 Thomas A. Ryan
Thomas A. Ryan David A. Tumbarello
David A. Tumbarello- Biological Sciences, University of Southampton, Southampton, United Kingdom
Optineurin is a multifunctional adaptor protein intimately involved in various vesicular trafficking pathways. Through interactions with an array of proteins, such as myosin VI, huntingtin, Rab8, and Tank-binding kinase 1, as well as via its oligomerisation, optineurin has the ability to act as an adaptor, scaffold, or signal regulator to coordinate many cellular processes associated with the trafficking of membrane-delivered cargo. Due to its diverse interactions and its distinct functions, optineurin is an essential component in a number of homeostatic pathways, such as protein trafficking and organelle maintenance. Through the binding of polyubiquitinated cargoes via its ubiquitin-binding domain, optineurin also serves as a selective autophagic receptor for the removal of a wide range of substrates. Alternatively, it can act in an ubiquitin-independent manner to mediate the clearance of protein aggregates. Regarding its disease associations, mutations in the optineurin gene are associated with glaucoma and have more recently been found to correlate with Paget’s disease of bone and amyotrophic lateral sclerosis (ALS). Indeed, ALS-associated mutations in optineurin result in defects in neuronal vesicular localisation, autophagosome–lysosome fusion, and secretory pathway function. More recent molecular and functional analysis has shown that it also plays a role in mitophagy, thus linking it to a number of other neurodegenerative conditions, such as Parkinson’s. Here, we review the role of optineurin in intracellular membrane trafficking, with a focus on autophagy, and describe how upstream signalling cascades are critical to its regulation. Current data and contradicting reports would suggest that optineurin is an important and selective autophagy receptor under specific conditions, whereby interplay, synergy, and functional redundancy with other receptors occurs. We will also discuss how dysfunction in optineurin-mediated pathways may lead to perturbation of critical cellular processes, which can drive the pathologies of number of diseases. Therefore, further understanding of optineurin function, its target specificity, and its mechanism of action will be critical in fully delineating its role in human disease.
Introduction
Optineurin, through a diverse set of interactions, regulates a number of crucial cellular processes, specifically those that require the coordinated trafficking of protein and membrane cargo. First isolated in 1998 in a yeast two-hybrid screen by its interaction with the adenoviral protein E3-14.7K, it was initially named 14.7K-interacting protein (FIP-2) (1). A later study identified that mutations in this gene, located on chromosome 10p14, were found to associate with normal tension glaucoma (NTG), a subtype of primary open-angle glaucoma (POAG) (2). Thus, it was designated OPTN, encoding the optineurin (for “optic neuropathy inducing”) protein.
Since then, optineurin has been implicated as a genetic risk factor in Paget’s disease of bone (3, 4), familial and sporadic forms of amyotrophic lateral sclerosis (ALS) (5–11) and Crohn’s disease (12). In addition, optineurin has also been found to localise to an array of intracellular structures and compartments, providing evidence of its ubiquitous distribution and potential multifunctional cellular role. As optineurin plays a critical function across several key pathways, its dysfunction is likely to lead to the disruption of mechanisms that aim to maintain cell homoeostasis and thus contribute to the development of a number of human pathologies.
Optineurin Protein Domain Structure and Interacting Partners
The human OPTN gene, containing three non-coding exons that makeup its 5′-untranslated region (UTR) and 13 exons that encode the 577 amino acid (66 kDa) protein, is ubiquitously expressed in most tissue and cell types (13). Four isoforms with identical open-reading frames have been reported to be generated through alternative splicing of the 5′-UTR (14).
OPTN originated from gene duplication of the NF-κB regulator NF-κB essential modulator (NEMO) (15) and contains two ubiquitin-binding motifs, which are the ubiquitin-binding domain (UBD) of ABIN proteins and NEMO (UBAN) domain and the zinc finger (ZF) domain (16). It has been previously suggested that the presence of two nearby ubiquitin-binding motifs within the protein may explain optineurin’s binding preference for longer polyubiquitin chains (17, 18). In addition to the aforementioned UBAN and ZF domains, optineurin also contains at least one leucine zipper (LZ), multiple coiled-coil (CC) domains, a NEMO-like domain, and a microtubule-associated protein 1 light chain 3 (LC3)-interacting region (LIR) (Figure 1A) (19).
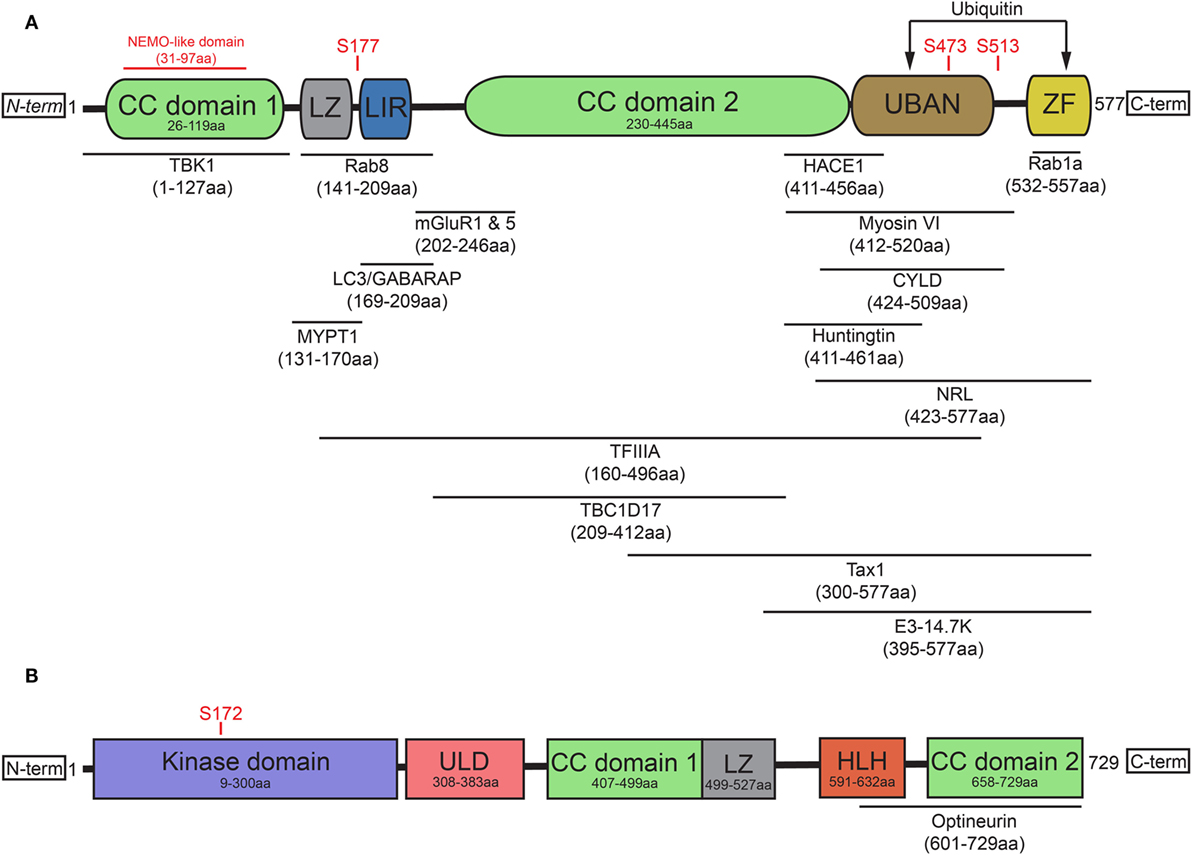
Figure 1. Optineurin and TBK1 both contain multiple structurally distinct domains associated with their regulation, binding, and activity. (A) Optineurin comprises two coiled-coil (CC) domains, a leucine zipper (LZ), an LC3-interacting region (LIR), UBAN domain, and a zinc finger (ZF) domain at it C-terminus. To date, a number of studies have identified the interacting regions of optineurin with its binding partners, defined in this figure. Serine phosphorylation sites are represented that regulate optineurin’s LC3-binding or ubiquitin-binding capacity. (B) TBK1 comprises a kinase domain, a ubiquitin-like domain (ULD), two CC domains, a LZ, and a helix-loop-helix (HLH) motif. Serine 172 represents the site that regulates TBK1’s kinase activity. TBK1 interacts with optineurin via its C-terminal HLH and CC domains.
The role of optineurin as an adaptor across many cellular processes is made possible by its ability to interact with a large number of proteins (Figure 1A). Through its functional interactions with TANK (TRAF family member-associated NF-κB activator)-binding kinase 1 (TBK1) (20–22), LC3 (22, 23), myosin VI (24–28), tax1 binding protein 1 (TAX1BP1) (29), Rab8 (25, 30), huntingtin (Htt) (30, 31), transferrin receptor (32), adenovirus E3-14.7K (1), receptor-interacting protein (RIP) (33), the bZIP transcription factor neural retina leucine zipper (34), myosin phosphatase targeting subunit 1 (35), transcription factor IIA (36), SOD1 (37), caspase 8 (38), HACE1 (39), CYLD (40), or metabotropic glutamate receptor 1 and 5 (41), optineurin can regulate a multitude of pathways. In addition to these interactions, optineurin can also oligomerise to form homo-hexameric structures (42), which are likely to have distinct roles from the monomeric form. The specific regulation, spatiotemporal dynamics, and cellular functions of many of these interactions will be discussed later in this review.
Post-translational modifications of optineurin also occur as part of its regulation. TBK1, a serine/threonine kinase, is one of the primary regulators of optineurin and mediates many of the optineurin-dependent cellular processes discussed in this review. To date, a number of disease-associated mutations, specifically in ALS and frontotemporal dementia (FTD), have been identified that perturb TBK1 binding with optineurin, resulting in dysfunction of trafficking pathways such as autophagy (43). Structurally, TBK1 contains an N-terminal kinase domain and ubiquitin-like domain (ULD), along with an α-helical scaffold dimerization domain (SDD) and adaptor binding (AB) domain within the C-terminal region (44, 45) (Figure 1B). Activation of TBK1 occurs through phosphorylation of the Ser172 residue within its kinase activation loop (46), inducing complete remodelling of this loop (47). Four dimerisation interfaces have been identified within TBK1, formed by the SDD interacting with either the N- or C-terminal lobes of the kinase domain, the ULD, or residues within the SDD itself (45). It may be the case that a dimeric form of TBK1 is maintained in an inactive state through prevention of Ser172 phosphorylation. Following specific stimuli, TBK1 is subsequently recruited to signalling scaffolds, whereby its clustering triggers the engagement of interdimeric interactions to promote Ser172 phosphorylation (47). Recruitment to discrete scaffolds, such as those that occur on the Golgi (48), or to polyubiquitylated optineurin, to regulate the interferon response (17, 49), may provide specificity in response to distinct stimuli, therefore allowing activation of specific pathways. TBK1 localisation is therefore critical in determining its activity and subsequently its impacts on optineurin function.
To date, less is understood about the spatiotemporal regulation of the TBK1/optineurin axis compared with the characterisation of their interactions. Indeed, TBK1 binding of optineurin, within the C-terminal CC domain (21) through polar and hydrophobic interactions (20), is required for TBK1-mediated phosphorylation of Ser177. This in turn has been shown to markedly enhance the LC3-binding capacity of the optineurin LIR (22, 50). Phosphorylation of optineurin by TBK1 at Ser473 and Ser513 also enhances its binding affinity for polyubiquitin chains via the UBAN domain (51, 52). These data demonstrate how the regulated dynamic binding capacity and post-translational modifications of optineurin are critical in modulating its function in cargo recognition during autophagy (Figure 1A). Throughout this review, we label optineurin as a receptor or an adaptor in accordance with either its function in cargo recognition within the lumen of the autophagosome or its ability to interact with cytosolic facing proteins on the external membrane of the autophagosome, respectively.
Role of Optineurin in Signalling and Intracellular Trafficking
Optineurin is associated with a number of signalling pathways. In particular, it has been shown to play an important role in the regulation of signalling cascades critical to the innate immune response. Several studies have shown optineurin to act upstream of NF-κB, negatively regulating its activity. Interleukin-1 receptor-associated kinase 1, along with tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6), activates the innate immune response (53, 54) and is degraded in a proteasome-dependent manner upon its phosphorylation (55). Optineurin directly binds IRAK1 and prevents TRAF6 polyubiquitination, which is critical for its mediation of NF-κB activation (56). Optineurin also inhibits NF-κB activation through another C-terminal-dependent interaction with the deubiquitinase CYLD. This interaction mediates a subsequent interaction between CYLD and RIP (40), the latter acting as an adaptor upon its ubiquitination of NEMO, which senses the polyubiquitination of RIP and activates downstream NF-κB signalling via IκB kinase complex (57, 58). Optineurin directly competes with NEMO for the binding to ubiquitylated RIP (33) and recruits CYLD, which deubiquitinates RIP to inhibit NF-κB activation (40). Recently, it was shown that activation of T-cell receptor signalling triggers the degradation of optineurin to overcome optineurin’s negative regulation of NF-κB signalling, which acts to suppress T-cell activation (59). Interestingly, NF-κB upregulates OPTN expression (60), suggesting a negative feedback loop exists to ensure proper regulation of NF-κB signalling. Furthermore, optineurin inhibits the antiviral innate immune response by targetting CYLD to TBK1 to suppress its kinase activity, subsequently inhibiting interferon production (61).
Along with its regulation of signal propagation, optineurin also plays an essential role in the maintenance of organelle structure and function. Optineurin associates with the Golgi complex (62, 63) and through an interaction with the multifunctional actin motor protein myosin VI, functions to maintain the structural organisation of this organelle (26, 64, 65). Loss or mutation of optineurin in cell lines leads to Golgi fragmentation (26, 66–68) and although this was not replicated in vivo in zebrafish embryos (69), increased cell death and vesicle trafficking defects were observed. However, since the loss-of-function zebrafish model retains a low level of optineurin mRNA and possibly a truncated version of optineurin protein, it remains to be determined the extent of this phenotype (67). Alternatively, the role of optineurin in Golgi maintenance may therefore be cell type specific, or alternative/compensatory pathways may exist that can maintain Golgi morphology but do not necessarily rescue vesicular trafficking defects.
In addition, optineurin associates with Htt and Rab8 at the Golgi, where it acts as part of a complex to regulate post-Golgi trafficking of proteins (26), sorted by clathrin adaptor protein complex 1B and myosin VI (70). Mutations in Htt can uncouple the optineurin/Rab8 complex at the late Golgi compartment, resulting in decreased trafficking to lysosomes (71). Htt also functions as part of a number of vesicular trafficking pathways (72–74), which suggests that Htt defects may have wide-ranging impacts on optineurin function along related trafficking pathways. Rab8 is a critical component of the trafficking along the biosynthetic pathway from the trans-Golgi network (75, 76) and it also functions along other discrete endosomal routes. In particular, it has been shown that an optineurin interaction with the Rab-activating protein TBC1D17 regulates Rab8-dependent endosomal tubule formation and recycling of the transferrin receptor (77). Furthermore, optineurin is phosphorylated by Plk1 at Ser177, which dissociates optineurin from the Golgi through abrogation of a Rab8 interaction, facilitating its translocation into the nucleus to promote mitotic progression through regulation of Plk1 activity (35). Optineurin also functions post-Golgi to facilitate secretory vesicle fusion at the plasma membrane via an interaction with myosin VI (24). Therefore, optineurin may participate as a ‘keystone’ adaptor protein within these complexes to maintain Golgi organisation and coordinate multiple routes of post-Golgi trafficking.
Interestingly, it has also been shown that optineurin is required for the recruitment of ubiquitylated TBK1 to the Golgi apparatus, a critical step in TBK1 activation following viral RNA sensing as part of the innate immune response (48). Therefore, it is likely that optineurin association with the Golgi through its interaction with Rab8 (26) also recruits ubiquitylated TBK1 through its UBD (48), thus acting as a necessary precursor to the activation of this heterodimeric complex (20). The stabilisation of the TBK1/optineurin complex via ubiquitin could in turn allow for the enhanced propagation of optineurin-mediated signalling, as well as increasing its affinity for LC3 to promote autophagy progression, a mechanism we discuss in detail later in this review.
Optineurin Regulation of Autophagy
The cellular mechanism to degrade cytosolic components is primarily carried out via the ubiquitin proteasome system (UPS) or autophagy. The latter process of autophagy, ‘cellular self-eating,’ acts to degrade proteins, organelles, and invading pathogens as part of a bulk process, whereas the UPS functions to degrade individual proteins (78). Indeed, both UPS and autophagic capacities are essential homeostatic pathways under basal conditions or in response to stress. Dysfunction in either is associated with the pathogenesis of a large number of disorders, ranging from neurodegenerative disease to cancer. Around 30% of newly synthesised proteins misfold (79), rendering them prone to aggregation. These aggregates cannot be efficiently degraded by the UPS, even resulting in inhibition of proteasomal functions (80, 81), and thus must be removed via autophagic mechanisms. It should, however, also be noted that significant cross-talk between the UPS and autophagy exists, despite the fact they are often considered as completely separate systems (82).
Autophagy is a catabolic process by which intracellular components are engulfed and degraded. There are three forms of autophagy that can be differentiated by their function and mechanism of cargo delivery. These are chaperone-mediated autophagy, microautophagy, and macroautophagy. In this review, we will exclusively discuss the implications of macroautophagy, which requires the formation of a distinct organelle, the autophagosome. Although non-selective, bulk macroautophagy (herein termed autophagy) can occur under conditions of nutrient starvation to recycle cytosolic content, cargo-specific autophagy (termed selective autophagy) is critical in the removal of potentially cytotoxic components, such as damaged organelles, protein aggregates, and invading pathogens. This process can be divided into five basic stages: cargo recognition, autophagosome nucleation, autophagosome elongation and maturation, fusion with the lysosome, and degradation of cargo (Figure 2).
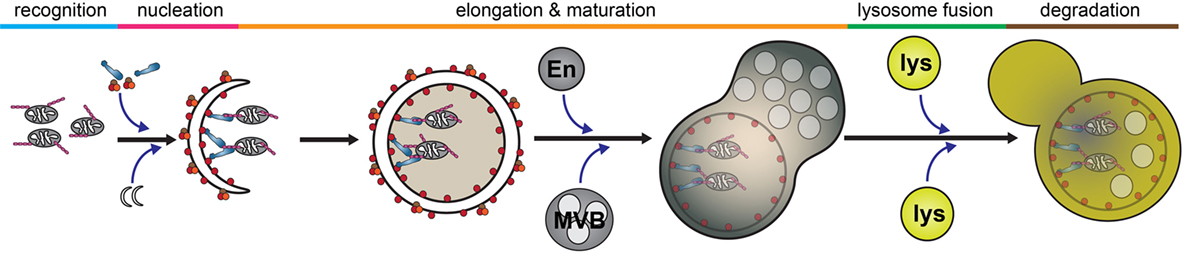
Figure 2. Selective autophagy. The autophagy pathway can be divided into five major steps: cargo recognition, phagophore nucleation, autophagosome elongation and maturation, fusion with the lysosome, and cargo degradation. Initial steps of cargo identification, as which occurs during mitochondrial capture, requires ubiquitination of a substrate and identification by autophagy receptors, such as optineurin, which facilitates the recruitment and nucleation of autophagosomal membrane to encapsulate the cargo. Subsequently, the autophagosome undergoes maturation following fusion with various endosomal vesicles and eventually fuses with the lysosome to facilitate cargo degradation. Abbreviations: En, endosome; MVB, multivesicular body; lys, lysosome.
To correctly engage selective forms of autophagy to mediate the degradation of specific substrates, autophagy receptor proteins such as optineurin, nuclear dot protein 52 (NDP52), TAX1BP1, neighbour of BRCA1 gene 1 (NBR1), or p62 are required (22, 28, 83–85). Substrates to be degraded are ubiquitylated and recognised by UBDs, specific for certain ubiquitin linkages types, present within the autophagy receptors. Through an additional LC3 interacting region (LIR), these receptors can directly interact with autophagosomal membrane, thus facilitating cargo recognition, trafficking, and degradation (86).
To date, over 30 autophagy-related (ATG) genes have been identified in the yeast, Saccharomyces cerevisiae (87, 88). In mammals, these have been shown to be involved in both ubiquitin-dependent and -independent mechanisms of autophagy (89). In yeast, Atg8, an ubiquitin-like protein, conjugates to phosphatidylethanolamine to be inserted into lipid membranes to mediate tethering and formation of the autophagosomal double membrane (90–92). The mammalian Atg8 homologues, LC3, γ-aminobutyric-acid-type-A-receptor-associated protein (GABARAP), and Golgi-associated ATPase enhancer of 16 kDa, were then later identified to undergo post-translational modifications to form species that can associate with autophagosomal membranes (93–96). p62 was subsequently shown to directly bind, via a LIR, to both LC3 and GABARAP (97) and ubiquitin-labelled proteins via its ubiquitin-associated (UBA) domain (98). Importantly, formation and clearance of ubiquitin-positive protein inclusions is ablated in p62-deficient cells (97, 99). Thus, p62 acts as a receptor protein between ubiquitylated protein aggregates and the LC3-positive autophagosomal membranes.
Similar to p62, other autophagy adaptors such as optineurin, TAX1BP1, NDP52, and NBR1 also directly bind ubiquitin and LC3 to coordinate the autophagosome-mediated engulfment of cargo. In particular, optineurin was first identified as an autophagic receptor through its interaction with Atg8-related proteins in a yeast two-hybrid assay and its localisation to LC3-positive autophagosomal membranes upon induction of xenophagy, the selective autophagy pathway for pathogens (22). Here, the authors identified that optineurin interacts with LC3 and GABARAP through an LIR located between its CC domains. Crucially, the demonstration that phosphorylation upstream of the optineurin LIR regulates its interaction with LC3, and thus its autophagic function, was a novel finding at the time showing a further level of regulation for autophagy receptors. In addition, optineurin’s ability to function as an autophagy receptor has relevance to distinct pathological mechanisms, as it was recently shown to directly interact with the endoplasmic reticulum stress protein IRE1α and function to suppress activation of the unfolded protein response via mediating the autophagic degradation of IRE1α (100).
The ‘ubiquitin code,’ which regulates signal transduction and degradation of labelled substrates, has an inherent complexity. This is due to the occurrence of both mono- and poly-ubiquitin chain types as well as the multiple layers of lysine-dependent heterotypic polyubiquitin chain linkages, such as those mediated by K6, K11, K27, K29, K33, K48, or K63 (101). Broadly, there are two routes of degradation for ubiquitylated substrates; UPS- or autophagic-mediated degradation. K63-linked polyubiquitin chains are thought to primarily determine autophagic clearance of a substrate (84, 102, 103). Optineurin contains two UBDs, an UBAN domain and ZF domain. The UBAN and ZF domains bind K63- but not K48-linked polyubiquitin chains (15, 16, 33) suggesting optineurin primarily functions along the autophagic degradation pathway or alternatively regulates signal propagation, as which occurs along the NF-κB pathway. However, optineurin, TAX1BP1, and NDP52 preferentially bind different types of ubiquitin chains (15), which may be critical in determining their cargo specificity during autophagy.
Intracellular pathogens, such as Salmonella enterica, which escape into the cytosol from a vacuolar compartment are targetted and degraded by the autophagy machinery (104). The capacity of optineurin to function as an autophagy receptor, which is enhanced by Ser177 phosphorylation, is critical to suppress the hyperproliferation of cytosolic S. enterica (22). TBK1, a critical regulator of autophagy (105), binds to optineurin (21) and induces phosphorylation within the N-terminal LC3-interacting motif of optineurin (22) (Figure 3). A similar axis has also been observed with TBK1-dependent modulation of NDP52 function, which promotes autophagy of S. enterica (85), suggesting the potential for synergism or functional redundancy between autophagy receptors in the innate immune response. Some suggestion of this has already been observed, whereby multiple receptors function cooperatively along the same pathway (22, 106). However, it has also been shown that for both xenophagy and the selective mitochondrial pathway, mitophagy, optineurin, and p62 are independently recruited to separate autophagosomal subdomains (22, 23), suggesting they function along parallel pathways to facilitate pathogen and mitochondrial degradation.
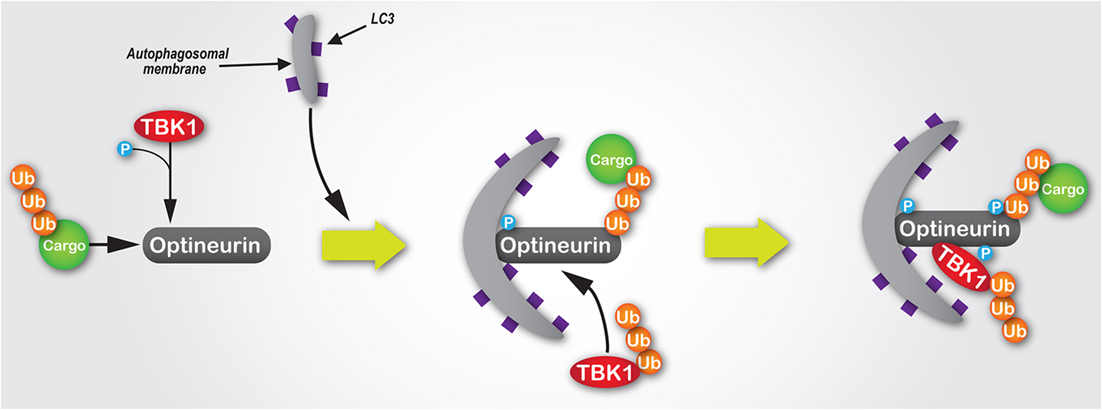
Figure 3. The mechanisms of the TBK1/optineurin complex during autophagy. Optineurin interacts with ubiquitylated cargo via its UBAN and zinc finger domains. TBK1 is then recruited via an interaction with optineurin to facilitate its phosphorylation at Ser177, which enhances its light chain 3 (LC3)-binding capacity and recruitment of autophagosomal membrane. Subsequently, TBK1-mediated phosphorylation of optineurin at Ser473 and Ser513 enhances its polyubiquitin-binding capacity, thus stabilising its interaction with ubiquitin-labelled cargo. Since K63-linked polyubiquitylation of TBK1 is required for its activation, as well as its recognition and recruitment by Golgi-localised optineurin, we would hypothesise that during autophagosome formation ubiquitylated TBK1 is recruited by optineurin, where it is activated and in turn phosphorylates optineurin, thus creating a positive signal amplification loop through the recruitment and stabilisation of the TBK1/optineurin heterodimeric complex on ubiquitylated cargo.
In the case of Listeria monocytogenes, upregulation of optineurin occurs in response to the bacterial expression of listeriolysin O (LLO) (107), a pore-forming cytolysin that allows the bacteria to escape from a vacuolar compartment into the cytosol following host entry (108). Here, TBK1 activity enhances optineurin-mediated clearance of the pathogen, whilst a reduction in optineurin expression results in less autophagosomal clearance of L. monocytogenes (107). These data together are indicative of the importance of the TBK1–optineurin axis in the clearance of several pathogenic bacteria. It also suggests that under these conditions, this optineurin-regulated immune defence system has specifically evolved to detect the LLO-mediated translocation of bacteria into the cytoplasm.
Further highlighting the importance of the TBK1–optineurin axis, pharmacological inhibition of TBK1 activation using BX795 (109) inhibits optineurin phosphorylation and subsequent LC3 recruitment (22). Moreover, activation of the TBK1–optineurin complex in mouse bone marrow-derived macrophages is perturbed by the ubiquitin-binding defective OPTND477N mutant (17, 110), suggesting that the binding of ubiquitin-tagged cargo by optineurin is a necessary precursor to its phosphorylation, and thus activation of this complex. Interestingly, TBK1-mediated phosphorylation of optineurin’s UBAN domain at S473 further enhances optineurin’s capacity to bind ubiquitin chains (52) (Figure 3). Indeed, optineurin has also been shown to directly regulate TBK1 activity (48). K63-linked polyubiquitination of TBK1 at residues K30 and K401 is required for TBK1 activation (111). These ubiquitin chains are sensed by optineurin localised at the Golgi apparatus via its interaction with Rab8 (26), which results in the formation of a complex between optineurin and TBK1, with the latter activated by trans-autophosphorylation (48).
Optineurin is also a key adaptor protein for the actin motor protein myosin VI (112). This interaction is critical for the spatiotemporal regulation of many optineurin-mediated functions, including autophagy and secretory vesicle fusion (24, 27, 28). There are around 40 different myosins expressed in humans (113) and due to the association of myosin dysfunction in a number of diseases, the development of small molecules to manipulate their function is a growing area of investigation (24). However, unlike other myosins, myosin VI movement is towards the pointed (minus) ends of actin filaments (114) using large powerstroke movements achieved through significant conformational rearrangement (115–117). Whilst the N-terminal motor domain, conserved across myosins, undergoes adenosine triphosphate (ATP)-dependent conformational changes to induce motor translocation (118), the C-terminal tail region is divergent across the myosin family and thus confers cargo specificity via direct interactions (112). Upon the binding of cargo, for example, via optineurin as an adaptor, myosin VI is able to dimerise and potentially function as a processive motor (119). To date, multiple binding motifs within the tail region of myosin VI have been identified, which allow specific interactions with a range of proteins that function in membrane trafficking (26, 28, 120–127). In particular, the RRL motif within the myosin VI tail is required for its interaction with optineurin, as well as the other autophagy receptors TAX1BP1 and NDP52 (26, 126, 127).
Mutation within, or deletion of, the optineurin UBD perturbs optineurin pull down of myosin VI, as well as target of myb protein 1 (Tom1) (128), highlighting the importance of this region in the interaction with the myosin cargo-binding tail and its potential to facilitate larger scale adaptor protein complexes. Recent data have shed further light on this. Within the C-terminal region of myosin VI, a motif interacting with ubiquitin domain exists (129). A second region, encompassing the RRL motif, was subsequently identified and termed the myosin VI ubiquitin-binding domain (MyUb) (123). Here, the authors found that residues R1117 (part of the RRL motif) and I1104 within the MyUb domain are critical for MyUb structural integrity and the binding of ubiquitin conjugated to optineurin, respectively. This may suggest that optineurin, separate to its function as a cargo-binding receptor that binds ubiquitin upon phosphorylation by TBK1, may act as an adaptor protein by interacting with the myosin VI MyUb domain or RRL motif to facilitate autophagosomal maturation.
The optineurin–myosin VI complex likely regulates a key aspect of autophagy, which is to facilitate the maturation of the autophagosome and its fusion with the lysosome (28, 130). In particular, myosin VI, through a direct interaction with optineurin via its RRL motif (26), delivers Tom1-positive endosomal membranes to autophagosomes, which is required for autophagosome–lysosome fusion (28). This holds significance because the origins of the autophagosomal membrane are wide-ranging and highly debated within the literature, with recruitment coming from the ER (131, 132), endosomal compartments (133–136), plasma membrane (137), mitochondria (138), and Golgi (139, 140) all contributing to nucleation and elongation of the phagophore membrane. It has also been demonstrated that autophagosomal membranes derive from ER–mitochondrial contact sites (141), as well as the ER–Golgi intermediate compartment (26, 142, 143). Tom1 is an alternative endosomal sorting complex required for transport (ESCRT) class 0 protein (144), a family of trafficking proteins required for cargo sorting along the endocytic route and in the autophagy pathway (145), and binds the WWY motif of myosin VI, unlike optineurin, NDP52, and TAX1BP1 which bind the RRL motif (28). Although multiple studies had previously shown Tom1 and myosin VI to interact (122), the more recent observations discussed here (28, 123, 129) may suggest how this specific and dynamic pathway is tightly regulated.
The capacity of optineurin to bind both ubiquitylated cargoes and autophagosomal LC3 via its UBD and LIR, respectively (19), and myosin VI in an ubiquitin-dependent (123) or -independent manner may represent distinct autophagic steps. In this paradigm, it may be that a specific stimulus results in TBK1 recruitment and subsequent phosphorylation of optineurin at sites of cargo recognition and autophagosome formation to enhance its binding to LC3 (22) and ubiquitin (52). Separately, the conjugation of cytosolic optineurin to ubiquitin may enhance its interaction with myosin VI, via the MyUb RRL motif (123), to recruit it to LC3-positive membranes and form an adaptor/membrane/motor complex to promote autophagosomal maturation. It is therefore important to note that optineurin likely has a dual function during autophagy, functioning as a cargo receptor in the lumen of the autophagosome and also functioning as an adaptor protein on the cytosolic face of the autophagosome. More recent data further implicate optineurin in autophagosomal maturation in neurons through an interaction with the GTPase Rab1a (146). Optineurin also mediates the recruitment of the Atg12-5-16L1 complex to promote autophagosomal elongation (147), suggesting a role distinct from its cargo-binding capacity. In addition, other autophagy receptors could play a cooperative role alongside optineurin. For example, NDP52 recruitment of TBK1 to autophagosomes via the formation of an ubiquitin-sensing complex with Nap1 and Sintbad (85) could stimulate the formation and stabilisation of the heterodimeric TBK1–optineurin axis. Moreover, optineurin, TAX1BP1, and NDP52 preferentially bind different types of ubiquitin chains (15), which may be critical in regulating their cargo specificity. Interestingly, the optineurin paralogue NEMO is negatively regulated by the E3 ubiquitin ligase TRIM29 via interactions within its CC domain, resulting in the ubiquitylation and degradation of NEMO (15, 148). Whether a similar mechanism exists to regulate optineurin function remains to be determined, but this may indicate the existence of a further mode of optineurin regulation.
Optineurin Function During Mitophagy
Mitochondria are a critical organelle in the eukaryotic cell, with most cellular ATP produced by oxidative phosphorylation (OXPHOS) within the mitochondrial matrix. Mitochondria provide the major source of intracellular cytotoxic reactive oxygen species (ROS) (149) as a by-product of OXPHOS, with ROS production increasing upon mitochondrial damage. It is therefore crucial that the accumulation of dysfunctional mitochondria is effectively prevented through homeostatic mitochondria quality control pathways, such as mitophagy. Failure of these mechanisms is strongly associated with a number of age-related diseases, such as Parkinson’s disease (150). Mitophagy, a term originally coined over a decade ago (151), is the selective autophagic removal of damaged mitochondria within a cell, although the UPS is also a critical component of this pathway (152–155). More recently, the role of receptors/adaptors such as optineurin in mitophagy has begun to emerge, which has resulted in their investigation in greater detail.
The most well studied form of mitophagy is regulated by the PTEN-induced putative kinase 1 (PINK1)/Parkin axis, although alternative pathways have been shown to exist. Under ‘normal’ or ‘healthy’ conditions, PINK1 is rapidly imported into mitochondria via TOM40 and translocation of inner membrane pores (156) in a mitochondrial membrane potential-dependent manner (157–159). Following its import, PINK1 undergoes intermembrane degradation by mitochondrial processing peptidase and presenilin-associated rhomboid-like protein (160–162), with the residual N-terminus then being exported into the cytosol for proteasomal turnover (163).
Upon mitochondrial damage, PINK1 is stabilised and selectively accumulates on the mitochondrial outer membrane (MOM), where it recruits and activates Parkin (158, 159, 164). PINK1 is critical for a number of post-translational modifications to Parkin (165, 166), MOM proteins (167), and ubiquitin (168–171), as well as promoting fission to isolate damaged mitochondria for degradation (172). Parkin subsequently ubiquitylates a number of MOM proteins (173–175), in addition to RHOT1/2 (Miro in Drosophila), a small GTPase involved in mitochondrial transport, resulting in the arrest of mitochondrial trafficking (176).
For mitophagy to correctly function and damaged mitochondria to be selectively degraded, autophagy receptors once again represent critical components of the pathway. The mitochondrial protein Nix has been identified as an autophagy receptor for the targetted clearance of mitochondria (177), which is regulated by its phosphorylation (178). Although p62 has been shown to act as a receptor during mitophagy (174), its importance has since been disputed (23, 179). We would suggest that both functional redundancy and cooperativity are likely to exist between autophagy receptors with respect to their role during mitophagy. It may be the case that specific receptors are critical at distinct points during mitophagy, or that they only function under different types of mitochondrial stress and in certain cell lines. For example, mitochondrial damage induced by oxidative stress may result in the activation of a different mitophagy pathway compared with pharmacological uncoupling of membrane potential. Although p62 is recruited to uncoupled mitochondria in HeLa cells (179), optineurin is also recruited under the same conditions (23) where it induces autophagosome assembly (180).
Optineurin, along with NDP52, is recruited by PINK1 to damaged mitochondria, but in a Parkin-independent manner (181). Optineurin then preferentially binds linear ubiquitin chains via its UBAN domain (38), with TBK1 activity regulating this interaction by phosphorylation of residues within this domain (51). Although the phosphorylation of ubiquitin has been suggested to be critical in PINK1/Parkin-dependent mitophagy (170) and TBK1-mediated phosphorylation of optineurin on Ser473 facilitates its binding of pSer65 ubiquitin chains on mitochondria (52), conflicting reports have also emerged on whether optineurin activity requires ubiquitin phosphorylation in the context of mitophagy (51, 181, 182). It may be the case that these phosphorylation events are dispensable for mitophagy under certain conditions, but not others.
The fact that p62 and optineurin are recruited to distinct domains on damaged mitochondria to facilitate the separate roles of mitochondrial aggregation and LC3 recruitment, respectively (23), demonstrates the functional divergence of autophagy receptors. More recently, the divergent pathways of the overall process of mitophagy have also become better understood. Degradation of mitochondrial proteins can occur via a pathway in which mitochondria-derived vesicles (MDVs) bud off from the organelle (183–185) in a Parkin/PINK1-dependent manner (186), with Syntaxin-17 mediating MDV fusion with endolysosomal compartments (187). This pathway is likely to represent both normal physiological recycling of mitochondrial proteins and the disposal of mitochondrial components damaged by low level stress. Although no direct assessments have been made to date, we would hypothesise that proteins such as optineurin may act as receptors and/or adaptors in this lysosomal degradation pathway as the loss of Parkin ubiquitin ligase activity perturbs the MDV pathway (186), suggesting that receptors with ubiquitin-binding capacity may be required downstream of Parkin to facilitate degradation. In addition, trafficking of MDVs containing mitochondrial proteins to lysosomes is likely to require adaptor protein interactions with molecular motors such as myosin VI to facilitate cargo delivery.
Such alternate mitophagic pathways could be activated only under specific stress conditions, whereby distinct autophagy receptors undergo mitochondrial recruitment. Some evidence of this has already been observed, whereby the receptor TAX1BP1 interacts with Parkin upon mitochondrial uncoupling, but only when fusion events are also inhibited by Bafilomycin A1 (175). This could suggest that specific autophagy receptors only play a role in this pathway if other stress conditions occur in parallel, or alternatively illustrate that these interactions are transient and the inhibition of other pathways leads to their retention. Indeed, this may even better represent actual physiological disease conditions, where cells are likely to be undergoing multiple stresses whilst trying to maintain homoeostasis. Extensive further work is therefore needed to delineate the specific role of receptors, such as optineurin, during mitophagy using physiologically relevant disease models.
Optineurin in Human Disease
Primary Open-Angle Glaucoma
As previously discussed, optineurin has been associated with a number of diseases across a wide range of genetic and functional-based studies. The first proven association with disease was over a decade ago when mutations in OPTN were shown to cause an autosomal dominant form of hereditary glaucoma (2). Here, the initial studies suggested that optineurin plays a neuroprotective role, a hypothesis that has been supported by numerous subsequent publications (188–191).
Glaucoma is a disease characterised by the progressive degeneration of the optic nerve. This optic neuropathy is the primary cause of irreversible blindness worldwide, with POAG being the most common subtype (192). Although often classed as a neurodegenerative disease, it has been hypothesised that it is a primary optic neuropathy with secondary pathogenic effects in the central nervous system (193). The bilateral blindness that results from glaucoma is a result of the progressive loss of retinal ganglion cells (RGCs) in the optic nerve head (194). A number of studies have suggested that mutations in optineurin that cause glaucoma are a result of defective autophagy (195). Furthermore, this pathology resulting from autophagic defects may be limited specifically to dysfunction in optineurin-mediated autophagy as a small-scale genetic study did not find mutations in the SQSTM1 gene encoding the autophagy receptor p62, also phosphorylated by TBK1 (105), in patients with NTG (196).
The optineurin E50K mutation, a primary cause of POAG-induced blindness (2), impairs autophagy. Indeed, in this initial study by Rezaie et al. to identify OPTN mutations as causative of glaucoma, the E50K mutation segregated with the NTG phenotype within a large family, providing solid evidence for their hypothesis and was associated with 16.7% of the familial NTG cases investigated. The extension of these data into E50K transgenic mouse models has further supported this hypothesis (190, 197, 198), with mice specifically exhibiting pathological features of POAG when physiological relevant levels of the transgene were expressed (199). Cell death is also induced in mouse photoreceptor cells derived from retinal tumours expressing either E50K or M98K glaucoma-associated variants (200) (Table 1).
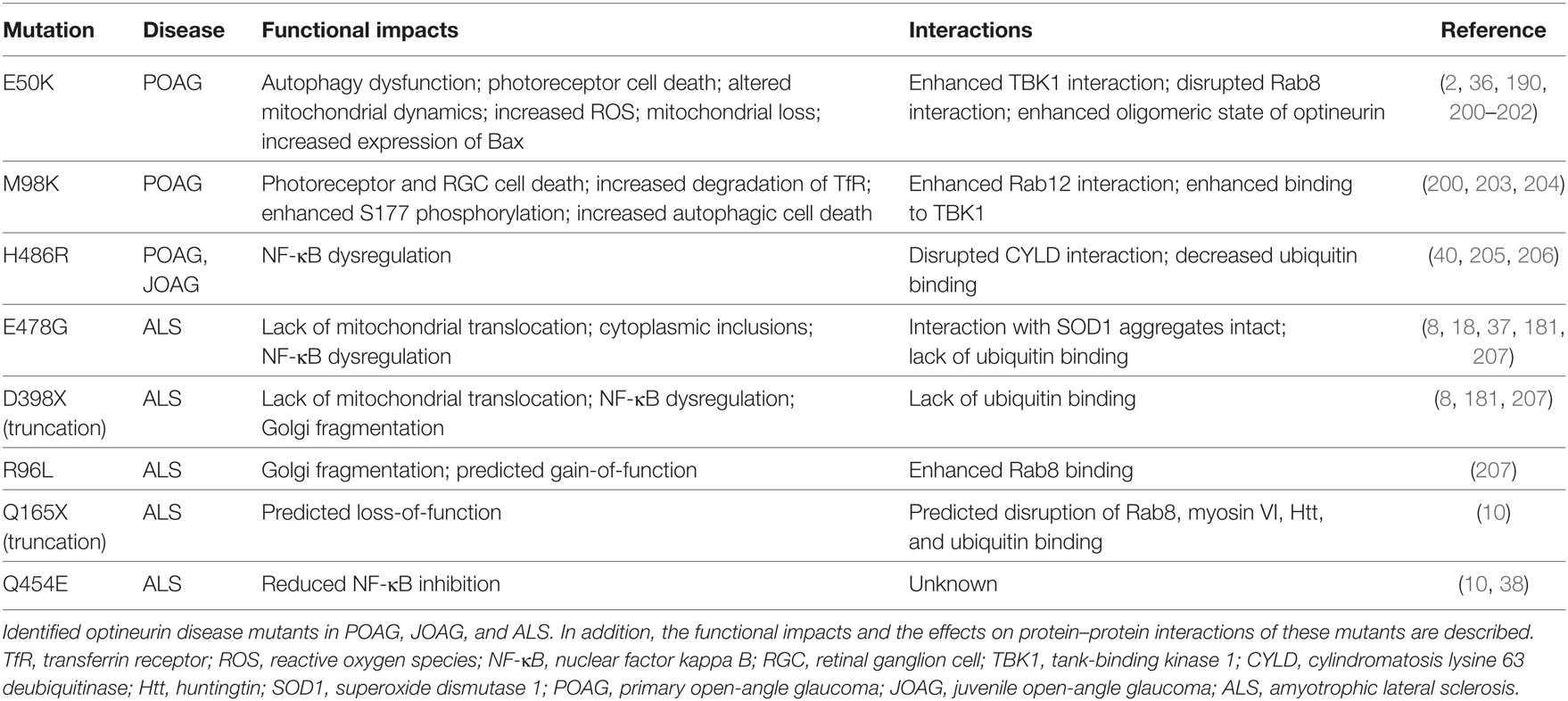
Table 1. Identified and characterised optineurin mutants associated with POAG and ALS.
At the subcellular level, the E50K mutation enhances its interaction with TBK1 (21), which disrupted proper oligomerisation resulting in its insolubility (201). This E50K mutation has also been shown to perturb optineurin’s interaction with Rab8 (77, 190, 208), a critical regulator of vesicular trafficking. The M98K mutation, found in 13.6% of NTG cases in one study (2), enhances the interaction of optineurin with Rab12 (203), a GTPase involved in vesicular trafficking and lysosomal degradation of the transferrin receptor (209). This enhanced interaction lead to the increased degradation of the transferrin receptor and RGC death (203). Furthermore, M98K demonstrates enhanced binding to TBK1, which in turn leads to enhanced Ser177 phosphorylation and thus optineurin activation in a TBK1-dependent manner, resulting in activation of autophagic cell death (204). In neuronal RGCs, the overexpression of wild-type or E50K optineurin compromises UPS-mediated turnover of optineurin leading to the accumulation of autophagosomes and apoptosis (202). It would appear that cells must maintain functional levels of optineurin and that the alteration of this homeostatic balance results in autophagic-induced cell death and/or autophagic dysfunction (Table 1).
The aberration of mitochondrial homoeostasis is also associated with glaucoma (210, 211). In transgenic mice or in vitro cultured RGCs, E50K expression alters mitochondrial dynamics and promotes expression of the proapoptotic protein Bax, leading to retinal cell death. In addition, this mutation resulted in mitochondrial loss through the induction of mitochondrial fission and the formation of mitochondrial-containing autophagosomes, as well as increased ROS production (212). This dysfunction in mitochondrial regulation may elucidate why oxidative stress-induced retinal cell death is associated with this particular optineurin mutation (189, 213).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is a neurodegenerative disease associated with mitochondrial dysfunction with respect to their function, morphology, transport, and turnover (214–217). Mutations in optineurin have been identified in ALS patients, as well as mutations in its complex binding partners TBK1 and p62, suggesting that autophagic dysfunction is the common pathway (8, 10, 18, 207). In support of this, optineurin and TBK1 mutations perturb the recruitment of LC3-positive membrane to damaged mitochondria, leading to less efficient mitophagy (50), which could account for some cases of mitochondrial dysfunction observed in ALS.
Disruption of the TBK1–optineurin interaction and their co-dependent regulatory mechanisms can be attributed to disease pathology. For example, whereas the glaucoma-associated E50K mutation in optineurin enhances its interaction with TBK1 resulting in impacts on the oligomeric state of optineurin, the ALS-associated E696K mutation of TBK1 abolishes its interaction with optineurin leading to a failure of mitochondrial translocation (20, 52). In addition, optineurin may be activated by TBK1-mediated Ser177 phosphorylation to induce autophagic clearance of protein aggregates in an ubiquitin-independent manner via its C-terminal CC domain. Interestingly, in this study, the optineurin UBAN mutant E478G still interacted with SOD1 protein aggregates, whereas depletion of optineurin in this ALS zebrafish model resulted in motor axonopathy (37). Importantly, these data have implications for both ALS and Huntington’s disease. Furthermore, mutations in TBK1 have more recently been associated with the development of FTD associated with ALS (218–221). SQSTM1 mutations in FTD and FTD with ALS have also been identified (222), which would indicate that autophagic dysfunction is at the heart of these diseases. It may therefore be the case that some TBK1 mutation-associated phenotypes in FTD/ALS occur through an optineurin-mediated action with resulting autophagic defects driving the degenerative pathology.
Many of the optineurin mutations associated with ALS are located within the UBAN domain, thus disrupting ubiquitin binding (8). ALS-associated optineurin mutations E478G and Q398X (both within the UBAN domain), as well as the ubiquitin-binding deficient D474N, do not translocate to mitochondria (181) (Table 1). However, the authors did find that the expression of the glaucoma-associated E50K mutation and the phospho-deficient S177A could marginally rescue mitophagy. This limited rescue may be explained by the fact that E50K and S177A optineurin mutants, unlike ubiquitin-binding deficient mutants, are still recruited to damaged mitochondria where they are still able to exhibit some activity, resulting in very low level recruitment of TBK1. These data are therefore indicative of an optineurin-mediated system in which its interaction with ubiquitin is most critical for mitophagy. A current hypothesis is therefore that mutations disrupting the ubiquitin-binding capacity of optineurin prevents efficient mitophagy in neurons and leads to the accumulation of cytotoxic dysfunctional mitochondria (180). p62 and optineurin are recruited to discrete domains on damaged mitochondria (23), suggesting distinct functional mechanisms exist. However, it would appear that disrupting just optineurin activity alone is enough to induce ALS pathology. The reason why neurodegeneration only occurs in specific neuronal subtypes carrying these ALS-associated familial mutations is likely due to these cells unique energetic demands and susceptibility to mitochondrial damage alongside their limited capacity for mitochondrial homeostatic pathways. Nevertheless, as wild-type optineurin binds and inactivates caspase-8 (38), mutations that result in a loss of this activity may potentiate the apoptotic pathway that occurs in ALS-associated pathologies following optineurin dysfunction.
Other Diseases
In addition to ALS and FTD, the TBK1/optineurin axis may also be implicated in the pathogenesis of other neurodegenerative disorders. Indeed, a patient carrying the optineurin E478G mutant was clinically diagnosed with both ALS and Parkinson’s disease, with autopsy analysis showing degeneration of the substantia nigra, as well as the presence of tau-positive neurofibrillary tangles and α-synuclein-positive Lewy bodies (223). As optineurin acts as a receptor during mitophagy (23), a pathway in which its dysfunction is known to cause Parkinson’s (224), it is possible that hereditary or somatic mutations in genes encoding optineurin or TBK1 may lead to a Parkinsonian progression through mitophagic perturbation.
Trinucleotide expansions within the HD gene encoding the Htt protein result in the progression of the devastating neurodegenerative disorder Huntington’s disease (225, 226). Due to its interaction with Htt (30), a protein known to regulate a number of vesicular trafficking pathways (71, 227, 228), optineurin is of significant interest in Huntington’s research. Although optineurin interacts with Rab8 and Htt at the Golgi (26), a localisation that is disrupted by mutant Htt resulting in lysosomal impairment (71), and is found in Htt protein inclusions observed in the cortex of Huntington’s patients (229), it is currently not known what role optineurin plays in the progression of the disease. Nevertheless, because optineurin is involved in the autophagic clearance of protein aggregates (37) and its abundance/neuronal distribution may confer susceptibility to Htt inclusions (230), its role in mediating clearance pathways may offer novel therapeutic targets as our understanding grows.
The impairment of vesicular trafficking and autophagy is not just associated with neurodegeneration, but has also been linked to a number of cancers (231–233). HACE1, an E3 ubiquitin ligase and potent tumour suppressor (234), ubiquitylates optineurin which promotes its interaction with p62 and induces autophagy (39). This accelerated degradation lead to a suppression of ROS and reduction of tumourigenicity of human lung cancer cells. Thus, optineurin-induced autophagy appears to represent a potential tumour suppressing pathway in some cancers.
Conclusion
Both autophagy and mitophagy have been implicated in cell survival and death pathways by a number of studies. The role of optineurin in these pathways currently remains relatively unexplored. Dysfunction in autophagy and mitophagy is associated with a number of neurodegenerative diseases and so questions therefore remain as to how optineurin-mediated autophagy, and its dysfunction, plays a role in directing neuronal death pathways under specific stress conditions. As multiple distinct pathways exist within each form of selective autophagy, which involves a number of distinct autophagy receptor and adaptor proteins, our understanding of which of these proteins play a role across each discrete pathway must be improved. For example, it is clear that the TBK1–optineurin complex plays a pivotal role during the innate immune response to target unwanted cellular pathogens, but how it spatially and temporally regulates this process with respect to related autophagy receptors has not yet been clearly defined. In addition, the outcome of TBK1 kinase activity may be regulated by the level and duration of activation, as well as by cross-talk between other kinase classes (235). Therefore, TBK1 regulation of autophagy may also occur in this manner, whereby only specific levels or discrete localisation of TBK1 activity leads to the activation of optineurin-dependent autophagy, thus allowing the cell to distinguish between different stimuli and mount the appropriate autophagic response. Nevertheless, disease-causing mutations in optineurin that result in the presentation of autophagic defects in patients highlights the central role that is played by this protein in the regulation of these cargo-specific membrane trafficking and recycling pathways.
Author Contributions
TR performed the literature research and wrote the manuscript. DT coordinated the study, performed the literature research, and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank the support of a Wellcome Trust Seed Award (205909/Z/17/Z). We would also like to thank Liam Ryan for his assistance with the figures.
Funding
TR and DT are supported by a Wellcome Trust Seed Award (205909/Z/17/Z). This work was also supported by the University of Southampton.
References
1. Li Y, Kang J, Horwitz MS. Interaction of an adenovirus E3 14.7-kilodalton protein with a novel tumor necrosis factor alpha-inducible cellular protein containing leucine zipper domains. Mol Cell Biol (1998) 18(3):1601–10. doi:10.1128/MCB.18.3.1601
2. Rezaie T, Child A, Hitchings R, Brice G, Miller L, Coca-Prados M, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science (2002) 295(5557):1077–9. doi:10.1126/science.1066901
3. Albagha OM, Visconti MR, Alonso N, Langston AL, Cundy T, Dargie R, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet (2010) 42(6):520–4. doi:10.1038/ng.562
4. Chung PY, Beyens G, Boonen S, Papapoulos S, Geusens P, Karperien M, et al. The majority of the genetic risk for Paget’s disease of bone is explained by genetic variants close to the CSF1, OPTN, TM7SF4, and TNFRSF11A genes. Hum Genet (2010) 128(6):615–26. doi:10.1007/s00439-010-0888-2
5. Beeldman E, van der Kooi AJ, de Visser M, van Maarle MC, van Ruissen F, Baas F. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener (2015) 16(5–6):410–1. doi:10.3109/21678421.2015.1066821
6. Iida A, Hosono N, Sano M, Kamei T, Oshima S, Tokuda T, et al. Optineurin mutations in Japanese amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry (2012) 83(2):233–5. doi:10.1136/jnnp.2010.234963
7. Iida A, Hosono N, Sano M, Kamei T, Oshima S, Tokuda T, et al. Novel deletion mutations of OPTN in amyotrophic lateral sclerosis in Japanese. Neurobiol Aging (2012) 33(8):1843.e19–24. doi:10.1016/j.neurobiolaging.2010.06.017
8. Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature (2010) 465(7295):223–6. doi:10.1038/nature08971
9. Tumer Z, Bertelsen B, Gredal O, Magyari M, Nielsen KC, Lucamp, et al. Novel heterozygous nonsense mutation of the OPTN gene segregating in a Danish family with ALS. Neurobiol Aging (2012) 33(1):208.e1–5. doi:10.1016/j.neurobiolaging.2011.07.001
10. van Blitterswijk M, van Vught PW, van Es MA, Schelhaas HJ, van der Kooi AJ, de Visser M, et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging (2012) 33(5):e1–7. doi:10.1016/j.neurobiolaging.2011.05.019
11. van Blitterswijk M, Vlam L, van Es MA, van der Pol WL, Hennekam EA, Dooijes D, et al. Genetic overlap between apparently sporadic motor neuron diseases. PLoS One (2012) 7(11):e48983. doi:10.1371/journal.pone.0048983
12. Smith AM, Sewell GW, Levine AP, Chew TS, Dunne J, O’Shea NR, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology (2015) 144(1):45–55. doi:10.1111/imm.12338
13. Slowicka K, Vereecke L, van Loo G. Cellular functions of optineurin in health and disease. Trends Immunol (2016) 37(9):621–33. doi:10.1016/j.it.2016.07.002
14. Rezaie T, Sarfarazi M. Molecular cloning, genomic structure, and protein characterization of mouse optineurin. Genomics (2005) 85(1):131–8. doi:10.1016/j.ygeno.2004.10.011
15. Tumbarello DA, Manna PT, Allen M, Bycroft M, Arden SD, Kendrick-Jones J, et al. The Autophagy receptor TAX1BP1 and the molecular motor myosin VI are required for clearance of Salmonella typhimurium by autophagy. PLoS Pathog (2015) 11(10):e1005174. doi:10.1371/journal.ppat.1005174
16. Wagner S, Carpentier I, Rogov V, Kreike M, Ikeda F, Lohr F, et al. Ubiquitin binding mediates the NF-kappaB inhibitory potential of ABIN proteins. Oncogene (2008) 27(26):3739–45. doi:10.1038/sj.onc.1211042
17. Gleason CE, Ordureau A, Gourlay R, Arthur JS, Cohen P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem (2011) 286(41):35663–74. doi:10.1074/jbc.M111.267567
18. Markovinovic A, Cimbro R, Ljutic T, Kriz J, Rogelj B, Munitic I. Optineurin in amyotrophic lateral sclerosis: multifunctional adaptor protein at the crossroads of different neuroprotective mechanisms. Prog Neurobiol (2017) 154:1–20. doi:10.1016/j.pneurobio.2017.04.005
19. Ying H, Yue BY. Cellular and molecular biology of optineurin. Int Rev Cell Mol Biol (2012) 294:223–58. doi:10.1016/B978-0-12-394305-7.00005-7
20. Li F, Xie X, Wang Y, Liu J, Cheng X, Guo Y, et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun (2016) 7:12708. doi:10.1038/ncomms12708
21. Morton S, Hesson L, Peggie M, Cohen P. Enhanced binding of TBK1 by an optineurin mutant that causes a familial form of primary open angle glaucoma. FEBS Lett (2008) 582(6):997–1002. doi:10.1016/j.febslet.2008.02.047
22. Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science (2011) 333(6039):228–33. doi:10.1126/science.1205405
23. Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A (2014) 111(42):E4439–48. doi:10.1073/pnas.1405752111
24. Bond LM, Peden AA, Kendrick-Jones J, Sellers JR, Buss F. Myosin VI and its binding partner optineurin are involved in secretory vesicle fusion at the plasma membrane. Mol Biol Cell (2011) 22(1):54–65. doi:10.1091/mbc.E10-06-0553
25. Chibalina MV, Roberts RC, Arden SD, Kendrick-Jones J, Buss F. Rab8-optineurin-myosin VI: analysis of interactions and functions in the secretory pathway. Methods Enzymol (2008) 438:11–24. doi:10.1016/S0076-6879(07)38002-6
26. Sahlender DA, Roberts RC, Arden SD, Spudich G, Taylor MJ, Luzio JP, et al. Optineurin links myosin VI to the Golgi complex and is involved in Golgi organization and exocytosis. J Cell Biol (2005) 169(2):285–95. doi:10.1083/jcb.200501162
27. Sundaramoorthy V, Walker AK, Tan V, Fifita JA, McCann EP, Williams KL, et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet (2015) 24(13):3830–46. doi:10.1093/hmg/ddv126
28. Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol (2012) 14(10):1024–35. doi:10.1038/ncb2589
29. Journo C, Filipe J, About F, Chevalier SA, Afonso PV, Brady JN, et al. NRP/optineurin cooperates with TAX1BP1 to potentiate the activation of NF-kappaB by human T-lymphotropic virus type 1 tax protein. PLoS Pathog (2009) 5(7):e1000521. doi:10.1371/journal.ppat.1000521
30. Hattula K, Peranen J. FIP-2, a coiled-coil protein, links huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol (2000) 10(24):1603–6. doi:10.1016/S0960-9822(00)00864-2
31. Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet (1998) 7(9):1463–74. doi:10.1093/hmg/7.9.1463
32. Park B, Ying H, Shen X, Park JS, Qiu Y, Shyam R, et al. Impairment of protein trafficking upon overexpression and mutation of optineurin. PLoS One (2010) 5(7):e11547. doi:10.1371/journal.pone.0011547
33. Zhu G, Wu CJ, Zhao Y, Ashwell JD. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol (2007) 17(16):1438–43. doi:10.1016/j.cub.2007.07.041
34. Wang C, Hosono K, Ohtsubo M, Ohishi K, Gao J, Nakanishi N, et al. Interaction between optineurin and the bZIP transcription factor NRL. Cell Biol Int (2014) 38(1):16–25. doi:10.1002/cbin.10174
35. Kachaner D, Filipe J, Laplantine E, Bauch A, Bennett KL, Superti-Furga G, et al. Plk1-dependent phosphorylation of optineurin provides a negative feedback mechanism for mitotic progression. Mol Cell (2012) 45(4):553–66. doi:10.1016/j.molcel.2011.12.030
36. Moreland RJ, Dresser ME, Rodgers JS, Roe BA, Conaway JW, Conaway RC, et al. Identification of a transcription factor IIIA-interacting protein. Nucleic Acids Res (2000) 28(9):1986–93. doi:10.1093/nar/28.9.1986
37. Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci (2013) 126(Pt 2):580–92. doi:10.1242/jcs.114926
38. Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun (2016) 7:12547. doi:10.1038/ncomms12547
39. Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, et al. Ubiquitylation of autophagy receptor optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell (2014) 26(1):106–20. doi:10.1016/j.ccr.2014.05.015
40. Nagabhushana A, Bansal M, Swarup G. Optineurin is required for CYLD-dependent inhibition of TNFalpha-induced NF-kappaB activation. PLoS One (2011) 6(3):e17477. doi:10.1371/journal.pone.0017477
41. Anborgh PH, Godin C, Pampillo M, Dhami GK, Dale LB, Cregan SP, et al. Inhibition of metabotropic glutamate receptor signaling by the huntingtin-binding protein optineurin. J Biol Chem (2005) 280(41):34840–8. doi:10.1074/jbc.M504508200
42. Ying H, Shen X, Park B, Yue BY. Posttranslational modifications, localization, and protein interactions of optineurin, the product of a glaucoma gene. PLoS One (2010) 5(2):e9168. doi:10.1371/journal.pone.0009168
43. Ahmad L, Zhang SY, Casanova JL, Sancho-Shimizu V. Human TBK1: a gatekeeper of neuroinflammation. Trends Mol Med (2016) 22(6):511–27. doi:10.1016/j.molmed.2016.04.006
44. Xu G, Lo YC, Li Q, Napolitano G, Wu X, Jiang X, et al. Crystal structure of inhibitor of kappaB kinase beta. Nature (2011) 472(7343):325–30. doi:10.1038/nature09853
45. Larabi A, Devos JM, Ng SL, Nanao MH, Round A, Maniatis T, et al. Crystal structure and mechanism of activation of TANK-binding kinase 1. Cell Rep (2013) 3(3):734–46. doi:10.1016/j.celrep.2013.01.034
46. Kishore N, Huynh QK, Mathialagan S, Hall T, Rouw S, Creely D, et al. IKK-i and TBK-1 are enzymatically distinct from the homologous enzyme IKK-2: comparative analysis of recombinant human IKK-i, TBK-1, and IKK-2. J Biol Chem (2002) 277(16):13840–7. doi:10.1074/jbc.M110474200
47. Ma X, Helgason E, Phung QT, Quan CL, Iyer RS, Lee MW, et al. Molecular basis of tank-binding kinase 1 activation by transautophosphorylation. Proc Natl Acad Sci U S A (2012) 109(24):9378–83. doi:10.1073/pnas.1121552109
48. Pourcelot M, Zemirli N, Silva Da Costa L, Loyant R, Garcin D, Vitour D, et al. The Golgi apparatus acts as a platform for TBK1 activation after viral RNA sensing. BMC Biol (2016) 14:69. doi:10.1186/s12915-016-0292-z
49. Meena NP, Zhu G, Mittelstadt PR, Giardino Torchia ML, Pourcelot M, Arnoult D, et al. The TBK1-binding domain of optineurin promotes type I interferon responses. FEBS Lett (2016) 590(10):1498–508. doi:10.1002/1873-3468.12176
50. Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci U S A (2016) 113(24):E3349–58. doi:10.1073/pnas.1523810113
51. Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell (2015) 60(1):7–20. doi:10.1016/j.molcel.2015.08.016
52. Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A (2016) 113(15):4039–44. doi:10.1073/pnas.1523926113
53. Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature (1996) 383(6599):443–6. doi:10.1038/383443a0
54. Gottipati S, Rao NL, Fung-Leung WP. IRAK1: a critical signaling mediator of innate immunity. Cell Signal (2008) 20(2):269–76. doi:10.1016/j.cellsig.2007.08.009
55. Yamin TT, Miller DK. The interleukin-1 receptor-associated kinase is degraded by proteasomes following its phosphorylation. J Biol Chem (1997) 272(34):21540–7. doi:10.1074/jbc.272.34.21540
56. Tanishima M, Takashima S, Honda A, Yasuda D, Tanikawa T, Ishii S, et al. Identification of optineurin as an interleukin-1 receptor-associated kinase 1-binding protein and its role in regulation of myd88-dependent signaling. J Biol Chem (2017) 292(42):17250–7. doi:10.1074/jbc.M117.813899
57. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell (2006) 22(2):245–57. doi:10.1016/j.molcel.2006.03.026
58. Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat Cell Biol (2006) 8(4):398–406. doi:10.1038/ncb1384
59. Montecalvo A, Watkins SC, Orange J, Kane LP. Inducible turnover of optineurin regulates T cell activation. Mol Immunol (2017) 85:9–17. doi:10.1016/j.molimm.2017.01.027
60. Sudhakar C, Nagabhushana A, Jain N, Swarup G. NF-kappaB mediates tumor necrosis factor alpha-induced expression of optineurin, a negative regulator of NF-kappaB. PLoS One (2009) 4(4):e5114. doi:10.1371/journal.pone.0005114
61. Genin P, Cuvelier F, Lambin S, Corte-Real Filipe J, Autrusseau E, Laurent C, et al. Optineurin regulates the interferon response in a cell cycle-dependent manner. PLoS Pathog (2015) 11(4):e1004877. doi:10.1371/journal.ppat.1004971
62. Schwamborn K, Weil R, Courtois G, Whiteside ST, Israel A. Phorbol esters and cytokines regulate the expression of the NEMO-related protein, a molecule involved in a NF-kappa B-independent pathway. J Biol Chem (2000) 275(30):22780–9. doi:10.1074/jbc.M001500200
63. Stroissnigg H, Repitz M, Miloloza A, Linhartova I, Beug H, Wiche G, et al. FIP-2, an IkappaB-kinase-gamma-related protein, is associated with the Golgi apparatus and translocates to the marginal band during chicken erythroblast differentiation. Exp Cell Res (2002) 278(2):133–45. doi:10.1006/excr.2002.5567
64. Buss F, Kendrick-Jones J, Lionne C, Knight AE, Cote GP, Paul Luzio J. The localization of myosin VI at the Golgi complex and leading edge of fibroblasts and its phosphorylation and recruitment into membrane ruffles of A431 cells after growth factor stimulation. J Cell Biol (1998) 143(6):1535–45. doi:10.1083/jcb.143.6.1535
65. Warner CL, Stewart A, Luzio JP, Steel KP, Libby RT, Kendrick-Jones J, et al. Loss of myosin VI reduces secretion and the size of the Golgi in fibroblasts from Snell’s waltzer mice. EMBO J (2003) 22(3):569–79. doi:10.1093/emboj/cdg055
66. Fifita JA, Williams KL, Sundaramoorthy V, McCann EP, Nicholson GA, Atkin JD, et al. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph Lateral Scler Frontotemporal Degener (2017) 18(1–2):126–33. doi:10.1080/21678421.2016.1218517
67. Park BC, Shen X, Samaraweera M, Yue BY. Studies of optineurin, a glaucoma gene: Golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am J Pathol (2006) 169(6):1976–89. doi:10.2353/ajpath.2006.060400
68. Sippl C, Zeilbeck LF, Fuchshofer R, Tamm ER. Optineurin associates with the podocyte Golgi complex to maintain its structure. Cell Tissue Res (2014) 358(2):567–83. doi:10.1007/s00441-014-1968-8
69. Paulus JD, Link BA. Loss of optineurin in vivo results in elevated cell death and alters axonal trafficking dynamics. PLoS One (2014) 9(10):e109922. doi:10.1371/journal.pone.0109922
70. Au JS, Puri C, Ihrke G, Kendrick-Jones J, Buss F. Myosin VI is required for sorting of AP-1B-dependent cargo to the basolateral domain in polarized MDCK cells. J Cell Biol (2007) 177(1):103–14. doi:10.1083/jcb.200608126
71. del Toro D, Alberch J, Lazaro-Dieguez F, Martin-Ibanez R, Xifro X, Egea G, et al. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol Biol Cell (2009) 20(5):1478–92. doi:10.1091/mbc.E08-07-0726
72. Singaraja RR, Hadano S, Metzler M, Givan S, Wellington CL, Warby S, et al. HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet (2002) 11(23):2815–28. doi:10.1093/hmg/11.23.2815
73. Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol (1998) 152(1):34–40. doi:10.1006/exnr.19986832
74. Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, et al. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet (2001) 10(17):1807–17. doi:10.1093/hmg/10.17.1807
75. Ang AL, Folsch H, Koivisto UM, Pypaert M, Mellman I. The Rab8 GTPase selectively regulates AP-1B-dependent basolateral transport in polarized Madin-Darby canine kidney cells. J Cell Biol (2003) 163(2):339–50. doi:10.1083/jcb.200307046
76. Huber LA, Pimplikar S, Parton RG, Virta H, Zerial M, Simons K. Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J Cell Biol (1993) 123(1):35–45. doi:10.1083/jcb.123.1.35
77. Vaibhava V, Nagabhushana A, Chalasani ML, Sudhakar C, Kumari A, Swarup G. Optineurin mediates a negative regulation of Rab8 by the GTPase-activating protein TBC1D17. J Cell Sci (2012) 125(Pt 21):5026–39. doi:10.1242/jcs.102327
78. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol (2015) 16(8):461–72. doi:10.1038/nrm4024
79. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature (2000) 404(6779):770–4. doi:10.1038/35008096
80. Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, et al. Global changes to the ubiquitin system in Huntington’s disease. Nature (2007) 448(7154):704–8. doi:10.1038/nature06022
81. Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem (2003) 278(14):11753–9. doi:10.1074/jbc.M208641200
82. Ji CH, Kwon YT. Crosstalk and interplay between the ubiquitin-proteasome system and autophagy. Mol Cells (2017) 40(7):441–9. doi:10.14348/molcells.2017.0115
83. Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol (2005) 171(4):603–14. doi:10.1083/jcb.200507002
84. Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell (2009) 34(3):259–69. doi:10.1016/j.molcel.2009.04.026
85. Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol (2009) 10(11):1215–21. doi:10.1038/ni.1800
86. Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem (2011) 80:125–56. doi:10.1146/annurev-biochem-052709-094552
87. Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol (2011) 27:107–32. doi:10.1146/annurev-cellbio-092910-154005
88. Suzuki K, Ohsumi Y. Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett (2007) 581(11):2156–61. doi:10.1016/j.febslet.2007.01.096
89. Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol (2016) 26(1):6–16. doi:10.1016/j.tcb.2015.08.010
90. Kirisako T, Baba M, Ishihara N, Miyazawa K, Ohsumi M, Yoshimori T, et al. Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J Cell Biol (1999) 147(2):435–46. doi:10.1083/jcb.147.2.435
91. Kirisako T, Ichimura Y, Okada H, Kabeya Y, Mizushima N, Yoshimori T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol (2000) 151(2):263–76. doi:10.1083/jcb.151.2.263
92. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell (2007) 130(1):165–78. doi:10.1016/j.cell.2007.05.021
93. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci (2004) 117(Pt 13):2805–12. doi:10.1242/jcs.01131
94. Sou YS, Tanida I, Komatsu M, Ueno T, Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. J Biol Chem (2006) 281(6):3017–24. doi:10.1074/jbc.M505888200
95. Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem (2004) 279(35):36268–76. doi:10.1074/jbc.M401461200
96. Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem (2004) 279(46):47704–10. doi:10.1074/jbc.M407016200
97. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem (2007) 282(33):24131–45. doi:10.1074/jbc.M702824200
98. Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol (2004) 24(18):8055–68. doi:10.1128/MCB.24.18.8055-8068.2004
99. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell (2007) 131(6):1149–63. doi:10.1016/j.cell.2007.10.035
100. Tschurtschenthaler M, Adolph TE, Ashcroft JW, Niederreiter L, Bharti R, Saveljeva S, et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J Exp Med (2017) 214(2):401–22. doi:10.1084/jem.20160791
101. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol (2016) 18(6):579–86. doi:10.1038/ncb3358
102. Shaid S, Brandts CH, Serve H, Dikic I. Ubiquitination and selective autophagy. Cell Death Differ (2013) 20(1):21–30. doi:10.1038/cdd.2012.72
103. Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet (2008) 17(3):431–9. doi:10.1093/hmg/ddm320
104. Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy (2006) 2(3):156–8. doi:10.4161/auto.2825
105. Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity (2012) 37(2):223–34. doi:10.1016/j.immuni.2012.04.015
106. Cemma M, Kim PK, Brumell JH. The ubiquitin-binding adaptor proteins p62/SQSTM1 and NDP52 are recruited independently to bacteria-associated microdomains to target Salmonella to the autophagy pathway. Autophagy (2011) 7(3):341–5. doi:10.4161/auto.7.3.14046
107. Puri M, La Pietra L, Mraheil MA, Lucas R, Chakraborty T, Pillich H. Listeriolysin O regulates the expression of optineurin, an autophagy adaptor that inhibits the growth of Listeria monocytogenes. Toxins (Basel) (2017) 9(9):E273. doi:10.3390/toxins9090273
108. Grundling A, Gonzalez MD, Higgins DE. Requirement of the Listeria monocytogenes broad-range phospholipase PC-PLC during infection of human epithelial cells. J Bacteriol (2003) 185(21):6295–307. doi:10.1128/JB.185.21.6295-6307.2003
109. Clark K, Plater L, Peggie M, Cohen P. Use of the pharmacological inhibitor BX795 to study the regulation and physiological roles of TBK1 and IkappaB kinase epsilon: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem (2009) 284(21):14136–46. doi:10.1074/jbc.M109.000414
110. Bakshi S, Taylor J, Strickson S, McCartney T, Cohen P. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon beta. Biochem J (2017) 474(7):1163–74. doi:10.1042/BCJ20160992
111. Tu D, Zhu Z, Zhou AY, Yun CH, Lee KE, Toms AV, et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep (2013) 3(3):747–58. doi:10.1016/j.celrep.2013.01.033
112. Tumbarello DA, Kendrick-Jones J, Buss F. Myosin VI and its cargo adaptors – linking endocytosis and autophagy. J Cell Sci (2013) 126(Pt 12):2561–70. doi:10.1242/jcs.095554
113. Berg JS, Powell BC, Cheney RE. A millennial myosin census. Mol Biol Cell (2001) 12(4):780–94. doi:10.1091/mbc.12.4.780
114. Wells AL, Lin AW, Chen LQ, Safer D, Cain SM, Hasson T, et al. Myosin VI is an actin-based motor that moves backwards. Nature (1999) 401(6752):505–8. doi:10.1038/46835
115. Lister I, Schmitz S, Walker M, Trinick J, Buss F, Veigel C, et al. A monomeric myosin VI with a large working stroke. EMBO J (2004) 23(8):1729–38. doi:10.1038/sj.emboj.7600180
116. Menetrey J, Llinas P, Mukherjea M, Sweeney HL, Houdusse A. The structural basis for the large powerstroke of myosin VI. Cell (2007) 131(2):300–8. doi:10.1016/j.cell.2007.08.027
117. Roberts R, Lister I, Schmitz S, Walker M, Veigel C, Trinick J, et al. Myosin VI: cellular functions and motor properties. Philos Trans R Soc Lond B Biol Sci (2004) 359(1452):1931–44. doi:10.1098/rstb.2004.1563
118. Geeves MA, Holmes KC. Structural mechanism of muscle contraction. Annu Rev Biochem (1999) 68:687–728. doi:10.1146/annurev.biochem.68.1.687
119. Phichith D, Travaglia M, Yang Z, Liu X, Zong AB, Safer D, et al. Cargo binding induces dimerization of myosin VI. Proc Natl Acad Sci U S A (2009) 106(41):17320–4. doi:10.1073/pnas.0909748106
120. Bunn RC, Jensen MA, Reed BC. Protein interactions with the glucose transporter binding protein GLUT1CBP that provide a link between GLUT1 and the cytoskeleton. Mol Biol Cell (1999) 10(4):819–32. doi:10.1091/mbc.10.4.819
121. Chibalina MV, Seaman MN, Miller CC, Kendrick-Jones J, Buss F. Myosin VI and its interacting protein LMTK2 regulate tubule formation and transport to the endocytic recycling compartment. J Cell Sci (2007) 120(Pt 24):4278–88. doi:10.1242/jcs.014217
122. Finan D, Hartman MA, Spudich JA. Proteomics approach to study the functions of Drosophila myosin VI through identification of multiple cargo-binding proteins. Proc Natl Acad Sci U S A (2011) 108(14):5566–71. doi:10.1073/pnas.1101415108
123. He F, Wollscheid HP, Nowicka U, Biancospino M, Valentini E, Ehlinger A, et al. Myosin VI contains a compact structural motif that binds to ubiquitin chains. Cell Rep (2016) 14(11):2683–94. doi:10.1016/j.celrep.2016.01.079
124. Inoue A, Sato O, Homma K, Ikebe M. DOC-2/DAB2 is the binding partner of myosin VI. Biochem Biophys Res Commun (2002) 292(2):300–7. doi:10.1006/bbrc.2002.6636
125. Morris SM, Arden SD, Roberts RC, Kendrick-Jones J, Cooper JA, Luzio JP, et al. Myosin VI binds to and localises with Dab2, potentially linking receptor-mediated endocytosis and the actin cytoskeleton. Traffic (2002) 3(5):331–41. doi:10.1034/j.1600-0854.2002.30503.x
126. Morriswood B, Ryzhakov G, Puri C, Arden SD, Roberts R, Dendrou C, et al. T6BP and NDP52 are myosin VI binding partners with potential roles in cytokine signalling and cell adhesion. J Cell Sci (2007) 120(Pt 15):2574–85. doi:10.1242/jcs.007005
127. Spudich G, Chibalina MV, Au JS, Arden SD, Buss F, Kendrick-Jones J. Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to disabled-2 and PtdIns(4,5)P2. Nat Cell Biol (2007) 9(2):176–83. doi:10.1038/ncb1531
128. Shen WC, Li HY, Chen GC, Chern Y, Tu PH. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy (2015) 11(4):685–700. doi:10.4161/auto.36098
129. Penengo L, Mapelli M, Murachelli AG, Confalonieri S, Magri L, Musacchio A, et al. Crystal structure of the ubiquitin binding domains of rabex-5 reveals two modes of interaction with ubiquitin. Cell (2006) 124(6):1183–95. doi:10.1016/j.cell.2006.02.020
130. Verlhac P, Gregoire IP, Azocar O, Petkova DS, Baguet J, Viret C, et al. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe (2015) 17(4):515–25. doi:10.1016/j.chom.2015.02.008
131. Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol (2008) 182(4):685–701. doi:10.1083/jcb.200803137
132. Perrotta I. The origin of the autophagosomal membrane in human atherosclerotic plaque: a preliminary ultrastructural study. Ultrastruct Pathol (2017) 41(5):327–34. doi:10.1080/01913123.2017.1349853
133. Knaevelsrud H, Soreng K, Raiborg C, Haberg K, Rasmuson F, Brech A, et al. Membrane remodeling by the PX-BAR protein SNX18 promotes autophagosome formation. J Cell Biol (2013) 202(2):331–49. doi:10.1083/jcb.201205129
134. Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol (2012) 197(5):659–75. doi:10.1083/jcb.201111079
135. Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. Diverse autophagosome membrane sources coalesce in recycling endosomes. Cell (2013) 154(6):1285–99. doi:10.1016/j.cell.2013.08.044
136. Razi M, Chan EY, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol (2009) 185(2):305–21. doi:10.1083/jcb.200810098
137. Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol (2010) 12(8):747–57. doi:10.1038/ncb2078
138. Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell (2010) 141(4):656–67. doi:10.1016/j.cell.2010.04.009
139. Geng J, Nair U, Yasumura-Yorimitsu K, Klionsky DJ. Post-Golgi Sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell (2010) 21(13):2257–69. doi:10.1091/mbc.E09-11-0969
140. van der Vaart A, Griffith J, Reggiori F. Exit from the Golgi is required for the expansion of the autophagosomal phagophore in yeast Saccharomyces cerevisiae. Mol Biol Cell (2010) 21(13):2270–84. doi:10.1091/mbc.E09-04-0345
141. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495(7441):389–93. doi:10.1038/nature11910
142. Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife (2013) 2:e00947. doi:10.7554/eLife.00947
143. Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. Elife (2014) 3:e04135. doi:10.7554/eLife.04135
144. Herman EK, Walker G, van der Giezen M, Dacks JB. Multivesicular bodies in the enigmatic amoeboflagellate Breviata anathema and the evolution of ESCRT 0. J Cell Sci (2011) 124(Pt 4):613–21. doi:10.1242/jcs.078436
145. Rusten TE, Stenmark H. How do ESCRT proteins control autophagy? J Cell Sci (2009) 122(Pt 13):2179–83. doi:10.1242/jcs.050021
146. Song GJ, Jeon H, Seo M, Jo M, Suk K. Interaction between optineurin and Rab1a regulates autophagosome formation in neuroblastoma cells. J Neurosci Res (2017) 96(3):407–415. doi:10.1002/jnr.24143
147. Bansal M, Moharir SC, Sailasree SP, Sirohi K, Sudhakar C, Sarathi DP, et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. J Biol Chem (2017) 293(1):132–147. doi:10.1074/jbc.M117.801944
148. Xing J, Weng L, Yuan B, Wang Z, Jia L, Jin R, et al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol (2016) 17(12):1373–80. doi:10.1038/ni.3580
149. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet (2005) 39:359–407. doi:10.1146/annurev.genet.39.110304.095751
150. Whitworth AJ, Pallanck LJ. PINK1/Parkin mitophagy and neurodegeneration—what do we really know in vivo? Curr Opin Genet Dev (2017) 44:47–53. doi:10.1016/j.gde.2017.01.016
151. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res (2005) 8(1):3–5. doi:10.1089/rej.2005.8.3
152. Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, et al. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol (2010) 191(7):1367–80. doi:10.1083/jcb.201007013
153. Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet (2011) 20(9):1726–37. doi:10.1093/hmg/ddr048
154. Morrison E, Thompson J, Williamson SJ, Cheetham ME, Robinson PA. A simple cell based assay to measure Parkin activity. J Neurochem (2011) 116(3):342–9. doi:10.1111/j.1471-4159.2010.07113.x
155. Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem (2011) 286(22):19630–40. doi:10.1074/jbc.M110.209338
156. Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell (2012) 22(2):320–33. doi:10.1016/j.devcel.2011.12.014
157. Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol (2010) 191(5):933–42. doi:10.1083/jcb.201008084
158. Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol (2010) 189(2):211–21. doi:10.1083/jcb.200910140
159. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8(1):e1000298. doi:10.1371/journal.pbio.1000298
160. Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet (2011) 20(5):867–79. doi:10.1093/hmg/ddq526
161. Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem (2011) 117(5):856–67. doi:10.1111/j.1471-4159.2011.07253.x
162. Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, et al. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep (2012) 13(4):378–85. doi:10.1038/embor.2012.14
163. Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy (2013) 9(11):1758–69. doi:10.4161/auto.24633
164. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183(5):795–803. doi:10.1083/jcb.200809125
165. Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol (2012) 2(5):120080. doi:10.1098/rsob.120080
166. Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, et al. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep (2012) 2:1002. doi:10.1038/srep01002
167. Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science (2013) 340(6131):471–5. doi:10.1126/science.1231031
168. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol (2014) 205(2):143–53. doi:10.1083/jcb.201402104
169. Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, et al. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J (2014) 460(1):127–39. doi:10.1042/BJ20140334
170. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature (2014) 510(7503):162–6. doi:10.1038/nature13392
171. Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, et al. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol (2015) 209(1):111–28. doi:10.1083/jcb.201410050
172. Pryde KR, Smith HL, Chau KY, Schapira AH. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol (2016) 213(2):163–71. doi:10.1083/jcb.201509003
173. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet (2010) 19(24):4861–70. doi:10.1093/hmg/ddq419
174. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12(2):119–31. doi:10.1038/ncb2012
175. Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, et al. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature (2013) 496(7445):372–6. doi:10.1038/nature12043
176. Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell (2011) 147(4):893–906. doi:10.1016/j.cell.2011.10.018
177. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep (2010) 11(1):45–51. doi:10.1038/embor.2009.256
178. Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep (2017) 7(1):1131. doi:10.1038/s41598-017-01258-6
179. Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy (2010) 6(8):1090–106. doi:10.4161/auto.6.8.13426
180. Wong YC, Holzbaur EL. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy (2015) 11(2):422–4. doi:10.1080/15548627.2015.1009792
181. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524(7565):309–14. doi:10.1038/nature14893
182. Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, et al. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A (2015) 112(21):6637–42. doi:10.1073/pnas.1506593112
183. Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, et al. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol (2008) 18(2):102–8. doi:10.1016/j.cub.2007.12.038
184. Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol (2012) 22(2):135–41. doi:10.1016/j.cub.2011.11.057
185. Cadete VJ, Deschenes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, et al. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol (2016) 594(18):5343–62. doi:10.1113/JP272703
186. McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J (2014) 33(4):282–95. doi:10.1002/embj.201385902
187. McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol (2016) 214(3):275–91. doi:10.1083/jcb.201603105
188. De Marco N, Buono M, Troise F, Diez-Roux G. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem (2006) 281(23):16147–56. doi:10.1074/jbc.M601467200
189. Chalasani ML, Radha V, Gupta V, Agarwal N, Balasubramanian D, Swarup G. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci (2007) 48(4):1607–14. doi:10.1167/iovs.06-0834
190. Chi ZL, Akahori M, Obazawa M, Minami M, Noda T, Nakaya N, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet (2010) 19(13):2606–15. doi:10.1093/hmg/ddq146
191. Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, et al. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem (2013) 126(6):699–704. doi:10.1111/jnc.12326
192. Weinreb RN, Leung CK, Crowston JG, Medeiros FA, Friedman DS, Wiggs JL, et al. Primary open-angle glaucoma. Nat Rev Dis Primers (2016) 2:16067. doi:10.1038/nrdp.2016.67
193. Danesh-Meyer HV, Levin LA. Glaucoma as a neurodegenerative disease. J Neuroophthalmol (2015) 35(Suppl 1):S22–8. doi:10.1097/WNO.0000000000000293
194. Agudo-Barriuso M, Villegas-Perez MP, de Imperial JM, Vidal-Sanz M. Anatomical and functional damage in experimental glaucoma. Curr Opin Pharmacol (2013) 13(1):5–11. doi:10.1016/j.coph.2012.09.006
195. Sirohi K, Swarup G. Defects in autophagy caused by glaucoma-associated mutations in optineurin. Exp Eye Res (2016) 144:54–63. doi:10.1016/j.exer.2015.08.020
196. Scheetz TE, Roos BR, Solivan-Timpe F, Miller K, DeLuca AP, Stone EM, et al. SQSTM1 mutations and glaucoma. PLoS One (2016) 11(6):e0156001. doi:10.1371/journal.pone.0156001
197. Li Y, Jin L, Dong A, Zhou X, Yuan H. Microarray expression profile analysis of long non-coding RNAs in optineurin E50K mutant transgenic mice. Mol Med Rep (2017) 16(2):1255–61. doi:10.3892/mmr.2017.6722
198. Meng Q, Xiao Z, Yuan H, Xue F, Zhu Y, Zhou X, et al. Transgenic mice with overexpression of mutated human optineurin(E50K) in the retina. Mol Biol Rep (2012) 39(2):1119–24. doi:10.1007/s11033-011-0840-0
199. Tseng HC, Riday TT, McKee C, Braine CE, Bomze H, Barak I, et al. Visual impairment in an optineurin mouse model of primary open-angle glaucoma. Neurobiol Aging (2015) 36(6):2201–12. doi:10.1016/j.neurobiolaging.2015.02.012
200. Sayyad Z, Sirohi K, Radha V, Swarup G. 661W is a retinal ganglion precursor-like cell line in which glaucoma-associated optineurin mutants induce cell death selectively. Sci Rep (2017) 7(1):16855. doi:10.1038/s41598-017-17241-0
201. Minegishi Y, Iejima D, Kobayashi H, Chi ZL, Kawase K, Yamamoto T, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet (2013) 22(17):3559–67. doi:10.1093/hmg/ddt210
202. Shen X, Ying H, Qiu Y, Park JS, Shyam R, Chi ZL, et al. Processing of optineurin in neuronal cells. J Biol Chem (2011) 286(5):3618–29. doi:10.1074/jbc.M110.175810
203. Sirohi K, Chalasani ML, Sudhakar C, Kumari A, Radha V, Swarup G. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy (2013) 9(4):510–27. doi:10.4161/auto.23458
204. Sirohi K, Kumari A, Radha V, Swarup GA. Glaucoma-associated variant of optineurin, M98K, activates Tbk1 to enhance autophagosome formation and retinal cell death dependent on Ser177 phosphorylation of optineurin. PLoS One (2015) 10(9):e0138289. doi:10.1371/journal.pone.0138289
205. Leung YF, Fan BJ, Lam DS, Lee WS, Tam PO, Chua JK, et al. Different optineurin mutation pattern in primary open-angle glaucoma. Invest Ophthalmol Vis Sci (2003) 44(9):3880–4. doi:10.1167/iovs.02-0693
206. Willoughby CE, Chan LL, Herd S, Billingsley G, Noordeh N, Levin AV, et al. Defining the pathogenicity of optineurin in juvenile open-angle glaucoma. Invest Ophthalmol Vis Sci (2004) 45(9):3122–30. doi:10.1167/iovs.04-0107
207. Turturro S, Shen X, Shyam R, Yue BY, Ying H. Effects of mutations and deletions in the human optineurin gene. Springerplus (2014) 3:99. doi:10.1186/2193-1801-3-99
208. Nagabhushana A, Chalasani ML, Jain N, Radha V, Rangaraj N, Balasubramanian D, et al. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. BMC Cell Biol (2010) 11:4. doi:10.1186/1471-2121-11-4
209. Matsui T, Itoh T, Fukuda M. Small GTPase Rab12 regulates constitutive degradation of transferrin receptor. Traffic (2011) 12(10):1432–43. doi:10.1111/j.1600-0854.2011.01240.x
210. Coughlin L, Morrison RS, Horner PJ, Inman DM. Mitochondrial morphology differences and mitophagy deficit in murine glaucomatous optic nerve. Invest Ophthalmol Vis Sci (2015) 56(3):1437–46. doi:10.1167/iovs.14-16126
211. Kim KY, Perkins GA, Shim MS, Bushong E, Alcasid N, Ju S, et al. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis (2015) 6:e1839. doi:10.1038/cddis.2015.180
212. Shim MS, Takihara Y, Kim KY, Iwata T, Yue BY, Inatani M, et al. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci Rep (2016) 6:33830. doi:10.1038/srep33830
213. Gao J, Ohtsubo M, Hotta Y, Minoshima S. Oligomerization of optineurin and its oxidative stress- or E50K mutation-driven covalent cross-linking: possible relationship with glaucoma pathology. PLoS One (2014) 9(7):e101206. doi:10.1371/journal.pone.0101206
214. Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron (1995) 14(6):1105–16. doi:10.1016/0896-6273(95)90259-7
215. Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci (1998) 18(9):3241–50. doi:10.1523/JNEUROSCI.18-09-03241.1998
216. Moller A, Bauer CS, Cohen RN, Webster CP, De Vos KJ. Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum Mol Genet (2017) 26(23):4668–79. doi:10.1093/hmg/ddx348
217. Rogers RS, Tungtur S, Tanaka T, Nadeau LL, Badawi Y, Wang H, et al. Impaired mitophagy plays a role in denervation of neuromuscular junctions in ALS mice. Front Neurosci (2017) 11:473. doi:10.3389/fnins.2017.00473
218. Caroppo P, Camuzat A, De Septenville A, Couratier P, Lacomblez L, Auriacombe S, et al. Semantic and nonfluent aphasic variants, secondarily associated with amyotrophic lateral sclerosis, are predominant frontotemporal lobar degeneration phenotypes in TBK1 carriers. Alzheimers Dement (Amst) (2015) 1(4):481–6. doi:10.1016/j.dadm.2015.10.002
219. Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci (2015) 18(5):631–6. doi:10.1038/nn.4000
220. Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P, et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging (2015) 36(11):3116.e5–e8. doi:10.1016/j.neurobiolaging.2015.08.009
221. Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science (2015) 347(6229):1436–41. doi:10.1126/science.aaa3650
222. Le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, Nicolas G, et al. SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol (2013) 70(11):1403–10. doi:10.1001/jamaneurol.2013.3849
223. Ayaki T, Ito H, Komure O, Kamada M, Nakamura M, Wate R, et al. Multiple proteinopathies in familial ALS cases with optineurin mutations. J Neuropathol Exp Neurol (2018) 77(2):128–38. doi:10.1093/jnen/nlx109
224. Deas E, Wood NW, Plun-Favreau H. Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta (2011) 1813(4):623–33. doi:10.1016/j.bbamcr.2010.08.007
225. Group HsDCR. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell (1993) 72(6):971–83. doi:10.1016/0092-8674(93)90585-E
226. Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat Genet (1993) 4(4):387–92. doi:10.1038/ng0893-387
227. DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron (1995) 14(5):1075–81. doi:10.1016/0896-6273(95)90346-1
228. del Toro D, Canals JM, Gines S, Kojima M, Egea G, Alberch J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci (2006) 26(49):12748–57. doi:10.1523/JNEUROSCI.3873-06.2006
229. Schwab C, Yu S, McGeer EG, McGeer PL. Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett (2012) 506(1):149–54. doi:10.1016/j.neulet.2011.10.070
230. Okita S, Morigaki R, Koizumi H, Kaji R, Nagahiro S, Goto S. Cell type-specific localization of optineurin in the striatal neurons of mice: implications for neuronal vulnerability in Huntington’s disease. Neuroscience (2012) 202:363–70. doi:10.1016/j.neuroscience.2011.11.059
231. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature (1999) 402(6762):672–6. doi:10.1038/45257
232. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell (2009) 137(6):1062–75. doi:10.1016/j.cell.2009.03.048
233. White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer (2012) 12(6):401–10. doi:10.1038/nrc3262
234. Zhang L, Anglesio MS, O’Sullivan M, Zhang F, Yang G, Sarao R, et al. The E3 ligase HACE1 is a critical chromosome 6q21 tumor suppressor involved in multiple cancers. Nat Med (2007) 13(9):1060–9. doi:10.1038/nm1621
Keywords: amyotrophic lateral sclerosis, autophagy, cell signalling, glaucoma, Golgi, membrane trafficking, mitophagy, xenophagy
Citation: Ryan TA and Tumbarello DA (2018) Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 9:1024. doi: 10.3389/fimmu.2018.01024
Received: 30 January 2018; Accepted: 24 April 2018;
Published: 15 May 2018
Edited by:
Andrew Mark Smith, University College London, United KingdomReviewed by:
Peter Monk, University of Sheffield, United KingdomJunji Xing, Houston Methodist Research Institute, United States
Copyright: © 2018 Ryan and Tumbarello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David A. Tumbarello, ZC5hLnR1bWJhcmVsbG9Ac290b24uYWMudWs=