 Wenting Qu1,2
Wenting Qu1,2 Yi Wang1,2
Yi Wang1,2 Yongjian Wu1,2
Yongjian Wu1,2 Yiting Liu1,2
Yiting Liu1,2 Kang Chen3
Kang Chen3 Xi Liu1Zhengyu Zou1,2
Xi Liu1Zhengyu Zou1,2 Xi Huang1,2
Xi Huang1,2 Minhao Wu1,2,4*
Minhao Wu1,2,4*
- 1Program of Pathobiology and Immunology, Fifth Affiliated Hospital, Zhongshan School of Medicine, Sun Yat-sen University, Guangdong, China
- 2Key Laboratory of Tropical Diseases Control, Ministry of Education, Sun Yat-sen University, Guangzhou, China
- 3Department of Laboratory Medicine, Zhongshan Hospital of Sun Yat-sen University, Zhongshan, China
- 4Guangdong Engineering & Technology Research Center for Disease-Model Animals, Sun Yat-sen University, Guangzhou, China
Triggering receptors expressed on myeloid cells 2 (TREM2) is a novel cell surface receptor and functions as an immunomodulatory receptor in infectious diseases. In this study, we investigated the function and regulatory mechanism of TREM2 in Pseudomonas aeruginosa (P. aeruginosa) keratitis. We found that P. aeruginosa keratitis was more severe in Trem2−/− versus wild type C57BL/6 mice as indicated by the increased clinical scores, bacterial load, and cornea pathology. The exacerbated disease progression caused by TREM2 deficiency was associated with boosted activation of caspase-1 and subsequent pyroptosis as well as increased expression of IL-1β. In addition, blockage of pyroptosis by caspase-1 inhibitor not only recovered the severe cornea pathology developed in Trem2−/− mice but also restored the P. aeruginosa clearance suppressed by TREM2 deficiency. Our study demonstrated that TREM2 promotes host resistance against P. aeruginosa keratitis by inhibiting caspase-1-dependent pyroptosis, which provides new insights of TREM2-mediated anti-bacterial immunity.
Introduction
Pseudomonas aeruginosa (P. aeruginosa) is one of the most common Gram-negative bacteria that cause diverse opportunistic infectious diseases, including keratitis in extended wear contact lens users (1). Clinically, P. aeruginosa keratitis progresses rapidly and results in severe corneal damage, including perforation and vision loss (1, 2). Both bacteria virulence factors and host inflammatory mediators contribute to the destruction of the cornea after infection (3).
Upon P. aeruginosa infection, diverse pathogen-associated molecular patterns were specifically recognized by corresponding pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) (4, 5) and Nod-like receptors (NLRs) (6). In P. aeruginosa-induced keratitis (corneal infection), the TLR-induced inflammatory response promotes bacteria clearance and repairs injured tissues. However, if out of control, the excessive inflammation also leads to tissue damage and corneal ulceration. Except for TLRs, P. aeruginosa infection was able to activate certain numbers of the NLR family, such as the NLRC4 (7) and NLRP3 (8), which function as core receptor molecules to initiate the assembly of multi-protein complexes named inflammasomes. Upon stimulation, the NLRs recruit the adaptor protein ASC via PYD–PYD domain association, and ASC further recruits precursors of caspase-1 through CARD–CARD domain interaction. After clustering and autocleavage, the activated caspase-1 is able to cleave the pro-IL-1β and pro-IL-18 into their mature forms, as well as initiate a type of rapid inflammatory cell death termed pyroptosis (9–11). Pyroptosis has been reported to play an important role in promoting the elimination of several intracellular bacteria, including Legionella pneumophila (12) and Francisella tularensis (13). Whereas in the setting of acute P. aeruginosa pneumonia, inhibition of inflammasome signaling significantly enhanced bacterial clearance via reducing cell death (14). However, whether pyroptosis is involved in the pathogenesis of P. aeruginosa keratitis is still uncertain.
Triggering receptors expressed on myeloid cells 2 (TREM2) is a novel member of PRRs and broadly existed on the surface of mononuclear phagocytes, including macrophages, microglia, and osteoclast precursors (15). TREM2 has been reported as an immune regulator and exerts important functions in bacteria clearance (16, 17). Recently, Holtzman’s group reported that TREM2 suppressed the apoptosis of lung macrophages during Sendai virus infection, then led to chronic inflammatory diseases (18). Colonna’s lab found that in Alzheimer’s disease (AD) patients and the 5xFAD transgenic murine model, deficiency in TREM2 caused autophagy of microglia in vivo (19). These results indicated the correlation between TREM2 and programmed cell death. Our previous studies demonstrated that TREM2 protected P. aeruginosa-infected corneas of BALB/c mice (which are Th2 responders and P. aeruginosa-resistant strain) by suppressing corneal inflammation and bacterial load (20). However, whether inflammasomes and pyroptosis are involved TREM2-mediated host defense against P. aeruginosa is still unclear.
In this study, we explored the relationship between TREM2 and caspase-1-dependent pyroptosis, and investigated the role of pyroptosis in P. aeruginosa keratitis using TREM2-deficient versus wild type C57BL/6 mice (which is a P. aeruginosa-susceptible strain). Our in vivo results indicated that TREM2 suppressed activation of caspase-1 and subsequent pyroptosis, thereby inhibiting cornea pathology as well as promoting the clearance of P. aeruginosa. These findings provide a better understanding of TREM2-mediated pyroptosis and bacterial eradication.
Materials and Methods
Mice and Reagents
Wild type (WT) C57BL/6 mice were purchased from the experimental animal center of Sun Yat-sen University. Trem2−/− C57BL/6 mice were generously provided by Marco Colonna (Washington University School of Medicine). P. aeruginosa (P. aeruginosa; strain 19660) was purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). Heat-killed P. aeruginosa (HK-PA) was prepared according to previous study (21). P. aeruginosa (ATCC 19660) was killed by heating 100 μl aliquots of bacteria solution [108 colony forming units (CFU)/ml] at 65°C for an hour. A sample of heat-killed PA was grown on PIA (BD Difco Laboratories) plates, and no live PA was observed after incubation at 37°C for 24 h. Ac-YVAD-CMK (caspase-1 inhibitor, Cat: SML0429), DMSO (Cat: D2650), and anti-β-actin (clone: AC-15; Cat: A1978) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Anti-mouse caspase-1 (p20) (clone: Casper-1; Cat: AG-20B-0042) and anti-mouse NLRP3 (clone: Cryo-2; Cat: AG-20B-0014) were ordered from Adipogen (San Diego, CA, USA). Anti-ASC (clone: N-15; Cat: sc-22514-R) was ordered from Santa Cruz biotechnology (San Diego, CA, USA). Anti-GSDMD (clone: EPR19828; Cat: ab209845) and Anti-TREM2 (clone: 6EP9; Cat: ab125117) were purchased from Abcam (Cambridge, MA, USA). Anti-caspase-11 (clone: 17D9; Cat: #14340) was purchased from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies against mouse and rabbit were obtained from Bio-Rad (Hercules, CA, USA).
Ocular Infection and Clinical Examination
The left cornea of the 6-week-old female WT or Trem2−/− B6 mice was infected by P. aeruginosa (ATCC 19660) as described before (22). Eyes were examined at 1, 3, and 5 days postinfection (p.i.) or at times described below. Corneal disease was graded using an established scale (23): 0, clear or slight opacity partially or fully covering the pupil; +1, slight opacity partially or fully covering the anterior segment; +2, dense opacity partially or fully covering the pupil; +3, dense opacity covering the entire anterior segment; and +4, corneal perforation or phthisis. A clinical score was recorded for each mouse after infection for statistical comparison of disease severity. Photography with a slit lamp was used to confirm and document the disease response. Animals were treated humanely and in compliance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Isolation of Bone Marrow-Derived Macrophages (BMDMs) and Cell Culture
Mouse BMDMs were isolated and differentiated as reported previously (20). In brief, WT or Trem2−/− mice were sacrificed and the bone marrow in femurs was flushed out and cultured in DMEM containing 10% FBS and 30% (vol/vol) L-929 fibroblast-conditioned medium as a source of macrophage colony-stimulated factor. BMDMs were obtained as a homogeneous population of adherent cells after 7 days culture. The BMDM purity was routinely >95% as assessed by flow cytometry. A6(1) corneal epithelial cell line was a gift from Dr. Peggy Zelenka (National Eye Institute/NIH) and cultured following their protocol (24). The A6(1) cells were derived from corneal epithelia of the 14-day-old immortomouse (Charles River Laboratories, Wilmington, MA, USA), then conditionally immortalized by a temperature sensitive SV40 T-antigen, under the control of an IFN-γ inducible promoter. Cells were cultured in Epilife™ media, supplemented with corneal epithelial growth supplement, interferon IFN-γ (5 units/ml), 1% penicillin–streptomycin, 1% l-glutamine, and 20% (v/v) FBS (all from Invitrogen) at the permissive temperature of 33°C, in a humidified atmosphere of 95% air and 5% CO2. For in vitro assays, subconfluent cultures were transferred to the nonpermissive temperature of 37°C and cultured in the same media, but without IFN-γ.
Real-Time PCR
Mice were sacrificed on 1 and 5 days p.i. and infected corneas of WT and Trem2−/− B6 mice (n = 5/group/time) were harvested. Total RNA was isolated from individual corneas using TRIzol (Invitrogen, Carlsbad, CA, USA) as described previously (23). Total RNA was quantitated using a NanoDrop 2000C Spectrophotometers (Thermo Scientific, West Palm Beach, FL, USA). 1 µg of total RNA was reversely transcribed to produce cDNA by using RevertAid First Strand cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA), and then amplified using SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s protocol. Quantitative real-time PCR reactions were performed using the CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA). The average threshold cycle (CT) values of samples were normalized to CT of β-actin gene. The relative expression was determined by the 2−ΔΔCT method. Primer sequences are listed in Table S1 in Supplementary Material.
Western Blot
To detect the corneal expression of NLRP3, ASC, caspase-11, caspase-1, and GSDMD, whole corneas (n = 5/group/time) were collected and pooled from normal uninfected and infected B6 mouse eyes at 1 and 5 days p.i. Pooled corneas were lysed and homogenized using a 1-ml glass tissue homogenizer in lysis buffer containing 1 mM phenylmethylsulfonyl fluoride, 1% (vol/vol) protease inhibitor cocktail, and 1 mM DTT (all from Sigma, St. Louis, MO, USA). Then, protein concentration of the supernatant was determined by Quick Start Bradford protein assay (Bio-Rad). 20 µg of each sample was loaded, separated on 12% SDS-PAGE, and then transferred to a supported nitrocellulose membrane (Pall Life Sciences, Ann Arbor, MI, USA). After blockage, blots were incubated overnight with the respective primary antibodies at 4°C, followed by incubation with appropriate HRP-conjugated secondary antibodies at room temperature for 1 h. Finally, blots were visualized with New-SUPER ECL (KeyGEN, Nanjing, China) according to the manufacturer’s protocol.
Hematoxylin-Eosin (HE) Staining and Immunohistochemistry
Infected eyes were enucleated (n = 3/group/time) at 1 and 5 days p.i. from WT and Trem2−/− B6 mice, embedded in Tissue-Tek OCT compound (Miles, Elkhart, IN, USA) and frozen in liquid nitrogen. 8 µm thick sections were cut and mounted to glass slides. For histopathology, sections were hematoxylin-eosin (HE) stained as described by others (25). Immunohistochemical staining was performed with the UltraSensitive SP Immunodetection Kit (Maixin, Inc., Fuzhou, China) according to the manufacturer’s instructions. Primary antibodies rabbit anti-mouse capase-1 (p10) (clone: M-20; Cat: sc-514) were purchased from Santa Cruz. All sections were visualized with a Carl Zeiss microscope (Carl Zeiss Inc.).
Terminal Deoxynucleotidyl Transferase-Mediated Uridine 5′-Triphosphate-Biotin Nick End Labeling (TUNEL) Staining
Infected eyes from the WT versus Trem2−/− B6 mice (n = 3/group/time) were enucleated at 5 days p.i. for TUNEL staining with a terminal deoxynucleotidyl transferase (TdT) kit (Promega, Madison, WI, USA) following the manufacturer’s instruction. Eyes were fixed in a 3.7% formaldehyde solution (Sigma) and embedded in paraffin. 8 µm thick sections were cut, deparaffinized, rehydrated, and rinsed with DNase-free water (Invitrogen). Sections were permeabilized using proteinase K solution (20 µg/ml, Sigma) for 15 min and then fixed again using 3.7% formaldehyde solution (Sigma). Each section was incubated with TdT incubation buffer, which contains 45 µl equilibration buffer, 5 µl nucleotide mix, and 1 µl TdT enzyme at 37°C for 1 h to label the DNA nick ends, and then incubated with 4,6-diamino-2-phenyl indole (DAPI, 1:10,000, Sigma) for nuclear staining. All sections were visualized with a Carl Zeiss microscope (Jena, Germany).
Bacterial Plate Counts
Corneas from WT versus Trem2−/− B6 mice (at 1 and 5 days p.i.) were harvested (n = 5/group/time) and the number of viable bacteria was quantitated as described before (26). Individual corneas were homogenized in normal saline solution containing 0.25% BSA. Serial 10-fold dilutions of the samples were plated on PIA (BD Difco Laboratories) in triplicate and plates were incubated overnight at 37°C. Results are reported as 105 CFU per cornea ± SEM.
Enzyme-Linked Immunosorbent Assay (ELISA)
Corneas from WT versus Trem2−/− B6 mice were individually collected (n = 5/group/time) at 1 and 5 days p.i. Corneas were homogenized in 0.5 ml of PBS with 0.1% Tween 20. All samples were centrifuged at 13,000 rpm for 5 min and an aliquot of each supernatant was assayed in duplicate for IL-1β protein by using ELISA kits from BD Biosciences (San Jose, CA, USA) following the manufacturer’s instructions. The reported sensitivity of these assays is <3.0 pg/ml for IL-1β.
Flow Cytometric Detection of Activated Caspase-1
Individual corneas from WT and Trem2−/− B6 mice (n = 5/group) were harvested at 5 days postinfection. Individual corneas were kept in sterile tubes containing 1 ml Liberase TL (Roche, Indianapolis, IN, USA, 2.5 mg/ml), followed by incubation at 37°C for 45 min. At the end of the incubation period, samples were triturated using a 2 ml syringe plunger, and passed through a 70 µm cell strainer. Finally, the single-cell suspension was washed with 1 ml RPMI 1640 containing 10% FBS and was pelleted at 500 × g for 8 min in a refrigerated centrifuge. Caspase-1 activation was detected in the corneas using a FAM-FLICA™ caspase-1 assay kit following the manufacturer’s instructions (Immunochemistry Technologies, Bloomington, MN, USA). Briefly, single-cell suspensions of infected corneas from WT and Trem2−/− B6 mice were incubated with FAM–FLICA caspase-1 at 37°C, 5% CO2 for 45 min. At the end of the incubation period, cells were washed twice with 1× wash buffer provided in the kit. Cell surface staining for CD11b, Gr1, F4/80, and CD11c molecules was carried out and after samples were immediately acquired using a LSRFortessa flow cytometer (Beckton Dickinson, San Jose, CA, USA). Data were analyzed using FlowJo software and presented as mean fluorescence intensity ± SEM. The following antibodies were used for cell surface staining: FITC-conjugated rat anti-mouse CD11b (M1/70, 1:200), APC-conjugated rat anti-mouse F4/80 (BM8, 1:200), PerCP-Cy5.5-conjugated rat anti-mouse Gr1 (RB6-8C5, 1:200), and PE-Cy7-conjugated rat anti-mouse CD11c (N418, 1:200). All antibodies were purchased from BD Biosciences (San Jose, CA, USA), eBiosciences (Waltham, MA, USA), or BioLegend (San Diego, CA, USA).
Immunoprecipitation
Individual uninfected and infected corneas from WT B6 mice were harvested at 5-day postinfection. Pooled corneas were lysed and homogenized using a 1-ml glass tissue homogenizer in Pierce IP lysis buffer. BMDMs from WT B6 mice were treated with HK-PA or stimulated with nigericin. After treatment or stimulation, cells were lysed with Pierce IP lysis buffer. The supernatant of cell lysates was incubated with protein A and anti-mouse TREM2 antibody overnight at 4°C to pull down protein complexes. The protein complexes were pelleted and washed three times with IP washing buffer. The immunoprecipitated protein complex was analyzed by immunoblot with anti-TREM2, anti-NLRP3, and anti-caspase-1. And β-actin, TREM2, NLRP3, and caspase-1 were also detected in input cell lysates by western blot analysis.
Statistical Analysis
The differences between two groups were analyzed by using an unpaired two-tailed Student’s t-test. Mann–Whitney U test was used to determine the difference in clinical score between two groups. Differences were considered statistically significant when the P value was <0.05.
Results
TREM-2 Promoted Host Resistance Against P. aeruginosa Keratitis
To confirm the role of TREM-2 in the pathogenesis of P. aeruginosa keratitis, wild type (WT) and Trem2−/− C57BL/6 mice were infected with P. aeruginosa routinely. Clinical scores were significantly higher in the corneas of Trem2-/mice compared with WT group at 3 and 5 days p.i. (Figure 1A). Representative photographs taken with a slit lamp of the infected corneas in WT (Figure 1B left) and Trem2−/− (Figure 1B right) mice were taken to illustrate disease severity. TREM2 deficiency resulted in corneal perforation (grade = + 4, Figure 1B right on the bottom), while the pupil of infected WT mice was partially or fully covered with dense opacity (grade = + 3, Figure 1B left on the bottom) at 5 days p.i. Since the disease pathogenesis of P. aeruginosa keratitis depends on both bacterial virulence and host inflammation, we first assessed the effects of TREM2 on bacterial load. As shown in Figure 1C, TREM2 deficiency elevated the number of viable bacteria in the infected corneas at 1 and 5 days p.i. We further compared the corneal inflammation in WT and Trem2−/− mice by enucleating the infected eyes for histopathology. Hematoxylin and eosin (H&E) staining results indicated that Trem2−/− corneas (Figure 1D right) were more swollen, with more infiltrated inflammatory cells and exhibited more severe tissue destruction, in contrast to the WT corneas (Figure 1D left). Together, these results suggested that TREM2 promoted host resistance to P. aeruginosa corneal infection in C57BL/6 mice.
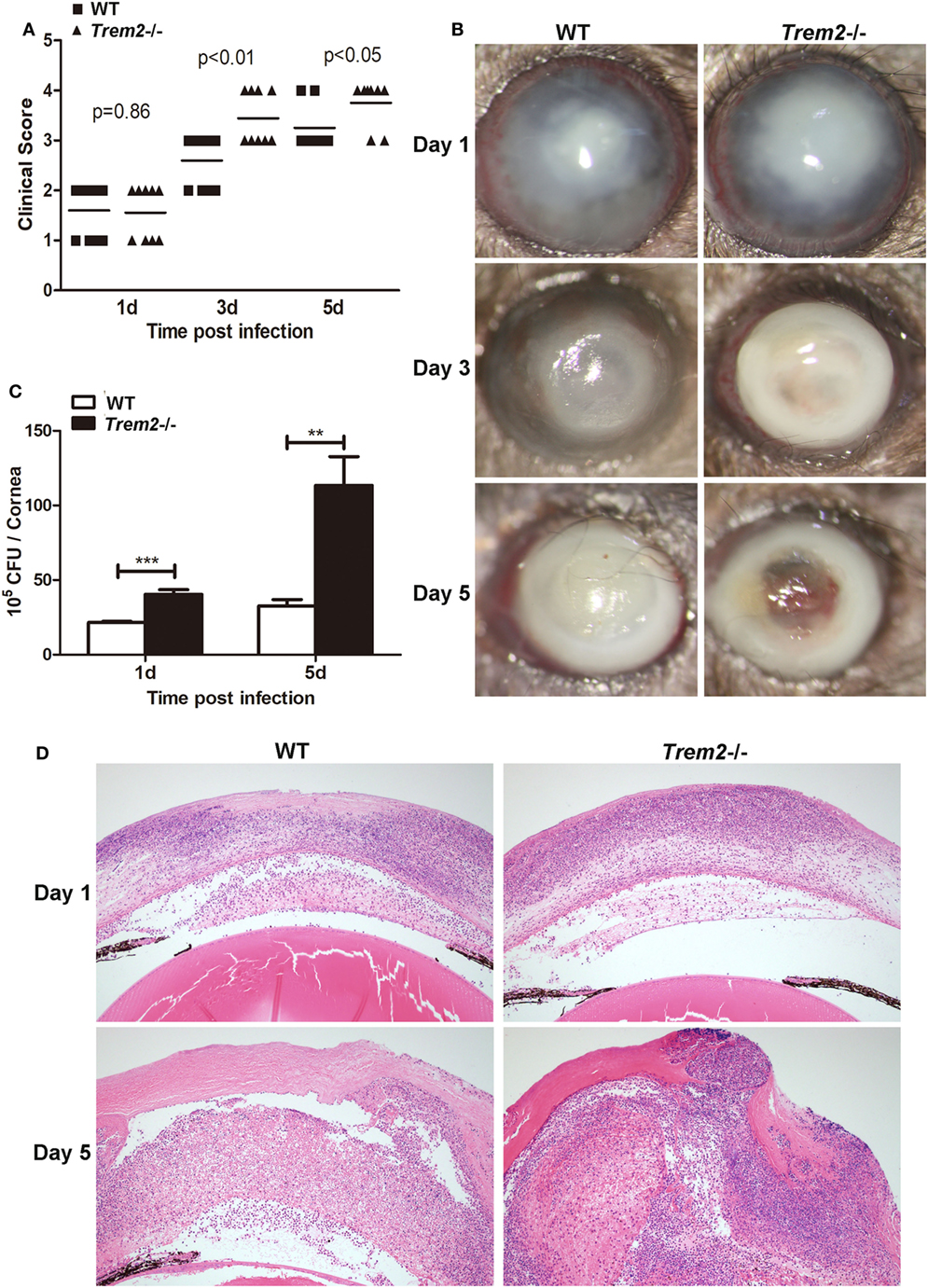
Figure 1. Triggering receptors expressed on myeloid cells 2 (TREM-2) promoted host resistance against Pseudomonas aeruginosa keratitis. Trem2−/− and wild type (WT) C57BL/6 mice were infected with P. aeruginosa. (A) Clinical score was recorded for each cornea at 1, 3, and 5 days after infection. (B) Representative slit-lamp photographs of infected eyes in WT (left) and Trem2−/− (right) mice were taken 1, 3, and 5 days after infection. (C) Bacterial load in the infected corneas was examined by plate count assay in WT versus Trem2−/− mice at 1 and 5 days after infection. (D) Hematoxylin and eosin staining was used to examine the histopathology of infected eyes in WT (left) and Trem2−/− (right) mice at 1 and 5 days after infection. Magnification = 100×. Data are the mean ± SEM and represent three individual experiments each with 10 mice per group (A,B) or five mice per group (C,D). *P < 0.05; **P < 0.01; ***P < 0.001.
TREM-2 Inhibited Pro-Inflammatory Cytokine Expression in P. aeruginosa Keratitis
As the inflammation was more severe in TREM2-deficient corneas (Figure 1D), we further determined the mechanism by which TREM-2 mediated the inflammatory response. We first checked the expression of pro-inflammatory cytokines and found that TREM2 deficiency markedly upregulated the mRNA levels of IL-1β (Figure 2A) and IL-18 (Figure 2C) as well as the protein levels of IL-1β (Figure 2B) at 1 and 5 days p.i. Expression and secretion of IL-1β can be promoted through either inflammasome-dependent or NF-κB pathway. However, the expression levels of tumor necrosis factor-alpha (TNF-α), a proinflammatory cytokine associated with NF-κB activation, were similar in these two groups (Figure 2D). Suggesting the increased IL-1β in Trem2−/− corneas was largely inflammasome-dependent. Furthermore, corneas of Trem2−/− mice appeared in higher expression of inflammasome-independent cytokines IFN-γ (Figure 2E) and MIP-2 (Figure 2F). To investigate whether the overall increase in these proinflammatory cytokines was due to the higher bacterial load in Trem2−/− corneas, we inoculated WT and Trem2−/− mice with HK-PA routinely. We found that expression of IL-1β and MIP-2 was also upregulated in Trem2−/− corneas while expression of IL-18 was comparable between the two groups (Figure 2G). Moreover, we observed similar results when WT and Trem2−/− BMDMs were treated with HK-PA (Figure 2H). These data indicated that TREM-2 downregulated the corneal inflammation via inhibiting expression of proinflammatory cytokine IL-1β and IL-18 in response to P. aeruginosa ocular infection.
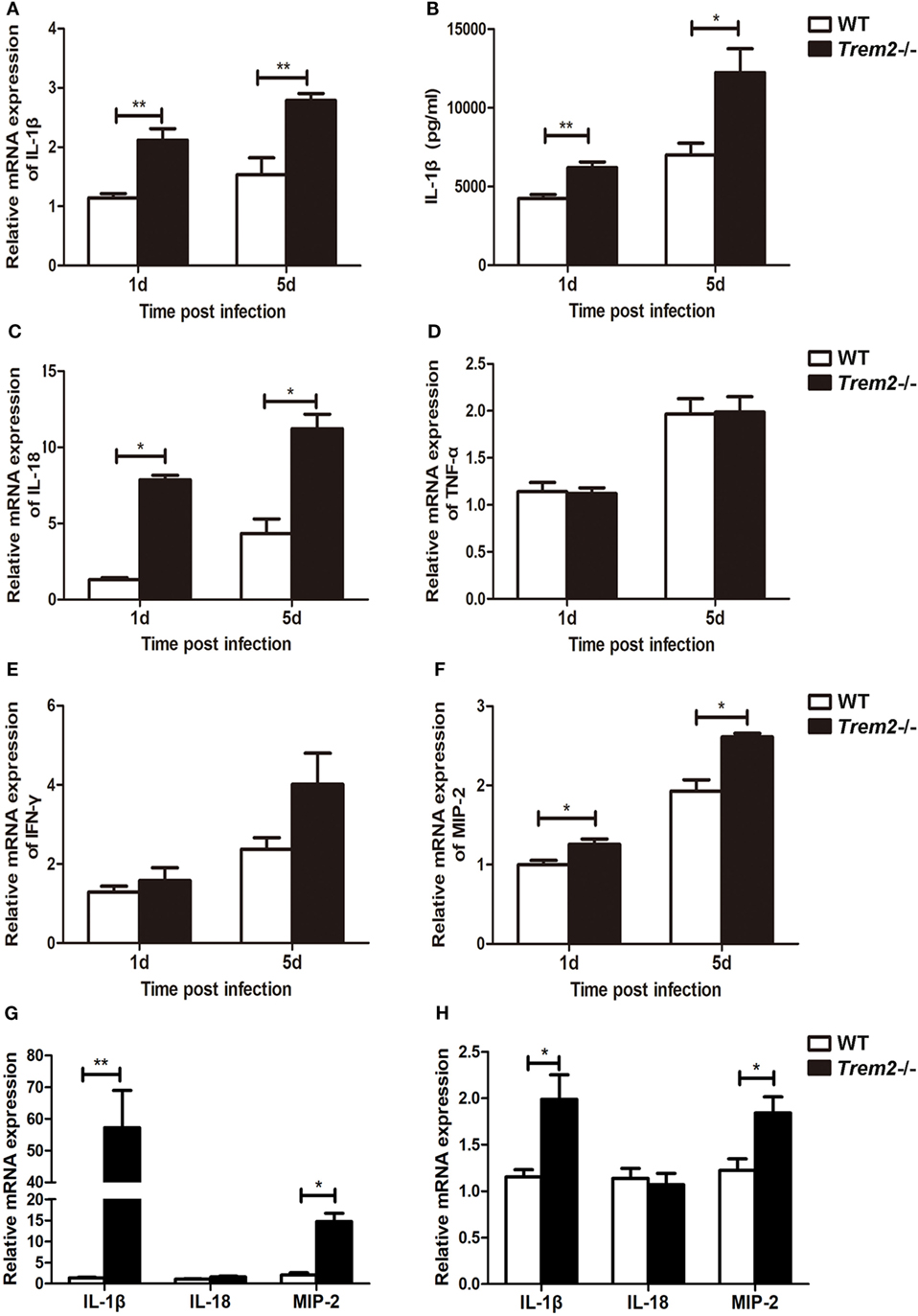
Figure 2. Triggering receptors expressed on myeloid cells 2 (TREM-2) inhibited pro-inflammatory cytokine expression after Pseudomonas aeruginosa infection. mRNA expression levels of pro-inflammatory cytokines, including IL-1β (A), IL-18 (C), tumor necrosis factor-alpha (D), IFN-γ (E), and MIP-2 (F) was examined by real-time PCR in infected wild type (WT) and Trem2−/− B6 corneas at 1 and 5 days after infection. (B) Protein level of IL-1β in WT and Trem2−/−-infected B6 corneas was tested by enzyme-linked immunosorbent assay at 1 and 5 days after infection. mRNA expression levels of IL-1β, IL-18, and MIP-2 was examined by real-time PCR in heat-killed P. aeruginosa (HK-PA)-treated WT and Trem2−/− B6 corneas at 5 days after treatment (G) and WT and Trem2−/− BMDM (H) which were treatment with HK-PA at an MOI of 5 for 6 h. Data are the mean ± SEM and represent three individual experiments each with five mice per group. *P < 0.05; **P < 0.01.
TREM-2 Inhibited the Activation of Caspase-1 and Subsequent Pyroptosis After P. aeruginosa Infection
As the expression and secretion of IL-1β were significantly increased in TREM2-deficient corneas after P. aeruginosa infection (Figures 2A–C), we further detected the expression and activation of caspase-1 in the corneas of WT and Trem2−/− mice. Indeed, Trem2−/− corneas appeared in higher mRNA expression of caspase-1 (Figure 3A). The expression level of caspase-1 was also higher in Trem2−/− corneas and BMDMs after HK-PA treatment (Figure S1A in Supplementary Material). As full catalytic activity of caspase-1 requires autoproteolytic processing of procaspase-1, we also detected caspase-1 cleavage by Western blot. Results showed that P. aeruginosa-induced caspase-1 cleavage (caspase-1 p20) as well as procaspase-1 expression was significantly increased in Trem2−/− corneas (Figure 3B). At the same time, the expression of NLRP3 was also higher in infected Trem2−/− corneas, while ASC levels were comparable between the two groups before and after P. aeruginosa infection (Figure 3B). Caspase-1 expression and activation after P. aeruginosa infection were further confirmed by immune-histochemistry. Higher level of positive staining for caspase-1 (depicted as brown dots) was detected in the infected Trem2−/− B6 corneas, mainly localizing in the corneal epithelium and stroma (Figure 3C). Since the noncanonical inflammasome activation mediated by caspase-11 can trigger both caspase-1-dependent and -independent production of IL-1β and IL-18, we also tested caspase-11 expression in the infected WT and Trem2−/− corneas and found its expression was comparable between the two groups (Figure S1B in Supplementary Material). To further determine what cell type plays important role in the severe pathology mediated by TREM2 deficiency, we investigated the caspase-1 activation on the major cell subsets in the infected WT and Trem2−/− corneas by flow cytometry. Our results showed that caspase-1 activation was enhanced in macrophages from Trem2−/− when compared to those from WT corneas, while its activation in polymorphonuclear neutrophils (PMN) and dendritic cells (DC) was comparable (Figures 3D,E). In addition, we also tested the role of TREM2 in A6(1), which is a mouse corneal epithelial cell line. Our data indicated that knockdown TREM2 in A6(1) did not affect the expression of caspase-1 as well as inflammatory cytokine, such as IL- β and MIP-2 after HK-PA treatment (Figure S1C in Supplementary Material). These findings support the hypothesis that the regulatory function of TREM2 in P. aeruginosa keratitis is exerted in macrophages, not other cell types, including PMNs, DCs, or corneal epithelial cells.
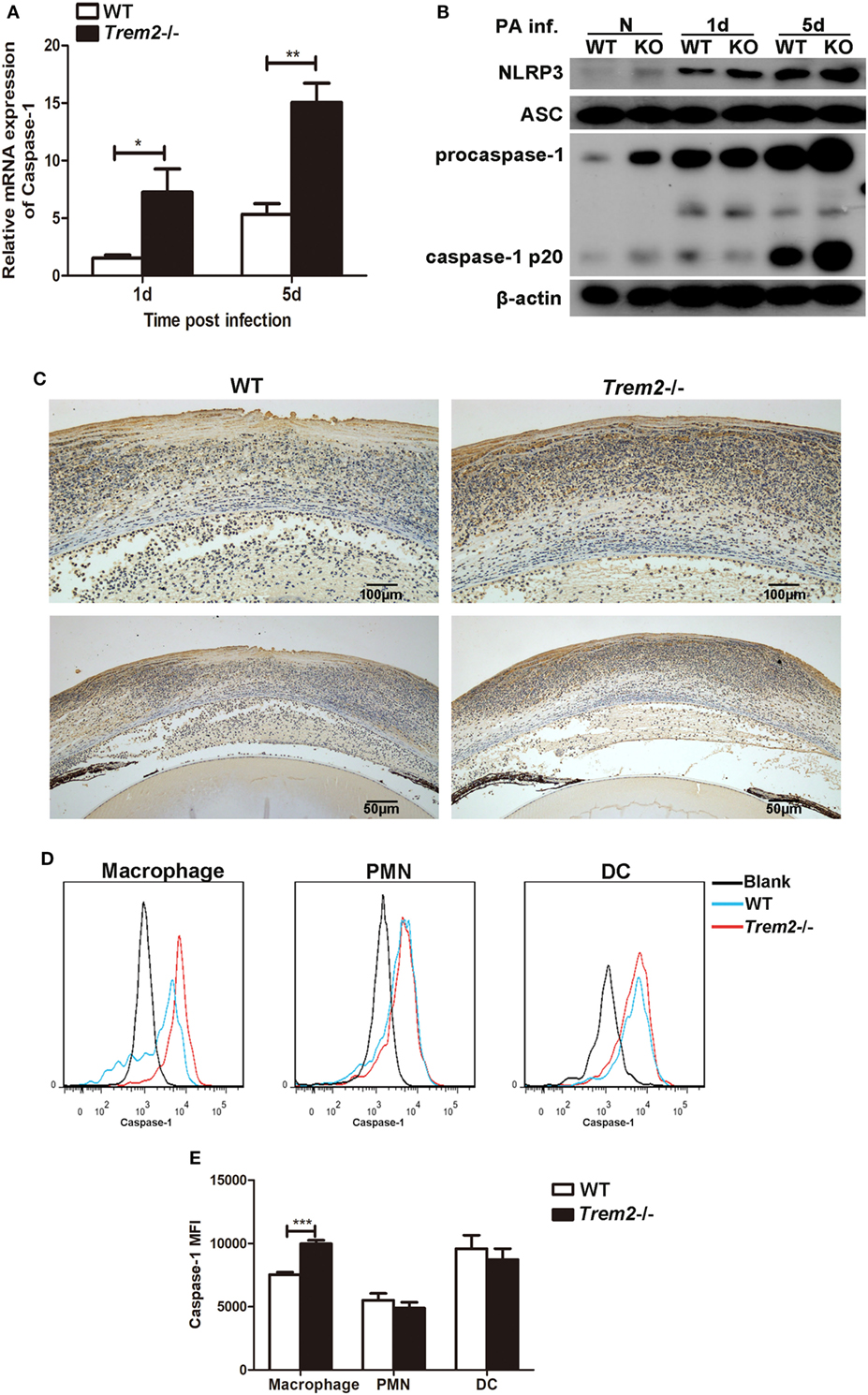
Figure 3. Triggering receptors expressed on myeloid cells 2 (TREM-2) inhibited the expression and activation of caspase-1 after Pseudomonas aeruginosa infection. mRNA expression level of caspase-1 (A) was examined by real-time PCR in infected wild type (WT) and triggering receptors expressed on myeloid cells 2 (Trem2)−/− B6 corneas at 1 and 5 days after infection. (B) The protein levels of total and cleaved caspase-1, NLRP3, and ASC in infected WT and Trem2−/− B6 corneas were detected with western blot at 1 and 5 days after infection. (C) Caspase-1 protein expression was also determined by using immune-histochemistry in WT (left) and Trem2−/− (right) B6 corneas after P. aeruginosa infection. Magnification was 100× at top panel and 200× at bottom panel, respectively. (D) FAM-FLICA staining was used to detect activated caspase-1 in macrophages (left), polymorphonuclear neutrophils (middle), and dendritic cells (right) from the infected WT and Trem2−/− corneas at 5 days postinfection. (E) The mean fluorescence intensity for activated caspase-1 was analyzed using FlowJo software. Data were the mean ± SEM and represent three individual experiments each with five mice per group. *P < 0.05; **P < 0.01.
Since caspase-1 activation is often associated with cell pyroptosis (a type of pro-inflammatory programmed cell death), here we also assessed cell death with TUNEL staining in WT and Trem2−/− corneas after P. aeruginosa infection. In contrast to the WT group (Figure 4A), the TREM2-deficient corneas (Figure 4B) showed more intense TUNEL-positive staining in the cornea stroma (magnification = 200×, 400×) after infection, suggesting that TREM2 suppressed the death of the infiltrating cells in P. aeruginosa-infected corneas. To confirm whether TREM2-modulated cell death is pyroptosis, we further detected the GSDMD and its N-terminal domain, which was recently identified as downstream of caspase-1 to cause pyroptosis. Western blot results showed that deficiency of TREM2 attenuated cleavage of GSDMD (Figure 4C), suggesting that TREM2 specifically suppressed P. aeruginosa-induced pyroptosis. Moreover, our results revealed that the number of macrophages in Trem2−/− corneas was lower than that in WT corneas after P. aeruginosa infection (Figure 4D), which is consistent with the in vivo data showing that TREM2 suppressed caspase-1-dependent pyroptosis in macrophages after P. aeruginosa infection (Figures 3D,E). Collectively, these results demonstrated that TREM2 inhibits caspase-1 activation and subsequent pyroptosis in P. aeruginosa keratitis.
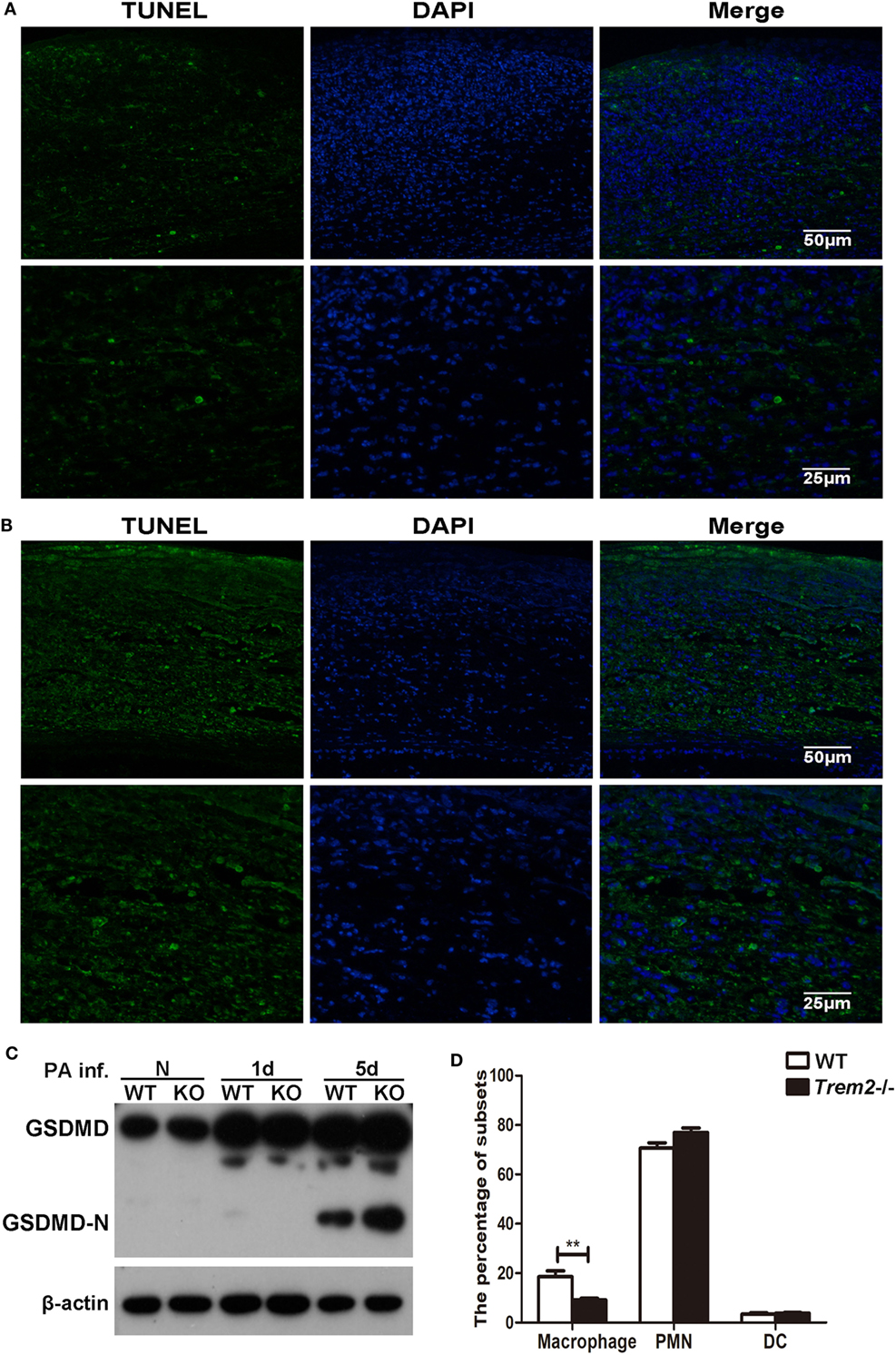
Figure 4. Triggering receptors expressed on myeloid cells 2 (TREM-2) inhibited pyroptosis of infiltrating cells in Pseudomonas aeruginosa keratitis. Pyroptosis in the infected cornea assessed with terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL) staining. TUNEL-positive staining (green) was detected in Trem2−/− (A) versus wild type (WT) (B) corneas at 5 days after infection. Cell nuclei were stained with 4,6-diamino-2-phenyl indole (DAPI; blue). Magnification was 200 and 400× respectively. (C) The protein levels of GSDMD and its N-terminal domain in Trem2−/− versus WT B6 corneas were detected with western blot. (D) The percentage of macrophages, polymorphonuclear neutrophils, and dendritic cells were detected by flow cytometry in the infected WT and Trem2−/− corneas at 5 days post infection. Data were shown to represent one of three individual experiments each with five mice per group.
Caspase-1 Was Essential for Cornea Pathology Develop and Bacterial Clearance in P. aeruginosa Keratitis
We have showed above that activation of caspase-1 and pyroptosis were markedly enhanced in Trem2−/− mice in response to P. aeruginosa ocular infection (Figures 3 and 4). To further determine whether TREM2 modulated host defense against P. aeruginosa through regulating caspase-1 activation and pyroptosis, C57BL/6 mice were subconjunctivally injected with caspase-1 siRNA or caspase-1-specific inhibitor YVAD, followed by routine P. aeruginosa corneal infection. The efficacy of silencing was confirmed by both mRNA (Figure 5A) and protein levels (Figure 5B) of caspase-1 which were decreased in the siCaspase-1-treated corneas after P. aeruginosa infection. Indeed, clinical scores showed that siCaspase-1 and YVAD-treated B6 corneas exhibited less disease severity (Figures 5C,G). Consistently, knockdown or inhibition of caspase-1 markedly suppress disease severity as demonstrated by less corneal opacity exhibited in the infected corneas (Figures 5E,I) compared with their control group (Figures 5D,H). Furthermore, silencing or inhibiting caspase-1 also resulted in reduced bacterial burden (Figures 5F,J). These data suggested that caspase-1 was required for corneal inflammation and bacterial elimination in P. aeruginosa keratitis.
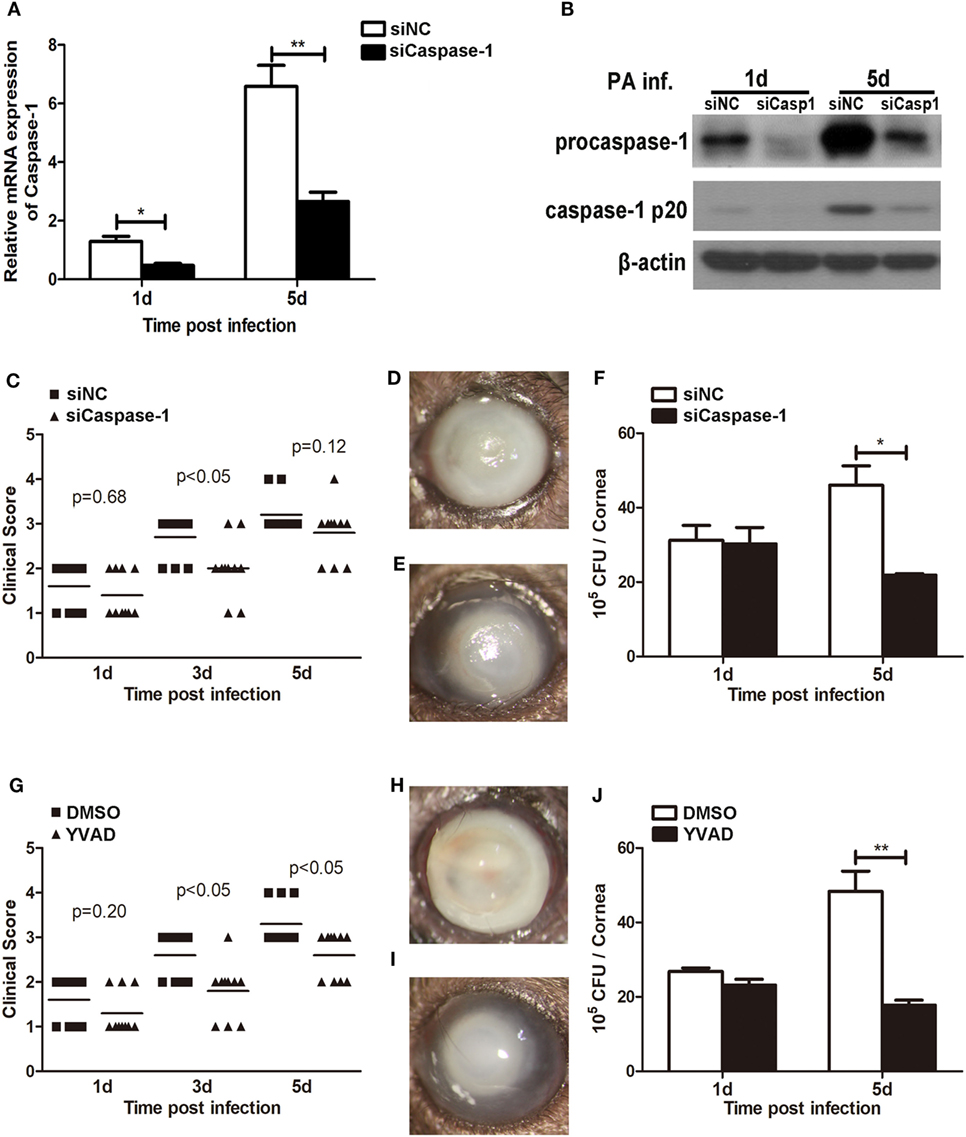
Figure 5. Caspase-1 was essential for disease progression and bacterial clearance in Pseudomonas aeruginosa keratitis. (A–F) C57BL/6 mice were subconjunctivally injected with caspase-1 siRNA or scrambled control, and then infected with PA routinely. (G–J) C57BL/6 mice were subconjunctivally injected with the caspase-1 inhibitor Ac-YVAD-CMK (YVAD) or vehicle control (DMSO), and then infected with P. aeruginosa routinely. (A) mRNA levels of caspase-1 was examined by real-time PCR in caspase-1 siRNA- versus control-treated corneas at 1 and 5 days after infection. (B) Protein levels of caspase-1 were detected with western blot in caspase-1 siRNA- versus control-treated corneas at 1 and 5 days after infection. (C,G) Clinical score was recorded for each cornea at 1, 3, and 5 days after infection. Representative photographs of infected eyes in caspase-1 siRNA- (E) versus control- (D), YVAD (I) versus DMSO (H) treated mice were taken at 5 days after infection. (F,J) Bacterial load in the infected corneas was examined by plate count assay at 1 and 5 days after infection. Data are the mean ± SEM and represent three individual experiments each with 10 mice per group (C–E,G–I) or five mice per group (A,B,F,J). *P < 0.05; **P < 0.01.
TREM-2 Promoted Host Resistance Against P. aeruginosa Keratitis by Inhibiting Caspase-1-Dependent Pyroptosis
We showed above that activated caspase-1 and pyroptosis were markedly enhanced in Trem2−/− corneas (Figures 3 and 4), and caspase-1 was involved in response to P. aeruginosa ocular infection (Figure 5). To determine whether caspase-1-dependent pyroptosis is responsible for the TREM2-mediated host resistance against P. aeruginosa keratitis, we treated TREM2-deficient mice with caspase-1 inhibitor YVAD or vehicle control DMSO by subconjunctival injection. We found that the increased disease severity in Trem2−/− mice was recovered after treating with YVAD (Figures 6A,B). Consistently, treatment with YVAD also restored the TREM2 deficiency-induced higher level of bacterial load and corneal inflammation (Figures 6C,D). Besides, treatment with YVAD markedly blocked enhanced IL-1β expression and secretion (Figures 7A,B), as well as IL-18 expression (Figure 7C) in Trem2−/− mice, but did not affect the expression of TNF-α in corneas of these groups (Figure 7D). However, YVAD treatment failed to restore enhanced IL-1β expression in HK-PA-treated Trem2−/− BMDMs (Figure 7E), suggesting the reduced mRNA expression of IL-1β in YVAD-treated Trem2−/− corneas is likely due to the decreased bacterial load and corneal inflammation. Furthermore, the enhanced pyroptosis in Trem2−/− corneas was also restored after YVAD treatment, as indicated by the change of protein levels of GSDMD N-terminal domain (Figure 7F). Taken together, these data indicated that TREM2 protected P. aeruginosa-infected corneas of C57BL/6 mice via suppressing caspase-1-dependent pyroptosis.
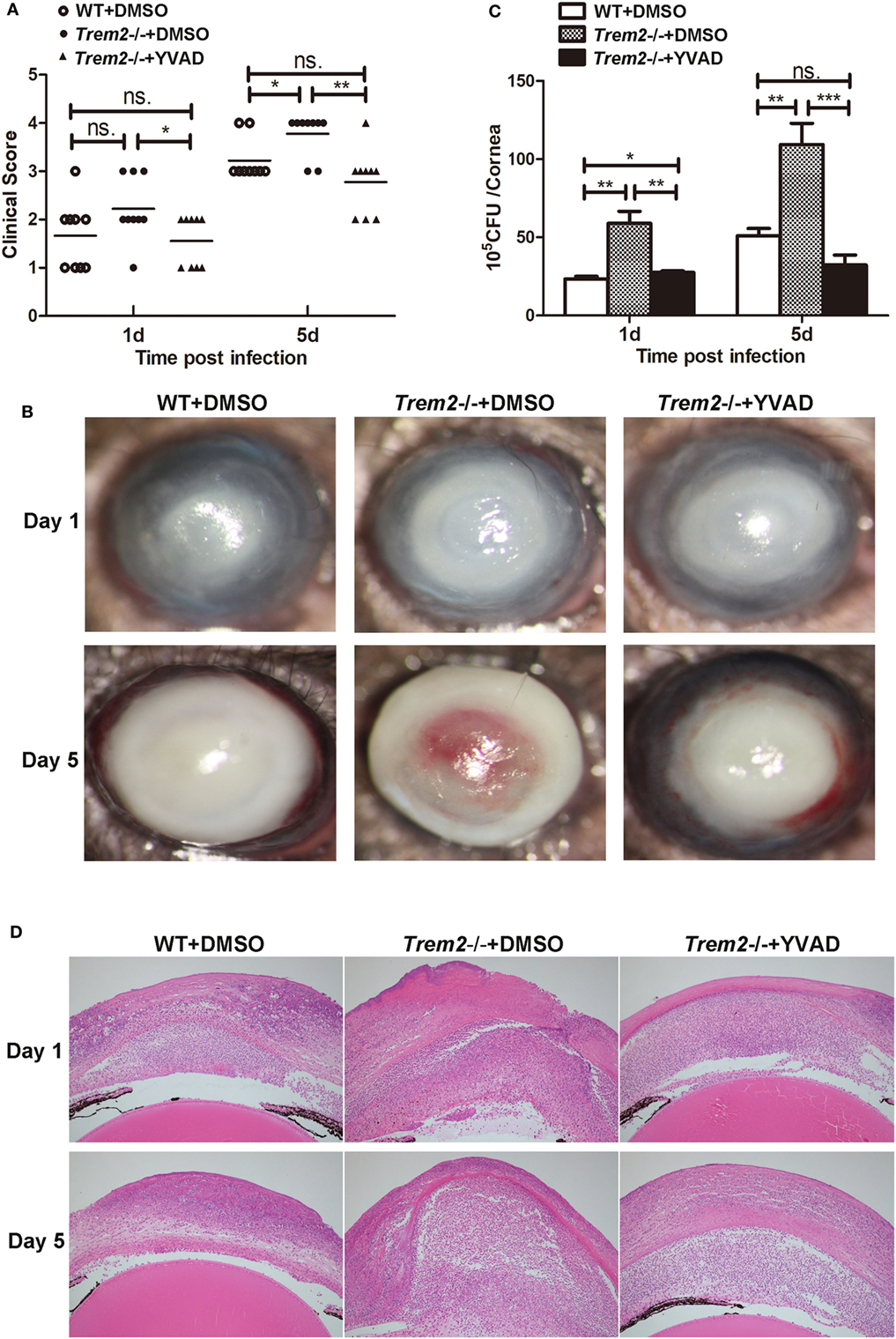
Figure 6. Triggering receptors expressed on myeloid cells 2 (TREM-2) decreased corneal inflammation and bacterial load in Pseudomonas aeruginosa keratitis via inhibiting caspase-1. Wild type (WT) mice were subconjunctivally injected with the vehicle control (DMSO) and Trem2−/− mice were subconjunctivally injected with the vehicle control (DMSO) or caspase-1 inhibitor YVAD, and then infected with P. aeruginosa routinely. (A) Clinical score was recorded for each cornea at 1 and 5 days after infection. (B) Representative photographs of infected eyes in YVAD- versus DMSO-treated Trem2−/− mice as well as DMSO-treated WT mice were taken at 1 and 5 days after infection. (C) Bacterial load in the infected corneas was examined by plate count in YVAD- versus DMSO-treated Trem2−/− corneas as well as DMSO-treated WT corneas at 1 and 5 days after infection. (D) Hematoxylin and eosin staining was used to examine the histopathology of infected eyes in DMSO-treated WT (left), DMSO-treated Trem2−/− (middle), and YVAD-treated Trem2−/− (right) mice at 1 and 5 days after infection. Magnification = 100×. Data are the mean ± SEM and represent three individual experiments each with 10 mice per group (A,B) or five mice per group (C,D). *P < 0.05; **P < 0.01; ***P < 0.001.
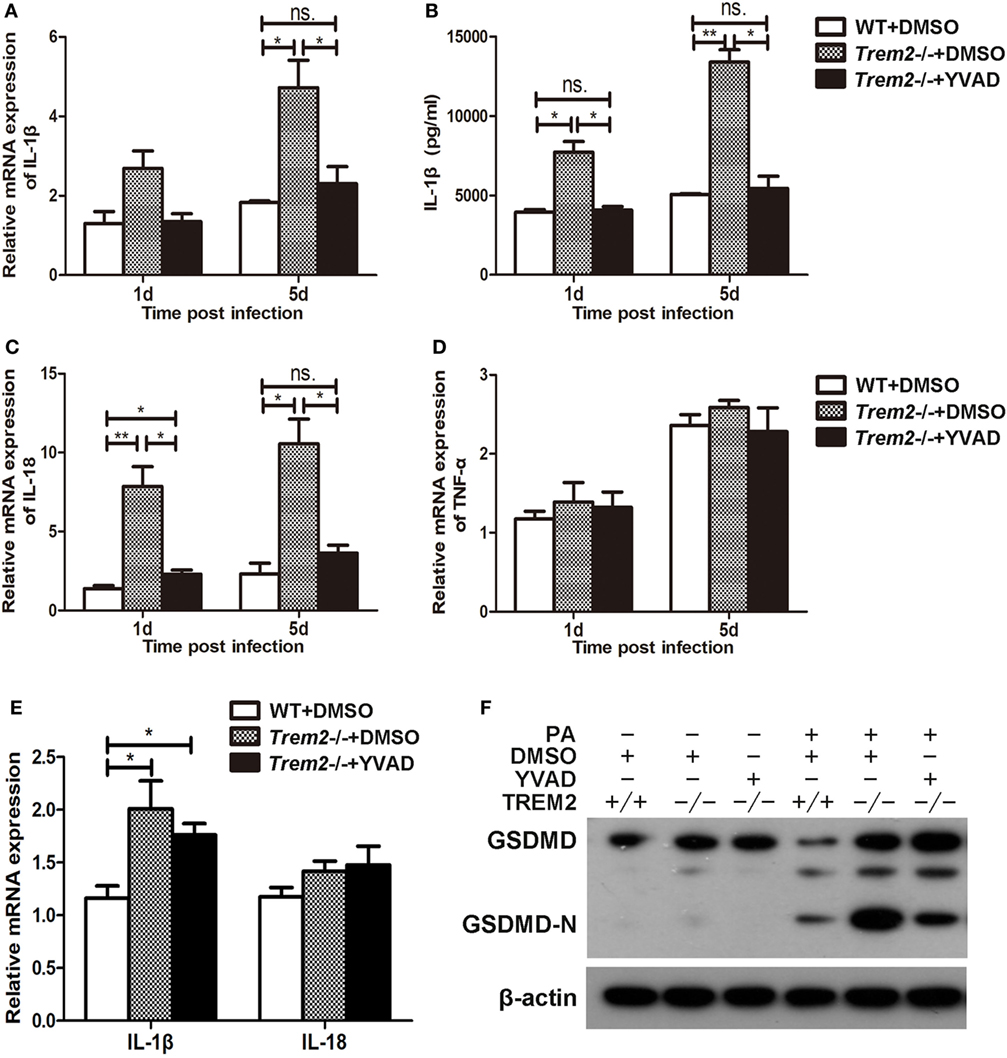
Figure 7. Triggering receptors expressed on myeloid cells 2 (TREM-2) promoted host resistance against Pseudomonas aeruginosa keratitis via inhibiting caspase-1-dependent pyroptosis. Wild type (WT) mice were subconjunctivally injected with the vehicle control (DMSO) and Trem2−/− mice were subconjunctivally injected with the vehicle control (DMSO) or caspase-1 inhibitor Ac-YVAD-CMK (YVAD), and then infected with P. aeruginosa routinely. mRNA expression levels of cytokines, including IL-1β (A), IL-18 (C), TNF-α (D) were examined by real-time PCR in the infected corneas at 1 and 5 days after infection. (B) Protein level of IL-1β in infected corneas was tested by enzyme-linked immunosorbent assay at 1 and 5 days after infection. (E) WT and Trem2−/− BMDM were treated with YVAD (40 µM) or DMSO for 1 h, followed by heat-killed P. aeruginosa treatment at MOI of 5 for 6 h. mRNA expression levels of IL-1β and IL-18 were examined by real-time PCR. (F) The protein levels of GSDMD and its N-terminal domain in corneas were detected by western blot. Data were the mean ± SEM and represent three individual experiments each with five mice per group. *P < 0.05; **P < 0.01.
TREM-2 Inhibited Caspase-1-Dependent Pyroptosis by Mediating the NLRP3 Inflammasome
As the results showed that TREM2 suppressed caspase-1-dependent pyroptosis in P. aeruginosa keratitis, we further investigated the mechanism by which TREM2 mediates caspase-1 activation and cleavage. Since the caspase-1 cleavage as well as the expression of NLRP3 was all upregulated in infected Trem2−/− corneas, we first examined the effect of TREM2 on NLRP3 inflammasome activation. We treated LPS-primed WT and Trem2−/− BMDMs with nigericin to activate canonical NLRP3 inflammasome, and found that caspase-1 cleavage and IL-1β maturation were indeed promoted in TREM2-deficient BMDMs (Figures 8A,B), suggesting TREM2 may suppress the NLRP3 inflammasome. Moreover, TREM2 co-immunoprecipitated with caspase-1 in BMDMs, and in this complex NLRP3 was also detected when BMDMs were treated with HK-PA or challenged with NLRP3 stimuli (Figure 8C). TREM2 also co-immunoprecipitated with caspase-1 and NLRP3 in P. aeruginosa-infected corneas (Figure 8D). Collectively, these results demonstrated that TREM2 inhibits caspase-1-dependent pyroptosis by reducing NLRP3 inflammasome activation.
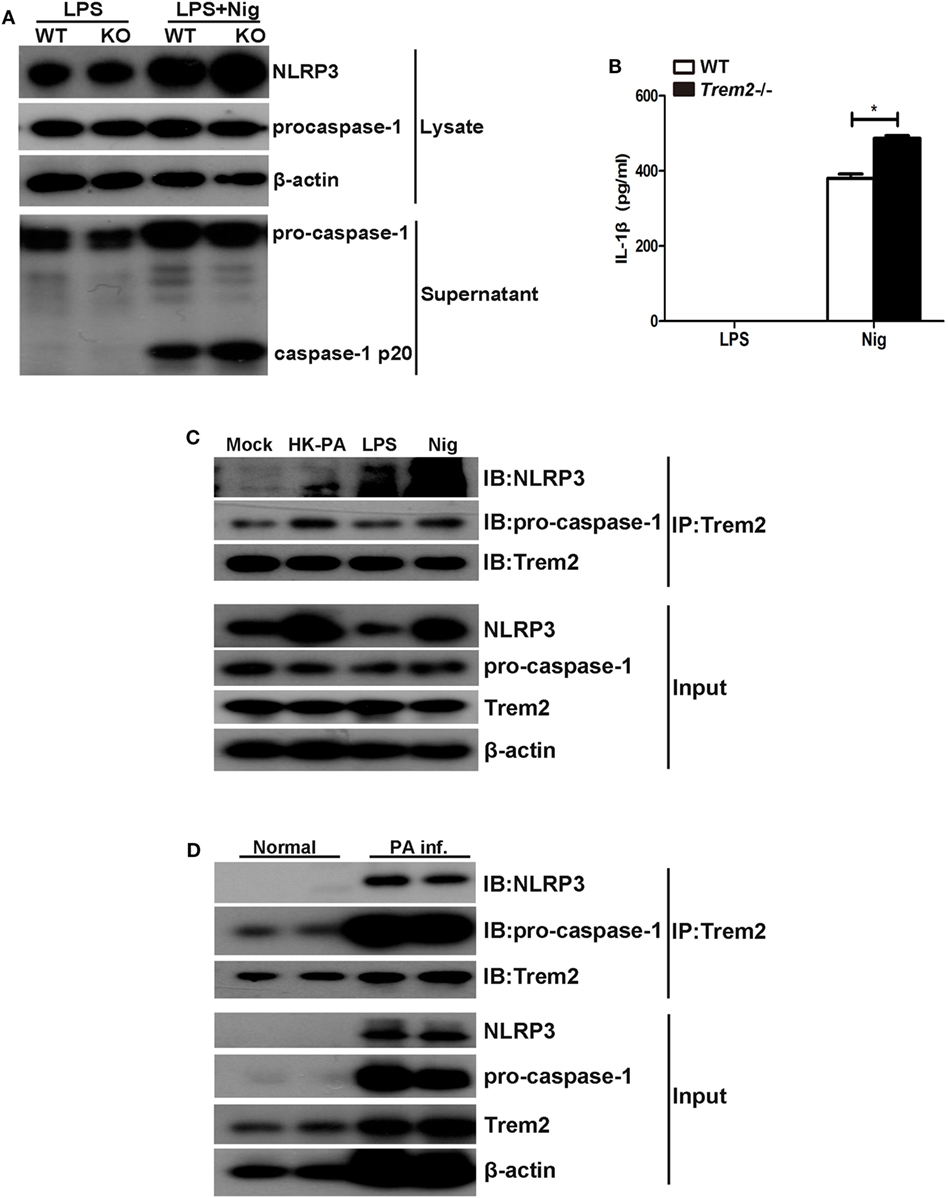
Figure 8. Triggering receptors expressed on myeloid cells 2 (TREM-2) inhibited caspase-1-dependent pyroptosis by mediating the NLRP3 inflammasome. (A) Immunoblot analysis of cleaved caspase-1 (p20) in culture supernatants of LPS-primed wild type (WT) and Trem2−/− bone marrow-derived macrophages (BMDMs) stimulated with nigericin, immunoblot analysis of the precursors of caspase-1, and NLRP3 in lysates of those cells (Input). (B) IL-1β in supernatants from LPS-primed WT and Trem2−/− BMDMs stimulated with nigericin were tested by enzyme-linked immunosorbent assay. (C) BMDMs from WT C57BL/6 mice were treated with heat-killed P. aeruginosa at MOI of 5 for 6 h or stimulated with nigericin, followed by immunoprecipitation using anti-TREM2 antibody. The immunoprecipitates were further immunoblotted with anti-TREM2, anti-caspase-1, and anti-NLRP3 antibodies. The blot shows the detection of caspase-1 and NLRP3 in the TREM2 immunoprecipitates. (D) Normal and infected corneas from WT C57BL/6 mice were harvested at 5 days after Pseudomonas aeruginosa infection, followed by immunoprecipitation using anti-TREM2 antibody. The immunoprecipitates were further immunoblotted with anti-TREM2, anti-caspase-1, and anti-NLRP3 antibodies. The blot shows the detection of caspase-1 and NLRP3 in the TREM2 immunoprecipitates. Data were the mean ± SEM and represent three individual experiments. *P < 0.05.
Discussion
Triggering receptors expressed on myeloid cells 2 is a novel member of PRRs expressed on myeloid cells, and has been reported to exert a critical role in regulating inflammation response and bacteria elimination (16, 17). However, its regulatory function is still controversial. Studies have demonstrated that TREM2 functions as a negative regulator in the immune response. However, Danese’s group reported that TREM-2 deficient DCs produced lower levels of inflammatory cytokines in a murine model of inflammatory bowel disease (27). Knapp’s lab also found that TREM-2 deficiency improved lung pathology and prevented systemic inflammation during pneumococcal pneumonia (17). These studies suggested a promotive role of TREM2 in regulating inflammatory response. Our previous study demonstrated that TREM2 suppressed P. aeruginosa-induced corneal inflammation and decreased bacterial loads in resistant BALB/c mice (which are Th2 responders), but the underlying bactericidal mechanism remains unclear (20). In this study, we found that TREM2 promotes host resistance against P. aeruginosa keratitis by inhibiting caspase-1-dependent pyroptosis in susceptible C57BL/6 mice (which are Th1 responders), providing evidence of a novel mechanism of TREM2-mediated immune defense against P. aeruginosa.
After infection, microbial pathogens and their virulence factors may impair normal organ function via inducing host cell death, such as apoptosis, necrosis, autophagy, and pyroptosis. Generally, pathogens may persist in infected hosts by causing the death of cells that required for host defense (28). While some intracellular pathogens may prevent cell death to escape immune clearance and disseminate to other host cells (29). Holtzman’s group demonstrated that the increased TREM2 expression induced by viral replication suppressed apoptosis of lung macrophages, and thus led to chronic inflammation disease in a murine model of Sendai virus infection (18). Colonna’s lab also found that autophagic-like vesicles accumulated in the microglia of TREM2-deficient mice during the development of AD (19). Moreover, Colonna’s lab reported that DAP12, an important adaptor of TREM2, was essential for the proliferation and survival of born marrow-derived macrophages (30). These results provide clues to the correlation between TREM2 and programmed cell death. Unlike viral infection, chronic infection or non-infective diseases, P. aeruginosa infection usually induces an acute inflammatory response, such as P. aeruginosa keratitis, which often leads to corneal perforation with 48 h postinfection if lacking proper treatment. It is reported that P. aeruginosa was able to trigger caspase-1-initiated pyroptosis, which was characterized by increased cell size due to osmotic swelling, and subsequent rupture of the cytoplasmic membrane as well as massive secretion of inflammatory cytokines IL-1β and IL-18 (31, 32). In this study, we found TREM2 deficiency promoted activation of caspase-1, secretion of IL-1β and subsequent cell death in P. aeruginosa keratitis. Recently, Shao F’s lab identified GSDMD as downstream of caspase-1 and revealed that after cleavage by caspase-1 and subsequent release of C-terminal domain, the N-terminal domain of GSDMD can accumulate on the plasma membrane to form pores and, therefore, result in pyroptosis (33). Consistently, our results showed that TREM2 deficiency promoted the cleavage of GSDMD to form its N-terminal domain. Besides, caspase-1-specific inhibitor YVAD restored TREM2-deficient-mediated increased cell death and IL-1β secretion. Collectively, these results indicated that TREM2 inhibited caspase-1-dependent pyroptosis induced by P. aeruginosa infection. To our knowledge, our study provides the first evidence that TREM2-mediated caspase-1 activation and pyroptosis. In addition, our data showed that caspase-1 activation in macrophage enhanced in infected Trem2−/− corneas, while its activation in PMN and DC were comparable in WT and Trem2−/− corneas. In the meantime, the number of macrophages was significantly decreased in Trem2−/− corneas compared with WT group after P. aeruginosa infection. Together, these findings support that the regulatory function of TREM2 in P. aeruginosa keratitis is primarily exerted in macrophages.
Pyroptosis was reported to restrict the survival of several intracellular pathogens, including L. pneumophila (12) and F. tularensis (13). These intracellular pathogens escape from host immune defense through residence and replication within macrophages, whereas pyroptosis exposes them to effective immune environment, lead to the subsequent bacterial phagocytosis and intracellular killing by neutrophils (12). However, the role of pyroptosis in P. aeruginosa elimination remains disputable. Prince’s group demonstrated that in a murine model of acute pneumonia, inflammasome activation impaired the ability of alveolar macrophages to eliminate P. aeruginosa by promoting macrophage pyroptosis (14). In addition, Hauser’s lab reported that the RhsT protein of P. aeruginosa promoted phagocytic cells undergoing inflammasome-mediated cell death with more IL-1β and IL-18 production and in favor of bacterial survival (34). To some extent, these results provide some clues that caspase-1-initiated pyroptosis may suppress P. aeruginosa clearance. In this study, we demonstrated that lack of TREM2 promoted bacterial survival, which could be restored by blockage of pyroptosis using caspase-1 inhibitor, suggesting that increased bacteria load is associated with enhanced caspase-1 activation and pyroptosis.
Along with caspase-1-dependent pyroptosis, plenty of pro-inflammatory mediators release from dead cells, such as IL-1β and IL-18. Both IL-1β and IL-18 belong to the IL-1 family cytokines. Similar to IL-1β, IL-18 is synthesized as an inactive precursor and can be processed by caspase-1 to generate an active form. But unlike IL-1β precursor, which is induced by microbial and inflammatory stimuli, the IL-18 precursor is constitutively expressed in most of the human and mouse cells (35). Our data showed that treatment with HK-PA did not induce IL-18 expression and no significant difference in IL-18 expression was detected in HK-PA-treated Trem2−/− and WT corneas and BMDMs. In contrast, IL-18 expression was enhanced in mouse corneas and BMDMs after P. aeruginosa infection, and TREM2 deficiency increased the PA-induced IL-18 expression in vivo. The underlying mechanism of different IL-18 expression in PA infection versus HK-PA treatment remains unknown, which needs further investigation.
In vivo studies proved that IL-1β impairs the P. aeruginosa clearance by using a murine model of P. aeruginosa pneumonia (36). Moreover, IL-1β has been reported as local inflammation mediator in bacterial keratitis (37, 38). IL-1β secretion may be promoted through inflammasome-dependent or -independent pathways. Previously, Hazlett’s group reported that knockout of caspase-1 (which is actually caspase-1 and caspase-11 double knockout), or treatment with caspase-1 inhibitor markedly decreases the ocular inflammation and bacterial load in P. aeruginosa keratitis (39, 40). However, Pearlman’s group found that in the P. aeruginosa-infected corneas, IL-1β cleavage was dependent on serine proteases of neutrophils but independent of caspase-1 and NLRC4 (41). In our study, TREM2 deficiency resulted in massive secretion of IL-1β which could be restored by treatment with caspase-1-specific inhibitor YVAD. Besides, knockout of TREM2 did not affect TNF-α, which is closely related to cell apoptosis and NF-κB signaling, suggesting TREM2 deficiency may promote IL-1β production in a caspase-1-dependent manner. To be noted, YVAD treatment blocked IL-1β and IL-18 transcription in Trem2−/− corneas but failed to restore enhanced IL-1β expression in HK-PA treated Trem2−/− BMDMs. Our data showed that inhibition of caspase-1 decreased bacterial load and inflammatory cell infiltration in the infected corneas. Since bacterial virulence factors can activate local and infiltrated inflammatory cells to induce the IL-1β and IL-18 production, we speculated the decreased bacterial load and corneal inflammation may be the reason of reduced IL-1β and IL-18 mRNA levels in YVAD-treated Trem2−/− corneas.
It is worth to mention that, in our previous study, silencing of TREM2 in corneas of BALB/c mice increased TNF-α expression, whereas in this study, lack of TREM2 failed to upregulate TNF-α in C57BL/6 mice. We also established a murine model of P. aeruginosa-induced sepsis using TREM2-deficient versus WT C57BL/6 mice, and found that TNF-α expression was comparable between the two groups (data not shown), which is consistent with our observation in this study. A recent study revealed that Th1 cells have more detectable amounts of NLRP3 protein than Th2 cells (42), suggesting the inflammasome activation may be different between these T helper subsets. Since C57BL/6 and BALB/c mice are known to preferentially demonstrate Th1 and Th2 responses, respectively, we speculate that the different effects of TREM2 on TNF-α expression in C57BL/6 and BALB/c mice may result from distinct responses (Th1- or Th2-favored).
Our in vivo studies indicated that TREM2 deficiency not only increased the production of IL-1β and IL-18, but also promoted expression of MIP-2. In susceptible C57BL/6 mice, IL-1β is able to upregulate the production of MIP-2, an important chemokine which attracts PMN influx into the infected cornea and, therefore, contributes to irreversible corneal tissue destruction (37). In this regard, the increased MIP-2 and severe inflammation are probably due to the high level of IL-1β in TREM2-deficient mice.
Caspase-1-dependent pyroptosis often results from activation of inflammasomes (43–45). Many Studies have demonstrated that P. aeruginosa infection mainly activated the NLRC4 inflammasome by its flagella. Recently, Hazlett’ group reported that the NLRC4 inflammasome in CD11blowLy6Glow cells contributed to the host resistance of BALB/c mice in response to P. aeruginosa infection, through regulating caspase-1 and IL-1β production (46). However, our previous research in human macrophages indicated that P. aeruginosa triggered the assembly of the NLRP3 inflammasomes, which suppressed the killing of P. aeruginosa by triggering autophagy of macrophages (47). Our study showed that caspase-1 cleavage and IL-1β maturation were promoted in TREM2-deficient BMDMs after treated with canonical NLRP3 stimuli nigericin (Figures 8A,B), suggesting TREM2 suppressed NLRP3 inflammasome to inhibit caspase-1 activation and pyroptosis. After P. aeruginosa infection, other inflammasomes (such as NLRC4 inflammasome or caspase-8/caspase-11-mediated noncanonical inflammasome) may also be activated. Loss of TREM2 delayed but did not halt the disease progression. Most of the wild type corneas were perforated at 5 days postinfection, while corneas of TREM2-deficient animals were perforated at 3 days postinfection.
Our endogenous Co-IP data showed that, TREM2 co-immunoprecipitated with procaspase-1 and NLRP3 in BMDMs treated with HK-PA, LPS, or nigericin, as well as P. aeruginosa-infected mouse corneas (Figures 8C,D). Since TREM2 is a transmembrane receptor with surface and cytoplasmic domains, the coimmunoprecipitation of NLRP3 with TREM2 suggests that TREM2 may regulate inflammasome activation by direct interaction.
In summary, we demonstrated that TREM2 deficiency increased host susceptibility to P. aeruginosa corneal infection in C57BL/6 mice. The increased sensitivity in Trem2−/− mice was associated with upregulated NLRP3 inflammasome activation and subsequent production of pro-inflammatory cytokines IL-1β. Furthermore, our data showed that caspase-1-dependent pyroptosis was responsible for more serious tissue pathological injuries and increased bacterial load. Together, our study revealed a novel mechanism by which TREM2 mediates the immune defense against P. aeruginosa.
Ethics Statement
The procedure was performed in accordance with the National Commission for the Protection of Subjects of Biomedical and Behavioral Research guidelines for animal experiments. All efforts were made to minimize suffering. Before the study was initiated, all experiment protocols were approved by the local Ethics Committee of Sun Yat-sen University.
Author Contributions
MW and WQ wrote the manuscript. MW designed experiments. WQ, YW, YW, YL, KC, XL, and ZZ performed experiments and analyzed data. XH provided scientific expertise. MW supervised the project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
The authors thank Dr. Feng Lianqiang of the Department of Immunology at Zhongshan School of Medicine, Sun Yat-sen University for his technical assistance.
Funding
This work was supported by grants from National Natural Science Foundation of China (31370868, 31670880, 31200662, 81261160323, 31470877), Guangdong Natural Science Fund for Distinguished Young Scholars (2016A030306004), Guangdong Special Support Program for Youth Science and Technology Innovation Talents (2015TQ01R473), Guangzhou Pearl River New Star Program (201610010064), Guangdong Innovative Research Team Program (2009010058, 2011Y035), Guangdong Natural Science Foundation (S2012040006680, 2015A030311009), Science and Technology Program of Guangdong (2015B090903063), the National Key Research and Development Program of China (2016YFC1200105), the National Science and Technology Key Projects for Major Infectious Diseases (2013ZX10003001 and 2017ZX10302301), and the 111 Project (No. B13037).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01121/full#supplementary-material.
Figure S1. (A) mRNA expression level of caspase-1 was examined by real-time PCR in heat-killed Pseudomonas aeruginosa (HK-PA) treated wild type (WT) and Trem2−/− B6 corneas at 5 days after treatment and WT and Trem2−/− BMDM which were treated with HK-PA at MOI of 5 for 6 h. (B) The protein level of caspase-11 in P. aeruginosa-infected WT and Trem2−/− B6 corneas were detected with western blot at 1 and 5 days after infection. (C) A6(1) cells were transfected with TREM2 siRNA versus control scrambled siRNA, followed by HK-PA treatment at MOI of 5 for 6 h. mRNA expression levels of TREM2, IL-1β, IL-18, MIP-2, and caspase-1 was examined by real-time PCR. Data were the mean ± SEM and represent three individual experiments. *P < 0.05.
References
1. Willcox MD. Pseudomonas aeruginosa infection and inflammation during contact lens wear: a review. Optom Vis Sci (2007) 84(4):273–8. doi:10.1097/OPX.0b013e3180439c3e
2. Hazlett LD. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res (2004) 23(1):1–30. doi:10.1016/j.preteyeres.2003.10.002
3. Fleiszig SM, Evans DJ. The pathogenesis of bacterial keratitis: studies with Pseudomonas aeruginosa. Clin Exp Optom (2002) 85(5):271–8. doi:10.1111/j.1444-0938.2002.tb03082.x
4. McIsaac SM, Stadnyk AW, Lin TJ. Toll-like receptors in the host defense against Pseudomonas aeruginosa respiratory infection and cystic fibrosis. J Leukoc Biol (2012) 92(5):977–85. doi:10.1189/jlb.0811410
5. Shin HS, Lee JH, Paek SH, Jung YW, Ha UH. Pseudomonas aeruginosa-dependent upregulation of TLR2 influences host responses to a secondary Staphylococcus aureus infection. Pathog Dis (2013) 69(2):149–56. doi:10.1111/2049-632X.12074
6. Travassos LH, Carneiro LA, Girardin SE, Boneca IG, Lemos R, Bozza MT, et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem (2005) 280(44):36714–8. doi:10.1074/jbc.M501649200
7. Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med (2007) 204(13):3235–45. doi:10.1084/jem.20071239
8. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun (2015) 6:6201. doi:10.1038/ncomms7201
9. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol (2013) 13(6):397–411. doi:10.1038/nri3452
10. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell (2014) 157(5):1013–22. doi:10.1016/j.cell.2014.04.007
11. de Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harb Perspect Biol (2014) 6(12):a016287. doi:10.1101/cshperspect.a016287
12. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol (2010) 11(12):1136–42. doi:10.1038/ni.1960
13. Dotson RJ, Rabadi SM, Westcott EL, Bradley S, Catlett SV, Banik S, et al. Repression of inflammasome by Francisella tularensis during early stages of infection. J Biol Chem (2013) 288(33):23844–57. doi:10.1074/jbc.M113.490086
14. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest (2013) 123(4):1630–7. doi:10.1172/JCI66142
15. Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol (2006) 7(12):1266–73. doi:10.1038/ni1411
16. N’Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol (2009) 184(2):215–23. doi:10.1083/jcb.200808080
17. Sharif O, Gawish R, Warszawska JM, Martins R, Lakovits K, Hladik A, et al. The triggering receptor expressed on myeloid cells 2 inhibits complement component 1q effector mechanisms and exerts detrimental effects during pneumococcal pneumonia. PLoS Pathog (2014) 10(6):e1004167. doi:10.1371/journal.ppat.1004167
18. Wu K, Byers DE, Jin X, Agapov E, Alexander-Brett J, Patel AC, et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J Exp Med (2015) 212(5):681–97. doi:10.1084/jem.20141732
19. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell (2017) 170(4):649–63.e13. doi:10.1016/j.cell.2017.07.023
20. Sun M, Zhu M, Chen K, Nie X, Deng Q, Hazlett LD, et al. TREM-2 promotes host resistance against Pseudomonas aeruginosa infection by suppressing corneal inflammation via a PI3K/Akt signaling pathway. Invest Ophthalmol Vis Sci (2013) 54(5):3451–62. doi:10.1167/iovs.12-10938
21. Narayanan S, Miller WL, McDermott AM. Expression of human beta-defensins in conjunctival epithelium: relevance to dry eye disease. Invest Ophthalmol Vis Sci (2003) 44(9):3795–801. doi:10.1167/iovs.02-1301
22. Wu M, McClellan SA, Barrett RP, Hazlett LD. Beta-defensin-2 promotes resistance against infection with P. aeruginosa. J Immunol (2009) 182(3):1609–16. doi:10.4049/jimmunol.182.3.1609
23. Wu M, Peng A, Sun M, Deng Q, Hazlett LD, Yuan J, et al. TREM-1 amplifies corneal inflammation after Pseudomonas aeruginosa infection by modulating toll-like receptor signaling and Th1/Th2-type immune responses. Infect Immun (2011) 79(7):2709–16. doi:10.1128/IAI.00144-11
24. Gao C, Negash S, Guo HT, Ledee D, Wang HS, Zelenka P. CDK5 regulates cell adhesion and migration in corneal epithelial cells. Mol Cancer Res (2002) 1(1):12–24.
25. Weihua Z, Makela S, Andersson LC, Salmi S, Saji S, Webster JI, et al. A role for estrogen receptor beta in the regulation of growth of the ventral prostate. Proc Natl Acad Sci U S A (2001) 98(11):6330–5. doi:10.1073/pnas.111150898
26. Huang X, Du W, Barrett RP, Hazlett LD. ST2 is essential for Th2 responsiveness and resistance to Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci (2007) 48(10):4626–33. doi:10.1167/iovs.07-0316
27. Correale C, Genua M, Vetrano S, Mazzini E, Martinoli C, Spinelli A, et al. Bacterial sensor triggering receptor expressed on myeloid cells-2 regulates the mucosal inflammatory response. Gastroenterology (2013) 144(2):346–56.e3. doi:10.1053/j.gastro.2012.10.040
28. Zychlinsky A, Sansonetti P. Perspectives series: host/pathogen interactions. Apoptosis in bacterial pathogenesis. J Clin Invest (1997) 100(3):493–5. doi:10.1172/JCI119557
29. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun (2005) 73(4):1907–16. doi:10.1128/IAI.73.4.1907-1916.2005
30. Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol (2009) 10(7):734–43. doi:10.1038/ni.1744
31. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol (2000) 38(1):31–40. doi:10.1046/j.1365-2958.2000.02103.x
32. Fink SL, Cookson BT. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol (2006) 8(11):1812–25. doi:10.1111/j.1462-5822.2006.00751.x
33. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi:10.1038/nature15514
34. Kung VL, Khare S, Stehlik C, Bacon EM, Hughes AJ, Hauser AR. An rhs gene of Pseudomonas aeruginosa encodes a virulence protein that activates the inflammasome. Proc Natl Acad Sci U S A (2012) 109(4):1275–80. doi:10.1073/pnas.1109285109
35. Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol (2013) 4:289. doi:10.3389/fimmu.2013.00289
36. Schultz MJ, Rijneveld AW, Florquin S, Edwards CK, Dinarello CA, van der Poll T. Role of interleukin-1 in the pulmonary immune response during Pseudomonas aeruginosa pneumonia. Am J Physiol Lung Cell Mol Physiol (2002) 282(2):L285–90. doi:10.1152/ajplung.00461.2000
37. Rudner XL, Kernacki KA, Barrett RP, Hazlett LD. Prolonged elevation of IL-1 in Pseudomonas aeruginosa ocular infection regulates macrophage-inflammatory protein-2 production, polymorphonuclear neutrophil persistence, and corneal perforation. J Immunol (2000) 164(12):6576–82. doi:10.4049/jimmunol.164.12.6576
38. Thakur A, Xue M, Stapleton F, Lloyd AR, Wakefield D, Willcox MD. Balance of pro- and anti-inflammatory cytokines correlates with outcome of acute experimental Pseudomonas aeruginosa keratitis. Infect Immun (2002) 70(4):2187–97. doi:10.1128/IAI.70.4.2187-2197.2002
39. Thakur A, Barrett RP, McClellan S, Hazlett LD. Regulation of Pseudomonas aeruginosa corneal infection in IL-1 beta converting enzyme (ICE, caspase-1) deficient mice. Curr Eye Res (2004) 29(4–5):225–33. doi:10.1080/02713680490516710
40. Thakur A, Barrett RP, Hobden JA, Hazlett LD. Caspase-1 inhibitor reduces severity of Pseudomonas aeruginosa keratitis in mice. Invest Ophthalmol Vis Sci (2004) 45(9):3177–84. doi:10.1167/iovs.04-0041
41. Karmakar M, Sun Y, Hise AG, Rietsch A, Pearlman E. Cutting edge: IL-1beta processing during Pseudomonas aeruginosa infection is mediated by neutrophil serine proteases and is independent of NLRC4 and caspase-1. J Immunol (2012) 189(9):4231–5. doi:10.4049/jimmunol.1201447
42. Bruchard M, Rebe C, Derangere V, Togbe D, Ryffel B, Boidot R, et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat Immunol (2015) 16(8):859–70. doi:10.1038/ni.3202
43. Case CL, Kohler LJ, Lima JB, Strowig T, de Zoete MR, Flavell RA, et al. Caspase-11 stimulates rapid flagellin-independent pyroptosis in response to Legionella pneumophila. Proc Natl Acad Sci U S A (2013) 110(5):1851–6. doi:10.1073/pnas.1211521110
44. Wellington M, Koselny K, Sutterwala FS, Krysan DJ. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell (2014) 13(2):329–40. doi:10.1128/EC.00336-13
45. Kovarova M, Hesker PR, Jania L, Nguyen M, Snouwaert JN, Xiang Z, et al. NLRP1-dependent pyroptosis leads to acute lung injury and morbidity in mice. J Immunol (2012) 189(4):2006–16. doi:10.4049/jimmunol.1201065
46. McClellan SA, Jerome A, Suvas S, Hazlett LD. NLRC4 regulates caspase-1 and IL-1beta production in a CD11blowLy6Glow population of cells required for resistance to Pseudomonas aeruginosa keratitis. PLoS One (2017) 12(9):e0185718. doi:10.1371/journal.pone.0185718
Keywords: Pseudomonas aeruginosa, triggering receptors expressed on myeloid cells 2, corneal infection, pyroptosis, inflammation, bacterial killing
Citation: Qu W, Wang Y, Wu Y, Liu Y, Chen K, Liu X, Zou Z, Huang X and Wu M (2018) Triggering Receptors Expressed on Myeloid Cells 2 Promotes Corneal Resistance Against Pseudomonas aeruginosa by Inhibiting Caspase-1-Dependent Pyroptosis. Front. Immunol. 9:1121. doi: 10.3389/fimmu.2018.01121
Received: 13 January 2018; Accepted: 03 May 2018;
Published: 25 May 2018
Edited by:
Joseph Alex Duncan, University of North Carolina at Chapel Hill, United StatesReviewed by:
Paras K. Anand, Imperial College London, United KingdomYu L. Lei, University of Michigan, United States
Copyright: © 2018 Qu, Wang, Wu, Liu, Chen, Liu, Zou, Huang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minhao Wu, d3VtaW5oYW9AbWFpbC5zeXN1LmVkdS5jbg==