 Paris Kosti
Paris Kosti John Maher
John Maher James N. Arnold
James N. Arnold- 1Faculty of Life Sciences and Medicine, School of Cancer and Pharmaceutical Sciences, King’s College London, Guy’s Hospital, London, United Kingdom
- 2Department of Immunology, Eastbourne Hospital, Eastbourne, East Sussex, United Kingdom
- 3Department of Clinical Immunology and Allergy, King’s College Hospital NHS Foundation Trust, London, United Kingdom
Chimeric antigen receptor (CAR) T-cell therapy entails the genetic engineering of a patient’s T-cells to express membrane spanning fusion receptors with defined specificities for tumor-associated antigens. These CARs are capable of eliciting robust T-cell activation to initiate killing of the target tumor cells. This therapeutic approach has produced unprecedented clinical outcomes in the treatment of “liquid” hematologic cancers, but to date has not produced comparable responses in targeting solid malignancies. Advances in our understanding of the immunobiology of solid tumors have highlighted several hurdles which currently hinder the efficacy of this therapy. These barriers include the insufficient accumulation of CAR T-cells in the tumor due to poor trafficking or physical exclusion and the exposure of infiltrating CAR T-cells to a panoply of immune suppressive checkpoint molecules, cytokines, and metabolic stresses that are not conducive to efficient immune reactions and can thereby render these cells anergic, exhausted, or apoptotic. This mini-review summarizes these hurdles and describes some recent approaches and innovations to genetically re-engineer CAR T-cells to counter inhibitory influences found in the tumor microenvironment. Novel immunotherapy drug combinations to potentiate the activity of CAR T-cells are also discussed. As our understanding of the immune landscape of tumors improves and our repertoire of immunotherapeutic drugs expands, it is envisaged that the efficacy of CAR T-cells against solid tumors might be potentiated using combination therapies, which it is hoped may lead to meaningful improvements in clinical outcome for patients with refractory solid malignancies.
Introduction
Chimeric antigen receptor (CAR) T-cell immunotherapy reached a significant milestone in 2017, receiving its first approval by the U.S. Food and Drug Administration for two CD19-targeted CAR T-cells, Tisagenlecleucel (1) and Axicabtagene Ciloleucel (2). This achievement paves the way for further expansion of this therapeutic approach in the treatment of cancer. CAR T-cell therapy involves the isolation and ex vivo expansion of the patient’s peripheral blood T-cells, followed by genetic engineering of these cells to express CAR molecules on the cell surface, which have specificity for non-HLA-restricted tumor antigens. The genetically modified and expanded T-cells are then re-infused back into the patient, often following the administration of lymphodepleting chemotherapy (3).
The CAR construct has become progressively more sophisticated over time as our knowledge of T-cell activation and the tumor microenvironment (TME) has improved. The endodomain of CAR molecules, which transmits the activation signal from the ectodomain, contains a variety of signaling and co-stimulatory moieties which are indicative of their “generation” and can include CD3ζ, CD28, CD27, 4-1BB, ICOS, and OX40 (4, 5) (Figure 1). As such, CAR molecules circumvent the requirement to engage with exogenous co-stimulatory molecules for T-cell activation, which can be lacking in the TME and compromise CD8+ T-cell responses (6). More recently, CAR vectors have been designed to co-express auxiliary receptors and cytokines to improve T-cell function, which will be discussed later in this review (Figure 1).
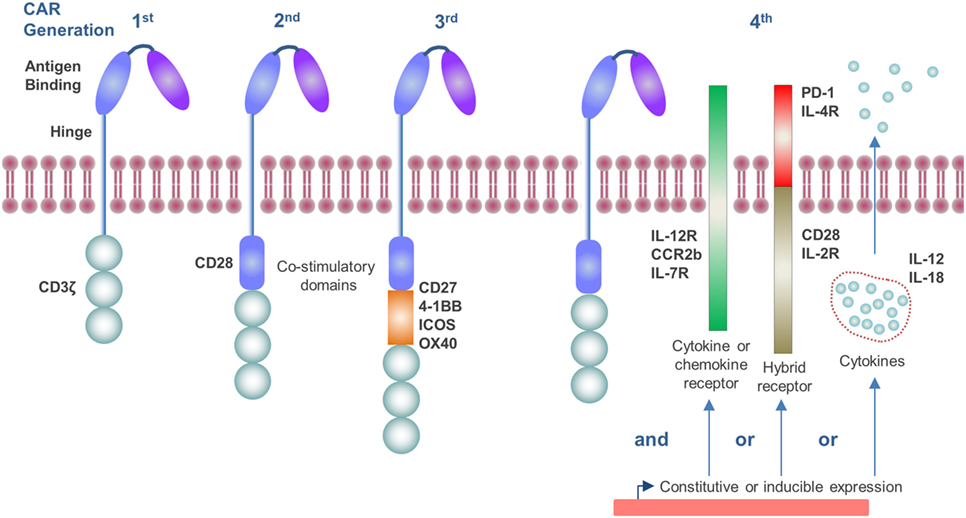
Figure 1. Generations of chimeric antigen receptor (CAR) molecules. First generation CARs contain a CD3ζ signaling endodomain. Second and third generation CARs, in addition to the CD3ζ domain, incorporate CD28 (second generation) or two or more additional co-stimulatory domains which may include CD27, 4-1BB, ICOS, or OX40 (third generation). Fourth generation CARs include constitutive or inducible expression of co-receptors or soluble cytokines alongside that of the CAR molecule which further promote T-cell activation.
Chimeric antigen receptor T-cell immunotherapy has achieved unprecedented clinical outcomes in patients with B-cell malignancies that previously had a very poor survival probability. At several centers, response rates consistently exceeding 80% have been reported in patients with relapsed/refractory B-cell acute lymphoblastic leukemia (ALL) (7–9) and lymphoma (10). Using anti-CD19 CAR T-cells in a Phase II trial involving 101 patients with B-cell lymphoma, 82% of patients had an overall objective response, and 54% had a complete response (2). Building on this highly impressive clinical data, CAR T-cells targeted against B-cell maturation antigen achieved a 89% overall response rate in 18 patients with evaluable multiple myeloma (11). Also, in a global multi-center Phase II trial, Tisagenlecleucel achieved an overall response rate of 81% in 75 pediatric and young adult patients with CD19+ relapsed or refractory B-cell ALL (12). With such impressive clinical responses, it is understandable that there has been significant interest in applying this therapy to solid malignancies, which account for the majority of cancer-related morbidity and mortality.
Clinical Evaluation of CAR T-Cell Immunotherapy for Solid Tumors
Chimeric antigen receptor T-cells have been evaluated for the treatment of a variety of solid tumors (13–17). However, the proportion of patients responding with a measurable objective clinical response in these trials has been variable. Anti-disialoganglioside GD2 CAR T-cells have been used to treat evaluable pediatric patients with neuroblastoma, where 3 of 11 patients with active disease achieved complete remission (13, 18). However, in a trial using epidermal growth factor receptor-targeted CAR T-cells in patients with non-small cell lung cancer, partial disease remission in 2 of 11 patients was the best clinical response (15). There are also instances, using other CAR targets, where stable disease was the best clinical response (19, 20) or no objective clinical responses (21–23) have been detected. Although clinical responses of CAR T-cell therapy in solid tumors to date have not paralleled the success seen in liquid cancers [extensively reviewed (16)], the fact that clinical responses have been observed provides some encouragement.
CAR Target Selection for Solid Tumors
A major hurdle in implementing CAR T-cell therapy against solid tumors is target selection. Since most solid tumors are of epithelial origin, the presence of tumor-specific antigens, which are absent on normal epithelial cells, is rare (24). This has resulted in instances of on-target off-tumor toxicities, such as was observed using Her2/neu targeted CAR T-cells in a patient with breast cancer (25). To expand the range of tumor-associated antigens (TAAs) that can be targeted, T-cell receptor (TCR)-mimetic CARs with specificity to HLA-presented antigens have been tested (26). CAR expression systems which exploit combinations of antigens, for example where ligation of a synthetic Notch receptor induces CAR expression (27), or tuning CAR affinity to preferentially target high density antigens (28), provide approaches to improve specificity. Dual-antigen targeting, where the CAR molecule can engage two separate TAAs can also be used to overcome antigen escape (29). Alternatively, the inclusion of inducible suicide switches which render CAR T-cells apoptotic have also been explored as a safety mechanism should toxicities arise (30, 31). The tumor stroma, which plays an important role in disease progression, has also been evaluated as a target. CAR T-cells targeting fibroblast activation protein alpha (FAP) which is expressed on the surface of cancer associated fibroblasts have shown efficacy in controlling tumor growth in preclinical models (32–34). However, as FAP+ stromal cells also play important roles in the periphery (35), off-tumor targeting of these populations by CAR T-cells results in cachexia and hematological toxicities in murine models, raising potential concern over FAP as a target (36).
Solid tumors contain various physical and environmental barriers which are not present in liquid cancers, and need to be considered. In the sections that follow, we consider the hurdles that T-cells encounter in the solid TME and how these may be overcome using novel innovations in CAR engineering or immunotherapeutic approaches.
Physical and Environmental Barriers to CAR T-Cell Therapy in Solid Tumors
Not Enough “Traffic” for CAR T-Cells
Unlike hematologic cancers where the infused CAR T-cells and tumor cells co-circulate in the blood, bone marrow, and lymphatics with ample opportunity for interaction, solid tumors represent discrete foci to which infused cells must migrate in order to contact the tumor cells and engage antigen. This involves chemotaxis in response to chemokines, such as CXCL-9, -10, and -11 (37). Since many human cancers display poor T-cell infiltration (“cold tumors”) (38), it is unsurprising that migration of CAR T-cells to the TME can be inefficient. Even the i.v. infusion of large numbers of CAR T-cells does not improve the clinical outcomes when tackling solid tumors, especially in patients with bulky disease; however, these trials were using the early first generation CARs (39, 40) (Figure 1). When clinically feasible, intra-tumoral injection has been demonstrated in vivo to circumvent poor trafficking of CAR T-cells, leading to tumor regression without systemic toxicity (41). In addition to the lack of T-cell chemokines being secreted by the TME, there are other chemokines, such as CXCL12, which actively inhibit T-cell migration into the tumor through engaging CXCR4 on their cell surface (42, 43). CXCL12 is highly expressed in variety of carcinomas, including pancreatic (44, 45), ovarian (46), and breast (47). However, pharmacologically blocking CXCR4 on the T-cell surface facilitated infiltration of T-cells into a spontaneous murine model of pancreatic ductal adenocarcinoma (43). As such, the CXCR4/CXCL12 axis may represent a therapeutic target to facilitate CAR T-cell infiltration in some solid tumors. Extravasation of T-cells into the tumor from the blood can also be inefficient due to the expression of molecules such as endothelin B receptor (ETBR) on the endothelium of the blood vessels within the tumor. High ETBR expression results in the nitric oxide-mediated deregulation of endothelial ICAM-1 expression, which reduces T-cell adhesion and compromises their ability to extravasate (48). Several attempts have been made to improve CAR T-cell trafficking to tumors (49–51). Kershaw and colleagues have demonstrated that expression of CXCR2 improved T-cell migration in a melanoma tumor which produced CXCL1, a chemokine commonly secreted by tumor cells (52). Similarly, CCR2b, the chemokine receptor for CCL2, has been co-expressed on CAR T-cells to exploit the CCR2/CCL2 axis, which facilitates myeloid cell recruitment into the tumor. CCR2b-expressing GD2-specific CAR T-cells had a 10-fold improvement in their migration to CCL2-producing neuroblastoma and demonstrated better in vivo antitumor activity in murine models (50). A similar observation was also achieved using mesothelin-directed CAR T-cells in murine models of mesothelioma (51). Others have improved CAR T-cell trafficking and the antitumor response in vivo using CAR T-cells that express a regulatory subunit I anchoring disrupter (RIAD) peptide that inhibits the protein kinase A-mediated suppression of the TCR, which can occur in the TME (53).
There are also physical barriers that restrict T-cell infiltration within solid tumors, where extracellular matrix proteins, such as proteoglycans and collagen, can restrict entry (54, 55). To overcome this, CAR T-cells have been engineered to express heparanase, which degrades heparin sulfate proteoglycans (56). This approach enhanced CAR T-cell infiltration within the tumor, leading to improved overall survival in xenograft tumor models.
Immune Checkpoint Molecules
Intra-tumoral T-cells are often functionally tolerant, or suppressed, displaying reduced effector functions compared to peripheral T-cell pools, including lowered cytokine, perforin, and granzyme-B expression (57, 58). This phenomenon has also been observed for CAR T-cells (59). Two receptors that have been of particular interest in inducing this anergic T-cell state are PD-1 and CTLA-4, which represent part of a family of regulatory receptor known as checkpoint molecules, which prevent inappropriate immune activation, but can be exploited by cancer. For example the ligands for PD-1, PDL-1 and PDL-2, can be found expressed on a variety of tumor and stromal cells (60–62). Antibody-mediated blockade of immune checkpoint receptors that are expressed on the T-cell surface have shown unprecedented clinical activity in the treatment of solid tumors by sustaining endogenous antitumor immune responses, most notably in melanoma (63). The checkpoint molecules offer significant scope as therapeutic targets for combination therapy, as these molecules behave in a hierarchical structure within each TME (43, 61). Indeed, combined PD-1 and CTLA-4 blockade (using nivolumab and ipilimumab, respectively) has improved response rates in patients with advanced melanoma, compared to that of the single therapies (64). Importantly, regulatory molecules such as PDL-1 are upregulated by effector molecules of T-cell activation, such as interferon (IFN)-γ (60, 65), suggesting that the greater the T-cell activity, the more suppressive the TME becomes. Anti-HER2 CAR T-cell therapy has an enhanced antitumor response in preclinical murine models when combined with PD-1 blockade (66). The combination of immune checkpoint blockade with CAR T-cell immunotherapy is now under study in clinical trials (67). Innovations to CAR T-cells to permit insensitivity to immune checkpoints are also under investigation at the preclinical stage, involving such approaches as genetic inactivation of the PD-1 gene (68), co-expressing dominant-negative variants of the inhibitory phosphatases, such as Src homology 2 phosphatase (SHP-2), which mediate the signaling of checkpoint receptors (69), or the expression of PD-1 receptors with no signaling moiety as decoy molecules (70). Others have attempted to positively exploit the interaction by fusing the PD-1 ectodomain to the CD28 cytoplasmic tail to induce a co-stimulatory, instead of inhibitory, signal upon PDL-1 binding (71). Brentjens et al. enhanced the antitumor efficacy of CD19-specific CAR T-cell in vivo by co-expressing the activating checkpoint molecule, CD40-ligand (72). CD40L co-expression resulted in an increased CAR T-cell proliferation, Th1 cytokine secretion, and increased cytotoxicity in vivo. The stroma, as well as the tumor cells, play a fundamental role in suppressing T-cells responses in the TME. Our group has shown that tumor-associated macrophages (TAMs) can inhibit immune-mediated tumor rejection in vivo through their expression of the heme-degrading enzyme heme oxygenase-1 (HO-1) (61, 73). Carbon monoxide is one of the by-products of heme catabolism by HO-1 and has been demonstrated to be capable of suppressing T-cell proliferation, IL-2 secretion (74), and T-cell effector function (61, 75, 76). The pharmacological inhibition of this enzyme within the TME of a murine model of breast cancer resulted in a rapid restoration of a chemotherapy-elicited antitumor CD8+ T-cell response and immunological control of tumor growth, leading us to propose that HO-1 should also be considered as an immune checkpoint molecule (61). As HO-1 is expressed in a variety of cancers (77), it may warrant therapeutic targeting in CAR T-cell therapy. Together, these considerations present a strong rationale for combining CAR T-cell and immune checkpoint blockade therapies. Appropriate combinations of these therapies may improve CAR T-cell efficacy in the TME and support more favorable clinical response rates.
The Anti-Inflammatory Cytokine Environment
The TME is a chronic inflammatory site and contains a variety of cytokines and chemokines, which influence the immune response. The TME is often skewed toward favoring pro-tumor Th2 cytokines, such as IL-4 and IL-13, rather than antitumor Th1 cytokines like IFN-γ and tumor necrosis factor-β (78). Cytotoxic T-cell function is depressed by cytokines, such as IL-4, IL-10, and transforming growth factor (TGF)-β, which are prevalent in the TME (79, 80). TAMs are an abundant stromal cell type in solid tumors and have been shown to secrete IL-10 as well as IL-6, which also suppresses T-cell cytolytic function and proliferation (81, 82). Similarly, regulatory T-cells (Tregs; CD4+ CD25+ FoxP3+) produce significant quantities of suppressive cytokines, including IL-10, TGF-β (83), and IL-35 (84). T-cells, which were engineered to express a dominant-negative form of the TGF-β receptor, have been demonstrated to have augmented antitumor responses in TGF-β producing tumors (85, 86). Alternatively, IL-4 signaling has been exploited to promote CAR T-cell expansion and activity. In one such approach, the IL-4 ectodomain has been fused to the common β chain of IL-2 and IL-15 receptors in order to achieve selective ex vivo amplification of CAR transduced T-cells. This approach renders it feasible to manufacture CAR T-cells from whole blood, circumventing the need for leukapheresis (87) and has the added benefit of augmenting CAR T-cell responses against tumors containing IL-4 in the TME (88, 89). Alternatively, the IL-4 receptor ectodomain was fused to the IL-7 receptor endodomain and co-expressed in prostate stem cell antigen-specific CAR T-cells in order to transduce a T-cell proliferation signal in response to IL-4 (90). The co-expression of this receptor led to improved in vivo CAR T-cell expansion and antitumor response. IL-12 is a potent inflammatory cytokine which enhances T-cell expansion and antitumor immune responses (91, 92). In murine preclinical tumor models, CAR T-cells which constitutively express IL-12 have been demonstrated to have enhanced proliferation, in vivo expansion, cytotoxicity, and antitumor efficacy (93–95). To circumvent off-target toxicities of this approach to expedite clinical translation, inducible IL-12 expression linked to CAR engagement has also been demonstrated to improve antitumor responses (96). CAR T-cells co-expressing IL-18 (97, 98), a constitutively active IL-7 cytokine receptor (99) or a tethered form of IL-15 (IL-15 peptide fused to IL-15Rα via flexible linker) (100) have also all augmented their antitumor response in vivo. It is clear that the cytokine environment within the TME is not conducive to permitting CD8+ T-cell activation and effector function. However, innovative ways to modulate and positively exploit the response of the CAR T-cells, such as through hybrid receptors, or IL-12 (Figure 1) show promising preclinical data and have started to progress through to clinical trials (NCT02498912 and NCT01818323).
Metabolism-Associated Immune Suppression in the TME
Tumor cells are highly metabolically active with increased glycolysis and glutaminolysis (101). These metabolic pathways result in a distinct accumulation of metabolites in the TME which can compromise CAR T-cell function. Lactate is a metabolite derived from the glycolytic-pathway that is highly produced by tumor cells and directly suppresses proliferation, cytokine production, and effector function of human cytotoxic T lymphocytes (102). Prostaglandins, which are derived from prostaglandin E2 synthase and cyclooxygenase (COX)-1/2-mediated catabolism of arachidonic acid, can also suppress T-cell function (103). In keeping with this, aspirin (a COX inhibitor), has been demonstrated to potentiate immune checkpoint therapy in improving CD8+ T-cell responses (104) and may warrant investigation in combination with CAR T-cell immunotherapy.
A number of amino acid-degrading enzymes that are commonly expressed in the TME can also suppress T-cell function. These include indoleamine-2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO) which degrade tryptophan, and arginase-1 and nitric oxide synthase (NOS) which degrade l-arginine. T-cells have been demonstrated to be particularly sensitive to the depletion of these amino acids, resulting in impaired proliferation and effector function (105–107) and increased T-cell apoptosis (108, 109). As well as the physical depletion of these amino acids, the catabolites of tryptophan degradation such as l-kynurenine and 3-hydroxyanthranilic acid have also been demonstrated to be immunosuppressive (110, 111). IDO inhibitors have been demonstrated to enhance CAR T-cell efficacy (111), and these are now being evaluated in clinical trials (112, 113). Arginase activity has also been demonstrated to inhibit proliferation and cytotoxity of GD2-specific CAR T-cells (114). The degradation of l-arginine by the NOS pathway also results in the generation of reactive nitrogen species (RNS). Myeloid-derived suppressor cells (Gr-1+ CD11b+ cells) and TAMs are potent sources of reactive oxygen species (ROS), which inhibit T-cell function (115). Both ROS and RNS are believed to induce T-cell tolerance by altering the flexibility of the TCR chains, which impairs the binding and responsiveness of CD8+ T-cells to peptide–MHC complexes (116). It would be interesting to consider how ROS/RNS modifications might also influence the antigen binding capabilities of the CAR. ROS/RNS may also inhibit T-cell infiltration into the tumor through inactivating CCL2 by nitration (117). Metabolites generated in the tumor, once regarded as by-products, are now accepted as crucial immune-modulatory molecules in their own right. As such, the metabolic pathways that facilitate immune-regulation may require therapeutic targeting alongside CAR T-cell therapy, such as IDO, which is currently under clinical investigation.
Conclusion
The success of CAR T-cell immunotherapy for hematological malignancies heralds a new era in the treatment of malignant disease. However, as this review has highlighted, the attainment of comparable clinical outcomes for patients with solid tumors will require considerable refinement of this therapeutic approach. Although there have been some encouraging recent case reports (118), CAR T-cells are subject to several additional constraints in patients with solid tumors which has hindered progress (Figure 2). The parallel success of immune checkpoint blockade therapies presents an opportunity for realignment of these distinct forms of immunotherapy through combinatorial therapeutic regimens. Similar pharmacologic opportunities are presented by the combination treatment with traditional cancer therapeutic modalities including radiotherapy (119) and chemotherapy (120), which have been demonstrated to sensitize tumors to CAR T-cell therapy, or molecular antagonists of inhibitory mechanisms that operate in the TME (e.g. IDO or HO-1 inhibitors). Equally, there are many opportunities for innovative re-engineering of CAR T-cells to deal with the physical and environmental barriers found in the solid TME. The configuration of these therapies represents the next challenge for the CAR T-cell field and will no doubt lead to meaningful improvements in the clinical response rates for the application of CAR T-cell therapy against solid malignancies.
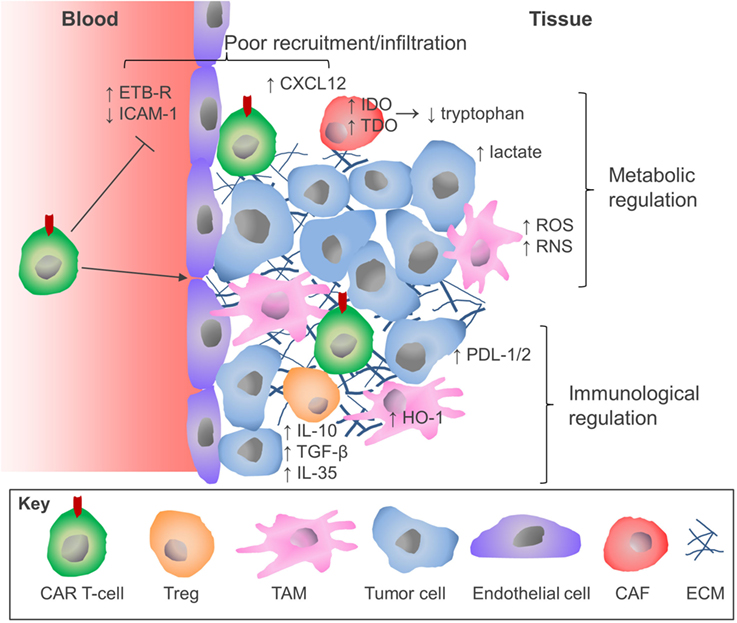
Figure 2. Overview of the barriers to CAR T-cells in solid tumors.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. David Marc Davies and Dr. Jonathan Caron for critically reviewing this article. JNA is funded by a grant from the European Research Council (335326). The authors acknowledge support from the Experimental Cancer Medicine Centre at King’s College London, the Cancer Research UK Centre at King’s Health Partners, and by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St. Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
References
1. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med (2017) 377:2545–54. doi:10.1056/NEJMoa1708566
2. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med (2017) 377(26):2531–44. doi:10.1056/NEJMoa1707447
3. Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol (2006) 6:383–93. doi:10.1038/nri1842
4. Maus MV, Thomas AK, Leonard DGB, Allman D, Addya K, Schlienger K, et al. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat Biotechnol (2002) 20:143–8. doi:10.1038/nbt0202-143
5. Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol (2009) 183:5563–74. doi:10.4049/jimmunol.0900447
6. Cederbom L, Hall H, Ivars F. CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur J Immunol (2000) 30:1538–43. doi:10.1002/1521-4141(200006)30:6<1538::AID-IMMU1538>3.0.CO;2-X
7. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med (2014) 371:1507–17. doi:10.1056/NEJMoa1407222
8. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5:ra38–177. doi:10.1126/scitranslmed.3005930
9. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood (2017) 129:3322–31. doi:10.1182/blood-2017-02-769208
10. Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol (2013) 10:267–76. doi:10.1038/nrclinonc.2013.46
11. Berdeja JG, Lin Y, Raje N, Munshi N, Siegel D, Liedtke M, et al. Durable clinical responses in heavily pretreated patients with relapsed/refractory multiple myeloma: updated results from a multicenter study of bb2121 anti-Bcma CAR T cell therapy. Blood (2017) 130:740.
12. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med (2018) 378:439–48. doi:10.1056/NEJMoa1709866
13. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi:10.1182/blood-2011-05-354449
14. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) – specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33:1688–96. doi:10.1200/JCO.2014.58.0225
15. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci (2016) 59:468–79. doi:10.1007/s11427-016-5023-8
16. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res (2017) 27:38–58. doi:10.1038/cr.2016.154
17. Lohmueller J, Finn OJ. Current modalities in cancer immunotherapy: immunomodulatory antibodies, CARs and vaccines. Pharmacol Ther (2017) 178:31–47. doi:10.1016/j.pharmthera.2017.03.008
18. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14:1264–70. doi:10.1038/nm.1882
19. Tanyi JL, Haas AR, Beatty GL, Morgan MA, Stashwick CJ, O’Hara MH, et al. Abstract CT105: safety and feasibility of chimeric antigen receptor modified T cells directed against mesothelin (CART-meso) in patients with mesothelin expressing cancers. Cancer Res (2015) 75:CT105. doi:10.1158/1538-7445.AM2015-CT105
20. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res (2014) 2:112–20. doi:10.1158/2326-6066.CIR-13-0170
21. Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol (2006) 24:e20–2. doi:10.1200/JCO.2006.05.9964
22. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21:904–12. doi:10.1038/mt.2013.17
23. Lamers CHJ, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans (2016) 44:951–9. doi:10.1042/BST20160037
24. Wang Y, Luo F, Yang J, Zhao C, Chu Y. New chimeric antigen receptor design for solid tumors. Front Immunol (2017) 8:1934. doi:10.3389/fimmu.2017.01934
25. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18:843–51. doi:10.1038/mt.2010.24
26. Rafiq S, Purdon TJ, Daniyan AF, Koneru M, Dao T, Liu C, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia (2017) 31:1788–97. doi:10.1038/leu.2016.373
27. Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell (2016) 164:770–9. doi:10.1016/j.cell.2016.01.011
28. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res (2015) 75:3505–18. doi:10.1158/0008-5472.CAN-15-0139
29. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi:10.1038/mtna.2013.32
30. Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med (2011) 365:1673–83. doi:10.1056/NEJMoa1106152
31. Stavrou M, Philip B, Traynor-White C, Davis CG, Onuoha S, Cordoba S, et al. A Rapamycin activated caspase 9 based suicide gene. Mol Ther (2018) 26:1266–76. doi:10.1016/j.ymthe.2018.03.001
32. Wang L-CS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res (2014) 2:154–66. doi:10.1158/2326-6066.CIR-13-0027
33. Kakarla S, Chow KK, Mata M, Shaffer DR, Song X-T, Wu M-F, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther (2013) 21:1611–20. doi:10.1038/mt.2013.110
34. Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med (2013) 11:187. doi:10.1186/1479-5876-11-187
35. Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med (2013) 210:1137–51. doi:10.1084/jem.20122344
36. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee C-CR, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med (2013) 210:1125–35. doi:10.1084/jem.20130110
37. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol (2011) 89:207–15. doi:10.1038/icb.2010.158
38. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol (2009) 183:937–44. doi:10.4049/jimmunol.0804253
39. Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther (2007) 15:825–33. doi:10.1038/sj.mt.6300104
40. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12:6106–15. doi:10.1158/1078-0432.CCR-06-1183
41. van der Stegen SJC, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, et al. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol (2013) 191:4589–98. doi:10.4049/jimmunol.1301523
42. Scadden DT, Poznansky MC, Olszak IT, Foxall R, Evans RH, Luster AD. Active movement of T cells away from a chemokine. Nat Med (2000) 6:543–8. doi:10.1038/75022
43. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110:20212–7. doi:10.1073/pnas.1320318110
44. Liang JJ, Zhu S, Bruggeman R, Zaino RJ, Evans DB, Fleming JB, et al. High levels of expression of human stromal cell-derived factor-1 are associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Cancer Epidemiol Biomarkers Prev (2010) 19:2598–604. doi:10.1158/1055-9965.EPI-10-0405
45. Thomas RM, Kim J, Revelo-Penafiel MP, Angel R, Dawson DW, Lowy AM. The chemokine receptor CXCR4 is expressed in pancreatic intraepithelial neoplasia. Gut (2008) 57:1555–60. doi:10.1136/gut.2007.143941
46. Guo L, Cui Z-M, Zhang J, Huang Y. Chemokine axes CXCL12/CXCR4 and CXCL16/CXCR6 correlate with lymph node metastasis in epithelial ovarian carcinoma. Chin J Cancer (2011) 30:336–43. doi:10.5732/cjc.010.10490
47. Liu F, Lang R, Wei J, Fan Y, Cui L, Gu F, et al. Increased expression of SDF-1/CXCR4 is associated with lymph node metastasis of invasive micropapillary carcinoma of the breast. Histopathology (2009) 54:741–50. doi:10.1111/j.1365-2559.2009.03289.x
48. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med (2008) 14:28–36. doi:10.1038/nm1699
49. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113:6392–402. doi:10.1182/blood-2009-03-209650
50. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother (2010) 33:780–8. doi:10.1097/CJI.0b013e3181ee6675
51. Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res (2011) 17:4719–30. doi:10.1158/1078-0432.CCR-11-0351
52. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther (2002) 13:1971–80. doi:10.1089/10430340260355374
53. Newick K, O’Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res (2016) 4:541–51. doi:10.1158/2326-6066.CIR-15-0263
54. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean M-C, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122:899–910. doi:10.1172/JCI45817
55. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol (2012) 196:395–406. doi:10.1083/jcb.201102147
56. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med (2015) 21:524–9. doi:10.1038/nm.3833
57. Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Liénard D, Lejeune F, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res (2004) 64:2865–73. doi:10.1158/0008-5472.CAN-03-3066
58. Mortarini R, Piris A, Maurichi A, Molla A, Bersani I, Bono A, et al. Lack of terminally differentiated tumor-specific CD8+ T cells at tumor site in spite of antitumor immunity to self-antigens in human metastatic melanoma. Cancer Res (2003) 63:2535–45.
59. Moon EK, Wang L-C, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res (2014) 20:4262–73. doi:10.1158/1078-0432.CCR-13-2627
60. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
61. Muliaditan T, Opzoomer JW, Caron J, Okesola M, Kosti P, Lall S, et al. Repurposing tin mesoporphyrin as an immune checkpoint inhibitor shows therapeutic efficacy in preclinical models of cancer. Clin Cancer Res (2018) 24(7):1617–28. doi:10.1158/1078-0432.CCR-17-2587
62. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161:205–14. doi:10.1016/j.cell.2015.03.030
63. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N Engl J Med (2012) 366:2443–54. doi:10.1056/NEJMoa1200690
64. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
65. Nazareth MR, Broderick L, Simpson-Abelson MR, Kelleher RJ, Yokota SJ, Bankert RB. Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells. J Immunol (2007) 178:5552–62. doi:10.4049/jimmunol.178.9.5552
66. John LB, Devaud C, Duong CPM, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified t cells. Clin Cancer Res (2013) 19:5636–46. doi:10.1158/1078-0432.CCR-13-0458
67. Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)–modified T cells: refueling the CAR. Blood (2017) 129:1039–41. doi:10.1182/blood-2016-09-738245
68. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res (2017) 23:2255–66. doi:10.1158/1078-0432.CCR-16-1300
69. Baldan V, Ghongane P, Kokalaki E, Lim WC, Onuoha S, Cordoba SP, et al. A dominant negative SHP-2 which abrogates PD-1 signalling pathways and restores function of cytotoxic CAR T cells. Blood (2017) 130:3190.
70. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest (2016) 126:3130–44. doi:10.1172/JCI83092
71. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res (2016) 76:1578–90. doi:10.1158/0008-5472.CAN-15-2524
72. Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther (2015) 23:769–78. doi:10.1038/mt.2015.4
73. Arnold JN, Magiera L, Kraman M, Fearon DT. Tumoral immune suppression by macrophages expressing fibroblast activation protein- and heme oxygenase-1. Cancer Immunol Res (2014) 2:121–6. doi:10.1158/2326-6066.CIR-13-0150
74. Pae H-O, Oh G-S, Choi B-M, Chae S-C, Kim Y-M, Chung K-R, et al. Carbon monoxide produced by heme oxygenase-1 suppresses T cell proliferation via inhibition of IL-2 production. J Immunol (2004) 172:4744–51. doi:10.4049/jimmunol.172.8.4744
75. Tardif V, Riquelme SA, Remy S, Carreño LJ, Cortés CM, Simon T, et al. Carbon monoxide decreases endosome-lysosome fusion and inhibits soluble antigen presentation by dendritic cells to T cells. Eur J Immunol (2013) 43:2832–44. doi:10.1002/eji.201343600
76. Mackern-Oberti JP, Obreque J, Méndez GP, Llanos C, Kalergis AM. Carbon monoxide inhibits T cell activation in target organs during systemic lupus erythematosus. Clin Exp Immunol (2015) 182:1–13. doi:10.1111/cei.12657
77. Chau L-Y. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci (2015) 22:22. doi:10.1186/s12929-015-0128-0
78. Roussel E, Gingras MC, Grimm EA, Bruner JM, Moser RP. Predominance of a type 2 intratumoural immune response in fresh tumour-infiltrating lymphocytes from human gliomas. Clin Exp Immunol (1996) 105:344–52. doi:10.1046/j.1365-2249.1996.d01-753.x
79. Seo N, Hayakawa S, Takigawa M, Tokura Y. Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4(+) T-regulatory cells and systemic collapse of antitumour immunity. Immunology (2001) 103:449–57. doi:10.1046/j.1365-2567.2001.01279.x
80. Jarnicki AG, Lysaght J, Todryk S, Mills KHG. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol (2006) 177:896–904. doi:10.4049/jimmunol.177.2.896
81. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol (2007) 179:977–83. doi:10.4049/jimmunol.179.2.977
82. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology (2014) 147:1393–404. doi:10.1053/j.gastro.2014.08.039
83. Strauss L, Bergmann C, Szczepanski M, Gooding W, Johnson JT, Whiteside TL. A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-1 mediates suppression in the tumor microenvironment. Clin Cancer Res (2007) 13:4345–54. doi:10.1158/1078-0432.CCR-07-0472
84. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature (2007) 450:566–9. doi:10.1038/nature06306
85. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. J Immunother (2008) 31:500–5. doi:10.1097/CJI.0b013e318177092b
86. Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther (2013) 20:575–80. doi:10.1038/gt.2012.75
87. van Schalkwyk MCII, Papa SE, Jeannon J-P, Guerrero Urbano T, Spicer JF, Maher J. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Hum Gene Ther Clin Dev (2013) 24:134–42. doi:10.1089/humc.2013.144
88. Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira ACP, Cleary S, van der Stegen SJC, et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem (2010) 285:25538–44. doi:10.1074/jbc.M110.127951
89. Klampatsa A, Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, et al. Intracavitary “T4 immunotherapy” of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett (2017) 393:52–9. doi:10.1016/j.canlet.2017.02.015
90. Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther (2017) 25:249–58. doi:10.1016/j.ymthe.2016.10.016
91. Steding CE, Wu S, Zhang Y, Jeng M-H, Elzey BD, Kao C. The role of interleukin-12 on modulating myeloid-derived suppressor cells, increasing overall survival and reducing metastasis. Immunology (2011) 133:221–38. doi:10.1111/j.1365-2567.2011.03429.x
92. Broderick L, Brooks SP, Takita H, Baer AN, Bernstein JM, Bankert RB. IL-12 reverses anergy to T cell receptor triggering in human lung tumor-associated memory T cells. Clin Immunol (2006) 118:159–69. doi:10.1016/j.clim.2005.09.008
93. Pegram HJ, Purdon TJ, van Leeuwen DG, Curran KJ, Giralt SA, Barker JN, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia (2015) 29:415–22. doi:10.1038/leu.2014.215
94. Koneru M, O’Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16ecto directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med (2015) 13:102. doi:10.1186/s12967-015-0460-x
95. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep (2017) 7:10541. doi:10.1038/s41598-017-10940-8
96. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71:5697–706. doi:10.1158/0008-5472.CAN-11-0103
97. Chmielewski M, Abken H. CAR T cells releasing IL-18 convert to T-bet high FoxO1 low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep (2017) 21:3205–19. doi:10.1016/j.celrep.2017.11.063
98. Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep (2017) 20:3025–33. doi:10.1016/j.celrep.2017.09.002
99. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov (2017) 7:1238–47. doi:10.1158/2159-8290.CD-17-0538
100. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A (2016) 113:E7788–97. doi:10.1073/pnas.1610544113
101. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2:e1600200. doi:10.1126/sciadv.1600200
102. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood (2007) 109:3812–9. doi:10.1182/blood-2006-07-035972
103. Chen JH, Perry CJ, Tsui Y-C, Staron MM, Parish IA, Dominguez CX, et al. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med (2015) 21:327–34. doi:10.1038/nm.3831
104. Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell (2015) 162:1257–70. doi:10.1016/j.cell.2015.08.015
105. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med (2002) 196:459–68. doi:10.1084/jem.20020121
106. Terness P, Bauer TM, Röse L, Dufter C, Watzlik A, Simon H, et al. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: mediation of suppression by tryptophan metabolites. J Exp Med (2002) 196:447–57. doi:10.1084/jem.20020052
107. Weber WP, Feder-Mengus C, Chiarugi A, Rosenthal R, Reschner A, Schumacher R, et al. Differential effects of the tryptophan metabolite3-hydroxyanthranilic acid on the proliferation of human CD8+ T cells induced by TCR triggering or homeostatic cytokines. Eur J Immunol (2006) 36:296–304. doi:10.1002/eji.200535616
108. Fallarino F, Grohmann U, Vacca C, Bianchi R, Orabona C, Spreca A, et al. T cell apoptosis by tryptophan catabolism. Cell Death Differ (2002) 9:1069–77. doi:10.1038/sj.cdd.4401073
109. Uyttenhove C, Pilotte L, Théate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med (2003) 9:1269–74. doi:10.1038/nm934
110. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest (2007) 117:1147–54. doi:10.1172/JCI31178
111. Ninomiya S, Narala N, Huye L, Yagyu S, Savoldo B, Dotti G, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood (2015) 125:3905–16. doi:10.1182/blood-2015-01-621474
112. Soliman HH, Minton SE, Han HS, Ismail-Khan R, Neuger A, Khambati F, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget (2016) 7:22928–38. doi:10.18632/oncotarget.8216
113. Gangadhar TC, Hamid O, Smith DC, Bauer TM, Wasser JS, Olszanski AJ, et al. Epacadostat plus pembrolizumab in patients with advanced melanoma and select solid tumors: updated phase 1 results from ECHO-202/KEYNOTE-037. Ann Oncol (2016) 27. doi:10.1093/annonc/mdw379.06
114. Mussai F, Egan S, Hunter S, Webber H, Fisher J, Wheat R, et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer Res (2015) 75:3043–53. doi:10.1158/0008-5472.CAN-14-3443
115. Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol (2004) 172:989–99. doi:10.4049/jimmunol.172.2.989
116. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med (2007) 13:828–35. doi:10.1038/nm1609
117. Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med (2011) 208:1949–62. doi:10.1084/jem.20101956
118. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375:2561–9. doi:10.1056/NEJMoa1610497
119. Weiss T, Weller M, Guckenberger M, Sentman CL, Roth P. NKG2D-based CAR-T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res (2018) 78:1031–43. doi:10.1158/0008-5472.CAN-17-1788
Keywords: chimeric antigen receptor, T-cells, immunotherapy, tumor microenvironment, solid tumor
Citation: Kosti P, Maher J and Arnold JN (2018) Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 9:1104. doi: 10.3389/fimmu.2018.01104
Received: 19 February 2018; Accepted: 02 May 2018;
Published: 22 May 2018
Edited by:
Rayne Rouce, Baylor College of Medicine, United StatesReviewed by:
Bipulendu Jena, University of Texas MD Anderson Cancer Center, United StatesPappanaicken R. Kumaresan, University of Texas MD Anderson Cancer Center, United States
Copyright: © 2018 Kosti, Maher and Arnold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Arnold, amFtZXMubi5hcm5vbGRAa2NsLmFjLnVr
†These authors have contributed equally to this work.