGCs) and degeneration of the neural axons that form the optic nerve fibre (32). It is the second leading cause of irreversible blindness, affecting over 57 million people worldwide (33). Primary open-angle glaucoma (POAG) is the most common type of glaucoma, where disease develops despite anatomically correct ocular structures. Increased intraocular pressure is a major risk factor for glaucoma (32). Normal tension glaucoma (NTG) is a subtype of POAG, where glaucoma arises in the absence of increased intraocular pressure (34). Mutations in the gene encoding myocilin are causative in 8–36% of juvenile open-angle glaucoma cases and 2–4% of adult-onset POAG cases (34). Missense mutations in OPTN (35) and copy number variations of TBK-1 (36) account for approximately 2–3% of NTG.
Optineurin Mutations in ALS, FTD, and Glaucoma
OPTN was first identified as an ALS causative gene in 2010, and since then, more than 20 mutations have been described. Sequencing of six Japanese individuals with consanguineous marriages first identified three mutations: homozygous deletion of exon 5, a homozygous mutation that results in a truncated protein (Q398X), and a heterozygous missense mutation (E478G) (2). Additional ALS mutations in OPTN were subsequently identified in ALS patients: R96L (37), Q165X (38), A136V (39), K395K (39), I451T (39), E516Q (39), and Q454E (38) (a full list of ALS-associated mutations in OPTN is detailed in Table 2). However, the involvement of optineurin in FTD is still under debate. One study identified OPTN variants in 4.8% of FTD patients without motor neuron involvement (31), but another study examining 371 FTD cases could not detect the presence of optineurin mutations (40). In contrast, optineurin mutations have been frequently reported in glaucoma. Analysis of 54 families with autosomal dominantly inherited adult-onset POAG revealed that 16.7% of patients possessed OPTN mutations (35). These mutations include E50K (35), M98K (35), H486R (35), R545Q (35), H26D (41), E322K (42), E103D (43), and V148V (43) [for a detailed recent review discussing OPTN mutations and glaucoma, see Ref. (33)].

Table 2. ALS-associated mutations in OPTN.
Functions of Optineurin
Optineurin is a highly conserved, 64 kDa hexameric protein, consisting of 577 amino acids (aa) (44). The putative domain structure of optineurin is illustrated in Figure 2. Optineurin is ubiquitously expressed in all human organs including heart, brain, liver, kidney, placenta, and pancreas, with exceptionally high expression in skeletal muscle (45). Optineurin normally undergoes post-translational modifications that impact significantly on its function, including phosphorylation and ubiquitination (44). One major kinase that is capable of phosphorylating optineurin on at least nine serine and two threonine residues is TBK-1 (46). Optineurin is ubiquitinated by either HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase-1 (HACE1) (47) or ERAD-associated E3 ubiquitin protein ligase-1 (Hrd1) (48).
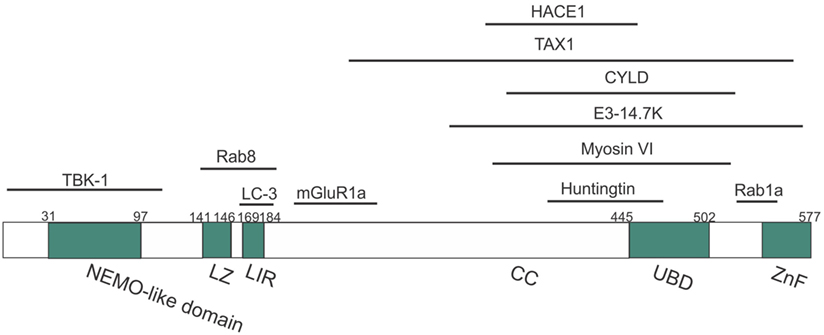
Figure 2. Domain structure of optineurin, illustrating possible regions that associate with its binding partners. Optineurin consists of a NEMO-like domain, a LC3-interacting (LIR) domain, a LZ domain, an ubiquitin-binding domain (UBD), coil-coiled (CC) domains, and a zinc-finger-like (ZnF) domain. TBK-1, Rab8, LC3, and mGlutR1a bind to the N-terminus whilst HACE1, TAX1, E3-14.7K, myosin VI, huntingtin, and Rab1a bind toward the C-terminus.
Optineurin is a multifunctional protein involved in several cellular processes. Importantly, it is an autophagy receptor (or adaptor) that is implicated in several forms of selective autophagy, including xenophagy (49), mitophagy (50), and aggrephagy (51). It also functions in vesicular trafficking (52), Golgi maintenance (53), secretion (54), and receptor recycling (55, 56). Furthermore, optineurin influences the innate immune response by negatively regulating the nuclear factor kappa B (NF-κB) pathway (45). This diversity in the functions of optineurin may originate from its numerous possible interacting partners (more than 20, Table 3; Figure 2).
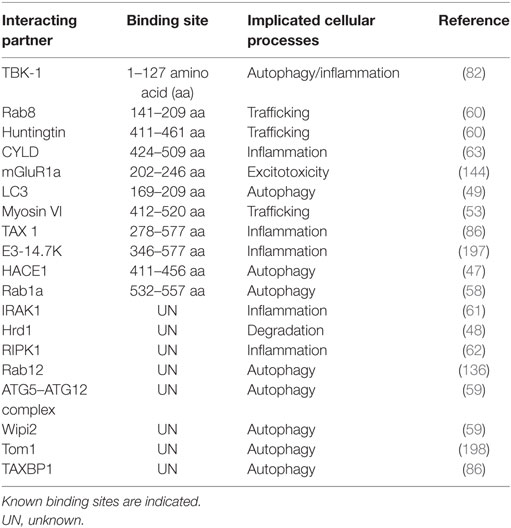
Table 3. Comprehensive list of putative optineurin-binding partners.
Optineurin was first discovered at the nuclear membrane as a binding partner for adenoviral protein E3-14.7K in a yeast-two hybrid system, and it was originally named “FIP2” (45). In this study, optineurin was found to suppress the protective effect of E3-14.7K against tumor necrosis factor alpha (TNFα)-induced cell death in HEK 293 cells. Optineurin was later found to also interact with several proteins involved in autophagy, including microtubule-associated proteins 1A/1B light chain 3B (LC3) (49), p62 (57), TBK-1 (49), Rab1a (58), and the ATG12-ATG5-ATG16L1 complex (59). During protein trafficking, optineurin forms a complex with myosin VI (53) and Rab8 (60) and it is also an interacting partner of huntingtin (htt) protein (60). In addition, during the inflammatory response, optineurin interacts with interleukin-1 receptor-associated kinase-1 (IRAK1) (61), receptor-interacting protein kinase-1 (RIPK-1) (62), and ubiquitin carboxylterminal hydrolase (CYLD) (63).
The Role of Optineurin in Neuroinflammation
Neuroinflammation
Neuroinflammation refers to chronic or acute central nervous system (CNS)-specific, inflammatory responses. It is a complex response mediated by activated microglia and/or astrocytes and is characterized by overproduction of inflammatory cytokines that may involve T-cell infiltration into the CNS. Whilst neuroinflammation is a normal response to CNS injury, it is increasingly accepted that neuroinflammatory processes are closely linked with neurodegenerative disorders, which are age-related conditions. TNFα and interleukin-1β (IL-1β) production can result from either normal aging or the neurodegenerative processes (64). This disturbed immune system in turn may exacerbate a favorable environment for neurodegeneration.
Microglia (65) are the innate immune cells of the CNS whose primary function is to monitor the environment to detect injury and infection. Depending on the signal and surrounding milieu, microglia may acquire different phenotypes. A simplified model describes two main phenotypes (66–69): M1 or “classically activated” microglia and M2 or “alternatively activated” microglia (69). M1 microglia produce pro-inflammatory cytokines including TNFα, IL-1β, IL-6, and IL-12, reactive oxygen species (ROS), and nitrogen-monoxide. This may lead to activation of CD8+ killer T cells, which aim to eradicate the infection. In contrast, M2 microglia produce anti-inflammatory cytokines including IL-4, IL-10, and insulin-growth factor-1 (IGF-1), which consequently activate CD4+ helper T cells. M2 microglia display enhanced phagocytosis compared to M1 microglia (70). Hence, depending on the phenotypes following activation, microglia can produce either cytotoxic or neuroprotective effects and the balance between M1 and M2 is thought to be essential for a healthy CNS.
Astrocytes (71) are specialized glial cells that are highly abundant in the CNS and they possess a wide range of essential functions. Astrocytes are also key players in neuroinflammation (72), and their response may also be either beneficial or harmful. During insults, their number and cellular volume can increase abnormally in a process known as ‘reactive astrogliosis’. Astrocytes also produce pro-inflammatory cytokines in response to inflammatory stimulators and they can also be activated via molecules secreted by activated microglia. Hence, during neuroinflammation, astrocytes can be exposed to a wide variety of stimuli, leading to a complex network of intracellular events (73–75).
The NF-κB family of transcription factors are key regulators of cytokine production, and they provide a mechanism to respond to a wide variety of stimuli associated with inflammation. NF-κB is ubiquitously expressed in mammalian cells, including neurons, and it can protect neurons against injury and regulate neuroinflammatory reactions, as well as contribute to neuronal degeneration (76–78). The family has the following five members: NF-κB1, NF-κB2, RelA, RelB, and c-Rel. NF-κB proteins bind to kB sites in the promoters of genes encoding inflammatory cytokines, thus inducing transcription (79). In basal conditions, NF-κB dimerizes in the cytoplasm and binds to the I kappa B (IKB) complex, consisting of IKB kinase α (IKKα), IKKβ, and NF-κB essential modulator (NEMO) kinases. Upon activation, the regulatory domain of IKB undergoes phosphorylation followed by ubiquitination, and this ultimately leads to degradation of the IKB complex. This then releases NF-κB, which translocates to the nucleus and promotes gene expression (79, 80) (Figure 3).
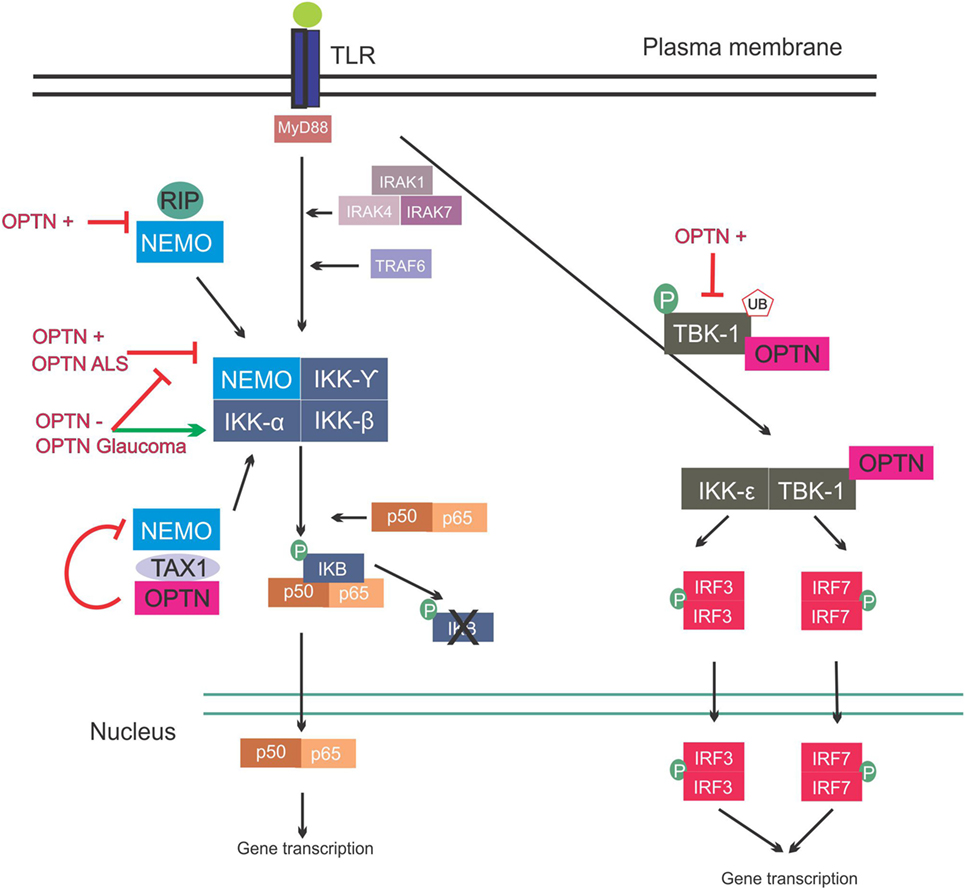
Figure 3. Involvement of optineurin in nuclear factor kappa B (NF-κB) signaling. Upon Toll-like receptor (TLR) signaling the NF-κB pathway is activated, leading to expression of inflammatory cytokines. This is mediated via interleukin-1 receptor-associated kinases (IRAKs) and TRAF. NEMO binds to the I kappa B (IKB) complex, leading to its phosphorylation and consequent degradation, and translocation of p50/p65 to the nucleus. NEMO also interacts with receptor-interacting protein (RIP) and trans-activating transcriptional regulatory protein of HTLV-1 (TAX1). Overexpression of wild-type or ALS mutant optineurin blocks NF-κB activation. Depletion of optineurin or overexpression of the glaucoma mutants leads to failure to inhibit NF-κB, leading to activation of this pathway. Optineurin binds to TAX1, blocking its interaction with NEMO and further inhibiting the pathway. Interferon regulatory factors are activated during TLR activation. IRF3 and IRF7 activation is mediated by TBK-1, and TBK-1 undergoes phosphorylation and ubiquitination during its activation. Optineurin binds to TBK-1, targeting it to CYLD, which inhibits its enzymatic activity. Abbreviations: OPTN−: optineurin depletion, OPTN+: optineurin overexpression, OPTN ALS: ALS mutant optineurin, OPTN Glaucoma: glaucoma mutant optineurin.
Optineurin’s Controversial Involvement in the NF-κB Pathway
It was initially hypothesized that optineurin would influence the NF-κB pathway because of its 53% aa similarity to NEMO (81). However, despite this similarity, optineurin did not bind to IKKβ, IKKα, or NEMO and it also lacked kinase activity (81). However, two other kinases were subsequently identified that associated with optineurin (81). Whilst these kinases were uncharacterized at the time, TBK-1 was later identified as a binding partner of optineurin (82). The first 127 aa of optineurin contain the TBK-1-binding site, and interestingly, the region 78–121 aa displays prominent homology to the TBK-1-binding site of TANK, another substrate of TBK-1 (Figure 2) (49). TBK-1 has a molecular weight of 84 kDa, similar to the unknown kinase, and it also belongs to the IKK-related family of kinases. Furthermore, as optineurin and TBK-1 are involved in immune responses, it is probable that this kinase was TBK-1. There has previously been controversy as to whether optineurin plays a role in NF-κB signaling. Whilst earlier studies failed to demonstrate a relationship between optineurin and NF-κB, more recent studies have provided evidence that optineurin negatively regulates this pathway. Depletion of optineurin abrogated the protective effect of E3-14.7K against receptor-interacting protein (RIP) and TNF receptor 1 (TNFR1)-induced death of HeLa cells (45). In contrast, depletion of optineurin did not influence p65 phosphorylation, pro-inflammatory IL-8 secretion, or the protective effect of E3-14.7K in H1299 cells, implying that optineurin was not required for NF-κB activation (83). In addition, optineurin did not associate with either NEMO or IKKβ and did not affect NF-κB signaling in murine pre-B-lymphocyte cell lines (81).
In contrast, other studies have concluded that optineurin is indeed involved in the NF-κB pathway. NEMO binds K63 poly-ubiquitinated RIP, which binds to TNFR1 during TNFα induced-NF-κB activation, and optineurin possesses a similar K63 ubiquitin-binding region to NEMO (Figure 3). This region competitively antagonized NEMO’s binding to RIP in HeLa cells, leading to decreased NF-κB activation (84). Similarly, in another study, optineurin was identified as a poly-ubiquitin-binding protein that regulates NF-κB activity (85). Another possible level of regulation of NF-κB by optineurin is through ubiquitin-binding domain (UBD)-dependent binding to trans-activating transcriptional regulatory protein of HTLV-1 (TAX1), which when ubiquitinated binds to NEMO, leading to NF-κB activation. This is mediated by binding of optineurin to TAX1-binding protein (TAXBP1), which regulates the ubiquitination of TAX1 (86) (Figure 3). Further homology with NEMO can be found in the zinc-finger domain of optineurin (87). The deubiquitinase CYLD also interacts with optineurin and is a negative regulator of NF-κB signaling through binding of RIP, similar to optineurin (88).
Despite the controversial involvement of optineurin in regulation of the NF-κB pathway, expression of optineurin itself is modulated through this pathway. The levels of optineurin increase upon treatment with either TNFα (45) or IFN (45, 81). Furthermore, the human optineurin promoter is activated by TNFα, and this is abolished by mutations in the putative NF-κB-binding site. Furthermore, overexpression of the NF-κB subunit p65 activates the optineurin promoter, providing evidence that optineurin expression is controlled by the NF-κB pathway (89).
Whilst not the focus of this review, it is important to note that optineurin is involved in antiviral and antibacterial immune responses outside of the CNS. Overexpression of optineurin inhibited IL-1β-, IRAK1-, and lipopolysaccharide (LPS)-induced NF-κB activation, whilst optineurin deficiency increased NF-κB activation in response to LPS (61). Optineurin has also been implicated in mediating the immune response to Salmonella infection (90). Interestingly, optineurin function in inflammation is highly dependent on its interaction with TBK-1. Pathogen associated molecular patterns (PAMPs) lead to the phosphorylation and ubiquitination of TBK-1, consequently leading to IFNβ production (91). TBK-1-mediated IRF3 activation is dependent on optineurin (92). Furthermore, optineurin was shown to dampen the antiviral innate immune response by targeting CYLD to TBK-1 to inhibit its enzymatic activity (93). Similarly, in mice expressing N-terminal truncated optineurin that lacks the TBK-1 interacting domain, PAMP activation led to decreased TBK-1 activity and low IFNβ production (91). Taken together, these findings indicate that the presence of functional optineurin is crucial for the normal innate immune response and dysfunctional optineurin can unbalance this response.
Based on the available evidence, it is likely that optineurin does indeed influence NF-κB activity, although this may happen in a cell type and stimulus-dependent manner. Furthermore, optineurin interacts with other proteins involved in NF-κB signaling including RIPK-1, which could affect its ubiquitination status via CYLD. Hence optineurin may influence the NF-κB signaling pathway by an indirect mechanism.
Role of Optineurin in Necroptosis
Necroptosis is a programed form of necrosis linked to inflammation (94). In contrast to apoptosis, necroptosis is caspase-independent and can be triggered by TNF treatment. Activation of TNFR1 by binding of TNF leads to either inflammation via activation of the NF-κB pathway or cell death, and RIPK-1 mediates this cellular response and dictates which pathway is activated (94). In most cells, TNFR1-bound TNF leads to activation of NF-κB signaling and the production of inflammatory cytokines (95). However, upon destabilization of the RIPK-1–TNFR1–TRADD complex, cell death is triggered. K63-mediated ubiquitination of RIPK-1 promotes cell survival, but upon de-ubiquitylation, the default pro-survival function is lost. Up-regulation of RIPK-1, RIPK-3, and mixed lineage kinase domain-like protein (MLKL) can trigger necroptosis (94).
A recent study identified a major role for optineurin in RIPK-1-mediated necroptosis (62). In optineurin-depleted mice, RIPK-1, RIPK-3, and MLK levels were increased in the spinal cord and increased cell death upon TNF treatment was observed, implying induction of necroptosis. Furthermore, ubiquitination and proteasomal turnover of RIPK-1 was decreased in these animals, implying that optineurin may influence the cellular sensitivity to necroptosis by this mechanism (62). The optineurin-depleted animals also displayed degeneration and swelling of motor neuronal axons, indicating a vital role for optineurin in axonal degeneration and neuroinflammation.
The Role of Optineurin in Vesicular Trafficking
Secretory proteins are first synthesized and folded within the ER, from where they bud via ER exit sites in membrane-bound vesicles that move towards the Golgi apparatus (96). The Rab family of small GTPases regulate all intracellular trafficking events, including recruitment of coatomer coat proteins that form the vesicular coat, vesicle budding, transport, docking, and fusion (96). The ER-derived vesicles arrive at the cis-Golgi and then transit to the trans-Golgi network (TGN), which sorts and packages proteins according to their destination: the plasma membrane, endosomes, or extracellular space (96). Motor proteins carry the transport vesicles along either actin filaments or microtubules to their destination, where they fuse with the donor membrane to release their cargo (96). Motor neurons are very long and vesicular trafficking could therefore have more severe consequences in these cells than in other types.
The important role of optineurin in vesicular trafficking in complex with myosin VI and Rab8 is now well established (Figure 4). Myosins are motor proteins that move cargo along actin tracks (97). The Rab8-binding site is localized within the N-terminal domain of optineurin (60), whereas the myosin VI-binding region is located at its C-terminus (Figure 2). Optineurin co-localizes with both Rab8 and myosin VI at the Golgi apparatus and in vesicles in the cytoplasm (53, 98). The co-localization between myosin VI and Rab8 is optineurin dependent (98), suggesting that optineurin controls the assembly of the whole complex. This complex mediates Golgi organization (99), post-Golgi trafficking, exocytosis (100), and the basolateral delivery of membrane proteins (101).
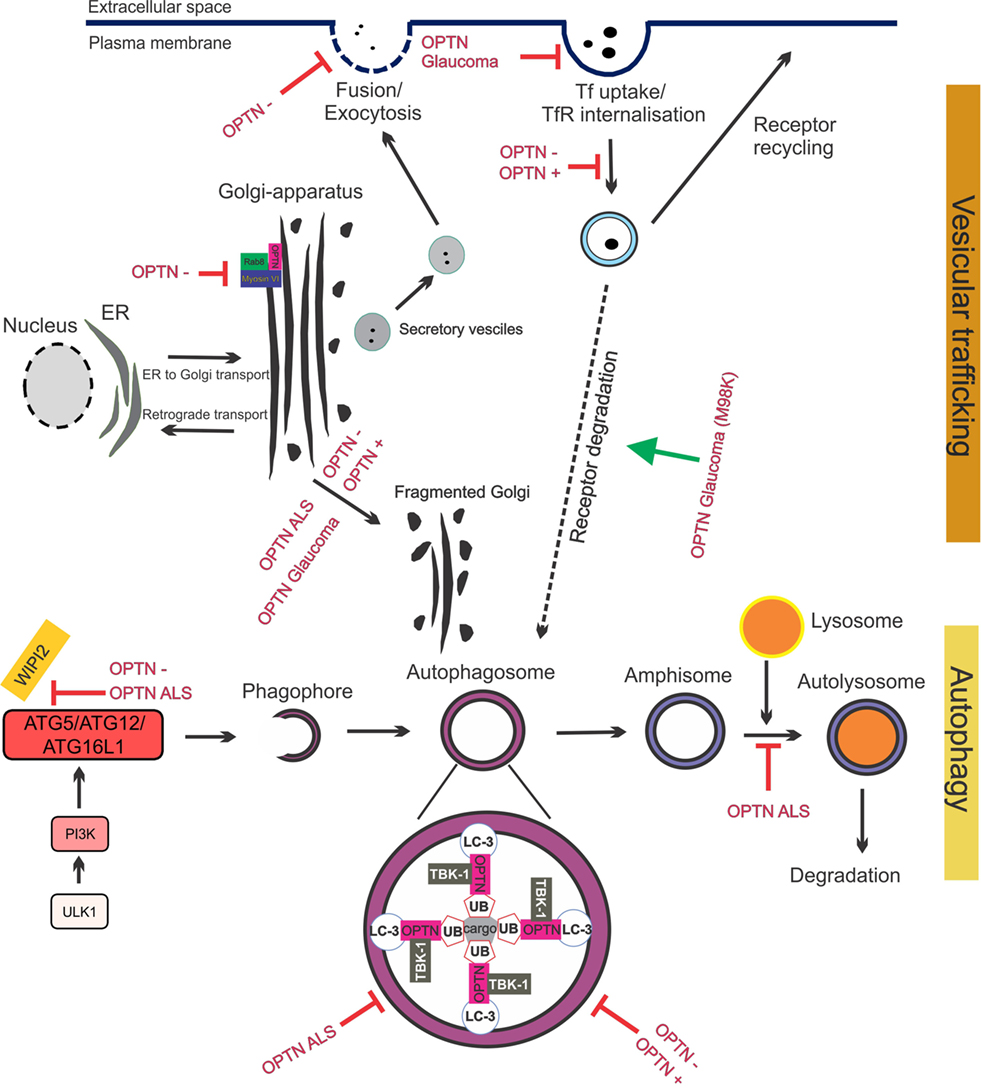
Figure 4. Optineurin mediates vesicular trafficking, receptor recycling and autophagy. (1) Optineurin controls Rab8 and myosin localization at the Golgi apparatus. Depletion of optineurin relocates the optineurin–Rab8–myosinVI complex from the Golgi. (2) Depletion of optineurin or overexpression of wild-type, ALS, or glaucoma mutant optineurin, results in Golgi fragmentation. (3) During vesicular trafficking, secretory vesicles travel from the Golgi to the plasma membrane where they fuse and undergo exocytosis. Depletion of optineurin abolishes the fusion of vesicles with the plasma membrane. (4) Transferrin receptor is recycled to the surface after transferrin uptake. E50K glaucoma mutant optineurin inhibits transferrin uptake, whilst depletion and overexpression of optineurin abolishes the recycling of transferrin receptor. M98K glaucoma mutant induces autophagic degradation of transferrin receptors by targeting them to autophagosomes. (5) Optineurin disrupts autophagy at several stages. The formation of the phagophore and autophagosome is inhibited by depletion of optineurin, as well as overexpression of ALS mutant optineurin, by inhibiting ATG5/ATG12/ATG6L1 complex formation. Optineurin links the ubiquitinated cargo to LC3, together facilitating autophagosomal engulfment of the cargo. Degradation of cargo, such as mitochondria and protein aggregates, is inhibited by both ALS and glaucoma mutant optineurin. Imbalance of optineurin expression, such as its overexpression or depletion, also interferes with autophagy, decreasing the clearance of cargo. ALS-associated mutant optineurin further blocks the fusion of the autophagosome to the lysosome, further inhibiting autophagy flux. Abbreviations: OPTN−: optineurin depletion, OPTN+: optineurin overexpression, OPTN ALS: ALS mutant optineurin, OPTN glaucoma: glaucoma mutant optineurin, Tf: transferrin, TfR: transferrin receptor, ER: endoplasmic reticulum.
In addition, optineurin mediates the binding between Rab8 and htt (60) and the latter is also thought to function in vesicular trafficking (60, 102). Htt interacts with the C-terminus of optineurin (Figure 2) and this interaction is dependent on the GTPase-activity of Rab8, as dominant-negative forms of Rab8 do not bind to optineurin (103). Furthermore, optineurin negatively controls Rab8 activity by interacting with its GTPase-activating protein (GAP) TBC1D17, thus perturbing Rab8-TBC1D17 interaction (103). Hence, optineurin provides the binding platform for myosin VI, Rab8, and htt, leading to their indirect interaction and regulation of vesicle trafficking. Hence together these studies establish an important regulatory function for optineurin in protein trafficking, via its interaction with key proteins implicated in vesicle formation and transport.
In most cell types, optineurin displays a vesicular, punctate pattern around the nucleus, Golgi, and plasma membrane (44, 53, 104–106). However, there is controversy as to how optineurin influences the Golgi apparatus and whether optineurin is a constant component of Golgi structure or is only transiently recruited there. Studies on perturbation of Golgi structure have suggested that optineurin plays a role in the function of the Golgi rather than in the maintenance of its structure (99). However, in a few cell types, such as retinal pigment epithelial cells (98) and normal rat kidney (NRK) cells (53), optineurin does not associate with the Golgi apparatus.
The Golgi normally undergoes extensive morphological changes during the cell cycle. Fragmented Golgi are produced during perturbed vesicular trafficking and in pathological conditions (107). Golgi fragmentation has been detected in cells overexpressing wild-type optineurin (98) and those depleted of optineurin (53, 54, 98), although this finding could not be reproduced in zebrafish (108). Depletion of optineurin from NRK cells results in loss of myosin VI from the Golgi and a disrupted Golgi ribbon structure (53). This implies that although optineurin is not located at the Golgi in NRK cells it still may influence Golgi function by directing myosin VI toward the Golgi. Nevertheless, this finding suggests that the levels of optineurin need to be tightly controlled to maintain Golgi morphology, suggesting a central role for optineurin in Golgi-mediated cellular processes. Whilst the absence of myosin VI inhibits ER-Golgi transport, depletion of optineurin had no effect on this process (100). However, there is evidence that optineurin functions in the post-Golgi part of the secretory pathway. Optineurin depletion affected vesicular fusion at the plasma membrane, which may involve other Rabs besides Rab8 (100).
There is also evidence that optineurin functions in the endocytic recycling of cell surface receptors. Depletion (55) or overexpression (56) of optineurin impairs endocytic trafficking of transferrin receptor (TfR) to the perinuclear region. Whilst optineurin did not co-localize with the early (Rab5 and Rab4) or late endosomal markers (Rab7) (56), a small portion co-localized with markers of recycling endosomes: TfR, Rab11 (55), and Rab8 (56). This appears to be mediated primarily by the UBD (55). These findings are consistent with those obtained from studies of classically secreted proteins. Depletion of optineurin led to reduced secretion of essential growth factors (neurotrophin 3 and ciliary neurotrophic factor) which induced apoptosis (54) and inhibited the delivery of epidermal growth factor receptor to the cell surface, leading to profound defects in lamellipodi formation. Again, depletion of myosin VI leads to similar defects, providing further evidence that optineurin and myosin VI possess a strong functional relationship (109). Hence together these data reveal that optineurin is important in secretory vesicle transport, endocytosis, and receptor recycling. Furthermore, recycling of cell surface receptors and efficient secretion of neurotransmitters are crucial for the conduction of action potentials at chemical synapses. Hence, dysfunction in these cellular processes could severely perturb important neuronal functions.
The Role of Optineurin in Autophagy
Related to its function in vesicular trafficking, optineurin also has an important role in autophagy. Autophagy is a lysosomal degradation system that maintains intracellular homeostasis by degrading and recycling proteins/organelles. The most common type of autophagy is macroautophagy, which is characterized by the formation of the autophagosome, a double membrane vesicle that forms around the cargo. Autophagosomes fuse with endosomes, creating amphisomes, and the fusion of amphisomes or autophagosomes with the lysosome creates the autolysosome, where acidic degradation of cellular components takes place (110). In this review, we focus only on macroautophagy, referring to it as “autophagy” from here onward.
LC3 is a central protein in the autophagy pathway, which functions in the selection of autophagy substrates and autophagosome biogenesis. Autophagy begins with activation of the autophagy initiation complex, which consequently interacts with the phosphoinositide 3-kinase (PI3K) complex, leading to phagophore formation (Figure 4) (110). This is controlled by the ATG12:ATG5:ATG16L1 complex, which is recruited to the phagophore by WD repeat domain phosphoinositide-interacting protein 2 (Wipi2) (59). The ATG12:ATG5:ATG16L1 complex facilitates the lipidation of LC3 by conjugating phosphatidylethanolamine (PE). LC3-PE/LC3-II recruitment to the phagophore leads to elongation of the phagophore and closure of the double membrane, forming the autophagosome (110).
Based on cargo selection, two subtypes of autophagy can be distinguished; selective (111) and non-selective autophagy. Nonselective autophagy is the cellular stress response to nutrient deprivation, which targets non-essential molecules and organelles to the autophagosome to compensate for lack of nutrients in the environment. During selective autophagy, a specific cargo is selectively targeted, hence it may be considered as a housekeeping function. Specific subtypes of selective autophagy exist depending on the cargo targeted for degradation, including xenophagy (bacteria), mitophagy (mitochondria), and aggrephagy (protein aggregates). Neurons are post-mitotic; hence, they are particularly dependent on autophagy to dilute dysfunctional proteins and organelles. Thus, highly precise protein quality control systems are essential for the survival of neurons and their specialized morphologies require specific adaptations of autophagy, as well as other cellular processes (111, 112).
Optineurin is an autophagy receptor; hence, it tethers cargo for degradation to the autophagosome. Furthermore, multiple studies have indicated that optineurin is an important regulator of autophagy. Optineurin interacts with LC3 via its LC3 interacting motif (LIR, Figure 2), and the importance of this binding site is demonstrated by the fact that mutation to the LIR domain (F178A) abrogates LC3 binding to optineurin (49). Depletion of optineurin leads to significantly lower levels of LC3-II and fewer autophagosomes and autolysosomes, suggesting that optineurin is necessary for autophagy induction or autophagosome formation (59). Furthermore, optineurin-depleted cells display impairment of ATG12-ATG5-ATG16L1 complex recruitment to Wipi2, implying that optineurin influences autophagy before the involvement of Wipi2 (59). Hence optineurin may be necessary for the formation of the phagophore and hence autophagosome, although there is also evidence that optineurin is involved in autophagosome-lysosome fusion (113). Post-translational modifications of optineurin are also known to influence its function during autophagy. Ubiquitination of optineurin affects its interaction with other autophagy receptors and also targets it for degradation. HACE1 promotes K48-linked ubiquitination (47), which occurs predominantly at Lys193 of optineurin, which interestingly, is outside of the LC3-binding region. This in turn promotes the interaction of optineurin with autophagy receptor p62, leading to formation of the optineurin-p62 complex and acceleration of autophagy flux (47).
The Role of Optineurin in Selective Autophagy
Optineurin is implicated in several different forms of selective autophagy including xenophagy, mitophagy, and aggrephagy. In this section, we focus on mitophagy and aggrephagy as the forms of selective autophagy that are the most relevant to neurodegenerative disorders.
Role of Optineurin During Mitophagy
Mitochondria that need to be degraded are first ubiquitinated, and this is the signal for autophagic engulfment. Damaged mitochondria are recognized by PTEN-induced kinase 1 (PINK1), which subsequently recruits parkin, an E3 ubiquitin ligase (114). Parkin ubiquitinates mitochondria, and then ubiquitin is recognized by optineurin and other autophagy receptors, such as p62 and NDP-52, via their UBDs, which recruit the autophagic machinery. Optineurin was first shown to be recruited to damaged mitochondria in a parkin-dependent manner in HeLa cells and its depletion resulted in inhibition of LC3 recruitment to these mitochondria (115). Optineurin also recruits TBK-1 to poly-ubiquitinated mitochondria, where in turn, TBK-1 phosphorylates optineurin, enhancing its binding to ubiquitin and facilitating LC3 recruitment and mitophagy (46). Interestingly, optineurin and p62, which also function in mitophagy, localize to different parts of the mitochondria, independently of each other. p62 was not required for optineurin/LC3 recruitment to damaged mitochondria in HeLa cells, implying that optineurin and p62 regulate mitophagy by different mechanisms (115). Despite this, TBK-1 phosphorylates both optineurin and p62 during mitophagy (116). In another study, optineurin and two other autophagy receptors, NDP-52 and TAX1BP1, were recruited to depolarized mitochondria in HeLa cells but only depletion of optineurin blocked efficient mitophagy (57). This finding provides evidence for the selective involvement of specific autophagy receptors in mitophagy, indicating that optineurin has a central role in this process.
Role of Optineurin in Aggrephagy
Protein inclusions are the pathological hallmark of neurodegenerative diseases and their presence implies dysfunction in protein degradation systems, either the UPS and/or autophagy. However, whilst the ubiquitin-proteasome system (UPS) degrades short-lived and soluble proteins, insoluble material, such as misfolded protein inclusions, are usually cleared by a subtype of selective autophagy, aggrephagy. Depletion of optineurin leads to increased aggregation of mutant G93C SOD1 and htt in HeLa cells, suggesting that it plays a role in aggrephagy (51). Similarly, overexpression of optineurin in Neuro2a cells prevented htt aggregation in an UBD-dependent manner (113). Interestingly, a relationship between optineurin and TBK-1 exists in aggrephagy and both proteins are mutated in fALS/sALS. Lack of TBK-1 expression and co-depletion of both optineurin and TBK-1 leads to the accumulation of protein aggregates to the same extent as depletion of optineurin alone, suggesting that optineurin function in aggrephagy is highly dependent on, and mediated by, TBK-1 (51).
Involvement of Optineurin in Neurodegeneration in ALS and Other Neurodegenerative Diseases
Whilst optineurin has not been well studied in relation to neurodegeneration, optineurin dysfunction is associated with several cellular mechanisms that are implicated in the pathogenesis of ALS and glaucoma.
The Role of Optineurin in Neuroinflammation in ALS
Increasing evidence suggests that neuroinflammation plays an important role in ALS pathogenesis. Overexpression of Toll-like receptor (TLR) 2/4 receptors in glial cells and TLR4 in motor neurons, as well as T-cell infiltration into the CNS (117, 118) and microglia activation, is observed in ALS patients (64). A recent study showed that the presence of regulatory T (Treg) cells slows down disease progression in SOD1 G93A transgenic mice. Furthermore, in ALS patients, there is an inverse correlation between the number of Treg cells and disease progression (117). However, the number of Treg cells decreases in rapidly progressing ALS patients (119). These findings highlight the importance of T cells in ALS progression. Importantly, microglia activation is an early event during ALS development, implying a role in disease pathogenesis (69, 120). In chimeric mice overexpressing wild-type and mSOD1 (G93A and G85R), non-neuronal cells that do not express mSOD1, including microglia, extend the survival of mSOD1 expressing motor neurons (121). Another study using the Cre/Lox system observed slower disease progression upon G37R SOD1 depletion in microglia (122). Furthermore, G93A SOD1 expressing microglia that are activated at the early stage of disease exhibit neuroprotective features that promote neuronal survival, whilst microglia from end stage animals display neurotoxic properties and induction of neuronal death (123). TLR4 antagonists are protective against the death of motor neurons co-cultured with microglia expressing mutant SOD1, and in a mouse model of spontaneous motor neuron degeneration, the wobbler mouse (124, 125). In addition, depletion of TLR4 prolongs the survival of G93A SOD1 mice (125). ATP release by dying motor neurons may activate microglia via ionotropic P2X and metabotropic P2Y purinergic receptors, leading to an inflammatory response. Interestingly, the levels of P2X increase in activated microglia in spinal cords of ALS patients and mutant G93A SOD1 transgenic mice (126, 127).
There is also evidence that neuroinflammation is involved in optineurin-associated ALS as well as in other forms of ALS. In sALS patients and fALS patients bearing optineurin mutations, NF-κB immunoreactivity in microglia is increased compared to controls and it is absent from the neuronal nucleus of patients (128). Depletion of optineurin increased NF-κB activity in neuronal cells leading to cell death, and wild-type overexpression of optineurin was able to rescue cell death, whilst ALS mutant optineurin could not (129) (Figure 3). Another study by Ito and colleagues demonstrated that in optineurin-depleted mice, severe axonopathy developed due to increased necroptosis (62). Furthermore, decompaction of myelin sheets due to oligodendrocyte death, and degeneration and swelling of motor neuron axons was observed. Interestingly, similar findings are observed in spinal cord of ALS patients (130). In the same study, optineurin depletion was carried out in specific cell types, including motor neurons, oligodendrocytes, astrocytes, and myeloid linages (62). This axonopathy and myelin abnormalities were only observed when optineurin was depleted from oligodendrocytes or microglia, and not from motor neurons or astrocytes, and it was linked to oligodendrocyte necroptosis. Interestingly, optineurin depletion in microglia only also led to axonopathy, suggesting that microglia contribute to oligodendrocyte health. Furthermore, depletion of optineurin did not result in motor neuron death in this model, even after 2 years. Whether depletion of optineurin in oligodendrocytes and microglia leads to a motor phenotype and ALS-like symptoms remains to be determined. Nonetheless, these data indicate that oligodendrocytes and microglia contribute to optineurin-mediated neurodegeneration.
The Role of Optineurin in NF-κB Signaling in Glaucoma
There have been few studies examining the role of optineurin in NF-κB signaling in glaucoma, and these studies have yielded conflicting findings. Expression of the glaucoma mutant E50K in RGCs revealed no difference in interferon regulatory transcription factor 3 (IRF3) activation compared to cells expressing wild-type optineurin (131), suggesting that this pathway may not be implicated in glaucoma. However, expression of another glaucoma mutant, H486R, resulted in an altered interaction of optineurin with CYLD compared to wild-type, resulting in a lack of NF-κB inhibition, similar to studies in which optineurin was knocked out. In cells expressing the same mutant, CYLD displayed decreased binding to RIP and deubiquitination of RIP was decreased, suggesting that CYLD binding and deubiqutination of RIP are optineurin-mediated (63). Mutant H486R also fails to inhibit NF-κB activation upon IRAK1-mediated activation (61). These findings suggest that whilst the link between optineurin, the NF-κB pathway, and glaucoma is not well established, the NF-κB pathway is dysfunctional and highly activated in glaucoma.
ER Stress and Secretory Defects in ALS and Glaucoma
Endoplasmic reticulum stress and fragmentation of the neuronal Golgi apparatus are now well-established cellular mechanisms associated with ALS (107, 132). The ER is the major site of protein folding, and if misfolded proteins accumulate, induction of the unfolded protein response occurs. Dysfunctional cellular trafficking is associated with ER stress and impaired cellular viability in all cell types, but neurons may be particularly susceptible to this type of stress, due to their extended and complex morphologies.
We have shown that expression of either wild-type or ALS-associated mutant optineurin (E478G and Q398X) induced ER stress in motor neuron-like cell lines (NSC-34), in contrast to the E50K glaucoma mutant, although this induction was significantly enhanced in the mutant expressing cells (52). Furthermore, this was accompanied by the inhibition of secretory protein transport from the Golgi to plasma membrane, Golgi fragmentation, and abnormal cytoplasmic distribution of optineurin in cells expressing the ALS-associated mutants. These findings were also associated with disruption of optineurin binding to myosin VI. Interestingly, optineurin binding to myosin VI was also decreased in spinal cord lysates from sALS patients compared to controls, implying that dysfunction to optineurin is also present in the much more common sporadic forms of ALS (52). Consistent with these findings, Golgi fragmentation was detected in 70% of anterior horn cells from an ALS patient bearing the E478G mutation (133) and also in another patient with the Q398X mutation, although optineurin immunoreactivity was absent from both the brain and spinal cord in the latter patient (134). Hence, together these data indicate that dysfunction to the ER and Golgi compartments are consistent features of ALS patients carrying optineurin mutations, as well as other forms of ALS.
Secretory defects have also been associated with optineurin in glaucoma. Expression of the E50K mutant reduced mir9 levels in RGC-5 cells, leading to increased levels of the transcriptional repressor RE1, and decreased levels of brain-derived neurotrophic factor (BDNF) (135). Hence, optineurin may mediate neural growth in RGCs through transcriptional control. The glaucoma mutant E50K forms larger and more granular structures compared to wild-type optineurin, inducing severe Golgi fragmentation (98). Furthermore, despite co-localizing with TfR, expression of E50K leads to significant inhibition of uptake of transferrin (Tf) (55, 56), indicating that whilst mutant optineurin can be recruited to TfR, uptake of Tf and recycling of TfR is abolished. E50K optineurin also leads to enhanced inhibition of Rab8 by TBC1D17, causing defective endocytic recycling of TfR (103) and linking this to RGC death. Furthermore, optineurin-positive vesicles displayed slower dynamics in E50K expressing cells compared to wild-type optineurin cells (55). Another glaucoma mutant, M98K, displayed enhanced interaction and co-localization with TfR, accompanied by decreased Tf uptake, similar to E50K optineurin (136). In contrast to E50K or wild-type optineurin overexpressing cells, the levels of TfR were lower in M98K overexpressing cells. This was due to increased autophagic degradation of TfR mediated by Rab12, leading to inhibition of cell death, which could be rescued by Rab12 depletion. Furthermore, M98K displays increased association with Rab12 compared to wild-type, suggesting that a toxic gain of function mechanism may exist (136).
Transgenic mice overexpressing E50K display a pronounced loss of RGCs and connecting synapses in the peripheral retina by 16 months of age, leading to thinning of the nerve fibre layer. This was associated with disrupted interaction between optineurin and Rab8 (137). Furthermore, in another mouse model, reactive gliosis was associated with deposition of mutant optineurin in the outer plexiform layer of the retina. In cell culture, E50K forms a stronger interaction with TBK-1 than with wild-type optineurin and this prohibited the self-oligomerization of E50K optineurin, inhibiting its intracellular degradation. In contrast, inhibition of TBK-1 abrogated the aberrant insolubility of E50K (138). Hence, together these data indicate that optineurin is involved in the pathogenesis of ALS and glaucoma. However, it remains unknown whether mutant forms of optineurin induce the same or distinct disease mechanisms in these disorders (Figure 1). It is tempting to speculate that the vesicular trafficking disturbances at least are shared mechanisms between both disorders, but further studies are required to state this conclusively.
Optineurin-Related Autophagy Defects in ALS, Glaucoma, and Alzheimer’s Disease
Similarly, ALS-associated mutations in optineurin are known to induce dysfunction to autophagy. The E478G mutant displays impaired ability to form ATG16L1-positive puncta as well as phagophores and autophagosomes, and it does not form a complex with Atg12-5 and Wipi2 as readily (59). Optineurin mutants lacking the UBD do not co-localize with ATG12, ATG16L1 or Wipi2 puncta, in contrast to other mutants (59), highlighting the importance of the UBD in phagophore formation. Comparable results were observed in E478G and UBD-deleted optineurin overexpressing cells, where diminished autophagy induction was evident by fewer LC3 puncta upon serum starvation (113). Similarly, we have shown that in NSC-34 cells, expression of either ALS mutants E478G or Q398X, but not the glaucoma mutant E50K, led to inhibited autophagosome maturation into autolysosomes and accumulation of autophagosomes (52). During mitophagy, unlike wild-type optineurin, overexpression of ALS mutant E478G could not rescue inhibition of LC3 recruitment to mitochondria in optineurin-depleted cells. Furthermore, ALS mutants E478G or Q398X failed to relocate to mitochondria or induce mitophagy, unlike wild-type optineurin (57). As the E478G mutation is present within the UBD, this indicates that LC3 and ubiquitin binding are equally important to optineurin-mediated autophagy (115).
Optineurin co-localizes with mutant SOD1 (G93C) aggregates in HeLa cells (51) via its C-terminal coil-coiled domain and this interaction is independent of the UBD. These mSOD1 aggregates are positive for autophagosome markers LC3 and p62 as well as ubiquitin, suggesting an ongoing autophagic event (51) and indicating that optineurin is involved in the autophagic clearance of SOD1 aggregates. Interestingly, there is evidence that Aβ fibrils associated with Alzheimer’s disease are degraded by microglia via autophagy, or more specifically, aggrephagy, involving optineurin (139). Depletion of specific autophagy genes (Atg7 and Map1lc3B) leads to increased levels of Aβ aggregates in microglia, without interfering with the uptake of Aβ fibrils. Furthermore, optineurin co-immunoprecipitates with Aβ fibrils, indicating that a direct or indirect interaction exists. Depletion of optineurin via siRNA from microglia also prevented Aβ fibril degradation (139).
Transgenic expression of the E50K mutant in mice leads to age-related loss of RGCs and elevated LC3-PE levels compared to mice expressing wild-type optineurin. Furthermore, in E50K transgenic mice, mitochondrial fragmentation, fewer mitochondria, and increased autophagosome formation, were detected in axons of the glial lamina, implying that death of these cells is associated with mitophagy in these animals. Alternatively, it is possible that the decreased numbers of mitochondria cannot support the significant energy needs of these neurons. Transport of mitochondria within the axon did not change in E50K mice, implying that the loss of mitochondria was due to increased mitophagy, and not redistribution of mitochondria (140).
In contrast, treatment with rapamycin, an inducer of autophagy via the mechanistic target of rapamycin (mTOR) pathway, rescues apoptosis in mouse RGCs expressing the glaucoma mutant E50K. It was suggested that this was due to the elimination of optineurin via autophagy (141). It should be recognized however that mTOR is linked to many cellular pathways as well as autophagy. In contrast, a more recent in vitro study involving wild-type and E50K provided evidence that optineurin is not degraded by autophagy (48). Optineurin can be poly-ubiquitinated at K63R and K48R by Hrd1, but this mediates the proteasomal degradation of optineurin rather than autophagosomal degradation. It remains unclear precisely how optineurin turnover is controlled, and whether autophagy or the UPS is the main quality control system (142).
Optineurin and the Balance Between Cytotoxity and Cytoprotection
Excitotoxicity is a pathological process by which neurons degenerate and die by excessive stimulation of receptors, due to binding of the excitatory neurotransmitter glutamate. These receptors include the metabotropic glutamate receptors (mGluRs) and the ionotropic AMPA and NMDA receptors (143). Optineurin functions as a negative regulator of mGlutR1a signaling by interacting with the mGluR1a receptor and attenuating its ability to couple to phospholipidase Cβ (144), which in turn decreases inositol phosphatase production and protein kinase C activation. Interestingly, htt with expanded glutamine regions (htt-G138) facilitates optineurin-mediated attenuation of mGluR1a signaling (144). However, whether optineurin-mediated inhibition of mGluR1a signaling results in beneficial or harmful consequences remains to be elucidated.
Interestingly, wild-type optineurin may exert protective effects on cellular survival. Overexpression of optineurin attenuates TNFα-induced cell death in HeLa cells (145) and ROS-mediated H2O2-induced cell death in NIH3T3 cells (105), although no protection was observed in RGC-5 cells (105, 145). This protection correlates with changes in the subcellular localization of optineurin, which translocates in a Rab8 GTPase-dependent manner from the Golgi apparatus to the nucleus. This translocation also occurs during cell division, and it is initiated by phosphorylation of optineurin by polo-like kinase 1 (Plk1) (146), which plays a pivotal role in cell-cycle progression through mitotic entry to cytokinesis (147). Depletion of optineurin increases Plk1 activity, which induces abscission failure and multi-nucleation of cells (146).
However, in some instances, wild-type optineurin can also induce toxicity, which is dependent on its ability to bind to Rab8 (148). In yeast, overexpression of wild-type optineurin-induced toxicity by a mitogen-activated protein kinase 25 (MAP2K5) kinase-dependent mechanism (148). This finding was confirmed in mammalian cells, where the formation of wild-type optineurin aggregates was reduced by treatment with a MAP2K5 inhibitor (148). The MAP2K/MAPK pathways are associated with both neuronal cell death (149) and survival (150). Whether MAP2K5-mediated cell death or survival is fine-tuned by optineurin remains to be elucidated. However, depletion of optineurin leads to inhibited cell growth in RGCs (151) and microarray analysis identified 112 genes that were differentially expressed in these cells compared to control cells expressing optineurin, including BDNF, neurotrophin 3, synaptosomal-associated protein 25 (SNAP25) and neurofilament light polypeptide. The lack of growth factors was associated with decreased growth in optineurin-depleted cells (151).
The ALS optineurin mutant E478G induced cell death specifically in motor neuron-like cells, compared to glaucoma mutants E50K or M98K, which did not affect cell survival. Furthermore, in contrast, the ALS mutants did not induce cell death in a photoreceptor cell line, 661W, whilst the glaucoma mutants did (152). Importantly, these data imply that cell death is induced by optineurin mutations only in the appropriate, specific cell type associated with each different neurodegenerative disease. Consistent with this notion, in an earlier study, M98K induced cell death in RGCs, but not in other neuronal or non-neuronal cells (IMR32, HeLa, and COS-1). Similarly, another glaucoma mutant, E50K, selectively induced ROS-associated-specific death of RGCs but not COS-1, HeLa or IMR32 cell lines, whilst overexpression of wild-type optineurin did not (136). However, other glaucoma-associated mutants (R545Q, H26D, and H486R) did not induce cell death, implying that E50K is more toxic than the other mutants (145, 153).
Overexpression of either ALS (E478G) or glaucoma optineurin mutants (E50K) resulted in a motor neuron axonopathy in zebrafish embryos, thus linking optineurin mutations to disease relevant phenotypes in vivo. Interestingly, knockout of MAPK2 rescued optineurin induced motor axonopathy in zebrafish (154), and the formation of E50K and E478G aggregates was inhibited by treatment with a MAP2K5 inhibitor in the absence of changes in expression level, suggesting that MAP2K5 has a direct effect on the formation of optineurin aggregates (154). Both wild-type and mutant E50K optineurin forms toxic, non-amyloid aggregates in yeast, and not surprisingly, E50K is more toxic than wild-type (148). This may be due to increased oligomerization of optineurin due to its enhanced binding to TBK-1 (82). Interestingly, wild-type and ALS mutant (E478G) optineurin display unique aggregation patterns in vitro compared to other aggregation prone proteins associated with ALS, including TDP-43 and FUS. Wild-type and E478G optineurin overexpression resulted in the formation of a single large inclusion in the majority of yeast cells, in contrast to cells expressing TDP-43 or FUS, that formed inclusions of multiple foci with heterogeneous size (148). This indicates that optineurin is an aggregation prone protein, consistent with the presence of inclusions.
Optineurin Pathology in Neurodegenerative Diseases
Whilst neurodegenerative diseases display a diverse range of symptoms and affect different subtypes of neurons, a common phenomenon is the presence of protein aggregates. Interestingly, optineurin is present and localized with in the aggregates in a wide range of neurodegenerative diseases. Almost all (97%) ALS patients display TDP-43-positive protein inclusions in degenerating motor neurons (155), and in some patients, SOD1, FUS, and C9orf72 aggregates may also be present. TDP-43 pathology (ubiquitinated, hyper-phosphorylated, and truncated TDP-43) is a feature of FTD also, and it is present in 45% of patients (155). Another 45% of FTD patients present with inclusions containing misfolded microtubule stabilizing protein tau without TDP-43 pathology (156).
One study identified optineurin-positive inclusions in both fALS and sALS patients that co-localized with TDP-43 and ubiquitin (2). Similarly, in fALS patients bearing mutations in SOD1, SOD1 inclusions were also immuno positive for optineurin (2). In contrast, however, another study examining 32 sALS cases observed that the majority of TDP-43 inclusions were negative for optineurin (3). Furthermore, whilst 33% of FTD-TDP cases examined displayed cytoplasmic optineurin neuronal inclusions in the brain (3), the majority of TDP-43 inclusions did not co-localize with optineurin (3). Similarly, in another study, the inclusions present in a patient bearing the ALS E478G mutation were positive for ubiquitin, p62, and TDP-43, but they were not immunoreactive with optineurin (133). Furthermore, optineurin-positive inclusions were absent from FTD patients manifesting with taupathies and α-synucleinopathies (3). In a recently published study (157), a fALS patient bearing an optineurin mutation (E478G) displayed features of several proteinopathies, including phosphorylated TDP-43, phosphorylated tau, and α-synuclein, although it was not examined whether optineurin is present in these inclusions. Hence together these data indicate that the involvement of optineurin in TDP-43 pathology in ALS and FTD remains controversial.
As well as ALS and FTD, optineurin pathology has also been detected in other neurodegenerative disorders. Optineurin-positive inclusions in neuronal and glial cells are occasionally observed in Alzheimer’s disease; however, they do not co-localize with tau or TDP-43 (158). Furthermore, the optineurin immunoreactivity displays a different pattern compared to phospho-TDP-43, providing further evidence that TDP-43 and optineurin pathology are independent of each other. Marinesco bodies are small, spherical intranuclear inclusions that are observed in neurons of the substantia nigra and locus coerelus in Alzheimer’s disease, Parkinson’s disease, and dementia with Lewy bodies (159). These bodies co-localize and display strong immunostaining for optineurin in Alzheimer’s disease (4). Furthermore, optineurin co-localizes with htt and frequently occurs in the neuronal intracellular inclusions in Huntington’s disease and, to a lesser extent, in inclusions of the neuropil and perikaryon. These inclusions are common in interneurons and are present but less frequent, in medium projection neurons of the cortex and striatum (5). Similarly, in dominant hereditary motor and sensory axonopathy patients, autopsy samples displayed optineurin-positive inclusions (158). In postmortem tissue from sporadic basophilic inclusion body disease (BIBD) and fALS with FUS mutations, all basophilic inclusions examined were positive and co-localized with optineurin and FUS, but not TDP-43 or SOD1 (160). In contrast, optineurin-positive inclusions were co-localized with TDP-43 in muscle tissue of sporadic inclusion body myositis (sIBM) patients (161). Hence, similar to ALS/FTD, the involvement of optineurin in the pathology of other neurodegenerative disorders remains unclear. Whether optineurin is misfolded and actively contributes to the formation of these aggregates and directly causes cell death, or alternatively, is passively recruited to the inclusions via interaction with aggregation prone proteins, is still under investigation. An intriguing alternative scenario is that optineurin is recruited to the inclusions to direct them towards the autophagic pathway. Nevertheless, these data indicate that optineurin is part of the pathology of a wide range of neurodegenerative diseases, highlighting a central role for optineurin in protein aggregation.
Conclusion
Dysfunctional optineurin is implicated in several neurodegenerative diseases, particularly ALS and glaucoma. In addition, the normal cellular functions of optineurin in neuroinflammation, autophagy, and vesicular trafficking, are important mechanisms that dysfunction in these disorders. Furthermore, mutations identified in familial forms of these disorders known to lead to aberrant autophagy, receptor recycling, and Golgi fragmentation, as well as unbalanced NF-κB signaling. However, these mechanisms are poorly defined and the relationship between optineurin and neurodegeneration has not been well studied in comparison to other proteins linked to ALS/glaucoma. Optineurin also has many putative binding partners. One possible scenario in neurodegenerative diseases is that these binding partners interact aberrantly with mutant optineurin. Previous studies have implicated a loss-of-function mechanism by reduced interaction with these proteins. However, it is also possible that mutant optineurin acquires additional, non-physiological binding partners in ALS, and/or glaucoma, resulting in a toxic gain-of-function mechanism. Further understanding of optineurin interactions is therefore needed to establish a conclusive link between neurodegeneration and altered interactions between optineurin and other proteins.
Author Contributions
Both authors contributed to this article. JDA and RPT co-wrote and edited the article throughout for content and style consistency. RPT prepared the tables and figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) Project grants (10305133 and 1086887), the MND Research Institute of Australia, and a NHMRC Dementia Team Research grant (1095215). RPT is supported by Macquarie University PhD scholarship.
References
1. Mirzaei M, Gupta VB, Chick JM, Greco TM, Wu Y, Chitranshi N, et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci Rep (2017) 7:12685. doi:10.1038/s41598-017-12858-7
2. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature (2010) 465:223–6. doi:10.1038/nature08971
3. Hortobágyi T, Troakes C, Nishimura AL, Vance C, van Swieten JC, Seelaar H, et al. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol (2011) 121:519–27. doi:10.1007/s00401-011-0813-3
4. Schwab C, Yu S, McGeer PL. Optineurin is colocalized with ubiquitin in Marinesco bodies. Acta Neuropathol (2012) 123:289–92. doi:10.1007/s00401-011-0915-y
5. Schwab C, Yu S, McGeer EG, McGeer PL. Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett (2012) 506:149–54. doi:10.1016/j.neulet.2011.10.070
6. Traxinger K, Kelly C, Johnson BA, Lyles RH, Glass JD. Prognosis and epidemiology of amyotrophic lateral sclerosis: analysis of a clinic population, 1997–2011. Neurol Clin Pract (2013) 3:313–20. doi:10.1212/CPJ.0b013e3182a1b8ab
7. Pupillo E, Messina P, Logroscino G, Beghi E; SLALOM Group. Long-term survival in amyotrophic lateral sclerosis: a population-based study. Ann Neurol (2014) 75:287–97. doi:10.1002/ana.24096
8. Marin B, Boumédiene F, Logroscino G, Couratier P, Babron MC, Leutenegger AL, et al. Variation in worldwide incidence of amyotrophic lateral sclerosis: a meta-analysis. Int J Epidemiol (2017) 46:57–74. doi:10.1093/ije/dyw061
9. Chesi A, Staahl BT, Jovičić A, Couthouis J, Fasolino M, Raphael AR, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci (2013) 16:851–5. doi:10.1038/nn.3412
10. van Doormaal PTC, Ticozzi N, Weishaupt JH, Kenna K, Diekstra FP, Verde F, et al. The role of de novo mutations in the development of amyotrophic lateral sclerosis. Hum Mutat (2017) 38:1534–41. doi:10.1002/humu.23295
11. Gibson SB, Downie JM, Tsetsou S, Feusier JE, Figueroa KP, Bromberg MB, et al. The evolving genetic risk for sporadic ALS. Neurology (2017) 89:226–33. doi:10.1212/WNL.0000000000004109
12. Keren N, Scott KM, Tsuda M, Barnwell J, Knibb JA, Ellis CM, et al. Evidence of an environmental effect on survival in ALS. Amyotroph Lateral Scler Frontotemporal Degener (2014) 15:528–33. doi:10.3109/21678421.2014.911326
13. Bozzoni V, Pansarasa O, Diamanti L, Nosari G, Cereda C, Ceroni M. Amyotrophic lateral sclerosis and environmental factors. Funct Neurol (2016) 31:7–19. doi:10.11138/FNeur/2016.31.1.007
14. Zhang M, Xi Z, Ghani M, Jia P, Pal M, Werynska K, et al. Genetic and epigenetic study of ALS-discordant identical twins with double mutations in SOD1 and ARHGEF28. J Neurol Neurosurg Psychiatry (2016) 87:1268–70. doi:10.1136/jnnp-2016-313592
15. Lam L, Chin L, Halder RC, Sagong B, Famenini S, Sayre J, et al. Epigenetic changes in T-cell and monocyte signatures and production of neurotoxic cytokines in ALS patients. FASEB J (2016) 30:3461–73. doi:10.1096/fj.201600259RR
16. Young PE, Kum Jew S, Buckland ME, Pamphlett R, Suter CM. Epigenetic differences between monozygotic twins discordant for amyotrophic lateral sclerosis (ALS) provide clues to disease pathogenesis. PLoS One (2017) 12:e0182638. doi:10.1371/journal.pone.0182638
17. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature (1993) 362:59–62. doi:10.1038/362059a0
18. Chia R, Chio A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol (2018) 17:94–102. doi:10.1016/S1474-4422(17)30401-5
19. Renton AE, Chiò A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci (2014) 17:17–23. doi:10.1038/nn.3584
20. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347:1436–41. doi:10.1126/science.aaa3650
21. Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron (2010) 68:857–64. doi:10.1016/j.neuron.2010.11.036
22. Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature (2011) 477:211–5. doi:10.1038/nature10353
23. Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun (2016) 7:11253. doi:10.1038/ncomms11253
24. Fifita JA, Williams KL, Sundaramoorthy V, Mccann EP, Nicholson GA, Atkin JD, et al. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph Lateral Scler Frontotemporal Degener (2017) 18:126–33. doi:10.1080/21678421.2016.1218517
25. Shahheydari H, Ragagnin A, Walker AK, Toth RP, Vidal M, Jagaraj CJ, et al. Protein quality control and the amyotrophic lateral sclerosis/frontotemporal dementia continuum. Front Mol Neurosci (2017) 10:119. doi:10.3389/fnmol.2017.00119
26. Young JJ, Lavakumar M, Tampi D, Balachandran S, Tampi RR. Frontotemporal dementia: latest evidence and clinical implications. Ther Adv Psychopharmacol (2018) 8:33–48. doi:10.1177/2045125317739818
27. Olney NT, Spina S, Miller BL. Frontotemporal dementia. Neurol Clin (2017) 35:339–74. doi:10.1016/j.ncl.2017.01.008
28. Bennion Callister J, Pickering-Brown SM. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp Neurol (2014) 262:84–90. doi:10.1016/j.expneurol.2014.06.001
29. Ji A-L, Zhang X, Chen W-W, Huang W-J. Genetics insight into the amyotrophic lateral sclerosis/frontotemporal dementia spectrum. J Med Genet (2017) 54:145. doi:10.1136/jmedgenet-2016-104271
30. Nguyen HP, Van Broeckhoven C, van der Zee J. ALS genes in the genomic era and their implications for FTD. Trends Genet (2018). doi:10.1016/j.tig.2018.03.001
31. Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol (2015) 130:77–92. doi:10.1007/s00401-015-1436-x
32. Weinreb RN, Aung T, Medeiros FA. The pathophysiology and treatment of glaucoma: a review. JAMA (2014) 311:1901–11. doi:10.1001/jama.2014.3192
33. Minegishi Y, Nakayama M, Iejima D, Kawase K, Iwata T. Significance of optineurin mutations in glaucoma and other diseases. Prog Retin Eye Res (2016) 55:149–81. doi:10.1016/j.preteyeres.2016.08.002
34. Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet (2017) 26:R21–7. doi:10.1093/hmg/ddx184
35. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science (2002) 295:1077–9. doi:10.1126/science.1066901
36. Fingert JH, Robin AL, Scheetz TE, Kwon YH, Liebmann JM, Ritch R, et al. Tank-binding kinase 1 (TBK1) gene and open-angle glaucomas (an American Ophthalmological Society thesis). Trans Am Ophthalmol Soc (2016) 114:T6.
37. Millecamps S, Boillée S, Chabrol E, Camu W, Cazeneuve C, Salachas F, et al. Screening of OPTN in French familial amyotrophic lateral sclerosis. Neurobiol Aging (2011) 32:557.e11–3. doi:10.1016/j.neurobiolaging.2010.11.005
38. van Blitterswijk M, van Vught PW, van Es MA, Schelhaas HJ, van der Kooi AJ, de Visser M, et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging (2012) 33:1016.e1–7. doi:10.1016/j.neurobiolaging.2011.05.019
39. Li C, Ji Y, Tang L, Zhang N, He J, Ye S, et al. Optineurin mutations in patients with sporadic amyotrophic lateral sclerosis in China. Amyotroph Lateral Scler Frontotemporal Degener (2015) 16:485–9. doi:10.3109/21678421.2015.1089909
40. Rollinson S, Bennion J, Toulson G, Halliwell N, Usher S, Snowden J, et al. Analysis of optineurin in frontotemporal lobar degeneration. Neurobiol Aging (2012) 33:425.e1–2. doi:10.1016/j.neurobiolaging.2010.10.002
41. Funayama T, Ishikawa K, Ohtake Y, Tanino T, Kurosaka D, Kimura I, et al. Variants in optineurin gene and their association with tumor necrosis factor-alpha polymorphisms in Japanese patients with glaucoma. Invest Ophthalmol Vis Sci (2004) 45:4359–67. doi:10.1167/iovs.03-1403
42. Xiao Z, Meng Q, Tsai JC, Yuan H, Xu N, Li Y. A novel optineurin genetic mutation associated with open-angle glaucoma in a Chinese family. Mol Vis (2009) 15:1649–54.
43. Leung YF, Fan BJ, Lam DS, Lee WS, Tam PO, Chua JK, et al. Different optineurin mutation pattern in primary open-angle glaucoma. Invest Ophthalmol Vis Sci (2003) 44:3880–4. doi:10.1167/iovs.02-0693
44. Ying H, Shen X, Park B, Yue BY. Posttranslational modifications, localization, and protein interactions of optineurin, the product of a glaucoma gene. PLoS One (2010) 5:e9168. doi:10.1371/journal.pone.0009168
45. Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol (1998) 18:1601–10. doi:10.1128/MCB.18.3.1601
46. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113:4039–44. doi:10.1073/pnas.1523926113
47. Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, et al. Ubiquitylation of autophagy receptor optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell (2014) 26:106–20. doi:10.1016/j.ccr.2014.05.015
48. Mao J, Xia Q, Liu C, Ying Z, Wang H, Wang G. A critical role of Hrd1 in the regulation of optineurin degradation and aggresome formation. Hum Mol Genet (2017) 26:1877–89. doi:10.1093/hmg/ddx096
49. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333:228–33. doi:10.1126/science.1205405
50. Moore AS, Holzbaur EL. Spatiotemporal dynamics of autophagy receptors in selective mitophagy. Autophagy (2016) 12:1956–7. doi:10.1080/15548627.2016.1212788
51. Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci (2013) 126:580–92. doi:10.1242/jcs.114926
52. Sundaramoorthy V, Walker AK, Tan V, Fifita JA, Mccann EP, Williams KL, et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet (2015) 24:3830–46. doi:10.1093/hmg/ddv126
53. Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol (2005) 169:285–95. doi:10.1083/jcb.200501162
54. Sippl C, Bosserhoff AK, Fischer D, Tamm ER. Depletion of optineurin in RGC-5 cells derived from retinal neurons causes apoptosis and reduces the secretion of neurotrophins. Exp Eye Res (2011) 93:669–80. doi:10.1016/j.exer.2011.08.011
55. Nagabhushana A, Chalasani ML, Jain N, Radha V, Rangaraj N, Balasubramanian D, et al. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol (2010) 11:4. doi:10.1186/1471-2121-11-4
56. Park B, Ying H, Shen X, Park JS, Qiu Y, Shyam R, et al. Impairment of protein trafficking upon overexpression and mutation of optineurin. PLoS One (2010) 5:e11547. doi:10.1371/journal.pone.0011547
57. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A (2016) 113:E3349–58. doi:10.1073/pnas.1523810113
58. Song GJ, Jeon H, Seo M, Jo M, Suk K. Interaction between optineurin and Rab1a regulates autophagosome formation in neuroblastoma cells. J Neurosci Res (2017) 96(3):407–15. doi:10.1002/jnr.24143
59. Bansal M, Moharir SC, Sailasree SP, Sirohi K, Sudhakar C, Sarathi DP, et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. J Biol Chem (2017) 293(1):132–47. doi:10.1074/jbc.M117.801944
60. Hattula K, Peranen J. FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol (2000) 10:1603–6. doi:10.1016/S0960-9822(00)00864-2
61. Tanishima M, Takashima S, Honda A, Yasuda D, Tanikawa T, Ishii S, et al. Identification of optineurin as an interleukin-1 receptor-associated kinase 1-binding protein and its role in regulation of MyD88-dependent signaling. J Biol Chem (2017) 292:17250–7. doi:10.1074/jbc.M117.813899
62. Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science (2016) 353:603–8. doi:10.1126/science.aaf6803
63. Nagabhushana A, Bansal M, Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFα-induced NF-κB activation. PLoS One (2011) 6:e17477. doi:10.1371/journal.pone.0017477
64. Rodriguez MJ, Mahy N. Neuron-microglia interactions in motor neuron degeneration. The inflammatory hypothesis in amyotrophic lateral sclerosis revisited. Curr Med Chem (2016) 23:4753–72. doi:10.2174/0929867324666161123091314
65. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi:10.1126/science.1194637
66. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19:987. doi:10.1038/nn.4338
67. Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol (2009) 4:389–98. doi:10.1007/s11481-009-9171-5
68. Brites D, Vaz AR. Microglia centered pathogenesis in ALS: insights in cell interconnectivity. Front Cell Neurosci (2014) 8:117. doi:10.3389/fncel.2014.00117
69. Geloso MC, Corvino V, Marchese E, Serrano A, Michetti F, D’Ambrosi N. The dual role of microglia in ALS: mechanisms and therapeutic approaches. Front Aging Neurosci (2017) 9:242. doi:10.3389/fnagi.2017.00242
70. Walker DG, Lue L-F. Immune phenotypes of microglia in human neurodegenerative disease: challenges to detecting microglial polarization in human brains. Alzheimers Res Ther (2015) 7:56. doi:10.1186/s13195-015-0139-9
71. Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A (2006) 103:17513–8. doi:10.1073/pnas.0602841103
72. Ben Haim L, Carrillo-de Sauvage MA, Ceyzeriat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci (2015) 9:278. doi:10.3389/fncel.2015.00278
73. Gonzalez-Reyes RE, Nava-Mesa MO, Vargas-Sanchez K, Ariza-Salamanca D, Mora-Munoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci (2017) 10:427. doi:10.3389/fnmol.2017.00427
74. Okada S, Hara M, Kobayakawa K, Matsumoto Y, Nakashima Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci Res (2018) 126:39–43. doi:10.1016/j.neures.2017.10.004
75. Vargas MR, Johnson JA. Astrogliosis in amyotrophic lateral sclerosis: role and therapeutic potential of astrocytes. Neurotherapeutics (2010) 7:471–81. doi:10.1016/j.nurt.2010.05.012
76. Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res (2005) 30:883–93. doi:10.1007/s11064-005-6961-x
77. Kaltschmidt B, Kaltschmidt C. NF-κB in the nervous system. Cold Spring Harb Perspect Biol (2009) 1:a001271. doi:10.1101/cshperspect.a001271
78. Shih R-H, Wang C-Y, Yang C-M. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci (2015) 8:77. doi:10.3389/fnmol.2015.00077
79. Lawrence T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1:a001651. doi:10.1101/cshperspect.a001651
80. Tilborghs S, Corthouts J, Verhoeven Y, Arias D, Rolfo C, Trinh XB, et al. The role of nuclear factor-kappa B signaling in human cervical cancer. Crit Rev Oncol Hematol (2017) 120:141–50. doi:10.1016/j.critrevonc.2017.11.001
81. Schwamborn K, Weil R, Courtois G, Whiteside ST, Israel A. Phorbol esters and cytokines regulate the expression of the NEMO-related protein, a molecule involved in a NF-kappa B-independent pathway. J Biol Chem (2000) 275:22780–9. doi:10.1074/jbc.M001500200
82. Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett (2008) 582:997–1002. doi:10.1016/j.febslet.2008.02.047
83. Klingseisen L, Ehrenschwender M, Heigl U, Wajant H, Hehlgans T, Schütze S, et al. E3-14.7K is recruited to TNF-receptor 1 and blocks TNF cytolysis independent from interaction with optineurin. PLoS One (2012) 7:e38348. doi:10.1371/journal.pone.0038348
84. Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha-induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol (2007) 17:1438–43. doi:10.1016/j.cub.2007.07.041
85. Fenner BJ, Scannell M, Prehn JH. Identification of polyubiquitin binding proteins involved in NF-kappaB signaling using protein arrays. Biochim Biophys Acta (2009) 1794:1010–6. doi:10.1016/j.bbapap.2009.02.013
86. Journo C, Filipe J, About F, Chevalier SA, Afonso PV, Brady JN, et al. NRP/optineurin cooperates with TAX1BP1 to potentiate the activation of NF-kappaB by human T-lymphotropic virus type 1 tax protein. PLoS Pathog (2009) 5:e1000521. doi:10.1371/journal.ppat.1000521
87. Laplantine E, Fontan E, Chiaravalli J, Lopez T, Lakisic G, Véron M, et al. NEMO specifically recognizes K63-linked poly-ubiquitin chains through a new bipartite ubiquitin-binding domain. EMBO J (2009) 28:2885–95. doi:10.1038/emboj.2009.241
88. Chalasani ML, Swarup G, Balasubramanian D. Optineurin and its mutants: molecules associated with some forms of glaucoma. Ophthalmic Res (2009) 42:176–84. doi:10.1159/000232400
89. Sudhakar C, Nagabhushana A, Jain N, Swarup G. NF-kappaB mediates tumor necrosis factor alpha-induced expression of optineurin, a negative regulator of NF-kappaB. PLoS One (2009) 4:e5114. doi:10.1371/journal.pone.0005114
90. Slowicka K, Vereecke L, Mc Guire C, Sze M, Maelfait J, Kolpe A, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur J Immunol (2016) 46:971–80. doi:10.1002/eji.201545863
91. Meena NP, Zhu G, Mittelstadt PR, Giardino Torchia ML, Pourcelot M, Arnoult D, et al. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett (2016) 590:1498–508. doi:10.1002/1873-3468.12176
92. Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem (2011) 286:35663–74. doi:10.1074/jbc.M111.267567
93. Pourcelot M, Zemirli N, Silva Da Costa L, Loyant R, Garcin D, Vitour D, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol (2016) 14:69. doi:10.1186/s12915-016-0292-z
94. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature (2015) 517:311–20. doi:10.1038/nature14191
96. van Vliet C, Thomas EC, Merino-Trigo A, Teasdale RD, Gleeson PA. Intracellular sorting and transport of proteins. Prog Biophys Mol Biol (2003) 83:1–45. doi:10.1016/S0079-6107(03)00019-1
97. Buss F, Spudich G, Kendrick-Jones J. Myosin VI: cellular functions and motor properties. Annu Rev Cell Dev Biol (2004) 20:649–76. doi:10.1146/annurev.cellbio.20.012103.094243
98. Park BC, Shen X, Samaraweera M, Yue BY. Studies of optineurin, a glaucoma gene: Golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am J Pathol (2006) 169:1976–89. doi:10.2353/ajpath.2006.060400
99. Stroissnigg H, Repitz M, Miloloza A, Linhartova I, Beug H, Wiche G, et al. FIP-2, an IkappaB-kinase-gamma-related protein, is associated with the Golgi apparatus and translocates to the marginal band during chicken erythroblast differentiation. Exp Cell Res (2002) 278:133–45. doi:10.1006/excr.2002.5567
100. Bond LM, Peden AA, Kendrick-Jones J, Sellers JR, Buss F. Myosin VI and its binding partner optineurin are involved in secretory vesicle fusion at the plasma membrane. Mol Biol Cell (2011) 22:54–65. doi:10.1091/mbc.E10-06-0553
101. Au JS, Puri C, Ihrke G, Kendrick-Jones J, Buss F. Myosin VI is required for sorting of AP-1B-dependent cargo to the basolateral domain in polarized MDCK cells. J Cell Biol (2007) 177:103–14. doi:10.1083/jcb.200608126
102. Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington’s disease pathology. Curr Trends Neurol (2011) 5:65–78.
103. Vaibhava V, Nagabhushana A, Chalasani ML, Sudhakar C, Kumari A, Swarup G. Optineurin mediates a negative regulation of Rab8 by the GTPase-activating protein TBC1D17. J Cell Sci (2012) 125:5026–39. doi:10.1242/jcs.102327
104. Li Y, Kang J, Horwitz MS. Interaction of an adenovirus 14.7-kilodalton protein inhibitor of tumor necrosis factor alpha cytolysis with a new member of the GTPase superfamily of signal transducers. J Virol (1997) 71:1576–82.
105. De Marco N, Buono M, Troise F, Diez-Roux G. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem (2006) 281:16147–56. doi:10.1074/jbc.M601467200
106. Sippl C, Zeilbeck LF, Fuchshofer R, Tamm ER. Optineurin associates with the podocyte Golgi complex to maintain its structure. Cell Tissue Res (2014) 358:567–83. doi:10.1007/s00441-014-1968-8
107. Sundaramoorthy V, Sultana JM, Atkin JD. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front Neurosci (2015) 9:400. doi:10.3389/fnins.2015.00400
108. Paulus JD, Link BA. Loss of optineurin in vivo results in elevated cell death and alters axonal trafficking dynamics. PLoS One (2014) 9:e109922. doi:10.1371/journal.pone.0109922
109. Chibalina MV, Poliakov A, Kendrick-Jones J, Buss F. Myosin VI and optineurin are required for polarized EGFR delivery and directed migration. Traffic (2010) 11:1290–303. doi:10.1111/j.1600-0854.2010.01101.x
110. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal (2014) 20:460–73. doi:10.1089/ars.2013.5371
111. Zaffagnini G, Martens S. Mechanisms of selective autophagy. J Mol Biol (2016) 428:1714–24. doi:10.1016/j.jmb.2016.02.004
112. Kulkarni VV, Maday S. Compartment-specific dynamics and functions of autophagy in neurons. Dev Neurobiol (2017) 78(3):298–310. doi:10.1002/dneu.22562
113. Shen WC, Li HY, Chen GC, Chern Y, Tu PH. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy (2015) 11:685–700. doi:10.4161/auto.36098
114. Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci (2012) 125:795. doi:10.1242/jcs.093849
115. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A (2014) 111:E4439–48. doi:10.1073/pnas.1405752111
116. Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet (2015) 24:4429–42. doi:10.1093/hmg/ddv179
117. Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, et al. Association of regulatory T-cell expansion with progression of amyotrophic lateral sclerosis: a study of humans and a transgenic mouse model. JAMA Neurol (2018). doi:10.1001/jamaneurol.2018.0035
118. Rentzos M, Evangelopoulos E, Sereti E, Zouvelou V, Marmara S, Alexakis T, et al. Alterations of T cell subsets in ALS: a systemic immune activation? Acta Neurol Scand (2012) 125:260–4. doi:10.1111/j.1600-0404.2011.01528.x
119. Beers DR, Zhao W, Wang J, Zhang X, Wen S, Neal D, et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight (2017) 2:e89530. doi:10.1172/jci.insight.89530
120. Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis (2004) 15:601–9. doi:10.1016/j.nbd.2003.12.012
121. Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science (2003) 302:113–7. doi:10.1126/science.1086071
122. Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science (2006) 312:1389–92. doi:10.1126/science.1123511
123. Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol (2012) 237:147–52. doi:10.1016/j.expneurol.2012.06.011
124. De Paola M, Mariani A, Bigini P, Peviani M, Ferrara G, Molteni M, et al. Neuroprotective effects of toll-like receptor 4 antagonism in spinal cord cultures and in a mouse model of motor neuron degeneration. Mol Med (2012) 18:971–81. doi:10.2119/molmed.2012.00020
125. Lee JY, Lee JD, Phipps S, Noakes PG, Woodruff TM. Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation (2015) 12:90. doi:10.1186/s12974-015-0310-z
126. Volonte C, Apolloni S, Parisi C, Amadio S. Purinergic contribution to amyotrophic lateral sclerosis. Neuropharmacology (2016) 104:180–93. doi:10.1016/j.neuropharm.2015.10.026
127. Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol (2006) 6:12. doi:10.1186/1471-2377-6-12
128. Sako W, Ito H, Yoshida M, Koizumi H, Kamada M, Fujita K, et al. Nuclear factor kappa B expression in patients with sporadic amyotrophic lateral sclerosis and hereditary amyotrophic lateral sclerosis with optineurin mutations. Clin Neuropathol (2012) 31:418–23. doi:10.5414/NP300493
129. Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, et al. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem (2013) 126:699–704. doi:10.1111/jnc.12326
130. Sasaki S, Tsutsumi Y, Yamane K, Sakuma H, Maruyama S. Sporadic amyotrophic lateral sclerosis with extensive neurological involvement. Acta Neuropathol (1992) 84:211–5. doi:10.1007/BF00311398
131. Sakaguchi T, Irie T, Kawabata R, Yoshida A, Maruyama H, Kawakami H. Optineurin with amyotrophic lateral sclerosis-related mutations abrogates inhibition of interferon regulatory factor-3 activation. Neurosci Lett (2011) 505:279–81. doi:10.1016/j.neulet.2011.10.040
132. Perri ER, Thomas CJ, Parakh S, Spencer DM, Atkin JD. The unfolded protein response and the role of protein disulfide isomerase in neurodegeneration. Front Cell Dev Biol (2015) 3:80. doi:10.3389/fcell.2015.00080
133. Ito H, Nakamura M, Komure O, Ayaki T, Wate R, Maruyama H, et al. Clinicopathologic study on an ALS family with a heterozygous E478G optineurin mutation. Acta Neuropathol (2011) 122:223–9. doi:10.1007/s00401-011-0842-y
134. Kamada M, Izumi Y, Ayaki T, Nakamura M, Kagawa S, Kudo E, et al. Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology (2014) 34:64–70. doi:10.1111/neup.12051
135. Jiang B, Gao L, Lei D, Liu J, Shao Z, Zhou X, et al. Decreased expression of miR9 due to E50K OPTN mutation causes disruption of the expression of BDNF leading to RGC5 cell apoptosis. Mol Med Rep (2016) 14:4901–5. doi:10.3892/mmr.2016.5810
136. Sirohi K, Chalasani ML, Sudhakar C, Kumari A, Radha V, Swarup G. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy (2013) 9:510–27. doi:10.4161/auto.23458
137. Chi ZL, Akahori M, Obazawa M, Minami M, Noda T, Nakaya N, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet (2010) 19:2606–15. doi:10.1093/hmg/ddq146
138. Minegishi Y, Iejima D, Kobayashi H, Chi ZL, Kawase K, Yamamoto T, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet (2013) 22:3559–67. doi:10.1093/hmg/ddt210
139. Cho MH, Cho K, Kang HJ, Jeon EY, Kim HS, Kwon HJ, et al. Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy (2014) 10:1761–75. doi:10.4161/auto.29647
140. Shim MS, Takihara Y, Kim KY, Iwata T, Yue BY, Inatani M, et al. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci Rep (2016) 6:33830. doi:10.1038/srep33830
141. Ying H, Turturro S, Nguyen T, Shen X, Zelkha R, Johnson EC, et al. Induction of autophagy in rats upon overexpression of wild-type and mutant optineurin gene. BMC Cell Biol (2015) 16:14. doi:10.1186/s12860-015-0060-x
142. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci (2009) 122:3589. doi:10.1242/jcs.051011
143. Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta (2006) 1762:1068–82. doi:10.1016/j.bbadis.2006.05.002
144. Anborgh PH, Godin C, Pampillo M, Dhami GK, Dale LB, Cregan SP, et al. Inhibition of metabotropic glutamate receptor signaling by the huntingtin-binding protein optineurin. J Biol Chem (2005) 280:34840–8. doi:10.1074/jbc.M504508200
145. Chalasani ML, Radha V, Gupta V, Agarwal N, Balasubramanian D, Swarup G. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci (2007) 48:1607–14. doi:10.1167/iovs.06-0834
146. Kachaner D, Filipe J, Laplantine E, Bauch A, Bennett KL, Superti-Furga G, et al. Plk1-dependent phosphorylation of optineurin provides a negative feedback mechanism for mitotic progression. Mol Cell (2012) 45:553–66. doi:10.1016/j.molcel.2011.12.030
147. Golsteyn RM, Mundt KE, Fry AM, Nigg EA. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J Cell Biol (1995) 129:1617–28. doi:10.1083/jcb.129.6.1617
148. Kryndushkin D, Ihrke G, Piermartiri TC, Shewmaker F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol Microbiol (2012) 86:1531–47. doi:10.1111/mmi.12075
149. Subramaniam S, Unsicker K. ERK and cell death: ERK1/2 in neuronal death. FEBS J (2010) 277:22–9. doi:10.1111/j.1742-4658.2009.07367.x
150. Parmar MS, Jaumotte JD, Wyrostek SL, Zigmond MJ, Cavanaugh JE. The role of ERK1, 2, and 5 in dopamine neuron survival during aging. Neurobiol Aging (2014) 35:669–79. doi:10.1016/j.neurobiolaging.2013.09.031
151. Li H, Ao X, Jia J, Wang Q, Zhang Z. Effects of optineurin siRNA on apoptotic genes and apoptosis in RGC-5 cells. Mol Vis (2011) 17:3314–25.
152. Sayyad Z, Sirohi K, Radha V, Swarup G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci Rep (2017) 7:16855. doi:10.1038/s41598-017-17241-0
153. Meng Q, Lv J, Ge H, Zhang L, Xue F, Zhu Y, et al. Overexpressed mutant optineurin(E50K) induces retinal ganglion cells apoptosis via the mitochondrial pathway. Mol Biol Rep (2012) 39:5867–73. doi:10.1007/s11033-011-1397-7
154. Jo M, Chung AY, Yachie N, Seo M, Jeon H, Nam Y, et al. Yeast genetic interaction screen of human genes associated with amyotrophic lateral sclerosis: identification of MAP2K5 kinase as a potential drug target. Genome Res (2017) 27(9):1487–500. doi:10.1101/gr.211649.116
155. Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron (2013) 79:416–38. doi:10.1016/j.neuron.2013.07.033
156. Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med (2008) 14:451–64. doi:10.2119/2007-00100.Irvine
157. Ayaki T, Ito H, Komure O, Kamada M, Nakamura M, Wate R, et al. Multiple proteinopathies in familial ALS cases with optineurin mutations. J Neuropathol Exp Neurol (2018) 77:128–38. doi:10.1093/jnen/nlx109
158. Fujita K, Yoshida M, Sako W, Maeda K, Hashizume Y, Goto S, et al. Brainstem and spinal cord motor neuron involvement with optineurin inclusions in proximal-dominant hereditary motor and sensory neuropathy. J Neurol Neurosurg Psychiatry (2011) 82:1402–3. doi:10.1136/jnnp-2011-300783
159. Beach TG, Walker DG, Sue LI, Newell A, Adler CC, Joyce JN. Substantia nigra Marinesco bodies are associated with decreased striatal expression of dopaminergic markers. J Neuropathol Exp Neurol (2004) 63:329–37. doi:10.1093/jnen/63.4.329
160. Ito H, Fujita K, Nakamura M, Wate R, Kaneko S, Sasaki S, et al. Optineurin is co-localized with FUS in basophilic inclusions of ALS with FUS mutation and in basophilic inclusion body disease. Acta Neuropathol (2011) 121:555–7. doi:10.1007/s00401-011-0809-z
161. Yamashita S, Kimura E, Tawara N, Sakaguchi H, Nakama T, Maeda Y, et al. Optineurin is potentially associated with TDP-43 and involved in the pathogenesis of inclusion body myositis. Neuropathol Appl Neurobiol (2013) 39:406–16. doi:10.1111/j.1365-2990.2012.01297.x
162. Figlewicz DA, Krizus A, Martinoli MG, Meininger V, Dib M, Rouleau GA, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet (1994) 3:1757–61. doi:10.1093/hmg/3.10.1757
163. Chance PF, Rabin BA, Ryan SG, Ding Y, Scavina M, Crain B, et al. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am J Hum Genet (1998) 62:633–40. doi:10.1086/301769
164. Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet (2001) 29:160–5. doi:10.1038/ng1001-160
165. Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, et al. Point mutations of the p150 subunit of (DCTN1) gene in ALS. Neurology (2004) 63:724–6. doi:10.1212/01.WNL.0000134608.83927.B1
166. Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet (2004) 75:822–31. doi:10.1086/425287
167. Greenway MJ, Alexander MD, Ennis S, Traynor BJ, Corr B, Frost E, et al. A novel candidate region for ALS on chromosome 14q11.2. Neurology (2004) 63:1936–8. doi:10.1212/01.WNL.0000144344.39103.F6
168. Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology (2006) 67:1074–7. doi:10.1212/01.wnl.0000231510.89311.8b
169. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (2006) 314:130–3. doi:10.1126/science.1134108
170. Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology (2009) 73:1180–5. doi:10.1212/WNL.0b013e3181bbff05
171. Simpson CL, Lemmens R, Miskiewicz K, Broom WJ, Hansen VK, van Vught PW, et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum Mol Genet (2009) 18:472–81. doi:10.1093/hmg/ddn375
172. Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM, et al. Deleterious variants of FIG 4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet (2009) 84:85–8. doi:10.1016/j.ajhg.2008.12.010
173. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron (2011) 72:245–56. doi:10.1016/j.neuron.2011.09.011
174. Fecto F, Yan J, Vemula SP, Liu E, Yang Y, Chen W, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol (2011) 68:1440–6. doi:10.1001/archneurol.2011.250
175. Couthouis J, Hart MP, Shorter J, DeJesus-Hernandez M, Erion R, Oristano R, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A (2011) 108:20881–90. doi:10.1073/pnas.1109434108
176. Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature (2010) 466:1069–75. doi:10.1038/nature09320
177. Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A, et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging (2012) 33:839.e5–9. doi:10.1016/j.neurobiolaging.2011.11.012
178. Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature (2012) 488:499–503. doi:10.1038/nature11280
179. Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet (2012) 21:2899–911. doi:10.1093/hmg/dds116
180. Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature (2013) 495:467–73. doi:10.1038/nature11922
181. Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet (2013) 93:900–5. doi:10.1016/j.ajhg.2013.09.008
182. Johnson JO, Glynn SM, Gibbs JR, Nalls MA, Sabatelli M, Restagno G, et al. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain (2014) 137:e311. doi:10.1093/brain/awu265
183. Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, et al. Mutations in the matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci (2014) 17:664–6. doi:10.1038/nn.3688
184. Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron (2014) 84:324–31. doi:10.1016/j.neuron.2014.09.027
185. Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet (2015) 24:1363–73. doi:10.1093/hmg/ddu545
186. van Rheenen W, Shatunov A, Dekker AM, McLaughlin RL, Diekstra FP, Pulit SL, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet (2016) 48:1043–8. doi:10.1038/ng.3622
187. Brenner D, Müller K, Wieland T, Weydt P, Böhm S, Lulé D, et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain (2016) 139:e28. doi:10.1093/brain/aww033
188. Thomas-Jinu S, Gordon PM, Fielding T, Taylor R, Smith BN, Snowden V, et al. Non-nuclear pool of splicing factor SFPQ regulates axonal transcripts required for normal motor development. Neuron (2017) 94:322–36.e5. doi:10.1016/j.neuron.2017.03.026
189. Brenner D, Yilmaz R, Müller K, Grehl T, Petri S, Meyer T, et al. Hot-spot KIF5A mutations cause familial ALS. Brain (2018) 141(3):688–97. doi:10.1093/brain/awx370
190. Belzil VV, Daoud H, Desjarlais A, Bouchard JP, Dupré N, Camu W, et al. Analysis of OPTN as a causative gene for amyotrophic lateral sclerosis. Neurobiol Aging (2011) 32:555.e13–4. doi:10.1016/j.neurobiolaging.2010.10.001
191. Del Bo R, Tiloca C, Pensato V, Corrado L, Ratti A, Ticozzi N, et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2011) 82:1239–43. doi:10.1136/jnnp.2011.242313
192. Naruse H, Takahashi Y, Kihira T, Yoshida S, Kokubo Y, Kuzuhara S, et al. Mutational analysis of familial and sporadic amyotrophic lateral sclerosis with OPTN mutations in Japanese population. Amyotroph Lateral Scler (2012) 13:562–6. doi:10.3109/17482968.2012.684213
193. Tumer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, Lucamp, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging (2012) 33:208.e201–5. doi:10.1016/j.neurobiolaging.2011.07.001
194. Beeldman E, van der Kooi AJ, de Visser M, van Maarle MC, van Ruissen F, Baas F. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener (2015) 16:410–1. doi:10.3109/21678421.2015.1066821
195. Bury JJ, Highley JR, Cooper-Knock J, Goodall EF, Higginbottom A, McDermott CJ, et al. Oligogenic inheritance of optineurin (OPTN) and C9ORF72 mutations in ALS highlights localisation of OPTN in the TDP-43-negative inclusions of C9ORF72-ALS. Neuropathology (2016) 36:125–34. doi:10.1111/neup.12240
196. Iida A, Hosono N, Sano M, Kamei T, Oshima S, Tokuda T, et al. Optineurin mutations in Japanese amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2012) 83:233–5. doi:10.1136/jnnp.2010.234963
197. Gooding LR, Sofola IO, Tollefson AE, Duerksen-Hughes P, Wold WS. The adenovirus E3-14.7K protein is a general inhibitor of tumor necrosis factor-mediated cytolysis. J Immunol (1990) 145:3080–6.
Keywords: optineurin, neuroinflammation, autophagy, vesicular trafficking, amyotrophic lateral sclerosis, glaucoma
Citation: Toth RP and Atkin JD (2018) Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 9:1017. doi: 10.3389/fimmu.2018.01017
Received: 02 March 2018; Accepted: 23 April 2018;
Published: 23 May 2018
Edited by:
Andrew Mark Smith, University College London, United KingdomReviewed by:
Christian Lobsiger, UMR7225 Institut du cerveau et de la moelle épinière (ICM), FranceAgustina Alaimo, Universidad de Buenos Aires, Argentina
Copyright: © 2018 Toth and Atkin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie D. Atkin, anVsaWUuYXRraW5AbXEuZWR1LmF1