 Takayuki Katsuyama
Takayuki Katsuyama George C. Tsokos
George C. Tsokos Vaishali R. Moulton
Vaishali R. Moulton- Division of Rheumatology, Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Systemic lupus erythematosus (SLE) is a chronic multi-organ debilitating autoimmune disease, which mainly afflicts women in the reproductive years. A complex interaction of genetics, environmental factors and hormones result in the breakdown of immune tolerance to “self” leading to damage and destruction of multiple organs, such as the skin, joints, kidneys, heart and brain. Both innate and adaptive immune systems are critically involved in the misguided immune response against self-antigens. Dendritic cells, neutrophils, and innate lymphoid cells are important in initiating antigen presentation and propagating inflammation at lymphoid and peripheral tissue sites. Autoantibodies produced by B lymphocytes and immune complex deposition in vital organs contribute to tissue damage. T lymphocytes are increasingly being recognized as key contributors to disease pathogenesis. CD4 T follicular helper cells enable autoantibody production, inflammatory Th17 subsets promote inflammation, while defects in regulatory T cells lead to unchecked immune responses. A better understanding of the molecular defects including signaling events and gene regulation underlying the dysfunctional T cells in SLE is necessary to pave the path for better management, therapy, and perhaps prevention of this complex disease. In this review, we focus on the aberrations in T cell signaling in SLE and highlight therapeutic advances in this field.
Introduction
Systemic lupus erythematosus (SLE) is a complex systemic autoimmune disease involving multiple organs leading to tissue damage and diverse clinical manifestations. Although the etiology of SLE is still unclear, a number of recent studies have advanced our understanding of disease pathogenesis. Clinical heterogeneity of SLE suggests that there are number of players in the immune system that contribute to the pathogenesis of SLE. B cells obviously are important in autoimmune diseases through the production of antibodies by plasma cells and presenting antigens to T cells. However, there is an increasing recognition and validation of the critical role of T cells in SLE pathogenesis (1–5). Historically, the T helper (Th)1/Th2 balance was considered to be important in the pathogenesis of SLE (6, 7). However, recent understanding of the detailed mechanisms of T cell differentiation and subsets have elucidated the more important and complicated role of T cells in the pathogenesis of this autoimmune disease. Many studies have shown abnormal cytokine production and aberrant cell signaling in SLE T cells, which dictate not only the abnormalities in T cell differentiation but also the excessive activation of B cells. It is expected that these abnormal signaling molecules can serve as therapeutic targets for the treatment of patients with SLE. In this review, we focus on signaling molecules and pathways in T cells from SLE patients and lupus-prone mice, and highlight those that can be exploited therapeutically (Figure 1).
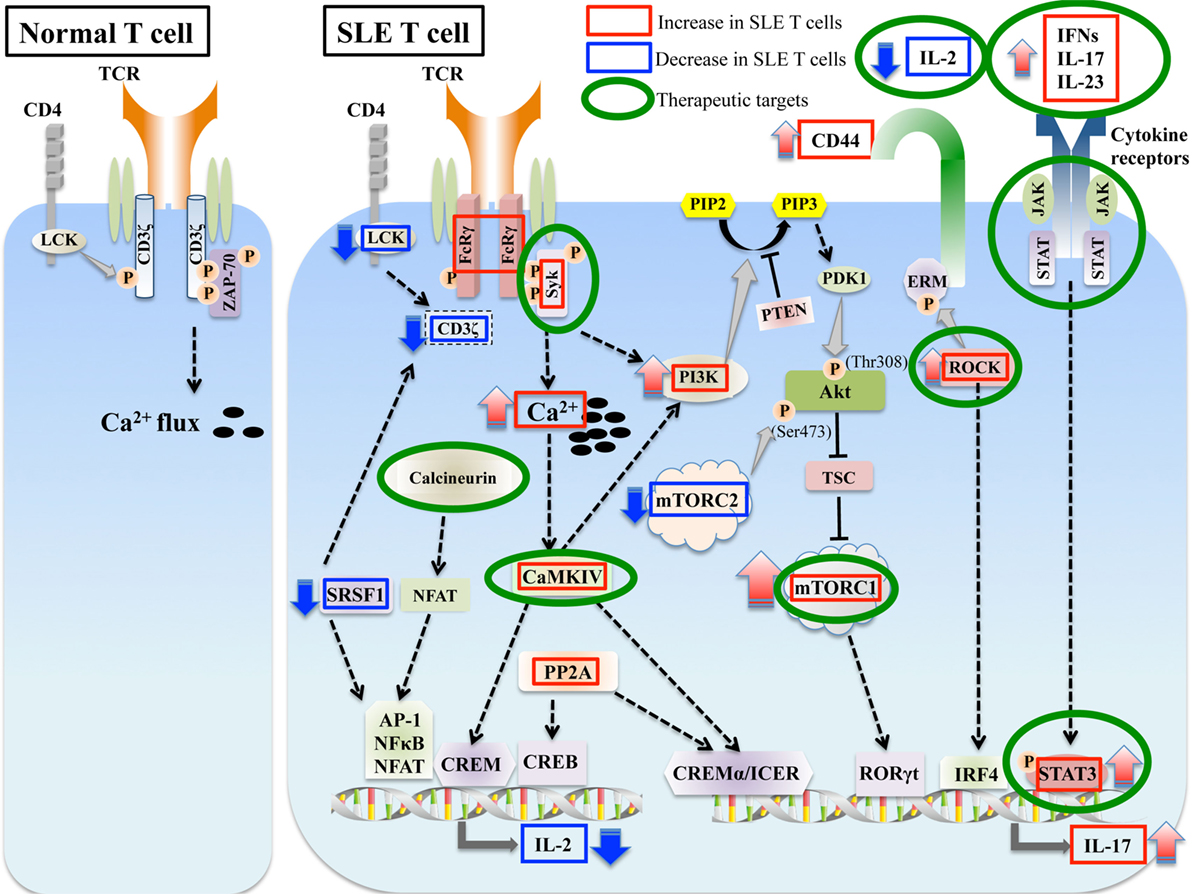
Figure 1. Aberrant signaling in systemic lupus erythematosus (SLE) T cells. SLE T cells are characterized by multiple aberrant signaling pathways, such as decreased CD3ζ, activated PI3K-Akt-mTORC1 pathway, Rho associated protein kinase (ROCK), calcium/calmodulin kinase IV (CaMKIV), and protein phosphatase 2A (PP2A). These are associated with abnormalities in T cell differentiation and production of proinflammatory cytokines such as IL-17 and decreased production of vital cytokines such as IL-2. Molecules aberrantly increased or decreased in SLE are indicated in red and blue boxes, respectively, and molecules that are potential therapeutic targets are in green circles.
T Cell Receptor (TCR)
The TCR is a heterodimer, consisting of the TCRα and TCRβ chains in most cells, which recognizes antigenic peptides presented by the major histocompatibility complex (MHC) on antigen presenting cells. The TCR is assembled with a complex of CD3 proteins (CD3δ, ε, γ, and ζ). CD3δ, ε, and γ are members of the immunoglobulin superfamily and genetically related to each other, whereas CD3ζ subunit is genetically and structurally unrelated to the other CD3 subunits (8–10). CD3ζ contains three immunoreceptor tyrosine-based activation motif (ITAM) domains, and the phosphorylation of ITAM residues is a key step in the complex process of TCR signaling. Following TCR recognition and engagement of the MHC—antigen complex, the Src kinase lymphocyte-specific protein tyrosine kinase (Lck) phosphorylates ITAMs of CD3ζ. Phosphorylated CD3 ITAMs recruit the spleen tyrosine kinase (Syk) family kinase ζ-associated protein kinase 70 (ZAP-70) via Src-homology 2 domain, and Lck phosphorylates the bound ZAP-70, resulting in the activation of ZAP-70 (11). Activated ZAP-70 phosphorylates tyrosine residues on the adaptor proteins linker for activation of T cells (LAT) and SLP-76, which bind and activate phospholipase Cγ (PLC-γ). Activated PLC-γ hydrolyzes phosphatidylinositol-4,5-bisphosphate (PIP2) to produce inositol 1,4,5-trisphosphate and diacylglycerol, resulting in the calcium flux and the activation of protein kinase C (PKC) and Ras-mitogen-activated protein kinase pathway through the recruitment of Ras guanine releasing protein 1 (12, 13).
The expression levels of CD3ζ chain are significantly decreased in T cells from SLE patients (14–16), and this defect coupled with a rewiring of the TCR complex, contributes to the aberrant signaling phenotype of SLE T cells. In association with the reduced levels of CD3ζ protein in SLE T cells, the TCR–CD3 complex bears a substitution by the homologous Fc receptor common gamma subunit chain (FcRγ), which is not normally expressed in resting T cells. Although FcRγ was identified as a component of the high affinity IgE receptor (FcεRI), it is now recognized as a common subunit of other Fc receptors (17, 18). FcRγ is upregulated upon activation in effector T cells (19–22). CD3ζ and FcRγ are structurally and functionally homologous (23). FcRγ recruits Syk instead of ZAP-70, which is normally recruited by CD3ζ. FcRγ–Syk interaction is significantly stronger than CD3ζ–ZAP-70 interaction, resulting in the higher calcium influx into T cells (14, 21). Reconstitution of CD3ζ in SLE T cells restores the aberrant signaling and calcium flux (24). Interestingly, CD3ζ-deficient mice spontaneously develop multi-organ tissue inflammation (25). Therefore, the reduced expression levels of CD3ζ are important in the aberrant T cell signaling phenotype, and understanding the mechanisms leading to its downregulation would help target those factors to correct the T cell signaling defect. A number of mechanisms for the downregulation of CD3ζ mRNA and protein in T cells from SLE patients have been elucidated. In addition to abnormalities in transcription (14, 26), aberrant alternative splicing (27–29) and stability (30, 31) of CD3ζ mRNA contribute to the decreased expression levels of CD3ζ protein in T cells from SLE patients. Serine/arginine-rich splicing factor 1 (SRSF1), also known as splicing factor 2/alternative splicing factor controls the alternative splicing (32) and contributes to the transcriptional activation (33) of CD3ζ, to promote normal expression of CD3ζ protein. Decreased SRSF1 expression in T cells from SLE patients correlates with worse SLE disease activity (34), and with reduced CD3ζ levels. Recently, it was reported that hypermethylation marks are present within the CD3ζ gene promoter in SLE patients (35). These findings suggest that CD3ζ hypermethylation may contribute to the downregulation of CD3ζ in T cells from SLE patients.
The serine/threonine protein phosphatase 2A (PP2A) is a ubiquitous serine-threonine phosphatase and composed of three distinct subunits; the scaffold A subunit (PP2AA), the regulatory B subunit (PP2AB), and the catalytic C subunit (PP2AC) (36). PP2A controls the expression of CD3ζ and FcRγ at the transcription level through the dephosphorylation of Elf-1 (37). In T cells from SLE patients, increased PP2Ac activity results in aberrant TCR signaling leading to abnormal T cell function.
Proximal TCR Signaling
TCR-CD3 engagement with antigens induces the phosphorylation of ITAM residues by Lck, a member of the Src kinase family. The expression levels of Lck are decreased in T cells from SLE patients (38–41). A potential mechanism for the reduced Lck expression is its degradation due to increased ubiquitination. Lipid rafts, microdomains in the plasma membrane enriched in cholesterol, sphingomyelin, and glycosphingolipids, play important role in TCR signaling (42, 43). Lck localizes to lipid rafts, and accumulation of lipid rafts induces the increased phosphorylation and signal transduction (44, 45). Freshly isolated SLE T cells express higher levels of ganglioside M1 and cholesterol, a component of raft domain, and aggregated lipid rafts (46–48). Atorvastatin, which reduces cholesterol synthesis, restores Lck expression and lipid raft-associated aberrant signaling in vitro in T cells from patients with SLE (49). Atorvastatin also reduces the production of IL-10 and IL-6 by activated T cells (49).
Phosphorylation of ITAM residues of the TCR-CD3 complex molecules following antigen recognition by the TCR leads to the recruitment and activation of downstream signaling molecules such as adaptor proteins and enzymes. As described above, phosphorylated ITAMs of CD3ζ serve as a recruitment site for tyrosine kinase ZAP-70, a member of the Syk kinase family (50). It is unclear whether ZAP-70 expression levels T cells from SLE patients are comparable to those in T cells from healthy individuals (51) or decreased (52).
In addition to its role in T cell signaling, Syk is also an important molecule downstream of the B cell receptor. Expression levels of Syk and phospho (p)-Syk in B cells from active SLE patients are increased compared with controls (53). Therefore, Syk inhibitors are promising therapeutics. Fostamatinib, also known as R788 is a small molecule pro-drug of the biologically active R406 (54, 55), which selectively inhibits Syk. Inhibition of Syk by fostamatinib prevents disease development including skin and renal involvement in MRL/lpr and BAK/BAX lupus-prone mice, and the discontinuation of the treatment results in extended suppression of renal disease for at least 4 weeks (56). The administration of fostamatinib after the development of disease also improves kidney damage in New Zealand black/white (NZB/NZW) lupus-prone mice (57). Further studies are required to assess the efficacy of Syk inhibitors in patients with SLE.
Enhanced early T cell signaling events and heightened calcium responses lead to increased activation of calcineurin. Calcineurin dephosphorylate inactive cytoplasmic nuclear factor of activated T cells (NFAT) and dephosphorylated NFAT translocates to the nucleus. Increased recruitment of NFATc2 is observed in the nuclei of activated T cells from SLE patients after CD3 stimulation compared with those from controls, and it binds and activates the promoters of CD154 (CD40L) and IL2 genes (58). CD40-CD40L signaling is also important for the differentiation of Th17 cells (59). Expression of NFATc1 is elevated in lupus-prone MRL/lpr mice (60). Dipyridamole, an inhibitor of the calcineurin-NFAT pathway, reduces CD154 expression and improves nephritis in MRL/lpr mice (60). Calcineurin inhibitors cyclosporine and tacrolimus are widely used for the treatment of SLE. They are known to be effective in the treatment of lupus nephritis as both remission induction and maintenance therapy (61).
CD44-ROCK-ERM Axis
CD44 is a cell surface glycoprotein involved in T cell activation, adhesion, and migration (62). Recent genome wide association studies (GWAS) have identified CD44 as a gene associated with SLE on meta-analysis of two SLE GWAS datasets by OASIS, a novel linkage disequilibrium clustering method (63). It was also reported that the expression levels of CD44 are increased in T cells from SLE patients (48, 64). The CD44 gene includes 10 variable (v) exons and there are numerous splice variants of CD44. CD44v3 and CD44v6 are expressed on T cells following activation (65, 66). The expression levels of CD44v3 and CD44v6 are increased and correlate with disease activity in patients with SLE (64).
The ezrin/radixin/moesin (ERM) proteins are important in linking plasma membrane proteins with actin filaments, and the interaction between ERM proteins and the intracellular domain of CD44 is associated with cell adhesion and migration function (67). T cells predominantly express ezrin and moesin (68). Moesin-deficient mice, which exhibit significantly lower levels of pERM (69), develop systemic autoimmune phenotype including glomerulonephritis (70), and exhibit reduced CD8+CD44+CD122+Ly49+ regulatory T (Treg) cells and defects in the signal transducer and activator of transcription (STAT) 5 activation by IL-15, which is known to regulate the development of CD8 Treg cells. The levels of ERM phosphorylation are increased in SLE T lymphocytes, and forced expression of constitutively active ezrin enhances the adhesion and migration in normal T cells, suggesting that phosphorylated ERM is responsible for increased adhesion and migration of SLE T cells (48).
Rho associated protein kinase (ROCK) is a serine/threonine kinase that phosphorylates ERM. The ROCKs play important roles in migration, activation, and differentiation of T cells (71). ROCKs are a family of two serine-threonine kinases, ROCK1 and ROCK2, which exhibit a high degree of identity in their kinase domains (72). ROCKs regulate the activity of cytoskeletal components including ERM and cell migration. ROCK activity is important for chemokine-mediated polarization and transendothelial migration of T cells (73). Recently, it was reported that ROCK also regulates the interstitial T cell migration (74). In addition to its role in T cell migration, ROCK2 plays an important role in the differentiation of Th17 cells by activation of interferon regulatory factor 4 (IRF4) and controls the production of IL-17 and IL-21 (75, 76). ROCK2 signaling is also required for the induction of T follicular helper cells (Tfh cells) (77). Peripheral blood mononuclear cells (PBMC) from patients with SLE express significantly higher levels of ROCK activity as compared with healthy controls (78, 79).
In accordance with these results, ROCK inhibitors are candidates to be used for the treatment of patients with SLE. KD025 is a selective ROCK2 inhibitor (80), whereas Y-27632 (81), and Simvastatin are broad non-isoform selective ROCK inhibitors (82). Oral administration of KD025 to healthy subjects in a randomized phase I clinical trial, decreased the production of IL-17 and IL-21 from human T cells (76). KD025 also reduced the number of Tfh cells and autoantibody production in MRL/lpr mice (77). Y-27632 decreased serum levels of IL-6, IL-1β, and TNF-α and increased serum levels of IL-10 and Treg cell proportions in spleen cells from MRL/lpr mice, whereas the improvement of clinical manifestations was not shown in the paper (83). Rozo et al. demonstrated that each Y-27632, KD025 or simvastatin inhibits the increased ROCK activity in Th17 cells from SLE patients. These agents also decreased the production of IL-17 and IL-21 from SLE T cells or Th17 cells (79).
Fasudil, a pan ROCK inhibitor, has been approved for clinical use in Japan and China for the improvement of cerebral vasospasm after surgery for subarachnoid hemorrhage (71, 84). Fasudil decreases the production of IL-17 and IL-21 and improve disease including production of autoantibody and proteinuria in MRL/lpr mice (75), and NZB/W F1 mice (85). These results indicate that ROCK signaling is a promising therapeutic target for patients with SLE.
Phosphoinositide-3 Kinases (PI3Ks) and Phosphatase and Tensin Homolog Deleted on Chromosome 10 (PTEN)
Class I PI3Ks, family members of lipid kinases, are classified as class IA and IB by activation mode. Class IA PI3Ks are activated by receptor tyrosine kinases including the TCR and costimulators, whereas Class IB PI3Ks are activated by G protein-coupled receptors such as chemokine receptors (86–88). Class I PI3Ks are composed of catalytic subunits p110 and regulatory subunits p85 or p87. There are three catalytic isoforms of Class IA PI3Ks (p110α, p110β, and p110δ), whereas only p110γ is a PI3K Class IB catalytic subunit. Compared with the ubiquitous expression of p110α and p110β, p110δ and p110γ are selectively expressed in lymphocytes (89). Class I PI3Ks phosphorylate PIP2 to form phosphatidylinositol-3,4,5-triphosphate (PIP3). Both Class IA and IB PI3Ks are expressed in leukocytes and play important roles in homeostasis, differentiation and function of T cells (88, 90, 91). PIP3 recruits phosphoinositide-dependent kinase 1 and activates Akt.
Phosphoinositide-3 kinase plays an important role in T cell differentiation (92). Transgenic mice expressing an active form of PI3K in T cells, p65PI3K Tg mice, develop lupus-like autoimmune phenotypes including kidney disease (93). Cleaved CD95 (Fas) ligand (CD95L/FasL) is increased in serum from patients with SLE and promotes cell migration through a c-yes/Ca2+/PI3K signal (94). Class I PI3K signaling is activated in lymphocytes of MRL/lpr mice, and treatment with AS605240, a PI3Kγ selective inhibitor, reduces the severity of glomerulonephritis and prolongs lifespan in these lupus-prone mice, indicating an important role of PI3K signaling in SLE pathogenesis (95). Activation of PI3Kp110δ is enhanced in T cells from SLE patients, and the activation of PI3K pathway is associated with the defect of activation-induced cell death (AICD) in SLE T cells (96). PI3Kδ inhibition by GS-9289, a selective inhibitor of p110δ subunit, prolongs life span and reduces kidney damage in MRL/lpr mice (97), and general PI3K inhibition by Ly294002 rescues the AICD defect in T cells from SLE patients (96), suggesting that PI3K inhibitors may be potentially important drugs to treat patients with SLE.
Phosphatase and tensin homolog deleted on chromosome 10 dephosphorylates PIP3 and regulates the PI3K/Akt pathway (98). PTEN was originally reported as a tumor suppressor gene in 1997 (99–101), and T-cell-specific PTEN deficient mice exhibit increases in thymic cells and develop T-cell-derived lymphomas (102, 103). Treg-specific PTEN deficient mice show autoimmune phenotypes by loss of Treg function and stability (104, 105). On the other hand, the role of PTEN in Th17 cell differentiation is controversial. Overexpression of PTEN inhibits STAT3 activation and Th17 differentiation, and ameliorates the development of collagen-induced arthritis (106). By contrast, Th17-specific PTEN deficient mice exhibit impaired in vitro Th17 cell differentiation and mitigated symptoms of experimental autoimmune encephalomyelitis (107). PTEN deficiency increases the production of IL-2 and phosphorylation of STAT5, but reduces STAT3 phosphorylation, suggesting that further studies are required to determine the exact role of PTEN in T cell differentiation and the activation of STAT signals.
There is limited evidence demonstrating how PTEN is associated with the pathogenesis of SLE. Overexpression of miR-148a-3p, which is increased in the glomeruli of patients with lupus nephritis, induces mesangial cell proliferation in glomeruli and reduces the expression level of PTEN (108). Also, SLE B cells exhibit decreased expression levels of PTEN, which inversely correlates with disease activity (109), whereas there is no clear evidence available to elucidate the role of PTEN in SLE T cells.
Mechanistic Target of Rapamycin (mTOR) Pathway
Mechanistic target of rapamycin, a ubiquitous serine-threonine kinase, integrates environmental cues from a variety of pathways to regulate various cellular processes including cellular survival, proliferation and differentiation, and cellular metabolism (110, 111). mTOR is a component of two distinct complexes, mTOR complex (C)1 and mTORC2. The components of mTORC1 are mTOR, regulatory protein associated with mTOR (Raptor), mammalian lethal with Sec13 protein 8 (mLST8) and inhibitory subunits proline-rich Akt substrate of 40 kDa and DEP domain containing mTOR-interacting protein (DEPTOR). mTORC2 also contains mTOR, mLST8, DEPTOR, whereas it is composed of rapamycin insensitive companion of mTOR (Rictor), instead of Raptor, and inhibitory subunits mammalian stress-activated protein kinase interacting protein 1 and Protor (protein observed with Rictor) 1/2 (112). mTORC1 phosphorylates two key effectors for protein synthesis; p70S6 kinase 1 (S6K1) and EIF4E binding protein, whereas mTORC2 phosphorylates serum- and glucocorticoid-induced kinase 1, Akt (Ser473), and PKC.
Mechanistic target of rapamycin plays an important role in cellular metabolism (113). mTORC1 increases the translation of the transcription factor hypoxia-inducible factor 1α, which induces glycolytic genes (114). Glycolysis is elevated in CD4+ T cells from lupus-prone (B6. Sle1.Sle2.Sle3 mice and B6. lpr mice) and SLE patients (115, 116). mTORC1 also regulates both general autophagy and mitophagy, which are important in maintaining mitochondrial function (117). T cells from SLE patients exhibit increased mitochondrial mass and mitochondria dysfunction, characterized by elevated mitochondrial transmembrane potential (118, 119). Increased mitochondrial metabolism in SLE T cells can contribute to aberrant T cell function (111). Along these lines, normalization of CD4+ T cell metabolism by mitochondrial metabolism inhibitor metformin and the glucose metabolism inhibitor 2-Deoxy-d-glucose reduced IFNγ production from CD4+ T cells in vitro and suppressed autoimmunity and nephritis in B6. Sle1.Sle2.Sle3 mice and NZB/W F1 mice (115).
Recent studies have proven the important role of mTOR in the polarization of T cells. Th1 and Th17 differentiation is selectively regulated by mTORC1 signaling (120), and the inhibition of mTOR in vivo reduces the proportion of Th1 cells and Th17 cells in the lamina propria and mesenteric lymph nodes (121). It is also reported that both mTORC1 and mTORC2 are essential for Tfh cell differentiation and germinal cell reaction under steady state and after antigen immunization and viral infection (122).
The role of mTOR in Treg differentiation is complicated. mTORC1 signaling is constitutively active in Treg cells and its disruption in Treg cells leads to profound loss of Treg suppressive activity, although mTORC1 does not directly impact the expression of Foxp3 (123). On the other hand, both mTORC1 and mTORC2 suppress induced-Treg generation in vitro (120, 124). PP2A activation induces the inhibition of the mTORC1 pathway but has no effect on the mTORC2 pathway, and Treg cell-specific ablation of the PP2A results in a severe systemic autoimmune disorder through Treg dysfunction (125).
Recently, it has been recognized that activation of the mTOR pathway plays an important role in the pathogenesis of autoimmune diseases including SLE (119). mTORC1 activity is increased in the livers of MRL/lpr mice (126). In SLE T cells, mTORC1 activity is increased while mTORC2 is reduced compared with T cells from healthy donors (127). Tuberous sclerosis complex (TSC), an autosomal dominant disorder, affects multiple organ systems resulting from mutations in either of TSC 1 or TSC2 genes, which negatively regulate mTORC1 activation (128). Singh et al. reported a fatal lupus patient complicated with TSC, suggesting that mTORC1 activation led to the development of unusually severe SLE (129). Therefore, mTOR has become a therapeutic target in SLE. Rapamycin, the best-known inhibitor of mTOR, has been approved by the FDA to preserve renal allografts (111). Recent studies have uncovered the effect of rapamycin on SLE T cells in vitro. Increased IL-17 expression in CD4+ T cells from SLE patients is suppressed and Treg cells are expanded by rapamycin (127, 130). SLE Treg cells exhibit increased mTORC1 and mTORC2, and IL21 stimulates mTORC1 and mTORC2 and blocks the differentiation of Treg cells (131). Rapamycin reduces both the activation of STAT3 and the number of IL-17 producing cells in patients with SLE (132), and decreases the severity of lupus nephritis and prolongs survival in MRL/lpr mice (133). There are reports of studies with small numbers of patients with SLE showing the efficacy of oral administration of rapamycin (22, 134). Importantly, the deficiency of the CD3ζ chain and upregulation of FcεRIγ chain and Syk in T cells from SLE patients in vitro are reversed by rapamycin treatment (22).
N-acetylcysteine (NAC), a precursor of glutathione, is another inhibitor of mTOR. A randomized double blind placebo-controlled study to assess the efficacy and the safety of NAC in SLE patients (135), demonstrated that 2.4 and 4.8 g daily NAC reduced disease activity and mTOR activity, reversed the expansion of CD3+ CD4-CD8- double negative (DN) T cells, and stimulated Foxp3 expression in CD4+CD25+ T cells. There are other reports showing the efficacy of NAC in SLE patients with lupus nephritis (136, 137).
Overall, mTOR inhibitors are accepted as a novel class of drugs that can target both cellular signaling and metabolism. To establish the efficacy of mTOR inhibitors in SLE patients and identify patients who respond to treatment, further studies with larger number of patients are necessary. Recently, results of a large prospective open-label, phase 1/2 trial of rapamycin (Sirolimus) in patients with active SLE were reported (138). During the course of 12 months of treatment, disease activity scores reduced in 16 (55%) of 29 patients treated with Sirolimus. Sirolimus treatment expanded Tregs and CD8+ memory T cell populations and inhibited IL-4 and IL-17 production by CD4+ and DN T cells. Although this study is a single-arm study and placebo-controlled clinical trials with increased number of patients are required, the trial suggests that mTOR blockade may be a promising therapeutic target in the treatment of SLE.
Cytokine Signaling
Cytokines play critical roles in the proliferation, activation, differentiation, and function of T cells. The Janus kinase (JAK)–STAT signaling pathway following cytokine-receptor activation is one of the most important pathways used by multiple cytokines. In humans, seven STAT family members have been identified (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) (139). Different cytokines can activate specific STATs, and STATs regulate transcription of various genes including master regulators of differentiated T cell subsets. STAT1/STAT4 activate Tbet, the transcription factor which drives Th1 cell differentiation, STAT6 induces GATA3 in Th2 differentiation, STAT3 activates RORγt which activates IL-17 and Th17 differentiation, STAT3 induces Bcl6 transcription factor of Tfh cells, and STAT5 activates Foxp3 which drives Treg differentiation (140). STAT proteins are, therefore, essential for the establishment of lineage-specific enhancer landscapes of differentiating T cells (141). A number of studies have shown that STAT signaling plays a critical role in autoimmune diseases including SLE (142).
STAT1 and Interferons
A number of studies have revealed that IFNs play important roles in lupus pathogenesis (143, 144). The phosphorylation of STAT1, which is activated by all types of IFNs, is increased in MRL/lpr mice (145, 146). Consistent with these results, it was observed that the expression levels of STAT1 are increased in leukocytes from SLE patients (147–149). The expression levels of miR-145, a suppressor of STAT1, are decreased in T cells from SLE patients, and increased levels of STAT1 in human SLE T cells are associated with lupus nephritis (150). Recently, it was reported that the levels of STAT1 protein were increased in CD4 T cells from SLE patients and positively correlated with disease activity (151), and high STAT1 phosphorylation responses were observed in activated Tregs, which were decreased in peripheral blood from SLE patients. These results suggest that STAT1 can be a therapeutic target in SLE. However, the involvement of STAT1 in SLE is complex because STAT1 deficient lupus-prone mice exhibit interstitial kidney inflammation associated with Th17 cells, by shunting to STAT3/4 activation (152).
IL-23—STAT3—IL-17 Axis
Th17 cells produce the IL-17 cytokines IL-17A and IL-17F. Increased numbers of Th17 cells and increased levels of IL-17 have been found in patients with SLE and in lupus-prone mice (153–155). IL-17-producing cells have been found in kidney biopsies of patients with lupus nephritis (156) and in kidneys and spleen of MRL/lpr lupus-prone mice (157), and levels of IL-17 correlate with SLE disease activity (153). DN T cells are a key source of IL-17 in MRL/lpr mice (156, 157), and more importantly they are present in the kidney tissue of patients with lupus nephritis (156).
Recent studies have uncovered aberrant mechanisms associated with Th17 differentiation and IL-17 production in SLE T cells. IL-23, a member of the IL-12 family, is important for the maintenance of Th17 cells. Serum levels of IL-23 are increased in patients with SLE with high disease activity (158). IL-23 induces the activation of STAT3 (159–161). STAT3 directly binds the promoters of IL-17A and IL-17F (162), and T cell-specific deletion of STAT3 reduces IL-17 expression and impairs RORγt expression (163). STAT3 is upregulated and activated in both lupus-prone mice (164, 165) and T cells from patients with SLE (166, 167).
In addition to its role in Th17 differentiation, STAT3 is also important for the development of follicular helper T cells (Tfh cells), which induce the differentiation of germinal center B cells into memory and antibody-secreting cells (168). Tfh cells are expanded in both patients with SLE and lupus-prone mice (169). STAT3 also plays a role in the production of other cytokines including IL-10, which promotes B-cell proliferation and antibody production, and is elevated in the serum and kidneys of patients with SLE (167, 170–172). STAT3 was shown to promote IL-10 expression through trans-activation and chromatin remodeling of the IL-10 locus in T cells from patients with SLE (167).
Therefore, STAT3 inhibitors could be promising therapeutic candidates to treat patients with SLE. Indeed, administration of a STAT3 inhibitor to MRL/lpr mice delays the onset of lupus nephritis in Ref. (173).
Janus kinase inhibitors are also promising therapeutic agents. JAK2 inhibitor AG490 suppressed the production of anti-histone/dsDNA antibodies in short-term culture (174). Tofacitinib is an oral JAK inhibitor, which inhibits JAK1, JAK3 (to a less extent), and JAK2, and has been approved for the treatment of rheumatoid arthritis. Tofacitinib improves disease activity of lupus-prone mice including nephritis, skin inflammation, and autoantibody production (175, 176). Baricitinib, another JAK inhibitor, is also under investigation for the treatment of SLE (177).
There are some reports indicating that IL-23 contributes to organ inflammation independent of its contribution to Th17 differentiation. IL-23 is important in the development of T cell-dependent colitis (178), yet IL-23-dependent colitis does not require IL-17 secretion by T cells, because CD4+ CD45RBhi T cells cannot induce colitis in Il23a−/− Rag1−/− recipients even though intestinal IL-17 is unaffected by the absence of IL-23 (179). Furthermore, although IL-23 is not essential for the expression of Foxp3, IL-23 can have an indirect effect on Treg cell generation. IL-23 receptor deficiency in lupus-prone mice results in decreased production of anti-dsDNA antibodies and proliferation of DN T cells (180, 181). Interestingly, IL-23 not only promotes IL-17 production but also decreases the production of IL-2 by impairing the Il2 gene enhancer NFκBp65 in mice (181). Also, IL-23 stimulation expands DN T cells from SLE patients in vitro (182). A phase IIa trial of Ustekinumab, targeting the p40 subunit common to IL-12 and IL-23, is underway in patients with SLE ((183). Inhibition of IL-23 signaling by an anti-IL-23p19 antibody ameliorates nephritis in MRL/lpr mice (184). Tildrakizumab (MK-3222), a monoclonal antibody targeting the p19 subunit, is under investigation for treatment of moderate-to-severe chronic plaque psoriasis (185, 186). Another monoclonal antibody targeting the p19 subunit, MEDI2070 (also known as AMG 139), improved clinical activity of Crohn’s disease in a phase IIa trial (187), although no data are available yet in patients with SLE.
There are other factors related to Th17 differentiation in SLE T cells. PP2A controls various signaling pathways, and CD4 T cells from transgenic mice that overexpress the catalytic subunit of PP2A in T cells produce increased amounts of IL-17 (188). The cAMP response element modulator (CREM) family of transcription factors also plays an important role in the differentiation of Th17 cells and IL-17 production. The suppressor isoform CREMα, which is increased in SLE T cells, reduces CpG-DNA methylation of the IL-17A locus, and controls IL-17A expression (189). Inducible cAMP early repressor (ICER), a transcriptional repressor isoform of CREM, is important for Th17 cell differentiation. ICER binds to the IL-17A promoter and enhances accumulation of the canonical IL-17 transcription factor RORγt (190). Calcium/calmodulin kinase IV (CaMKIV) is activated in T cells from SLE patients and MRL/lpr mice (191–193), and promotes the differentiation of Th17 cells and IL-17 production by activating the Akt/mTOR pathway (130). In MRL/lpr mice, genetic deletion of CaMKIV prolongs survival, and CaMKIV inhibitor KN-93 leads the suppression of nephritis and skin disease (192, 193). Moreover, as described above, ROCK is also associated with Th17 differentiation and production of IL-17 through the activation of IRF4 (75, 76).
Secukinumab and Ixekizumab are monoclonal antibodies targeting IL-17A while Brodalumab targets the IL-17A receptor, thus inhibiting the IL-17 signaling pathway. Although the evidence is clear for the efficacy and safety of these agents in the treatment of psoriasis and ankylosing spondylitis (194), there are no data showing efficacy of inhibition of IL-17 in SLE patients so far. Despite the overwhelming evidence that IL-17 contributes to lupus pathology, IL-17A deficiency in lupus-prone MRL/lpr mice or IL-17A neutralization in NZB/NZW mice did not affect the course of nephritis (195). Further work is needed to dissect the role of this signaling pathway in lupus pathogenesis in order to target it effectively.
STAT5 and IL-2
IL-2 is a key cytokine important in the proliferation, activation, and differentiation of T cells (196). Importantly, IL-2 plays a vital role in the homeostasis of Treg cells. Mice and humans deficient in IL-2, IL-2Rα (CD25), or IL-2Rβ (CD122) develop systemic autoimmunity due to impaired Treg cells (197–203). Also, IL-2 negatively regulates IL-17 production in vivo and in vitro (204, 205). In addition, IL-2 inhibits the differentiation of Tfh cells through the activation of Akt-mTORC1 signaling, and instead promotes the differentiation of Th1 cells (206). IL-2 also plays a critical role in the induction of AICD, a key process responsible for the deletion of autoreactive cells (207, 208).
It has been known for a long time that the insufficient production of IL-2 from T cells is one of the most important characteristic features of both SLE patients and lupus-prone mice (209–211). The molecular mechanisms of the decreased IL-2 production from SLE T cells have not completely been elucidated, whereas a number of studies have identified several mechanisms. Various transcription factors binding to the IL-2 promoter affect the expression of IL-2. NF-κB and activator protein 1 (c-fos/c-jun heterodimer) are downregulated in T cells from SLE patients, and linked to decreased IL-2 transcription (212–214). PP2A, a ubiquitous phosphatase, is increased in SLE T cells. PP2A dephosphorylates cyclic AMP-responsive element-binding protein 1, which can directly bind to the IL-2 promoter and reduce IL-2 production (215). CaMKIV plays a role in the shortage of IL-2 in SLE T cells as well. CaMKIV is increased in SLE T cells, and phosphorylates CREM to suppress IL-2 transcription (191). As described above, it was recently reported that PTEN deficiency increases the production of IL-2 and phosphorylation of STAT5 (107), suggesting a novel mechanism of the IL-2 deficiency in SLE T cells, whereas the role of PTEN in SLE T cells remains unclear. SRSF1 is a multifunctional protein, which contributes to the transcriptional activation of IL-2. SRSF1 levels are decreased in T cells from SLE patients, and overexpression of SRSF1 into SLE T cells, rescues IL-2 production (34). It was demonstrated that increased expression of miR-200a-3p is associated with the decreased production of IL-2 through zinc finger E-box binding homeobox–C-terminal binding protein 2 in MRL/lpr mice (216).
Although the molecules that contribute to the decreased production of IL-2 can serve as therapeutic targets for the treatment of patients with SLE, strategies to restore IL-2 levels have been exploited. Recently, the safety and efficacy of low-dose IL-2 therapy for patients with graft-versus-host disease (217, 218), type 1 diabetes (219), and cryoglobulinemia associated with HCV infection have been reported (220). There are uncontrolled reports indicating the efficacy of low-dose IL-2 therapy in patients with SLE (221–223). Treatment of MRL/lpr lupus-prone mice with an IL-2-expressing recombinant adeno-associated virus resulted in reduced pathology, decreased DN cell numbers and increased Treg cell numbers (224). Subcutaneous injection of low-dose IL-2 on five consecutive days in a small number of patients with SLE, achieved decreases in SLE Disease Activity Index (SLEDAI) and increased peripheral Treg cells (221, 225). An uncontrolled study of 37 consenting patients with SLE claims that subcutaneous administration of recombinant IL-2 every other day for 2 weeks decreased SLEDAI, Th17, Tfh, and DN T cells, and increased Treg cell numbers (222). Further studies are required to overcome the challenges of maintaining IL-2 levels due to a very short half-life of the cytokine. It is important to note that not only the production of IL-2 by T cells from patients with SLE impaired, but also the response to exogenous IL-2 is impaired in CD4 T cells compared with healthy controls (226). These results suggest that we should also consider strategies to restore IL-2 sensitivity of T cells during low-dose IL-2 therapy. Indeed, the engagement of SLAMF3 in T cells from normal subjects and patients with SLE increased their IL-2-initiated signaling strength (227).
Transforming Growth Factor-β (TGF-β) Signaling
Transforming growth factor-β has three different isoforms (TGF-β1, 2, and 3), and regulates cell growth and differentiation. TGF-β signaling is essential for the differentiation of Treg cells. TGF-β signaling induces the expression of Foxp3 (228), and T cell-specific loss of TGF-β results in the defect in the differentiation of thymic Treg cells in mice (229). In addition, TGF-β also acts as a direct regulator against autoreactive T cells in part through the regulation of GM-CSF production (230, 231). Moreover, TGF-β also contributes to the differentiation of Th17 cells (232), whereas Th17 cells also can be generated without TGF-β signaling but with IL-6, IL-1β, and IL-23 (233).
The role of TGF-β in SLE patients remains unclear. It was reported that serum levels of TGF-β are decreased in active SLE patients (234, 235). On the other hand, some reports demonstrated that TGF-β1 production is increased from SLE PBMC (236). Impaired response of peripheral blood cells to TGF-β1 in patients with active SLE has been reported (237). CD4+CD25-Lag3+ Treg cells expressing early growth response gene (Egr)2 and Egr3 exhibit immune suppressive capacity by secreting TGF-β3, and mice with T cell-specific deletion of Egr2/3 mice develop lupus-like disease (238, 239). Further studies are required to uncover the role of TGF-β in the pathogenesis of lupus.
Conclusion
A great effort has been made to delineate specific abnormalities in immune cells from SLE patients, and a dramatic expansion has been achieved in our understanding of cellular and molecular phenotypes in the pathogenesis of SLE. Here, we have reviewed the important features of aberrant signaling pathways in SLE T cells. T cells have a vital role in the immune response, whereas other immune cells such as B cells, dendritic cells, macrophages, and neutrophils cannot be ignored in the development of autoimmune diseases. Abnormal activation of the TCR and PI3K-Akt-mTOR signaling pathways and various molecules including PP2A, CaMKIV, CD44, ROCK, mTOR, and SRSF1 affect the function and the differentiation of T cells. Moreover, aberrant cytokine production and the activation of JAK–STAT pathways are also involved in the differentiation of pathogenic effector T cells and impaired Treg cells. In addition to the aberrant pathways described above, alterations in metabolism of immune cells have been recently recognized in patients with autoimmune diseases (113, 117).
Clinical manifestations including symptoms, severities, and clinical response are extremely variable in SLE patients, indicating that no single mediator or pathway can account for the complex pathogenesis. For example, decreased expression levels of CD3ζ are found in many but not all SLE patients (240). The more we understand and elucidate cellular and molecular aberrations in SLE, the more we realize the complexities of the pathogenesis of SLE. However, each aberration has the possibility to be a promising therapeutic target (Table 1). In addition, the analysis of various molecular phenotypes may contribute to patient stratification leading the development of more personalized strategies in SLE treatment.
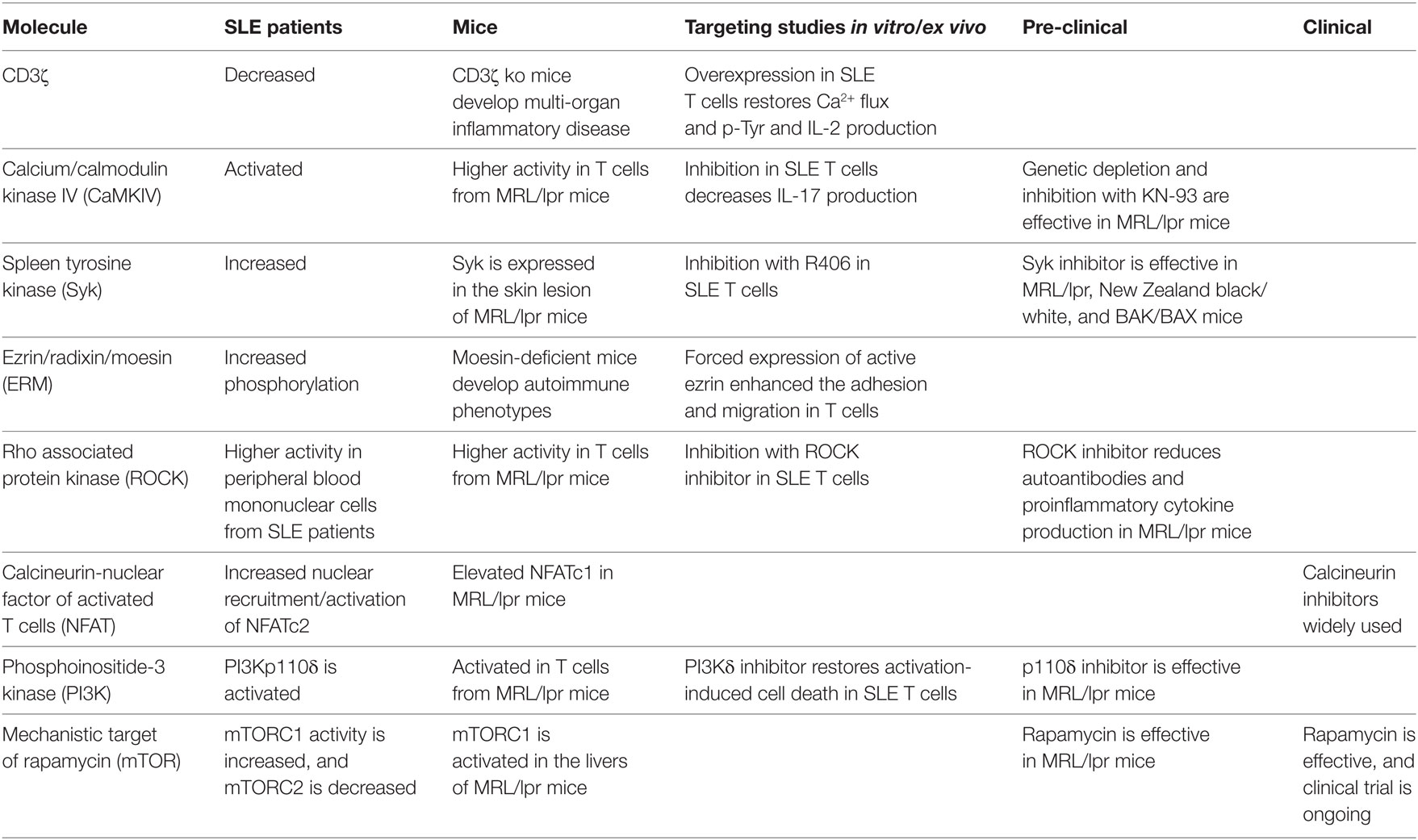
Table 1. Signaling molecules as potential therapeutic targets for systemic lupus erythematosus (SLE).
Author Contributions
TK, VM, and GT conceptualized the article, reviewed the literature, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Authors acknowledge the funding from the United States National Institutes of Health grants R01AR068974, R01AI42269, R37AI49954, R01AI65687, R01AI085567, and R01 AR064350.
References
1. Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol (2001) 1:147–53. doi:10.1038/35100573
2. Tsokos GC. Systemic lupus erythematosus. N Engl J Med (2011) 365:2110–21. doi:10.1056/NEJMra1100359
3. Moulton VR, Tsokos GC. T cell signaling abnormalities contribute to aberrant immune cell function and autoimmunity. J Clin Invest (2015) 125:2220–7. doi:10.1172/JCI78087
4. Rother N, van der Vlag J. Disturbed T cell signaling and altered Th17 and regulatory T cell subsets in the pathogenesis of systemic lupus erythematosus. Front Immunol (2015) 6:610. doi:10.3389/fimmu.2015.00610
5. Moulton VR, Suarez-Fueyo A, Meidan E, Li H, Mizui M, Tsokos GC. Pathogenesis of human systemic lupus erythematosus: a cellular perspective. Trends Mol Med (2017) 23:615–35. doi:10.1016/j.molmed.2017.05.006
6. Klinman DM, Steinberg AD. Inquiry into murine and human lupus. Immunol Rev (1995) 144:157–93. doi:10.1111/j.1600-065X.1995.tb00069.x
7. Akahoshi M, Nakashima H, Tanaka Y, Kohsaka T, Nagano S, Ohgami E, et al. Th1/Th2 balance of peripheral T helper cells in systemic lupus erythematosus. Arthritis Rheum (1999) 42:1644–8. doi:10.1002/1529-0131(199908)42:8<1644::AID-ANR12>3.0.CO;2-L
8. van den Elsen P, Shepley BA, Borst J, Coligan JE, Markham AF, Orkin S, et al. Isolation of cDNA clones encoding the 20K T3 glycoprotein of human T-cell receptor complex. Nature (1984) 312:413–8. doi:10.1038/312413a0
9. Krissansen GW, Owen MJ, Verbi W, Crumpton MJ. Primary structure of the T3 gamma subunit of the T3/T cell antigen receptor complex deduced from cDNA sequences: evolution of the T3 gamma and delta subunits. EMBO J (1986) 5:1799–808.
10. Alcover A, Alarcon B, Di Bartolo V. Cell biology of T cell receptor expression and regulation. Annu Rev Immunol (2017) 36:103–25. doi:10.1146/annurev-immunol-042617-053429
11. Thill PA, Weiss A, Chakraborty AK. Phosphorylation of a tyrosine residue on Zap70 by Lck and its subsequent binding via an SH2 domain may be a key gatekeeper of T cell receptor signaling in vivo. Mol Cell Biol (2016) 36:2396–402. doi:10.1128/MCB.00165-16
12. Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol (2002) 20:371–94. doi:10.1146/annurev.immunol.20.092601.111357
13. Krishna S, Zhong X. Role of diacylglycerol kinases in T cell development and function. Crit Rev Immunol (2013) 33:97–118. doi:10.1615/CritRevImmunol.2013006696
14. Liossis SN, Ding XZ, Dennis GJ, Tsokos GC. Altered pattern of TCR/CD3-mediated protein-tyrosyl phosphorylation in T cells from patients with systemic lupus erythematosus. Deficient expression of the T cell receptor zeta chain. J Clin Invest (1998) 101:1448–57. doi:10.1172/JCI1457
15. Brundula V, Rivas LJ, Blasini AM, Paris M, Salazar S, Stekman IL, et al. Diminished levels of T cell receptor zeta chains in peripheral blood T lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum (1999) 42:1908–16. doi:10.1002/1529-0131(199909)42:9<1908::AID-ANR17>3.0.CO;2-7
16. Pang M, Setoyama Y, Tsuzaka K, Yoshimoto K, Amano K, Abe T, et al. Defective expression and tyrosine phosphorylation of the T cell receptor zeta chain in peripheral blood T cells from systemic lupus erythematosus patients. Clin Exp Immunol (2002) 129:160–8. doi:10.1046/j.1365-2249.2002.01833.x
17. Suzuki K, Hirose T, Matsuda H, Hasegawa S, Okumura K, Ra C. The Fc receptor (FcR) gamma subunit is essential for IgE-binding activity of cell-surface expressed chimeric receptor molecules constructed from human high-affinity IgE receptor (Fc epsilon RI) alpha and FcR gamma subunits. Mol Immunol (1998) 35:259–70. doi:10.1016/S0161-5890(98)00047-9
18. Takai T. Fc receptors and their role in immune regulation and autoimmunity. J Clin Immunol (2005) 25:1–18. doi:10.1007/s10875-005-0353-8
19. Enyedy EJ, Nambiar MP, Liossis SN, Dennis G, Kammer GM, Tsokos GC. Fc epsilon receptor type I gamma chain replaces the deficient T cell receptor zeta chain in T cells of patients with systemic lupus erythematosus. Arthritis Rheum (2001) 44:1114–21. doi:10.1002/1529-0131(200105)44:5<1114::AID-ANR192>3.0.CO;2-B
20. Krishnan S, Warke VG, Nambiar MP, Wong HK, Tsokos GC, Farber DL. Generation and biochemical analysis of human effector CD4 T cells: alterations in tyrosine phosphorylation and loss of CD3zeta expression. Blood (2001) 97:3851–9. doi:10.1182/blood.V97.12.3851
21. Krishnan S, Warke VG, Nambiar MP, Tsokos GC, Farber DL. The FcR gamma subunit and Syk kinase replace the CD3 zeta-chain and ZAP-70 kinase in the TCR signaling complex of human effector CD4 T cells. J Immunol (2003) 170:4189–95. doi:10.4049/jimmunol.170.8.4189
22. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol (2009) 182:2063–73. doi:10.4049/jimmunol.0803600
23. Liu CP, Lin WJ, Huang M, Kappler JW, Marrack P. Development and function of T cells in T cell antigen receptor/CD3 zeta knockout mice reconstituted with Fc epsilon RI gamma. Proc Natl Acad Sci U S A (1997) 94:616–21. doi:10.1073/pnas.94.2.616
24. Nambiar MP, Fisher CU, Warke VG, Krishnan S, Mitchell JP, Delaney N, et al. Reconstitution of deficient T cell receptor zeta chain restores T cell signaling and augments T cell receptor/CD3-induced interleukin-2 production in patients with systemic lupus erythematosus. Arthritis Rheum (2003) 48:1948–55. doi:10.1002/art.11072
25. Deng GM, Beltran J, Chen C, Terhorst C, Tsokos GC. T cell CD3zeta deficiency enables multiorgan tissue inflammation. J Immunol (2013) 191:3563–7. doi:10.4049/jimmunol.1300634
26. Juang YT, Tenbrock K, Nambiar MP, Gourley MF, Tsokos GC. Defective production of functional 98-kDa form of Elf-1 is responsible for the decreased expression of TCR zeta-chain in patients with systemic lupus erythematosus. J Immunol (2002) 169:6048–55. doi:10.4049/jimmunol.169.10.6048
27. Nambiar MP, Enyedy EJ, Warke VG, Krishnan S, Dennis G, Kammer GM, et al. Polymorphisms/mutations of TCR-zeta-chain promoter and 3’ untranslated region and selective expression of TCR zeta-chain with an alternatively spliced 3’ untranslated region in patients with systemic lupus erythematosus. J Autoimmun (2001) 16:133–42. doi:10.1006/jaut.2000.0475
28. Tsuzaka K, Fukuhara I, Setoyama Y, Yoshimoto K, Suzuki K, Abe T, et al. TCR zeta mRNA with an alternatively spliced 3’-untranslated region detected in systemic lupus erythematosus patients leads to the down-regulation of TCR zeta and TCR/CD3 complex. J Immunol (2003) 171:2496–503. doi:10.4049/jimmunol.171.5.2496
29. Tsuzaka K, Setoyama Y, Yoshimoto K, Shiraishi K, Suzuki K, Abe T, et al. A splice variant of the TCR zeta mRNA lacking exon 7 leads to the down-regulation of TCR zeta, the TCR/CD3 complex, and IL-2 production in systemic lupus erythematosus T cells. J Immunol (2005) 174:3518–25. doi:10.4049/jimmunol.174.6.3518
30. Chowdhury B, Tsokos CG, Krishnan S, Robertson J, Fisher CU, Warke RG, et al. Decreased stability and translation of T cell receptor zeta mRNA with an alternatively spliced 3’-untranslated region contribute to zeta chain down-regulation in patients with systemic lupus erythematosus. J Biol Chem (2005) 280:18959–66. doi:10.1074/jbc.M501048200
31. Moulton VR, Kyttaris VC, Juang YT, Chowdhury B, Tsokos GC. The RNA-stabilizing protein HuR regulates the expression of zeta chain of the human T cell receptor-associated CD3 complex. J Biol Chem (2008) 283:20037–44. doi:10.1074/jbc.M710434200
32. Moulton VR, Tsokos GC. Alternative splicing factor/splicing factor 2 regulates the expression of the zeta subunit of the human T cell receptor-associated CD3 complex. J Biol Chem (2010) 285:12490–6. doi:10.1074/jbc.M109.091660
33. Moulton VR, Gillooly AR, Perl MA, Markopoulou A, Tsokos GC. Serine arginine-rich splicing factor 1 (SRSF1) contributes to the transcriptional activation of CD3zeta in human T cells. PLoS One (2015) 10:e0131073. doi:10.1371/journal.pone.0131073
34. Moulton VR, Grammatikos AP, Fitzgerald LM, Tsokos GC. Splicing factor SF2/ASF rescues IL-2 production in T cells from systemic lupus erythematosus patients by activating IL-2 transcription. Proc Natl Acad Sci U S A (2013) 110:1845–50. doi:10.1073/pnas.1214207110
35. Hong KM, Kim HK, Park SY, Poojan S, Kim MK, Sung J, et al. CD3Z hypermethylation is associated with severe clinical manifestations in systemic lupus erythematosus and reduces CD3zeta-chain expression in T cells. Rheumatology (Oxford) (2017) 56:467–76. doi:10.1093/rheumatology/kew405
36. Sharabi A, Kasper IR, Tsokos GC. The serine/threonine protein phosphatase 2A controls autoimmunity. Clin Immunol (2018) 186:38–42. doi:10.1016/j.clim.2017.07.012
37. Juang YT, Wang Y, Jiang G, Peng HB, Ergin S, Finnell M, et al. PP2A dephosphorylates Elf-1 and determines the expression of CD3zeta and FcRgamma in human systemic lupus erythematosus T cells. J Immunol (2008) 181:3658–64. doi:10.4049/jimmunol.181.5.3658
38. Blasini AM, Brundula V, Paris M, Rivas L, Salazar S, Stekman IL, et al. Protein tyrosine kinase activity in T lymphocytes from patients with systemic lupus erythematosus. J Autoimmun (1998) 11:387–93. doi:10.1006/jaut.1998.0230
39. Matache C, Stefanescu M, Onu A, Tanaseanu S, Matei I, Frade R, et al. p56lck activity and expression in peripheral blood lymphocytes from patients with systemic lupus erythematosus. Autoimmunity (1999) 29:111–20. doi:10.3109/08916939908995380
40. Matache C, Onu A, Stefanescu M, Tanaseanu S, Dragomir C, Dolganiuc A, et al. Dysregulation of p56lck kinase in patients with systemic lupus erythematosus. Autoimmunity (2001) 34:27–38. doi:10.3109/08916930108994123
41. Jury EC, Kabouridis PS, Abba A, Mageed RA, Isenberg DA. Increased ubiquitination and reduced expression of LCK in T lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum (2003) 48:1343–54. doi:10.1002/art.10978
42. Ike H, Kosugi A, Kato A, Iino R, Hirano H, Fujiwara T, et al. Mechanism of Lck recruitment to the T-cell receptor cluster as studied by single-molecule-fluorescence video imaging. Chemphyschem (2003) 4:620–6. doi:10.1002/cphc.200300670
43. Kabouridis PS. Lipid rafts in T cell receptor signalling. Mol Membr Biol (2006) 23:49–57. doi:10.1080/09687860500453673
44. Gaus K, Chklovskaia E, Fazekas De St Groth B, Jessup W, Harder T. Condensation of the plasma membrane at the site of T lymphocyte activation. J Cell Biol (2005) 171:121–31. doi:10.1083/jcb.200505047
45. Jury EC, Flores-Borja F, Kabouridis PS. Lipid rafts in T cell signalling and disease. Semin Cell Dev Biol (2007) 18:608–15. doi:10.1016/j.semcdb.2007.08.002
46. Jury EC, Kabouridis PS, Flores-Borja F, Mageed RA, Isenberg DA. Altered lipid raft-associated signaling and ganglioside expression in T lymphocytes from patients with systemic lupus erythematosus. J Clin Invest (2004) 113:1176–87. doi:10.1172/JCI200420345
47. Krishnan S, Nambiar MP, Warke VG, Fisher CU, Mitchell J, Delaney N, et al. Alterations in lipid raft composition and dynamics contribute to abnormal T cell responses in systemic lupus erythematosus. J Immunol (2004) 172:7821–31. doi:10.4049/jimmunol.172.12.7821
48. Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol (2007) 178:1938–47. doi:10.4049/jimmunol.178.3.1938
49. Jury EC, Isenberg DA, Mauri C, Ehrenstein MR. Atorvastatin restores Lck expression and lipid raft-associated signaling in T cells from patients with systemic lupus erythematosus. J Immunol (2006) 177:7416–22. doi:10.4049/jimmunol.177.10.7416
50. Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell (1992) 71:649–62. doi:10.1016/0092-8674(92)90598-7
51. Krishnan S, Juang YT, Chowdhury B, Magilavy A, Fisher CU, Nguyen H, et al. Differential expression and molecular associations of Syk in systemic lupus erythematosus T cells. J Immunol (2008) 181:8145–52. doi:10.4049/jimmunol.181.11.8145
52. Pamuk ON, Gurkan H, Pamuk GE, Tozkir H, Duymaz J, Yazar M. BLK pathway-associated rs13277113 GA genotype is more frequent in SLE patients and associated with low gene expression and increased flares. Clin Rheumatol (2017) 36:103–9. doi:10.1007/s10067-016-3475-7
53. Iwata S, Yamaoka K, Niiro H, Jabbarzadeh-Tabrizi S, Wang SP, Kondo M, et al. Increased Syk phosphorylation leads to overexpression of TRAF6 in peripheral B cells of patients with systemic lupus erythematosus. Lupus (2015) 24:695–704. doi:10.1177/0961203314560424
54. Sheridan C. Small molecule challenges dominance of TNF-alpha inhibitors. Nat Biotechnol (2008) 26:143–4. doi:10.1038/nbt0208-143
55. Sweeny DJ, Li W, Clough J, Bhamidipati S, Singh R, Park G, et al. Metabolism of fostamatinib, the oral methylene phosphate prodrug of the spleen tyrosine kinase inhibitor R406 in humans: contribution of hepatic and gut bacterial processes to the overall biotransformation. Drug Metab Dispos (2010) 38:1166–76. doi:10.1124/dmd.110.032151
56. Deng GM, Liu L, Bahjat FR, Pine PR, Tsokos GC. Suppression of skin and kidney disease by inhibition of spleen tyrosine kinase in lupus-prone mice. Arthritis Rheum (2010) 62:2086–92. doi:10.1002/art.27452
57. Bahjat FR, Pine PR, Reitsma A, Cassafer G, Baluom M, Grillo S, et al. An orally bioavailable spleen tyrosine kinase inhibitor delays disease progression and prolongs survival in murine lupus. Arthritis Rheum (2008) 58:1433–44. doi:10.1002/art.23428
58. Kyttaris VC, Wang Y, Juang YT, Weinstein A, Tsokos GC. Increased levels of NF-ATc2 differentially regulate CD154 and IL-2 genes in T cells from patients with systemic lupus erythematosus. J Immunol (2007) 178:1960–6. doi:10.4049/jimmunol.178.3.1960
59. Iezzi G, Sonderegger I, Ampenberger F, Schmitz N, Marsland BJ, Kopf M. CD40-CD40L cross-talk integrates strong antigenic signals and microbial stimuli to induce development of IL-17-producing CD4+ T cells. Proc Natl Acad Sci U S A (2009) 106:876–81. doi:10.1073/pnas.0810769106
60. Kyttaris VC, Zhang Z, Kampagianni O, Tsokos GC. Calcium signaling in systemic lupus erythematosus T cells: a treatment target. Arthritis Rheum (2011) 63:2058–66. doi:10.1002/art.30353
61. Mok CC. Pro: the use of calcineurin inhibitors in the treatment of lupus nephritis. Nephrol Dial Transplant (2016) 31:1561–6. doi:10.1093/ndt/gfw289
62. Jordan AR, Racine RR, Hennig MJ, Lokeshwar VB. The role of CD44 in disease pathophysiology and targeted treatment. Front Immunol (2015) 6:182. doi:10.3389/fimmu.2015.00182
63. Saeed M. Novel linkage disequilibrium clustering algorithm identifies new lupus genes on meta-analysis of GWAS datasets. Immunogenetics (2017) 69:295–302. doi:10.1007/s00251-017-0976-8
64. Crispin JC, Keenan BT, Finnell MD, Bermas BL, Schur P, Massarotti E, et al. Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis Rheum (2010) 62:1431–7. doi:10.1002/art.27385
65. Seiter S, Schmidt DS, Zoller M. The CD44 variant isoforms CD44v6 and CD44v7 are expressed by distinct leukocyte subpopulations and exert non-overlapping functional activities. Int Immunol (2000) 12:37–49. doi:10.1093/intimm/12.1.37
66. Forster-Horvath C, Bocsi J, Raso E, Orban TI, Olah E, Timar J, et al. Constitutive intracellular expression and activation-induced cell surface up-regulation of CD44v3 in human T lymphocytes. Eur J Immunol (2001) 31:600–8. doi:10.1002/1521-4141(200102)31:2<600::AID-IMMU600>3.0.CO;2-8
67. Mori T, Kitano K, Terawaki S, Maesaki R, Fukami Y, Hakoshima T. Structural basis for CD44 recognition by ERM proteins. J Biol Chem (2008) 283:29602–12. doi:10.1074/jbc.M803606200
68. Shcherbina A, Bretscher A, Kenney DM, Remold-O’donnell E. Moesin, the major ERM protein of lymphocytes and platelets, differs from ezrin in its insensitivity to calpain. FEBS Lett (1999) 443:31–6. doi:10.1016/S0014-5793(98)01674-3
69. Hirata T, Nomachi A, Tohya K, Miyasaka M, Tsukita S, Watanabe T, et al. Moesin-deficient mice reveal a non-redundant role for moesin in lymphocyte homeostasis. Int Immunol (2012) 24:705–17. doi:10.1093/intimm/dxs077
70. Satooka H, Nagakubo D, Sato T, Hirata T. The ERM protein moesin regulates CD8(+) regulatory T cell homeostasis and self-tolerance. J Immunol (2017) 199:3418–26. doi:10.4049/jimmunol.1700074
71. Pernis AB, Ricker E, Weng CH, Rozo C, Yi W. Rho kinases in autoimmune diseases. Annu Rev Med (2016) 67:355–74. doi:10.1146/annurev-med-051914-022120
72. Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton (Hoboken) (2010) 67:545–54. doi:10.1002/cm.20472
73. Heasman SJ, Ridley AJ. Multiple roles for RhoA during T cell transendothelial migration. Small GTPases (2010) 1:174–9. doi:10.4161/sgtp.1.3.14724
74. Mrass P, Oruganti SR, Fricke GM, Tafoya J, Byrum JR, Yang L, et al. ROCK regulates the intermittent mode of interstitial T cell migration in inflamed lungs. Nat Commun (2017) 8:1010. doi:10.1038/s41467-017-01032-2
75. Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest (2010) 120:3280–95. doi:10.1172/JCI42856
76. Zanin-Zhorov A, Weiss JM, Nyuydzefe MS, Chen W, Scher JU, Mo R, et al. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc Natl Acad Sci U S A (2014) 111:16814–9. doi:10.1073/pnas.1414189111
77. Weiss JM, Chen W, Nyuydzefe MS, Trzeciak A, Flynn R, Tonra JR, et al. ROCK2 signaling is required to induce a subset of T follicular helper cells through opposing effects on STATs in autoimmune settings. Sci Signal (2016) 9:ra73. doi:10.1126/scisignal.aad8953
78. Isgro J, Gupta S, Jacek E, Pavri T, Duculan R, Kim M, et al. Enhanced rho-associated protein kinase activation in patients with systemic lupus erythematosus. Arthritis Rheum (2013) 65:1592–602. doi:10.1002/art.37934
79. Rozo C, Chinenov Y, Maharaj RK, Gupta S, Leuenberger L, Kirou KA, et al. Targeting the RhoA-ROCK pathway to reverse T-cell dysfunction in SLE. Ann Rheum Dis (2017) 76:740–7. doi:10.1136/annrheumdis-2016-209850
80. Lee JH, Zheng Y, Von Bornstadt D, Wei Y, Balcioglu A, Daneshmand A, et al. Selective ROCK2 inhibition in focal cerebral ischemia. Ann Clin Transl Neurol (2014) 1:2–14. doi:10.1002/acn3.19
81. Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature (1997) 389:990–4. doi:10.1038/40187
82. Lee MH, Cho YS, Han YM. Simvastatin suppresses self-renewal of mouse embryonic stem cells by inhibiting RhoA geranylgeranylation. Stem Cells (2007) 25:1654–63. doi:10.1634/stemcells.2006-0753
83. Wang Y, Lu Y, Chai J, Sun M, Hu X, He W, et al. Y-27632, a Rho-associated protein kinase inhibitor, inhibits systemic lupus erythematosus. Biomed Pharmacother (2017) 88:359–66. doi:10.1016/j.biopha.2017.01.069
84. Feng Y, Lograsso PV, Defert O, Li R. Rho kinase (ROCK) inhibitors and their therapeutic potential. J Med Chem (2016) 59:2269–300. doi:10.1021/acs.jmedchem.5b00683
85. Stirzaker RA, Biswas PS, Gupta S, Song L, Bhagat G, Pernis AB. Administration of fasudil, a ROCK inhibitor, attenuates disease in lupus-prone NZB/W F1 female mice. Lupus (2012) 21:656–61. doi:10.1177/0961203312436862
86. Vanhaesebroeck B, Ali K, Bilancio A, Geering B, Foukas LC. Signalling by PI3K isoforms: insights from gene-targeted mice. Trends Biochem Sci (2005) 30:194–204. doi:10.1016/j.tibs.2005.02.008
87. Fruman DA, Bismuth G. Fine tuning the immune response with PI3K. Immunol Rev (2009) 228:253–72. doi:10.1111/j.1600-065X.2008.00750.x
88. Han JM, Patterson SJ, Levings MK. The role of the PI3K signaling pathway in CD4(+) T cell differentiation and function. Front Immunol (2012) 3:245. doi:10.3389/fimmu.2012.00245
89. So L, Fruman DA. PI3K signalling in B- and T-lymphocytes: new developments and therapeutic advances. Biochem J (2012) 442:465–81. doi:10.1042/BJ20112092
90. Sasaki T, Irie-Sasaki J, Jones RG, Oliveira-Dos-Santos AJ, Stanford WL, Bolon B, et al. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science (2000) 287:1040–6. doi:10.1126/science.287.5455.1040
91. Barber DF, Bartolome A, Hernandez C, Flores JM, Fernandez-Arias C, Rodriguez-Borlado L, et al. Class IB-phosphatidylinositol 3-kinase (PI3K) deficiency ameliorates IA-PI3K-induced systemic lupus but not T cell invasion. J Immunol (2006) 176:589–93. doi:10.4049/jimmunol.176.1.589
92. Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol (2012) 12:649–61. doi:10.1038/nri3278
93. Borlado LR, Redondo C, Alvarez B, Jimenez C, Criado LM, Flores J, et al. Increased phosphoinositide 3-kinase activity induces a lymphoproliferative disorder and contributes to tumor generation in vivo. FASEB J (2000) 14:895–903. doi:10.1096/fasebj.14.7.895
94. Tauzin S, Chaigne-Delalande B, Selva E, Khadra N, Daburon S, Contin-Bordes C, et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol (2011) 9:e1001090. doi:10.1371/journal.pbio.1001090
95. Barber DF, Bartolome A, Hernandez C, Flores JM, Redondo C, Fernandez-Arias C, et al. PI3Kgamma inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat Med (2005) 11:933–5. doi:10.1038/nm1291
96. Suarez-Fueyo A, Barber DF, Martinez-Ara J, Zea-Mendoza AC, Carrera AC. Enhanced phosphoinositide 3-kinase delta activity is a frequent event in systemic lupus erythematosus that confers resistance to activation-induced T cell death. J Immunol (2011) 187:2376–85. doi:10.4049/jimmunol.1101602
97. Suarez-Fueyo A, Rojas JM, Cariaga AE, Garcia E, Steiner BH, Barber DF, et al. Inhibition of PI3Kdelta reduces kidney infiltration by macrophages and ameliorates systemic lupus in the mouse. J Immunol (2014) 193:544–54. doi:10.4049/jimmunol.1400350
98. Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, et al. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science (2002) 295:2088–91. doi:10.1126/science.1068094
99. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res (1997) 57:2124–9.
100. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science (1997) 275:1943–7. doi:10.1126/science.275.5308.1943
101. Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet (1997) 15:356–62. doi:10.1038/ng0497-356
102. Suzuki A, Yamaguchi MT, Ohteki T, Sasaki T, Kaisho T, Kimura Y, et al. T cell-specific loss of Pten leads to defects in central and peripheral tolerance. Immunity (2001) 14:523–34. doi:10.1016/S1074-7613(01)00134-0
103. Buckler JL, Liu X, Turka LA. Regulation of T-cell responses by PTEN. Immunol Rev (2008) 224:239–48. doi:10.1111/j.1600-065X.2008.00650.x
104. Huynh A, Dupage M, Priyadharshini B, Sage PT, Quiros J, Borges CM, et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat Immunol (2015) 16:188–96. doi:10.1038/ni.3077
105. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol (2015) 16:178–87. doi:10.1038/ni.3076
106. Lee SH, Park JS, Byun JK, Jhun J, Jung K, Seo HB, et al. PTEN ameliorates autoimmune arthritis through down-regulating STAT3 activation with reciprocal balance of Th17 and Tregs. Sci Rep (2016) 6:34617. doi:10.1038/srep34617
107. Kim HS, Jang SW, Lee W, Kim K, Sohn H, Hwang SS, et al. PTEN drives Th17 cell differentiation by preventing IL-2 production. J Exp Med (2017) 214:3381–98. doi:10.1084/jem.20170523
108. Qingjuan L, Xiaojuan F, Wei Z, Chao W, Pengpeng K, Hongbo L, et al. miR-148a-3p overexpression contributes to glomerular cell proliferation by targeting PTEN in lupus nephritis. Am J Physiol Cell Physiol (2016) 310:C470–8. doi:10.1152/ajpcell.00129.2015
109. Wu XN, Ye YX, Niu JW, Li Y, Li X, You X, et al. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci Transl Med (2014) 6:246ra299. doi:10.1126/scitranslmed.3009131
110. Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell (2007) 12:487–502. doi:10.1016/j.devcel.2007.03.020
111. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol (2016) 12:169–82. doi:10.1038/nrrheum.2015.172
112. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168:960–76. doi:10.1016/j.cell.2017.02.004
113. Perl A. Review: metabolic control of immune system activation in rheumatic diseases. Arthritis Rheumatol (2017) 69:2259–70. doi:10.1002/art.40223
114. Lum JJ, Bui T, Gruber M, Gordan JD, Deberardinis RJ, Covello KL, et al. The transcription factor HIF-1alpha plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes Dev (2007) 21:1037–49. doi:10.1101/gad.1529107
115. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med (2015) 7:274ra218. doi:10.1126/scitranslmed.aaa0835
116. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol (2016) 196:80–90. doi:10.4049/jimmunol.1501537
117. Morel L. Immunometabolism in systemic lupus erythematosus. Nat Rev Rheumatol (2017) 13:280–90. doi:10.1038/nrrheum.2017.43
118. Gergely P Jr, Grossman C, Niland B, Puskas F, Neupane H, Allam F, et al. Mitochondrial hyperpolarization and ATP depletion in patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:175–90. doi:10.1002/1529-0131(200201)46:1<175::AID-ART10015>3.0.CO;2-H
119. Oaks Z, Winans T, Huang N, Banki K, Perl A. Activation of the mechanistic target of rapamycin in SLE: explosion of evidence in the last five years. Curr Rheumatol Rep (2016) 18:73. doi:10.1007/s11926-016-0622-8
120. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol (2011) 12:295–303. doi:10.1038/ni.2005
121. Hu S, Chen M, Wang Y, Wang Z, Pei Y, Fan R, et al. mTOR inhibition attenuates dextran sulfate sodium-induced colitis by suppressing T cell proliferation and balancing TH1/TH17/Treg profile. PLoS One (2016) 11:e0154564. doi:10.1371/journal.pone.0154564
122. Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 kinase signaling and glucose metabolism drive follicular helper T cell differentiation. Immunity (2016) 45:540–54. doi:10.1016/j.immuni.2016.08.017
123. Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature (2013) 499:485–90. doi:10.1038/nature12297
124. Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity (2009) 30:832–44. doi:10.1016/j.immuni.2009.04.014
125. Apostolidis SA, Rodriguez-Rodriguez N, Suarez-Fueyo A, Dioufa N, Ozcan E, Crispin JC, et al. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat Immunol (2016) 17:556–64. doi:10.1038/ni.3390
126. Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol (2016) 68:2728–39. doi:10.1002/art.39791
127. Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol (2014) 192:4134–44. doi:10.4049/jimmunol.1301859
128. Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers (2016) 2:16035. doi:10.1038/nrdp.2016.35
129. Singh N, Birkenbach M, Caza T, Perl A, Cohen PL. Tuberous sclerosis and fulminant lupus in a young woman. J Clin Rheumatol (2013) 19:134–7. doi:10.1097/RHU.0b013e318289c033
130. Koga T, Hedrich CM, Mizui M, Yoshida N, Otomo K, Lieberman LA, et al. CaMK4-dependent activation of AKT/mTOR and CREM-alpha underlies autoimmunity-associated Th17 imbalance. J Clin Invest (2014) 124:2234–45. doi:10.1172/JCI73411
131. Kato H, Perl A. The IL-21-mTOR axis blocks treg differentiation and function by suppression of autophagy in patients with systemic lupus erythematosus. Arthritis Rheumatol (2018) 70(3):427–38. doi:10.1002/art.40380
132. Kshirsagar S, Riedl M, Billing H, Tonshoff B, Thangavadivel S, Steuber C, et al. Akt-dependent enhanced migratory capacity of Th17 cells from children with lupus nephritis. J Immunol (2014) 193:4895–903. doi:10.4049/jimmunol.1400044
133. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, et al. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY) (2016) 8:1102–14. doi:10.18632/aging.100925
134. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum (2006) 54:2983–8. doi:10.1002/art.22085
135. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum (2012) 64:2937–46. doi:10.1002/art.34502
136. Tewthanom K, Janwitayanujit S, Totemchockcyakarn K, Panomvana Na Ayudhya D. The effect of high dose of N-acetylcysteine in lupus nephritis: a case report and literature review. J Clin Pharm Ther (2010) 35:483–5. doi:10.1111/j.1365-2710.2009.01108.x
137. Li M, Gao W, Ma J, Zhu Y, Li X. Early-stage lupus nephritis treated with N-acetylcysteine: a report of two cases. Exp Ther Med (2015) 10:689–92. doi:10.3892/etm.2015.2510
138. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (2018) 391:1186–96. doi:10.1016/S0140-6736(18)30485-9
139. O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, Mcinnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med (2015) 66:311–28. doi:10.1146/annurev-med-051113-024537
140. Christie D, Zhu J. Transcriptional regulatory networks for CD4 T cell differentiation. Curr Top Microbiol Immunol (2014) 381:125–72. doi:10.1007/82_2014_372
141. Vahedi G, Takahashi H, Nakayamada S, Sun HW, Sartorelli V, Kanno Y, et al. STATs shape the active enhancer landscape of T cell populations. Cell (2012) 151:981–93. doi:10.1016/j.cell.2012.09.044
142. Goropevsek A, Holcar M, Avcin T. The role of STAT signaling pathways in the pathogenesis of systemic lupus erythematosus. Clin Rev Allergy Immunol (2017) 52:164–81. doi:10.1007/s12016-016-8550-y
143. Gottschalk TA, Tsantikos E, Hibbs ML. Pathogenic inflammation and its therapeutic targeting in systemic lupus erythematosus. Front Immunol (2015) 6:550. doi:10.3389/fimmu.2015.00550
144. Bengtsson AA, Ronnblom L. Role of interferons in SLE. Best Pract Res Clin Rheumatol (2017) 31:415–28. doi:10.1016/j.berh.2017.10.003
145. Hadj-Slimane R, Chelbi-Alix MK, Tovey MG, Bobe P. An essential role for IFN-alpha in the overexpression of Fas ligand on MRL/lpr lymphocytes and on their spontaneous Fas-mediated cytotoxic potential. J Interferon Cytokine Res (2004) 24:717–28. doi:10.1089/1079990042722882
146. Dong J, Wang QX, Zhou CY, Ma XF, Zhang YC. Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus (2007) 16:101–9. doi:10.1177/0961203306075383
147. Karonitsch T, Feierl E, Steiner CW, Dalwigk K, Korb A, Binder N, et al. Activation of the interferon-gamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum (2009) 60:1463–71. doi:10.1002/art.24449
148. Dominguez-Gutierrez PR, Ceribelli A, Satoh M, Sobel ES, Reeves WH, Chan EK. Elevated signal transducers and activators of transcription 1 correlates with increased C-C motif chemokine ligand 2 and C-X-C motif chemokine 10 levels in peripheral blood of patients with systemic lupus erythematosus. Arthritis Res Ther (2014) 16:R20. doi:10.1186/ar4448
149. Dominguez-Gutierrez PR, Ceribelli A, Satoh M, Sobel ES, Reeves WH, Chan EK. Positive correlation of STAT1 and miR-146a with anemia in patients with systemic lupus erythematosus. J Clin Immunol (2014) 34:171–80. doi:10.1007/s10875-013-9973-3
150. Lu MC, Lai NS, Chen HC, Yu HC, Huang KY, Tung CH, et al. Decreased microRNA(miR)-145 and increased miR-224 expression in T cells from patients with systemic lupus erythematosus involved in lupus immunopathogenesis. Clin Exp Immunol (2013) 171:91–9. doi:10.1111/j.1365-2249.2012.04676.x
151. Goropevsek A, Gorenjak M, Gradisnik S, Dai K, Holc I, Hojs R, et al. Increased levels of STAT1 protein in blood CD4 T cells from systemic lupus erythematosus patients are associated with perturbed homeostasis of activated CD45RA(-)FOXP3(hi) regulatory subset and follow-up disease severity. J Interferon Cytokine Res (2017) 37:254–68. doi:10.1089/jir.2016.0040
152. Yiu G, Rasmussen TK, Ajami B, Haddon DJ, Chu AD, Tangsombatvisit S, et al. Development of Th17-associated interstitial kidney inflammation in lupus-prone mice lacking the gene encoding STAT-1. Arthritis Rheumatol (2016) 68:1233–44. doi:10.1002/art.39535
153. Yang XY, Wang HY, Zhao XY, Wang LJ, Lv QH, Wang QQ. Th22, but not Th17 might be a good index to predict the tissue involvement of systemic lupus erythematosus. J Clin Immunol (2013) 33:767–74. doi:10.1007/s10875-013-9878-1
154. Konya C, Paz Z, Apostolidis SA, Tsokos GC. Update on the role of Interleukin 17 in rheumatologic autoimmune diseases. Cytokine (2015) 75:207–15. doi:10.1016/j.cyto.2015.01.003
155. Koga T, Ichinose K, Tsokos GC. T cells and IL-17 in lupus nephritis. Clin Immunol (2017) 185:95–9. doi:10.1016/j.clim.2016.04.010
156. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol (2008) 181:8761–6. doi:10.4049/jimmunol.181.12.8761
157. Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol (2009) 183:3160–9. doi:10.4049/jimmunol.0900385
158. Zickert A, Amoudruz P, Sundstrom Y, Ronnelid J, Malmstrom V, Gunnarsson I. IL-17 and IL-23 in lupus nephritis – association to histopathology and response to treatment. BMC Immunol (2015) 16:7. doi:10.1186/s12865-015-0070-7
159. Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity (2000) 13:715–25. doi:10.1016/S1074-7613(00)00070-4
160. Parham C, Chirica M, Timans J, Vaisberg E, Travis M, Cheung J, et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J Immunol (2002) 168:5699–708. doi:10.4049/jimmunol.168.11.5699
161. Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol (2008) 181:5948–55. doi:10.4049/jimmunol.181.9.5948
162. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, et al. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A (2006) 103:8137–42. doi:10.1073/pnas.0600666103
163. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem (2007) 282:9358–63. doi:10.1074/jbc.C600321200
164. Nakou M, Bertsias G, Stagakis I, Centola M, Tassiulas I, Hatziapostolou M, et al. Gene network analysis of bone marrow mononuclear cells reveals activation of multiple kinase pathways in human systemic lupus erythematosus. PLoS One (2010) 5:e13351. doi:10.1371/journal.pone.0013351
165. Liu Q, Du Y, Li K, Zhang W, Feng X, Hao J, et al. Anti-OSM antibody inhibits tubulointerstitial lesion in a murine model of lupus nephritis. Mediators Inflamm (2017) 2017:3038514. doi:10.1155/2017/3038514
166. Harada T, Kyttaris V, Li Y, Juang YT, Wang Y, Tsokos GC. Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity (2007) 40:1–8. doi:10.1080/08916930601095148
167. Hedrich CM, Rauen T, Apostolidis SA, Grammatikos AP, Rodriguez-Rodriguez N, Ioannidis C, et al. Stat3 promotes IL-10 expression in lupus T cells through trans-activation and chromatin remodeling. Proc Natl Acad Sci U S A (2014) 111:13457–62. doi:10.1073/pnas.1408023111
168. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood (2012) 119:3997–4008. doi:10.1182/blood-2011-11-392985
169. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16:142–52. doi:10.1038/ni.3054
170. Uhm WS, Na K, Song GW, Jung SS, Lee T, Park MH, et al. Cytokine balance in kidney tissue from lupus nephritis patients. Rheumatology (Oxford) (2003) 42:935–8. doi:10.1093/rheumatology/keg255
171. Mellor-Pita S, Citores MJ, Castejon R, Yebra-Bango M, Tutor-Ureta P, Rosado S, et al. Monocytes and T lymphocytes contribute to a predominance of interleukin 6 and interleukin 10 in systemic lupus erythematosus. Cytometry B Clin Cytom (2009) 76:261–70. doi:10.1002/cyto.b.20468
172. Zhao M, Tang J, Gao F, Wu X, Liang Y, Yin H, et al. Hypomethylation of IL10 and IL13 promoters in CD4+ T cells of patients with systemic lupus erythematosus. J Biomed Biotechnol (2010) 2010:931018. doi:10.1155/2010/931018
173. Edwards LJ, Mizui M, Kyttaris V. Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin Immunol (2015) 158:221–30. doi:10.1016/j.clim.2015.04.004
174. Liu K, Liang C, Liang Z, Tus K, Wakeland EK. Sle1ab mediates the aberrant activation of STAT3 and Ras-ERK signaling pathways in B lymphocytes. J Immunol (2005) 174:1630–7. doi:10.4049/jimmunol.174.3.1630
175. Ripoll E, De Ramon L, Draibe Bordignon J, Merino A, Bolanos N, Goma M, et al. JAK3-STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res Ther (2016) 18:134. doi:10.1186/s13075-016-1034-x
176. Furumoto Y, Smith CK, Blanco L, Zhao W, Brooks SR, Thacker SG, et al. Tofacitinib ameliorates murine lupus and its associated vascular dysfunction. Arthritis Rheumatol (2017) 69:148–60. doi:10.1002/art.39818
177. Markham A. Baricitinib: first global approval. Drugs (2017) 77:697–704. doi:10.1007/s40265-017-0723-3
178. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, Mckenzie BS, et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J Exp Med (2006) 203:2473–83. doi:10.1084/jem.20061099
179. Izcue A, Hue S, Buonocore S, Arancibia-Carcamo CV, Ahern PP, Iwakura Y, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity (2008) 28:559–70. doi:10.1016/j.immuni.2008.02.019
180. Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol (2010) 184:4605–9. doi:10.4049/jimmunol.0903595
181. Dai H, He F, Tsokos GC, Kyttaris VC. IL-23 limits the production of IL-2 and promotes autoimmunity in lupus. J Immunol (2017) 199:903–10. doi:10.4049/jimmunol.1700418
182. Shaltout AS, Sayed D, Badary MS, Nafee AM, El Zohri MH, Bakry R, et al. Effect of IL6 and IL23 on double negative T cells and anti ds-DNA in systemic lupus erythematosus patients. Hum Immunol (2016) 77:937–43. doi:10.1016/j.humimm.2016.06.007
183. Van Vollenhoven RF, Hahn BH, Tsokos GC, Wagner C, Lipsky P, Hsu B, et al. Efficacy and safety of ustekinumab, an interleukin 12/23 inhibitor, in patients with active systemic lupus erythematosus: results of a phase 2, randomized placebo-controlled study [abstract]. Arthritis Rheumatol (2017) 69:(suppl 10).
184. Kyttaris VC, Kampagianni O, Tsokos GC. Treatment with anti-interleukin 23 antibody ameliorates disease in lupus-prone mice. Biomed Res Int (2013) 2013:861028. doi:10.1155/2013/861028
185. Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, et al. Clinical improvement in psoriasis with specific targeting of interleukin-23. Nature (2015) 521:222–6. doi:10.1038/nature14175
186. Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet (2017) 390:276–88. doi:10.1016/S0140-6736(17)31279-5
187. Sands BE, Chen J, Feagan BG, Penney M, Rees WA, Danese S, et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, in patients with moderate to severe Crohn’s disease: a phase 2a study. Gastroenterology (2017) 153:77–86.e6. doi:10.1053/j.gastro.2017.03.049
188. Crispin JC, Apostolidis SA, Rosetti F, Keszei M, Wang N, Terhorst C, et al. Cutting edge: protein phosphatase 2A confers susceptibility to autoimmune disease through an IL-17-dependent mechanism. J Immunol (2012) 188:3567–71. doi:10.4049/jimmunol.1200143
189. Hedrich CM, Crispin JC, Rauen T, Ioannidis C, Apostolidis SA, Lo MS, et al. cAMP response element modulator alpha controls IL2 and IL17A expression during CD4 lineage commitment and subset distribution in lupus. Proc Natl Acad Sci U S A (2012) 109:16606–11. doi:10.1073/pnas.1210129109
190. Yoshida N, Comte D, Mizui M, Otomo K, Rosetti F, Mayadas TN, et al. ICER is requisite for Th17 differentiation. Nat Commun (2016) 7:12993. doi:10.1038/ncomms12993
191. Juang YT, Wang Y, Solomou EE, Li Y, Mawrin C, Tenbrock K, et al. Systemic lupus erythematosus serum IgG increases CREM binding to the IL-2 promoter and suppresses IL-2 production through CaMKIV. J Clin Invest (2005) 115:996–1005. doi:10.1172/JCI22854
192. Ichinose K, Juang YT, Crispin JC, Kis-Toth K, Tsokos GC. Suppression of autoimmunity and organ pathology in lupus-prone mice upon inhibition of calcium/calmodulin-dependent protein kinase type IV. Arthritis Rheum (2011) 63:523–9. doi:10.1002/art.30085
193. Koga T, Ichinose K, Mizui M, Crispin JC, Tsokos GC. Calcium/calmodulin-dependent protein kinase IV suppresses IL-2 production and regulatory T cell activity in lupus. J Immunol (2012) 189:3490–6. doi:10.4049/jimmunol.1201785
194. Kurschus FC, Moos S. IL-17 for therapy. J Dermatol Sci (2017) 87:221–7. doi:10.1016/j.jdermsci.2017.06.010
195. Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol (2015) 67:475–87. doi:10.1002/art.38955
196. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12:180–90. doi:10.1038/nri3156
197. Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell (1993) 75:253–61. doi:10.1016/0092-8674(93)80067-O
198. Sadlack B, Lohler J, Schorle H, Klebb G, Haber H, Sickel E, et al. Generalized autoimmune disease in interleukin-2-deficient mice is triggered by an uncontrolled activation and proliferation of CD4+ T cells. Eur J Immunol (1995) 25:3053–9. doi:10.1002/eji.1830251111
199. Suzuki H, Kundig TM, Furlonger C, Wakeham A, Timms E, Matsuyama T, et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science (1995) 268:1472–6. doi:10.1126/science.7770771
200. Willerford DM, Chen J, Ferry JA, Davidson L, Ma A, Alt FW. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity (1995) 3:521–30. doi:10.1016/1074-7613(95)90180-9
201. Malek TR, Yu A, Vincek V, Scibelli P, Kong L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity (2002) 17:167–78. doi:10.1016/S1074-7613(02)00367-9
202. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6:1142–51. doi:10.1038/ni1263
203. Caudy AA, Reddy ST, Chatila T, Atkinson JP, Verbsky JW. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol (2007) 119:482–7. doi:10.1016/j.jaci.2006.10.007
204. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity (2007) 26:371–81. doi:10.1016/j.immuni.2007.02.009
205. Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol (2012) 13:770–7. doi:10.1038/ni.2363
206. Ray JP, Staron MM, Shyer JA, Ho PC, Marshall HD, Gray SM, et al. The interleukin-2-mTORc1 kinase axis defines the signaling, differentiation, and metabolism of T helper 1 and follicular B helper T cells. Immunity (2015) 43:690–702. doi:10.1016/j.immuni.2015.08.017
207. Lenardo MJ. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature (1991) 353:858–61. doi:10.1038/353858a0
208. Refaeli Y, Van Parijs L, London CA, Tschopp J, Abbas AK. Biochemical mechanisms of IL-2-regulated Fas-mediated T cell apoptosis. Immunity (1998) 8:615–23. doi:10.1016/S1074-7613(00)80566-X
209. Altman A, Theofilopoulos AN, Weiner R, Katz DH, Dixon FJ. Analysis of T cell function in autoimmune murine strains. Defects in production and responsiveness to interleukin 2. J Exp Med (1981) 154:791–808. doi:10.1084/jem.154.3.791
210. Alcocer-Varela J, Alarcon-Segovia D. Decreased production of and response to interleukin-2 by cultured lymphocytes from patients with systemic lupus erythematosus. J Clin Invest (1982) 69:1388–92. doi:10.1172/JCI110579
211. Linker-Israeli M, Bakke AC, Kitridou RC, Gendler S, Gillis S, Horwitz DA. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE). J Immunol (1983) 130:2651–5.
212. Rothenberg EV, Ward SB. A dynamic assembly of diverse transcription factors integrates activation and cell-type information for interleukin 2 gene regulation. Proc Natl Acad Sci U S A (1996) 93:9358–65. doi:10.1073/pnas.93.18.9358
213. Kyttaris VC, Juang YT, Tenbrock K, Weinstein A, Tsokos GC. Cyclic adenosine 5’-monophosphate response element modulator is responsible for the decreased expression of c-fos and activator protein-1 binding in T cells from patients with systemic lupus erythematosus. J Immunol (2004) 173:3557–63. doi:10.4049/jimmunol.173.5.3557
214. Katsiari CG, Tsokos GC. Transcriptional repression of interleukin-2 in human systemic lupus erythematosus. Autoimmun Rev (2006) 5:118–21. doi:10.1016/j.autrev.2005.08.009
215. Katsiari CG, Kyttaris VC, Juang YT, Tsokos GC. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J Clin Invest (2005) 115:3193–204. doi:10.1172/JCI24895
216. Katsuyama E, Yan M, Watanabe KS, Matsushima S, Yamamura Y, Hiramatsu S, et al. Downregulation of miR-200a-3p, targeting CtBP2 complex, is involved in the hypoproduction of IL-2 in systemic lupus erythematosus-derived T cells. J Immunol (2017) 198:4268–76. doi:10.4049/jimmunol.1601705
217. Koreth J, Matsuoka K, Kim HT, Mcdonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365:2055–66. doi:10.1056/NEJMoa1108188
218. Matsuoka K, Koreth J, Kim HT, Bascug G, Mcdonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Transl Med (2013) 5:179ra143. doi:10.1126/scitranslmed.3005265
219. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1:295–305. doi:10.1016/S2213-8587(13)70113-X
220. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365:2067–77. doi:10.1056/NEJMoa1105143
221. Humrich JY, Von Spee-Mayer C, Siegert E, Alexander T, Hiepe F, Radbruch A, et al. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann Rheum Dis (2015) 74:791–2. doi:10.1136/annrheumdis-2014-206506
222. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22:991–3. doi:10.1038/nm.4148
223. Mizui M, Tsokos GC. Low-dose IL-2 in the treatment of lupus. Curr Rheumatol Rep (2016) 18:68. doi:10.1007/s11926-016-0617-5
224. Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4-CD8- IL-17-producing T cells. J Immunol (2014) 193:2168–77. doi:10.4049/jimmunol.1400977
225. von Spee-Mayer C, Siegert E, Abdirama D, Rose A, Klaus A, Alexander T, et al. Low-dose interleukin-2 selectively corrects regulatory T cell defects in patients with systemic lupus erythematosus. Ann Rheum Dis (2016) 75:1407–15. doi:10.1136/annrheumdis-2015-207776
226. Comte D, Karampetsou MP, Kis-Toth K, Yoshida N, Bradley SJ, Kyttaris VC, et al. Brief report: CD4+ T cells from patients with systemic lupus erythematosus respond poorly to exogenous interleukin-2. Arthritis Rheumatol (2017) 69:808–13. doi:10.1002/art.40014
227. Comte D, Karampetsou MP, Kis-Toth K, Yoshida N, Bradley SJ, Mizui M, et al. Engagement of SLAMF3 enhances CD4+ T-cell sensitivity to IL-2 and favors regulatory T-cell polarization in systemic lupus erythematosus. Proc Natl Acad Sci U S A (2016) 113:9321–6. doi:10.1073/pnas.1605081113
228. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi:10.1084/jem.20030152
229. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A critical function for TGF-beta signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat Immunol (2008) 9:632–40. doi:10.1038/ni.1607
230. Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ, et al. Excessive Th1 responses due to the absence of TGF-beta signaling cause autoimmune diabetes and dysregulated Treg cell homeostasis. Proc Natl Acad Sci U S A (2013) 110:6961–6. doi:10.1073/pnas.1304498110
231. Oh SA, Liu M, Nixon BG, Kang D, Toure A, Bivona M, et al. Foxp3-independent mechanism by which TGF-beta controls peripheral T cell tolerance. Proc Natl Acad Sci U S A (2017) 114:E7536–44. doi:10.1073/pnas.1706356114
232. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi:10.1016/j.immuni.2006.01.001
233. Ghoreschi K, Laurence A, Yang XP, Tato CM, Mcgeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature (2010) 467:967–71. doi:10.1038/nature09447
234. Becker-Merok A, Eilertsen GO, Nossent JC. Levels of transforming growth factor-beta are low in systemic lupus erythematosus patients with active disease. J Rheumatol (2010) 37:2039–45. doi:10.3899/jrheum.100180
235. Edelbauer M, Kshirsagar S, Riedl M, Billing H, Tonshoff B, Haffner D, et al. Activity of childhood lupus nephritis is linked to altered T cell and cytokine homeostasis. J Clin Immunol (2012) 32:477–87. doi:10.1007/s10875-011-9637-0
236. Yuan Y, Yang M, Wang K, Sun J, Song L, Diao X, et al. Excessive activation of the TLR9/TGF-beta1/PDGF-B pathway in the peripheral blood of patients with systemic lupus erythematosus. Arthritis Res Ther (2017) 19:70. doi:10.1186/s13075-017-1238-8
237. Elbeldi-Ferchiou A, Ben Ahmed M, Smiti-Khanfir M, Houman MH, Abdeladhim M, Belhadj Hmida N, et al. Resistance to exogenous TGF-beta effects in patients with systemic lupus erythematosus. J Clin Immunol (2011) 31:574–83. doi:10.1007/s10875-011-9531-9
238. Okamura T, Sumitomo S, Morita K, Iwasaki Y, Inoue M, Nakachi S, et al. TGF-beta3-expressing CD4+CD25(-)LAG3+ regulatory T cells control humoral immune responses. Nat Commun (2015) 6:6329. doi:10.1038/ncomms7329
239. Morita K, Okamura T, Inoue M, Komai T, Teruya S, Iwasaki Y, et al. Egr2 and Egr3 in regulatory T cells cooperatively control systemic autoimmunity through Ltbp3-mediated TGF-beta3 production. Proc Natl Acad Sci U S A (2016) 113:E8131–40. doi:10.1073/pnas.1611286114
240. Nambiar MP, Enyedy EJ, Fisher CU, Krishnan S, Warke VG, Gilliland WR, et al. Abnormal expression of various molecular forms and distribution of T cell receptor zeta chain in patients with systemic lupus erythematosus. Arthritis Rheum (2002) 46:163–74. doi:10.1002/1529-0131(200201)46:1<163::AID-ART10065>3.0.CO;2-J
Keywords: SLE, autoimmunity, signaling, T cells, Autoimmune disease
Citation: Katsuyama T, Tsokos GC and Moulton VR (2018) Aberrant T Cell Signaling and Subsets in Systemic Lupus Erythematosus. Front. Immunol. 9:1088. doi: 10.3389/fimmu.2018.01088
Received: 05 February 2018; Accepted: 01 May 2018;
Published: 17 May 2018
Edited by:
Massimo Gadina, National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), United StatesReviewed by:
Johan Van Der Vlag, Radboud University Nijmegen, NetherlandsCaroline Jefferies, Cedars-Sinai Medical Center, United States
Copyright: © 2018 Katsuyama, Tsokos and Moulton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vaishali R. Moulton, dm1vdWx0b25AYmlkbWMuaGFydmFyZC5lZHU=