 Anna Prossomariti
Anna Prossomariti Harry Sokol
Harry Sokol Luigi Ricciardiello
Luigi Ricciardiello- 1Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
- 2Center for Applied Biomedical Research (CRBA), S. Orsola-Malpighi Hospital, University of Bologna, Bologna, Italy
- 3Sorbonne Université, École normale supérieure, PSL Research University, CNRS, INSERM, AP-HP, Hôpital Saint-Antoine, Laboratoire de biomolécules, LBM, Paris, France
- 4INRA, UMR1319 Micalis & AgroParisTech, Jouy en Josas, France
The nucleotide-binding domain leucine-rich repeat containing (NLR) proteins play a fundamental role in innate immunity and intestinal tissue repair. A dysbiotic intestinal microbiota, developed as a consequence of alterations in NLR proteins, has recently emerged as a crucial hit for the development of ulcerative colitis (UC) and colitis-associated cancer (CAC). The concept of the existence of functional axes interconnecting bacteria with NLR proteins in a causal role in intestinal inflammation and CAC aroused a great interest for the potential development of preventive and therapeutic strategies against UC and CAC. However, the most recent scientific evidence, which highlights many confounding factors in studies based on microbiota characterization, underlines the need for an in-depth reconsideration of the data obtained until now. The purpose of this review is to discuss the recent findings concerning the cross talk between the NLR signaling and the intestinal microbiota in UC and CAC development, and to highlight the open issues that should be explored and addressed in future studies.
Introduction
Under physiological conditions, a delicate balance between immune activation and self-tolerance, resulting from a beneficial mutualistic relationship with the host gut microbial community, is responsible for maintaining a healthy intestinal homeostasis. Indeed, the ability of the host immune system to discriminate between potential harmful stimuli (i.e., pathogenic microbes, exogenous dangerous substances, and general cellular stress signals), and harmless signals (i.e., commensal gut microbiota and normal host-derived molecules), represents a sophisticated mechanism for preserving the intestinal epithelial integrity (1). The impairment of this host defense, allowing the overgrowth of pathogenic bacteria and weakening the intestinal barrier function, favors the onset of a pro-inflammatory and pro-tumorigenic microenvironment (2). To avoid the development of unfavorable pathological conditions, the host innate immune system is naturally able to recognize specific microbial conserved structures, named pathogen-associated molecular patterns (PAMPs), and to sense microbial molecules that characterize macrophages, neutrophils, monocyte, dendritic cells, and epithelial cells through a subset of pattern recognition receptors (PRRs) (3). PRRs families can be classified as transmembrane receptors, including toll-like receptors and C-type lectins receptors, responsible for recognizing extracellular and endosomal-derived PAMPs; and cytosolic receptors including retinoic acid-inducible gene-I-like receptors and nucleotide-binding domain leucine-rich repeat containing (NLRs) proteins, involved in the intracellular surveillance toward infections and recognition of self-derived damage-associated molecular patterns (DAMPs) (4). Alterations of the NLR family members have recently emerged as critically involved in the gut microbiota regulation, as well as in the genesis of colonic inflammation and colitis-associated tumorigenesis (5, 6). Here, we describe the recent knowledge on the relationship between NLR alterations and microbiota in the pathogenesis of colitis and colitis-associated cancer (CAC).
NLR Family: An Overview
The NLR family comprises more than 20 intracellular immune receptors sharing structural domains with functional specialization. The central nucleotide-binding domain (NACHT), common to all NLR family members, is responsible for the activation of signaling complexes mediating the essential self ATP-dependent oligomerization (7). Differently, the C-terminal leucine-rich repeats domains and the N-terminal pyrin domains (PYD), or Caspase recruitment domain (CARD) are implicated in the recognition of PAMPs, and in autoregulation or in homotypic protein–protein interactions, respectively. According to their N-terminal domains, NLRs can be classified in different subfamilies: (a) the NLRA subfamily consisting of CIITA, which have an N-terminal CARD domain followed by an acid transactivation domain (AD), (b) the NLRB subfamily represented by NAIP which has a baculoviral inhibitory repeat-like domain, (c) the NLRP subfamily including NLRP1–14 which have an N-terminal PYD, and (d) the NLRC/X subfamily which includes proteins having an N-terminal CARD domain, also known as NODs (NOD1/NLRC1, NOD2/NLRC2) and NLRC4 (or IPAF) or an undefined domain (NOD3/NLRC3, NOD4/NLRC5, NOD5/NLRX1) (6). NLR receptors expression in the gastrointestinal tract was often recognized in both hematopoietic and non-hematopoietic cell lineages. All the members of NLR family cooperate in the innate immunity through different mechanisms including NF-kB and MAPK induction (8), or by triggering the Caspase-1 (Casp1) activation through the generation of multimeric signaling platforms named “inflammasomes” whose final effect is the processing and the maturation of IL-1β, IL-18, and IL-33 (9, 10). NOD1 and NOD2 contribute to the cytosolic surveillance in the presence of specific peptidoglycan fragments originating during cytosolic bacterial cell divisions in infected immune cells or as a result of bacterial wall degradation in host lysozymes, and act mainly through NF-kB or MAPK signaling activation (11). To date, the knowledge of NOD3/NLRC3 and NOD4/NLRC4 functions is very poor. On the other hand, NOD5/NLRX1 represents the only member of NLR family with a mitochondrial localization and a putative role in mitochondrial antiviral immunity (12) and in the modulation of the mitochondrial reactive oxygen species production (13). As mentioned above, NLR signaling does not evolve exclusively through the activation of NF-kB and MAPK pathways but also through the formation, under appropriate stimuli, of inflammasomes. Several NLR family members seem to be able to have potential inflammasome activity, however, the best-characterized NLRs inflammasomes are NLRP1, NLRP3, NLRC4 (or IPAF), NLRP6, and NLRP12. A multiplicity of different PAMPs or host-derived DAMPs are able to stimulate and activate NLRP3 inflammasome (14). Under these conditions, the NLRP3 oligomerization recruits the ASC (PYCARD) adaptor protein through the homotypic interaction between their PYDs. Similarly, the ASC protein, acting as a molecular bridge, makes its CARD domain available for the interaction with the same domain present in pro-Casp1. This event allows the autocleavage of pro-Casp1 and the subsequent formation of mature and active Casp1 which promotes the proteolitic processing and subsequently maturation of pro-inflammatory cytokines IL-1β and IL-18 in their biologically active state and their externalization (15). An important role for NLRP3 inflammasome and its dysregulation has been defined not only in microbial host-response but also in metabolic dysfunction (16, 17), inflammatory bowel disease (IBD), and colorectal cancer (18). Differently from NLRP3, NLRP1-mediated Casp1 activation does not require the recruitment of ASC: this can occur directly through the physical interaction between a CARD domain present in a C-terminal extension of NLRP1 and a classical CARD domain in pro-Casp1, although it is known that the involvement of ASC in the complex is able to enhance this inflammasome activity (19). The precise mechanisms underlying NLRP1 activation are still partially known. Differently from other inflammasomes, the NLRC4 can induce Casp1 activity with ASC-dependent or -independent (8) mechanisms. NLRC4 is generally activated by Gram-negative bacteria and specifically induced by cytosolic flagellin (20) and components of type III or IV secretion systems (21). More recently, NLRP6, another member of NLR family, has been found to be able to generate inflammasomes (22). It has been reported that NLRP6 can induce the maturation of IL-1β and the activation of NF-kB signaling assembling ASC and pro-Casp1 proteins in a classical molecular platform in response to DAMPs (23). Finally, the function of NLRP12, also known as Monarch1 protein, is recently emerging as a crucial negative regulator of canonical and non-canonical NF-kB signaling (24, 25) and a positive regulator of dendritic and myeloid cell migration (26).
Alterations of NLR Proteins as Drivers of Intestinal Inflammation and CAC: Evidence from Pre-Clinical Models
Although it is widely accepted that a defective intestinal barrier function coupled with an inappropriate host–microbial interactions could promote colonic inflammation and CAC (2), the contribution of the NLR signaling to the intestinal homeostasis, colitis, and CAC is emerging in the latest years. In particular, a protective role of different inflammasome components on colitis and CAC has been observed. One of the first evidence supporting this hypothesis was provided by Dupaul-Chicoine and colleagues. They showed that Casp1−/− and Asc−/− mice were more susceptible to dextran sulfate sodium (DSS)-induced colitis (an acute model of epithelial injury) than wild-type (wt) mice and that exogenous administration of Il-18 rescued this phenotype, proving a fundamental role of Il-18 for tissue repair after injury (27). At the same time Allen et al. showed a critical disease outcome and increased tumor burden in the same Casp1−/− and Asc−/− mice in the DSS and azoxymethane (AOM)-DSS models of colitis and CAC (28). Interestingly, the enhanced tumor incidence encountered in Asc−/− mice was associated with a significant attenuation of Il-1β and Il-18 levels in the colon within the tumor tissue. Moreover, compared to wt mice, a significant increase in inflammatory features, tumor multiplicity, and max tumor size was found in AOM-DSS-treated Nlrp3−/− mice, but not in Nlrc4−/− mice. These results led the authors to hypothesize a negative regulator function of Nlrp3 inflammasome during colitis and CAC (28). The role of Asc, Nlrp3, and Casp1 deficiency, in the increased susceptibility to CAC was also simultaneously investigated by Zaki et al. In all three genotypes (Asc−/−, Nlrp3−/−, and Casp1−/−), the tumor burden was significantly higher compared to wt mice. The increased tumor incidence found in Nlrp3−/− and Casp1−/− mice correlated with an important local reduction of Il-18 levels and an increased colonic infiltration of macrophages. Importantly, the authors also found that the protective effect of Il-18 against CAC were associated with its ability to induce the tumor suppressors Ifn-γ and Stat3 (29). In agreement with the previously reported data, Hu et al. observed an enhanced tumor formation in Casp1 deficient mice subjected to AOM-DSS through a modulation of proliferation and apoptosis in colonic epithelial cells (30). However, differently from Allen et al., they also demonstrated that Nlrc4−/− mice showed significantly increased tumor numbers and load compared to wt mice, with tumors characterized by increased aggressiveness and invasive potential. Importantly, the authors did not observe differences in colonic inflammation severity induced by DSS (30). These results suggest a role of Nlrc4 in the protection against CAC. By contrast, a recent study by Zhou and colleagues showed that Nlrp3 inhibition by the flavonoid oroxylin A may have beneficial effects against colitis in DSS-treated mice (31).
More recent evidence also supports a protective role of Nlrc3 on CAC onset. Karki and colleagues found a significant increased number of colonic lesions in Nlrc3−/− mice treated with AOM-DSS compared to wt mice. The increased tumor number was associated with a colonic upregulation of inflammatory mediators (including Il-1β, Il-6, Tnf, G-csf, Mcp1, and Mip1α, but not Il-18) and an early hyperactivation of mTOR pathway (32). In addition, it has been shown that Nlrc3 is able to counteract CRC development by reducing c-Myc and the mTOR downstream targets FoxO3a and FoxO1 and inducing apoptosis (33). Moreover, Nlrp12 deficiency significantly increased the colonic inflammation and tumorigenesis susceptibility through an increased production of pro-inflammatory cytokines and colonic activation of NF-kB, Erk, and Stat3 (34). In particular, generating bone marrow chimeras, the authors showed that Nlrp12 in immune, rather than in epithelial cells, is critical for the protection against colitis and CAC and this effect appeared to be related to the Nlrp12-mediated suppression of canonical NF-kB and Erk in macrophages (34). Although a major role of canonical NF-kB signaling for CAC induction in Nlrp12 deficient mice was proposed by Allen et al., a subsequent study emphasized the enhancement of non-canonical NF-kB signaling and MAPK activation as drivers of colonic inflammation and tumor-prone microenvironment derived from both hematopoietic and non-hematopoietic compartments (35). To explain these discrepancies, the authors speculated on the possibility that Nlrp12 may indirectly mediate canonical NF-kB signaling through the non-canonical pathway with a mechanism dependent on Ikkα and Traf3 (36). The importance of innate immunity in the regulation of inflammation and intestinal tumorigenesis was also extended to other non-forming inflammasomes NLR components, including Nod1. Chen et al. investigated the role of Nod1 deficiency in models of colitis- and familial adenomatous polyposis-related carcinogenesis. The authors showed that Nod1 deficiency alone or in combination with mutations in the Wnt signaling component Apc, in the context of inflammation, enhances tumor formation and progression by enhancing an inflammatory response that drives cell proliferation and malignant transformation (37). These results support the importance of NLR proteins in the prevention of intestinal inflammation and carcinogenesis.
Role of NLR Proteins in Human IBD
Although functional studies aiming at clarifying the role of NLR proteins in the development of colitis and CAC have been mainly performed on pre-clinical models, observational and association studies conducted in humans partially support a protective role of some NLR proteins on IBD onset and progression. In particular, polymorphisms in genes encoding for NLR family members would confer a genetic predisposition or protection to IBD. Among NLR proteins, NOD2 (CARD15) has been investigated for a long time as susceptibility locus for Crohn’s disease (CD). Indeed, many studies led to establish that NOD2 loss-of-function variants are associated with an increased CD risk [for review see Ref. (38)]. In addition, the NOD2 missense variant R702W would enhance the CRC susceptibility in this population (39). Furthermore, NLRP3 has emerged as a predisposing gene for CD development, although, not all the studies give rise to robust results in different populations (40). Recently, further studies in humans have found low NLRP12 levels in ulcerative colitis (UC) patients (41), while NLRP6 upregulation has been observed in biopsy specimens from ileal CD patients (42). Importantly, despite studies on animal models suggested that reduced IL-18 levels, as downstream effect of NLR proteins alterations, might be critically responsible of increased susceptibility to colitis and CAC, data on humans showed opposite results. Indeed, increased IL-18 and IL-1β levels have been found in colonic tissues and cells from hematopoietic lineage from IBD patients [for review see Ref. (43)]. These observations led to hypothesize a causative role of the inflammasome hyperactivation in IBD pathogenesis and to question the translatability of the results obtained in inflammatory murine models in humans. Regarding the dual role of NLR proteins during inflammation, it has been suggested that increased IL-18 and IL-1β levels in the intestinal epithelial cells may be relevant to ensure a proper intestinal tissue regeneration and repair, while their induction in the lamina propria might trigger pro-inflammatory and deleterious responses (44).
Starting from the assumption that an aberrant activation of inflammasome may have a pivotal role in the development of IBD, therapeutic strategies aimed at repressing the activation of NLR proteins and thus reducing the levels of its functional mediators, have been proposed (45, 46). However, to date, data available on humans are limited to provide conclusive results.
Thus, to clarify the effective role of NLR system in the onset and the evolution of IBD, and to identify patients who can obtain a real benefit from therapies based on NLR proteins regulation, more studies will be necessary. For this purpose, it would be clinically relevant to stratify patients with IBD distinguishing between the type (CD vs. UC) and the duration (early- vs. long-term disease), to correlate colonic levels of inflammatory mediators with other markers of intestinal inflammation during IBD (i.e., fecal calprotectin) and to conduct more functional studies on intestinal organoids derived from IBD patients.
NLR Proteins, Microbiota, and Risk for Colitis and Inflammatory-Driven CRC
The interest on the role of NLR proteins in the regulation of intestinal microbiota and its impact on the onset of intestinal inflammation and CAC have increased significantly over the past few years. Elinav and colleagues have first described this intriguing interplay. They demonstrated that, upon DSS treatment, mice with Nlrp6, Asc, or Casp1 defect restricted to colonic epithelial cells were characterized by higher inflammation, intestinal hyperplasia, and reduced Il-18 levels. Intriguingly, this increased colitis susceptibility resulted to be transferable to co-housed wt mice, through a colitogenic microbiota characterized by a high prevalence of Prevotellaceae and TM7 bacterial taxa, particularly present in Nlrp6 deficient mice (22). Moreover, wt mice co-housed with Il-18 but not with Il-1β or I1-1R deficient mice had an increased susceptibility to DSS-induced colitis suggesting that the colonic defect in Il-18 is the major downstream event responsible of enhanced colitogenic microbiota in Nrp6−/− mice. Moreover, a recent published work demonstrated that Nlrp6 was able to prevent colitis development in Il-10−/− mice by modulating the abundance of Akkermansia muciniphila which may act as a pro-inflammatory bacterium (47). However, the role of Akkermansia in the pathogenesis of UC and CAC remains controversial. Indeed, we previously found an increase in the relative abundance of Akkermansia muciniphila in mice upon AOM-DSS treatment (48), while its depletion has been observed in long-term UC patients compared to healthy controls (49). Importantly, further studies, based on germ-free, antibiotics treated mice, as well as knockout animal models, have highlighted the importance of both commensal bacteria, microbial metabolites, and functional Nlrp6 for the activation of the inflammasome, the induction of Il-18 and the consequent production of anti-microbial peptides which is necessary to maintain a normal microbiota. Noteworthy, the administration of taurine to DSS-exposed mice, restoring an eubiotic microbiota, was associated with mucosal healing and improved survival (50). Accordingly, Asc−/−, Nlrp6−/−, and Il-18−/−, but not Nlrc4−/− and Il-1R−/− mice were found to be more prone to AOM-DSS-driven CAC (51).
Further studies have demonstrated that Nlrp1b deficiency in mice was associated with reduced colonic levels of Il-18 and Il-1β with a concomitant increased colitis susceptibility and CAC development upon DSS- and AOM-DSS treatment. Importantly, treatment with a broad-spectrum antibiotic mixture led to an amelioration of the phenotype (52).
Similarly, Aim2−/− mice showed an impaired Il-1β and Il-18 synthesis associated with an overgrowth of colonic E. coli and an increased risk of developing colitis which may be counteracted by Il-18 infusion (53). In addition, a recent work demonstrated that deficiency for Nlrp12 led to an increased colonic inflammation and caused a microbiota imbalance in mice by favoring the overgrowth of inflammatory bacteria, including members of Erysipelothricaceae family and inducing a concomitant reduction of beneficial strains such as members of Lachnospiraceae (41).
The protective effects of NLR signaling are also mediated by members of the NLR family that exert their function in immunity beyond the inflammmasome. Indeed, a Nod2-dependent protective effect of the strain Lactobacillus salivarius Ls33 has been described in a mouse model of 2,4,6-trinitrobenzene induced colitis (54). Simultaneously, a transmissible risk of colitis and CAC in Nod2-deficient mice transiently co-housed with wt mice in the context of AOM-DSS treatment has been observed in another study. Noteworthy, the Nod2 mediated dysbiosis, following chemically induced injuries, impaired colonic epithelium integrity and generated an intestinal pro-inflammatory milieu with a critical role in the enhanced epithelial dysplasia. Importantly, this transmissible colitis risk was enhanced in germ-free wt hosts recolonized with dysbiotic fecal microbiota from Nod2-deficient mice, and conversely colonizing germ-free Nod2-deficient mice with wt microbiota reduced colitis severity, suggesting a reversible primary role of dysbiotic microbiota in intestinal tumorigenesis driven by inflammation (55). To support the idea that Nod2 alterations could be directly linked with an abnormal composition of the microbiota, Ramanan et al. recently demonstrated that Nod2 deficiency was responsible for the commensal Bacteroides vulgatus expansion in the small intestine which is transmissible to the next generation and responsible for goblet cells dysfunction, overproduction of Ifn-γ by intraepithelial lymphocytes and increased inflammation (56). A substantially altered microbial community structure in the terminal ileum of Nod2-deficient mice was also observed in another work, which contextually demonstrated that Nod2 genotypes were associated with the microbial composition in humans (57). This effect in humans has also been pointed out by Knights et al., showing particularly an association between NOD2 risk allele count and increased relative abundance of Enterobacteriaceae (58). Conversely, a recent work demonstrated that the increased susceptibility to CAC observed in Nod2-deficient mice, might be due to a dysregulation of MAPK and NF-kB activation during inflammation, rather than the intestinal dysbiosis (59). Deficiency for Card9 gene, an adaptor protein, acting downstream of Nod2 on the MAPK pathway, confers an increased susceptibility to colitis and shows an altered gut microbiota profile and metabolism (60, 61).
Many studies that proposed a causal role of the inflammasome components in the induction of intestinal dysbiosis were conducted using non-littermate controls. Recent studies performed using littermate controls demonstrated that Nlrp6 deficiency does not directly affect the gut microbiota composition (62). Accordingly, previous studies aimed at evaluating the effect of Nod1 and Nod2 on microbiota using littermate controls revealed no direct effect of the genetic background on the gut microbiota composition (63, 64).
Although this is a complex issue in which both littermate status and mice separation need to be taken into account, these emerging findings suggest that non-genetic confounding factors such as maternal inheritance and the housing conditions may have critically influenced previous results obtained using non-littermate controls. Moreover, despite the fact that the pathogenic role of a dysbiotic microbiota in IBD has been widely demonstrated, only few studies investigated the hypothesis that changes in the relative abundance of specific bacterial populations could be driven by alterations of NLR proteins in humans. Indeed, to date, a causal role of NOD2 in the determination of ileal microbiota composition in CD patients has been proved [for review see Ref. (65)], while no consistent data regarding the impact of other NLR family components on microbiota structure, are currently available.
Conclusion
To date, there is growing evidence indicating that the NLR family components work as tumor suppressors in inflammation-induced tumorigenesis. Contextually, the crucial role of the intestinal microbiota during the onset and development of CAC has been proved (66–68). The important emerging concept is that alterations of NLR protein functions and a dysbiotic microbiota are not distinct phenomena during the genesis of colitis and CAC, but rather closely connected events in a causal mutual relationship (see Figure 1 for a schematic model on effects of NLR proteins deficiency on CAC development). Improper activity in the intestinal immune system due to the loss of function of specific NLR receptors during the exposure to inflammatory stimuli might support (or lead to) the overgrowth of inflammatory bacteria enhancing intestinal tumorigenesis. This observation indicates that by modulating the composition of the gut microbiota we could possibly counteract the deleterious effects of an unfavorable genetic predisposition. However, it is important to underline that this experimental evidence is mainly derived from animal models and the translation of these observations in patients with IBD or CAC will be necessary. In addition, some of these results were not confirmed in studies conducted using littermate controls. In light of this new evidence, it will be necessary to reconsider previous obtained findings reducing the impact of confounding factors. Moreover, a number of relevant questions is still open: (1) What is the impact of NLR proteins on human intestinal microbiota in patients with IBD? (2) Can we really define a “colitogenic” microbiota during defective NLR signaling? (3) Could we use microbial changes to early detect alterations in NLR protein families? and (4) Can we prevent CAC development improving the protective NLR functions during long-standing IBD?
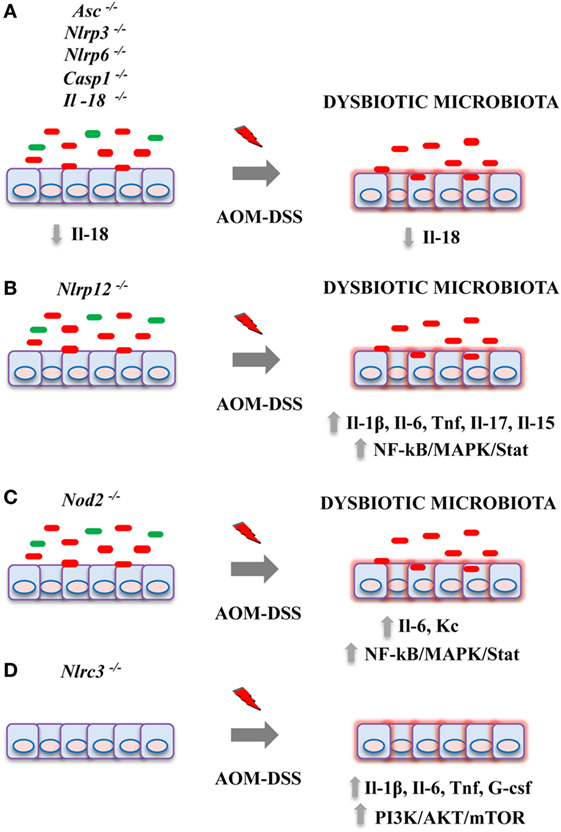
Figure 1. Effects of nucleotide-binding domain leucine-rich repeat containing (NLR) proteins deficiency on colitis-associated cancer (CAC) development in pre-clinical models. Genetic alterations in NLR proteins result in an increased tumor burden in azoxymethane (AOM)-dextran sulfate sodium (DSS) mouse model of CAC by acting on multiple mechanisms. (A) Deficiency for Asc, Nlrp3, Nlrp6, Casp1, or Il-18 genes led to a dysbiotic microbiota, reduced colonic Il-18 levels, and an impairment of intestinal tissue repair upon injury; (B) Nlrp12; and (C) Nod2 deficiency have been associated with an overgrowth of inflammatory bacteria, enhanced cytokine production, and activation of pro-tumorigenic pathways including NF-kB, MAPK, and Stat3. (D) Knockout for Nlrc3 led to increased colonic inflammation and mTOR signaling activation.
In conclusion, although the role of the microbiota in supporting a tumor-prone microenvironment in a context where the protective immunity is defective seems crucial, it is necessary to best characterize the microbial species able to exacerbate intestinal inflammation and cancer when specific NLR family components were defective. Moreover, the validation of the functional effects of microbiota changes discovered through high-throughput analysis may clarify its role in the diseases allowing the development of promising approaches for the prevention and treatment of colitis and CAC.
Author Contributions
AP and LR retrieved and analyzed concerned literatures and wrote the manuscript. HS revised the manuscript for important intellectual content. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
LR has received an unrestricted research grant by SLA Pharma and Takeda. No potential conflict of interest was disclosed by the other authors.
Funding
This work was supported by Associazione Italiana perla Ricerca sul Cancro (Award number: IG14281 to LR).
Abbreviations
AOM, azoxymethane; CAC, colitis-associated cancer; Casp1, Caspase1; CLRs, C-type lectins receptors; CD, Crohn’s disease; DAMPs, damage-associated molecular patterns; DSS, dextran sulfate sodium; IBD, inflammatory bowel disease; NLR, nucleotide-binding domain leucine-rich repeat containing; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; RLRs, retinoic acid-inducible gene (RIG)-I-like receptors; TLRs, toll-like receptors; UC, ulcerative colitis; wt, wild-type.
References
1. Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science (2001) 292:1115–8. doi:10.1126/science.1058709
2. Waldner MJ, Neurath MF. Mechanisms of immune signaling in colitis-associated cancer. Cell Mol Gastroenterol Hepatol (2015) 1:6–16. doi:10.1016/j.jcmgh.2014.11.006
3. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell (2010) 140:805–20. doi:10.1016/j.cell.2010.01.022
4. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi:10.1128/CMR.00046-08
5. Biswas A, Kobayashi KS. Regulation of intestinal microbiota by the NLR protein family. Int Immunol (2013) 25:207–14. doi:10.1093/intimm/dxs116
6. Stutz A, Golenbock DT, Latz E. Inflammasomes: too big to miss. J Clin Invest (2009) 119:3502–11. doi:10.1172/JCI40599
7. Koonin EV, Aravind L. The NACHT family – a new group of predicted NTPases implicated in apoptosis and MHC transcription activation. Trends Biochem Sci (2000) 25:223–4. doi:10.1016/S0968-0004(00)01577-2
8. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature (2004) 430:213–8. doi:10.1038/nature02664
9. Elinav E, Strowig T, Henao-Mejia J, Flavell RA. Regulation of the antimicrobial response by NLR proteins. Immunity (2011) 34:665–79. doi:10.1016/j.immuni.2011.05.007
10. Martinon F, Burns K, Tschopp J. The inflammasome. Mol Cell (2002) 10:417–26. doi:10.1016/S1097-2765(02)00599-3
11. Park J-H, Kim Y-G, McDonald C, Kanneganti T-D, Hasegawa M, Body-Malapel M, et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol (2007) 178:2380–6. doi:10.4049/jimmunol.178.4.2380
12. Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature (2008) 451:573–7. doi:10.1038/nature06501
13. Tattoli I, Carneiro LA, Jéhanno M, Magalhaes JG, Shu Y, Philpott DJ, et al. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep (2008) 9:293–300. doi:10.1038/sj.embor.7401161
14. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
15. Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe (2008) 4:198–208. doi:10.1016/j.chom.2008.08.007
16. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol (2010) 10:210–5. doi:10.1038/nri2725
17. Dixit VD. Nlrp3 inflammasome activation in type 2 diabetes: is it clinically relevant? Diabetes (2013) 62:22–4. doi:10.2337/db12-1115
18. Zaki MH, Lamkanfi M, Kanneganti T-D. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol (2011) 32:171–9. doi:10.1016/j.it.2011.02.002
19. Faustin B, Lartigue L, Bruey J-M, Luciano F, Sergienko E, Bailly-Maitre B, et al. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell (2007) 25:713–24. doi:10.1016/j.molcel.2007.01.032
20. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat Immunol (2006) 7:569–75. doi:10.1038/ni1344
21. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci U S A (2010) 107:3076–80. doi:10.1073/pnas.0913087107
22. Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell (2011) 145:745–57. doi:10.1016/j.cell.2011.04.022
23. Grenier JM, Wang L, Manji GA, Huang WJ, Al-Garawi A, Kelly R, et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett (2002) 530:73–8. doi:10.1016/S0014-5793(02)03416-6
24. Lich JD, Williams KL, Moore CB, Arthur JC, Davis BK, Taxman DJ, et al. Cutting edge: monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J Immunol (2007) 178:1256–60. doi:10.4049/jimmunol.178.3.1256
25. Williams KL, Lich JD, Duncan JA, Reed W, Rallabhandi P, Moore C, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor alpha-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem (2005) 280:39914–24. doi:10.1074/jbc.M502820200
26. Arthur JC, Lich JD, Ye Z, Allen IC, Gris D, Wilson JE, et al. Cutting edge: NLRP12 controls dendritic and myeloid cell migration to affect contact hypersensitivity. J Immunol (2010) 185:4515–9. doi:10.4049/jimmunol.1002227
27. Dupaul-Chicoine J, Yeretssian G, Doiron K, Bergstrom KSB, McIntire CR, LeBlanc PM, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity (2010) 32:367–78. doi:10.1016/j.immuni.2010.02.012
28. Allen IC, TeKippe EM, Woodford R-MT, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207:1045–56. doi:10.1084/jem.20100050
29. Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M, Kanneganti T-D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol (2010) 185:4912–20. doi:10.4049/jimmunol.1002046
30. Hu B, Elinav E, Huber S, Booth CJ, Strowig T, Jin C, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A (2010) 107:21635–40. doi:10.1073/pnas.1016814108
31. Zhou W, Liu X, Zhang X, Tang J, Li Z, Wang Q, et al. Oroxylin A inhibits colitis by inactivating NLRP3 inflammasome. Oncotarget (2017) 8:58903–17. doi:10.18632/oncotarget.19440
32. Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, et al. NLRC3 is an inhibitory sensor of PI3K–mTOR pathways in cancer. Nature (2016) 540:583–7. doi:10.1038/nature20597
33. Karki R, Malireddi RKS, Zhu Q, Kanneganti T-D. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle (2017) 16:1243–51. doi:10.1080/15384101.2017.1317414
34. Zaki MH, Vogel P, Malireddi RKS, Body-Malapel M, Anand PK, Bertin J, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell (2011) 20:649–60. doi:10.1016/j.ccr.2011.10.022
35. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity (2012) 36:742–54. doi:10.1016/j.immuni.2012.03.012
36. Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-kappaB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A (2008) 105:3503–8. doi:10.1073/pnas.0707959105
37. Chen GY, Shaw MH, Redondo G, Núñez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res (2008) 68:10060–7. doi:10.1158/0008-5472.CAN-08-2061
38. Caruso R, Warner N, Inohara N, Núñez G. NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity (2014) 41:898–908. doi:10.1016/j.immuni.2014.12.010
39. Roberts RL, Gearry RB, Allington MDE, Morrin HR, Robinson BA, Frizelle FA. Caspase recruitment domain-containing protein 15 mutations in patients with colorectal cancer. Cancer Res (2006) 66:2532–5. doi:10.1158/0008-5472.CAN-05-4165
40. Davis BK, Philipson C, Hontecillas R, Eden K, Bassaganya-Riera J, Allen IC. Emerging significance of NLRs in inflammatory bowel disease. Inflamm Bowel Dis (2014) 20:2412–32. doi:10.1097/MIB.0000000000000151
41. Chen L, Wilson JE, Koenigsknecht MJ, Chou W-C, Montgomery SA, Truax AD, et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol (2017) 18:541–51. doi:10.1038/ni.3690
42. Ranson N, Veldhuis M, Mitchell B, Fanning S, Cook AL, Kunde D, et al. Nod-like receptor pyrin-containing protein 6 (NLRP6) is up-regulated in ileal Crohn’s disease and differentially expressed in goblet cells. Cell Mol Gastroenterol Hepatol (2018). doi:10.1016/j.jcmgh.2018.03.001
43. Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol (2010) 28:573–621. doi:10.1146/annurev-immunol-030409-101225
44. Siegmund B. Interleukin-18 in intestinal inflammation: friend and foe? Immunity (2010) 32:300–2. doi:10.1016/j.immuni.2010.03.010
45. Perera AP, Kunde D, Eri R. NLRP3 inhibitors as potential therapeutic agents for treatment of inflammatory bowel disease. Curr Pharm Des (2017) 23:2321–7. doi:10.2174/1381612823666170201162414
46. Kanai T, Kamada N, Hisamatsu T. Clinical strategies for the blockade of IL-18 in inflammatory bowel diseases. Curr Drug Targets (2013) 14:1392–9. doi:10.2174/13894501113149990006
47. Seregin SS, Golovchenko N, Schaf B, Chen J, Pudlo NA, Mitchell J, et al. NLRP6 protects Il10−/− mice from colitis by limiting colonization of Akkermansia muciniphila. Cell Rep (2017) 19:733–45. doi:10.1016/j.celrep.2017.03.080
48. Piazzi G, D’Argenio G, Prossomariti A, Lembo V, Mazzone G, Candela M, et al. Eicosapentaenoic acid free fatty acid prevents and suppresses colonic neoplasia in colitis-associated colorectal cancer acting on Notch signaling and gut microbiota. Int J Cancer (2014) 135:2004–13. doi:10.1002/ijc.28853
49. Prossomariti A, Scaioli E, Piazzi G, Fazio C, Bellanova M, Biagi E, et al. Short-term treatment with eicosapentaenoic acid improves inflammation and affects colonic differentiation markers and microbiota in patients with ulcerative colitis. Sci Rep (2017) 7:7458. doi:10.1038/s41598-017-07992-1
50. Levy M, Thaiss CA, Zeevi D, Dohnalová L, Zilberman-Schapira G, Mahdi JA, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell (2015) 163:1428–43. doi:10.1016/j.cell.2015.10.048
51. Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A (2013) 110:9862–7. doi:10.1073/pnas.1307575110
52. Williams TM, Leeth RA, Rothschild DE, Coutermarsh-Ott SL, McDaniel DK, Simmons AE, et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol (2015) 194:3369–80. doi:10.4049/jimmunol.1402098
53. Hu S, Peng L, Kwak Y-T, Tekippe EM, Pasare C, Malter JS, et al. The DNA sensor AIM2 maintains intestinal homeostasis via regulation of epithelial antimicrobial host defense. Cell Rep (2015) 13:1922–36. doi:10.1016/j.celrep.2015.10.040
54. Macho Fernandez E, Valenti V, Rockel C, Hermann C, Pot B, Boneca IG, et al. Anti-inflammatory capacity of selected lactobacilli in experimental colitis is driven by NOD2-mediated recognition of a specific peptidoglycan-derived muropeptide. Gut (2011) 60:1050–9. doi:10.1136/gut.2010.232918
55. Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest (2013) 123:700–11. doi:10.1172/JCI62236
56. Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K. Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity (2014) 41:311–24. doi:10.1016/j.immuni.2014.06.015
57. Rehman A, Sina C, Gavrilova O, Häsler R, Ott S, Baines JF, et al. Nod2 is essential for temporal development of intestinal microbial communities. Gut (2011) 60:1354–62. doi:10.1136/gut.2010.216259
58. Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med (2014) 6:107. doi:10.1186/s13073-014-0107-1
59. Udden SMN, Peng L, Gan J-L, Shelton JM, Malter JS, Hooper LV, et al. NOD2 suppresses colorectal tumorigenesis via downregulation of the TLR pathways. Cell Rep (2017) 19:2756–70. doi:10.1016/j.celrep.2017.05.084
60. Sokol H, Conway KL, Zhang M, Choi M, Morin B, Cao Z, et al. Card9 mediates intestinal epithelial cell restitution, T-helper 17 responses, and control of bacterial infection in mice. Gastroenterology (2013) 145:591–601.e3. doi:10.1053/j.gastro.2013.05.047
61. Lamas B, Richard ML, Leducq V, Pham H-P, Michel M-L, Da Costa G, et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med (2016) 22:598–605. doi:10.1038/nm.4102
62. Mamantopoulos M, Ronchi F, Van Hauwermeiren F, Vieira-Silva S, Yilmaz B, Martens L, et al. Nlrp6- and ASC-dependent inflammasomes do not shape the commensal gut microbiota composition. Immunity (2017) 47:339–48.e4. doi:10.1016/j.immuni.2017.07.011
63. Robertson SJ, Zhou JY, Geddes K, Rubino SJ, Cho JH, Girardin SE, et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes (2013) 4:222–31. doi:10.4161/gmic.24373
64. Robertson SJ, Geddes K, Maisonneuve C, Streutker CJ, Philpott DJ. Resilience of the intestinal microbiota following pathogenic bacterial infection is independent of innate immunity mediated by NOD1 or NOD2. Microbes Infect (2016) 18:460–71. doi:10.1016/j.micinf.2016.03.014
65. Sidiq T, Yoshihama S, Downs I, Kobayashi KS. Nod2: a critical regulator of ileal microbiota and Crohn’s disease. Front Immunol (2016) 7:367. doi:10.3389/fimmu.2016.00367
66. Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One (2009) 4:e6026. doi:10.1371/journal.pone.0006026
67. Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science (2012) 338:120–3. doi:10.1126/science.1224820
Keywords: nucleotide-binding domain leucine-rich repeat containing signaling, microbiota, intestinal inflammation, ulcerative colitis, colitis-associated cancer
Citation: Prossomariti A, Sokol H and Ricciardiello L (2018) Nucleotide-Binding Domain Leucine-Rich Repeat Containing Proteins and Intestinal Microbiota: Pivotal Players in Colitis and Colitis-Associated Cancer Development. Front. Immunol. 9:1039. doi: 10.3389/fimmu.2018.01039
Received: 26 February 2018; Accepted: 26 April 2018;
Published: 14 May 2018
Edited by:
Alessandra Mortellaro, Istituto San Raffaele Telethon per la Terapia Genica (SR-Tiget), ItalyReviewed by:
Federica Laudisi, Università degli Studi di Roma Tor Vergata, ItalyRajendra Karki, St. Jude Children’s Research Hospital, United States
Copyright: © 2018 Prossomariti, Sokol and Ricciardiello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anna Prossomariti, YW5uYS5wcm9zc29tYXJpdGlAZ21haWwuY29t;
Luigi Ricciardiello, bHVpZ2kucmljY2lhcmRpZWxsb0B1bmliby5pdA==