 Francesca A. Ververs
Francesca A. Ververs Eric Kalkhoven
Eric Kalkhoven Belinda van’t Land
Belinda van’t Land Marianne Boes
Marianne Boes Henk S. Schipper
Henk S. Schipper- 1Laboratory for Translational Immunology, University Medical Center Utrecht, Utrecht, Netherlands
- 2Department of Molecular Cancer Research, Center for Molecular Medicine, University Medical Center Utrecht, University Utrecht, Utrecht, Netherlands
- 3Department of Immunology, Nutricia Research, Utrecht, Netherlands
- 4Department of Pediatric Immunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands
- 5Department of Pediatric Cardiology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands
Invariant natural killer T (iNKT) cells are lipid-reactive T cells with profound immunomodulatory potential. They are unique in their restriction to lipid antigens presented in CD1d molecules, which underlies their role in lipid-driven disorders such as obesity and atherosclerosis. In this review, we discuss the contribution of iNKT cell activation to immunometabolic disease, metabolic programming of lipid antigen presentation, and immunometabolic activation of iNKT cells. First, we outline the role of iNKT cells in immunometabolic disease. Second, we discuss the effects of cellular metabolism on lipid antigen processing and presentation to iNKT cells. The synthesis and processing of glycolipids and other potential endogenous lipid antigens depends on metabolic demand and may steer iNKT cells toward adopting a Th1 or Th2 signature. Third, external signals such as toll-like receptor ligands, adipokines, and cytokines modulate antigen presentation and subsequent iNKT cell responses. Finally, we will discuss the relevance of metabolic programming of iNKT cells in human disease, focusing on their role in disorders such as obesity and atherosclerosis. The critical response to metabolic changes places iNKT cells at the helm of immunometabolic disease.
Invariant Natural Killer T (iNKT) Cells Center Stage in Immunometabolic Disease
Immunometabolic diseases such as obesity, type 2 diabetes, and cardiovascular disease (CVD) are the major health burdens of our time and illustrate the intricate web between metabolic dysregulation and inflammation (1). The links between metabolism and inflammation may be explained from an evolutionary perspective. An effective immune defense critically depends on efficient energy storage and release, as reflected by the co-evolution of the immune system and metabolism in Drosophila fat bodies, and the reminiscent immune cell functions of adipocytes in humans and other higher organisms (2). Unfortunately, evolution could not foresee the endemic nutritional overload in 21st century Western societies, causing glucotoxicity and lipotoxicity, and propagating local and systemic inflammation (3).
NKT cells were identified as important players in immunometabolism due to their unique response to lipid antigens and hybrid qualities of both the innate and adaptive immune system (4). NKT cells readily produce copious amounts of Th1, Th2, and/or Th17 cytokines upon activation, which resembles an innate activation scheme (5). Similar to T cells, NKT cells develop in the thymus and undergo positive and negative thymic selection. However, instead of interacting with MHC class 2 molecules, iNKT cells are selected by CD1d-expressing thymocytes. Two NKT cell subtypes have been defined: type 1 signifies CD1d-restricted iNKT cells carrying an invariant T cell receptor that recognizes the prototypic ligand alpha-galactosylceramide, while type 2 signifies CD1d-restricted iNKT cells carrying different T cell receptors not recognizing alpha-galactosylceramide (6). This review focuses on type 1 NKT cells, also known as iNKT cells, which represent the most studied NKT cell subset.
Invariant natural killer T cell frequency in peripheral blood is low, but they are highly enriched in adipose tissue (AT) in mice and humans (7, 8). Functionally, AT-resident iNKT cells have an anti-inflammatory phenotype by secreting IL-4, which contributes to prevention of insulin resistance and AT inflammation (7, 9). In obesity, the protective IL-4 production by iNKT cells is lost, and total iNKT cell numbers in AT and peripheral blood decrease, making leeway for adipose tissue inflammation, insulin resistance, and type 2 diabetes to develop (7–10). The same phenomenon is observed in other metabolic disorders. When comparing human identical twins, of which only one sibling developed type 1 diabetes, diabetic siblings show lower frequencies of iNKT cells. When multiple iNKT clones were compared from the twins, all clones isolated from diabetic siblings produced only IFN-γ upon stimulation, while all clones isolated from the healthy twin produced both IL-4 and IFN-γ (11). In atherosclerosis, a similar decrease in iNKT cell numbers and production of IL-4 is observed in established CVD (12). Notably, iNKT cell numbers in peripheral blood seem to increase in the earliest phase of atherosclerosis, accompanied by an increase in IL-4 production, GATA3- and CD69 expression, and increased proliferative capacity (13). This model, in which iNKT cells play an anti-inflammatory or pro-homeostatic role early in disease development, seems widely applicable for human disease (14), and begs the question: what do iNKT cells see when trouble starts stirring?
iNKT Cell Activation by Sphingolipid Ligands
In the early 1990s, it was discovered that iNKT cells can be activated by glycosphingolipids (GSL) following identification of alpha-galactosylceramide, a potent marine sponge sphingolipid antigen identified in a cancer antigen screen (15). Since then, endogenous sphingolipids have been scrutinized as potential lipid antigens for iNKT cells.
Sphingolipids are synthesized either via the salvage pathway, by degradation and re-usage of existing sphingolipids, or via de novo synthesis in the endoplasmic reticulum (ER), by attachment of a fatty acid to a sphingosine base (16). Spingomyelinases and glucosidases are important enzymes in the salvage pathway, converting membrane sphingomyelin and glucosylceramides back to ceramide within the lysosome (17). Serine palmitoyl transferase (SPT) and ceramide synthases are important for de novo synthesis. De novo synthesis is orchestrated by six different ceramide synthases (CerS), which determine the length of the fatty acid chain attached to the sphingosine base. Sphingosine with one fatty acid attached is called ceramide, which is the central metabolite in sphingolipid metabolism. More complex sphingolipids such as GSL are generated in the Golgi by addition of different headgroups by UDP-glucose ceramide glucosyltransferase (UGCG) and other glycosyltransferases (18). Translocation to the Golgi is facilitated by ceramide transfer proteins (CERT) (17). The simplest glycosphingolipid has only one sugar residue attached, either glucose or galactose. The sugar headgroup can be attached to ceramide in a beta- or alpha-anomeric fashion. To date, only beta-anomeric GSL have been identified in humans. Some studies reported iNKT cell reactivity to beta-linked GSL, but this was disputed later as contamination of alpha-linkages was found in the preparations (19–22). The alpha-anomeric linkage remains one of the key determinants for antigenicity (20, 23, 24). Enter the search for endogenous lipid ligands continues as, unfortunately, developing a robust method for isolation of these ligands is technically challenging. In the meantime, extensive studies on the effect of various synthetic alpha-galcer analogs on iNKT function were performed, including analogs with truncated alkyl chains, varying saturation status, or the presence of aromatic structures (24–26). These efforts revealed that analogs with a shorter alkyl chain can elicit an IL-4 response without prior IFN-γ induction in mice in vivo (alpha-GalCer C10:0, alpha-GalCer C20:2, alpha-GalCer C20:4, OCH, alpha-GalCer-PGB1) (26). However, in human iNKT cells, even though almost all glycolipid analogs elicit a potent cytokine response, there is hardly any Th2-polarization (24, 26). Enter differences between mouse and human ligand-mediated activation are abound: there are differences in potential endogenous ligands and where the ligands derive from, considering that human CD1d and mouse CD1d1 travel to different subcellular compartments for endogenous ligand extraction (27–32). The secretory route from the ER, via the Golgi, to the plasma membrane is similar for human CD1d and mouse CD1d1. Upon folding in the ER and association with beta-2-microglobulin, lipid transfer proteins such as microsomal transfer protein mediate loading of chaperone lipids in the ER and/or endogenous lipid antigens in the Golgi (33–35). The endolysosomal recycling route, however, is different for human CD1d and mouse CD1d1. On the basal side of the membrane, CD1d has a short cytoplasmic tail carrying a sorting motif. The sorting motif binds to the adaptor protein complex 2 upon which membrane internalization is mediated to enter the early endosome (36). Only mouse CD1d1, but not human CD1d, can also bind adaptor protein 3, which then targets late endosomes and lysosomes (31). Considering the observed co-localization of human CD1d with the lysosomal membrane protein LAMP1, the lack of a lysosomal sorting motif does not preclude lysosomal transportation of human CD1d (31, 32). Nevertheless, differences in endolysosomal trafficking may result in loading of different lipid antigens. LDL receptor (LDLR)-mediated uptake of GSL for example, is processed in the endosomal compartment (37), while the salvage pathway of plasma membrane GSL starts in the lysosomal compartment (38) (Figure 1). These differences are important to keep in mind when studying iNKT cells and Th1/Th2 skewing in mouse models.
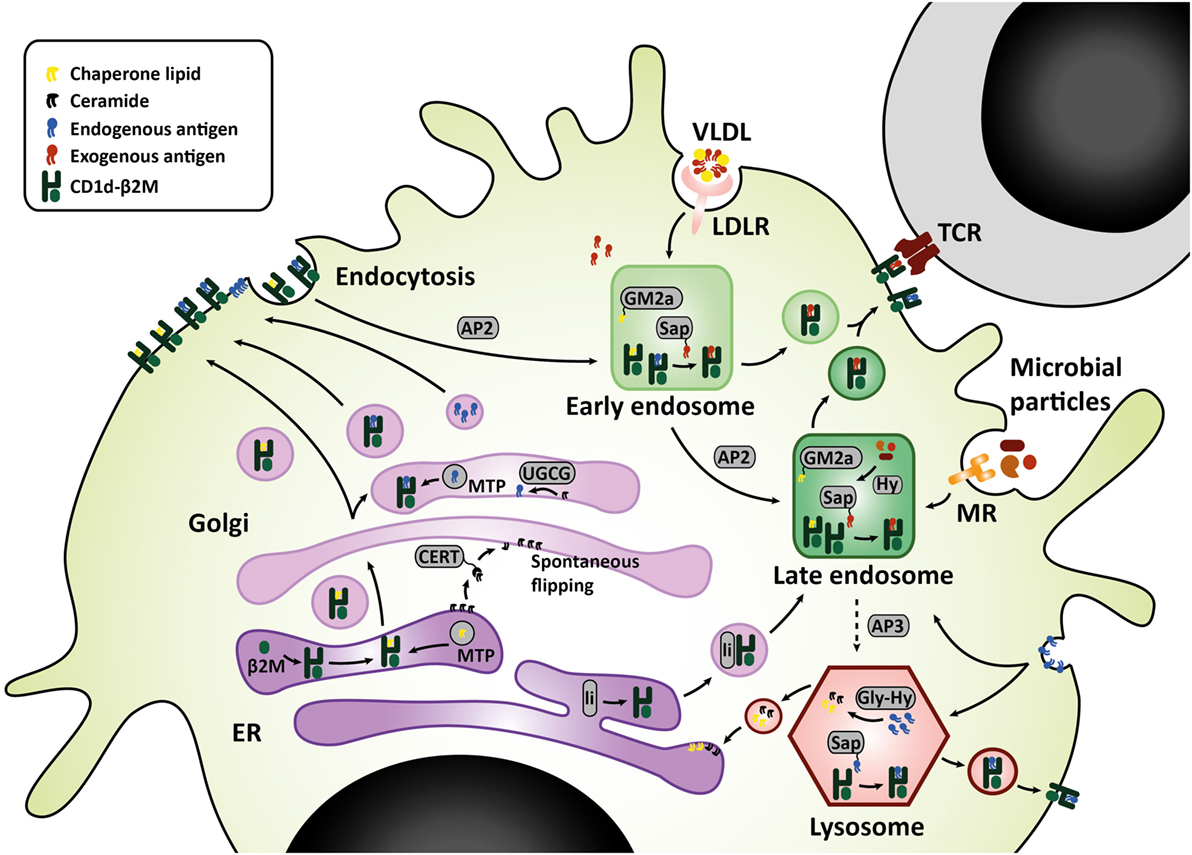
Figure 1. CD1d lipid loading at the crossroads of glycosphingolipid metabolism. In the ER–Golgi pathway that is similar for mouse and human, CD1d heavy chains assemble with β2M and chaperone lipids in the ER before transit to the cell surface. Alternatively, the Golgi complex produces GSL that are loaded onto CD1d by microsomal transfer protein, and may serve as endogenous lipid antigens. Ceramide precursors are transported to the Golgi by ceramide transfer protein (CERT). In the Golgi, UGCG and other glycosyltransferases convert ceramide into GSL. These GSL endproducts can be loaded onto CD1d, or transported to the plasma membrane in membrane-bound transport carriers. In the endolysosomal pathway, mouse CD1d1 may target the endosome and lysosome directly via interaction with sorting motifs adaptor protein 2 (AP2), which targets the endosomal compartment, and adaptor protein 3 (AP3), which targets the lysosomal compartment. Human CD1d is internalized via AP2 but cannot bind AP3. However, human CD1d can still be found in the lysosomal compartment. Possibly, ER-resident CD1d proteins gain access to the endolysosomal compartment via an auxiliary pathway, in conjunction with MHC class II-associated invariant chain (li). In the endolysosomal compartments, CD1d proteins are loaded with exogenous or endogenous lipid antigens, orchestrated by a variety of lipid transfer proteins including GM2 activator (GM2a), saposins A-D (Sap), and Niemann-Pick type C2 protein. Exogenous lipid antigens are delivered to the endosomal compartment via endocytosis of LDLR-associated glycolipids, MR-associated microbial lipids, and other scavenger receptors. Some exogenous lipids require processing into antigenic lipids before CD1d-loading, for example, through lipid hydrolases (Hy). Endogenous lipid antigens are delivered to the endolysosomal compartment via endocytosis of membrane-associated GSL, which can be loaded onto CD1d or degraded in lysosomes by glycohydrolases (Gly-Hy) and accessory proteins, before recycling to the ER (salvage pathway). Upon lipid antigen loading in the endolysosomal compartments, CD1d–lipid complexes recycle back to the cell surface for interaction with the invariant TCR on invariant natural killer T cells. Abbreviations: AP, adaptor protein; β2M, β2-microglobulin; ER, endoplasmic reticulum; Gly-Hy, glycohydrolase; GM2a, GM2 activator; GSL, glycosphingolipids; Hy, hydrolase; LDLR, LDL-receptor; Li, MHC class II-associated invariant chain; MR, mannose receptor; Sap, saposins; TCR, T-cell receptor; UGCG, UDP-glycose ceramide glucosyltransferase; VLDL, very-low-density lipoprotein.
Sphingolipids in Immunometabolic Disease
Sphingolipids play a key role in immunometabolic disease, which supports their potential relevance as iNKT cell antigens (17, 18, 39). The six CerS involved in de novo sphingolipid synthesis are differentially expressed, allowing for tissue- and cell type-dependent variation in ceramide acyl chain length profiles (16). Importantly, differences in sphingolipid chain length may orchestrate glucose metabolism and mitochondrial homeostasis, and play a key role in obesity and type 2 diabetes. For example, reduction in C16 sphingolipid levels increases beta-oxidation and improves glucose metabolism, with a 30–50% reduction in C16 levels being sufficient to prevent diet-induced obesity and insulin resistance (40). Furthermore, acyl chain length may determine cell fate. While C22-24 ceramides prevent apoptosis, C16 ceramides can induce apoptosis via activation of the intrinsic mitochondrial apoptotic pathway (16, 41–43). In CVD, C16 ceramides are considered harmful, as Cer(d18:1/16:0)/Cer(d18:1/24:0) ratios predict cardiovascular death (44). Intriguingly, sphingolipids such as glucosylceramide, lactosylceramide, ceramide, dihydroceramide, sphingomyelin, and sphingosine-1-phosphate (S1P) amass in human atherosclerotic plaques. All except S1P induce apoptosis in vitro, and are associated with plaque instability (39, 45, 46). Consequently, D-PDMP, an inhibitor of glucosylceramide synthase and lactosylceramide synthase, has an astounding protective effect on atherosclerosis development in ApoE−/− mice. Treatment led to complete prevention of intima media thickening and arterial stiffening measured as aortic pulse-wave velocity (47). Likewise, treatment with the SPT inhibitor myriocin was shown to ameliorate insulin resistance and atherosclerosis in mouse and rat models (18).
It is tempting to speculate that the pathophysiological role of sphingolipids in immunometabolic disease is partly explained by their role as iNKT cell ligands. In order to identify sphingolipid antigens potentially involved in immunometabolic disease, several approaches may be explored. First, the intracellular crossing between the sphingolipid metabolism and the iNKT cell lipid loading pathways can be scrutinized (48, 49). The Golgi and lysosomal compartment facilitate exchange of chaperone lipids bound to CD1d for antigenic lipids and are, therefore, important crossroads in iNKT cell lipid antigen loading and sphingolipid metabolism (Figure 1). Alternatively, animal or cellular models of naturally occurring disorders in sphingolipid metabolism may be exploited to identify metabolic intermediates or end products in lipid antigen presentation. For example, the mouse model for Fabry disease, alpha-galactosidase A knock out, combined with globoside 3 synthase- or isogloboside 3 synthase double knock out, revealed that globosides, but not isoglobosides, are responsible for iNKT cell deficiency in Fabry disease (50). Hexb knock out mice, a model for Tay Sach and Sandoff disease, also show severe iNKT cell deficiency (51). The iNKT cell deficiency in these lysosomal storage disease mouse models suggests that the glycosphingolipid synthetic pathways involved may contain endogenous lipid antigens for iNKT cells. Alternatively, glycosphingolipid accumulation may hinder antigen presentation similarly to acLDL accumulation or cholesterol accumulation following NPC1 deficiency (50), and possibly NPC2 deficiency (52), regardless of the glycosphingolipid involved (50, 53). The latter model aligns with the lipid raft hypothesis, which proposes that iNKT cell activating lipids may either function as bona fide lipid antigens, or may impact CD1d loading, stabilization or clustering on the cell membrane, and in that way enforce iNKT cell activation (50, 54–56). Finally, immunometabolic diseases may serve as a starting point to identify sphingolipid antigens (18). In CVD, for example, lipoprotein particles that enter the cell via LDL receptor- and scavenger receptor-mediated uptake are important carriers of glycosphingolipid species (39). The increased uptake of oxidized lipoproteins via class A scavenger receptors in atherosclerosis may potentially induce a different iNKT cell effector response due to co-transported glycosphingolipid species (37). In conclusion, sphingolipids are promising candidate antigens from an immunometabolic perspective. However, translation of the changes in sphingolipid metabolism to iNKT cell activation remains technically challenging.
Indirect Activation of iNKT Cells
At present, two principal ways of iNKT cell activation have been described. As discussed before, high affinity lipid antigens may induce a strong T-cell receptor (TCR) signal and activate iNKT cells directly. Alternatively, innate activation of an antigen-presenting cell (APC) leads to presentation of an endogenous lipid ligand with low affinity, followed by a weak TCR signal that can fully activate iNKT cells in combination with cytokine co-stimulation secreted by the activated APC (5, 57–60). Innate activation of the APC can either be due to inflammatory or metabolic cues. For example, LPS can trigger iNKT-cell activation. This activation is CD1d- and APC dependent. However, this activation is also IL-12 dependent, in both mice and in human in vitro models (57, 58). The current model is that iNKT cells are first triggered via their TCR to upregulate CD40L, to enhance APC–iNKT cell interaction, and maintain proximity for paracrine IL-12 co-stimulation, which is induced by CD40:CD40L interaction (23, 61, 62). It was postulated that the duration of TCR triggering determines CD40L upregulation. Duration of TCR triggering, in turn, depends on the alkyl chain length and stabilization of the CD1d–glycosphingolipid-complex (24, 25). Furthermore, IL-12 ultimately drives a Th1-biased iNKT cell response (62). In vivo, IL-4 can be detected 2 h after intra-peritoneal lipid agonist injection, while IFN-γ is measured after 6 h, as is IL-12 (61). The relatively slow IFN-γ response, which also requires prolonged and enhanced APC–iNKT cell interaction, suggests that the IFN-γ response requires de novo IFN-γ protein synthesis, while IL-4 is pre-synthesized and can, therefore, be released instantly even upon weak or short TCR stimulation. In fact, binding affinity of glycolipids to CD1d correlates very well with IFN-γ production but not at all with IL-4 production by human iNKT cells (63). Transgenic mouse studies revealed that the Notch and RBP-J pathway might be responsible for the IL-4 response by iNKT cells, mainly regulated by the conserved noncoding sequence-2 enhancer (CNS-2). As Notch- and TCR signaling synergistically contribute to T cell activation, this could explain why a weak TCR signal still allows for a relatively high IL-4 production by iNKT cells (64). This leaves us with a model in which iNKT cells are potent effector memory IL-4 producers upon homeostatic, weak antigenic stimulation, which can become highly inflammatory IFN-γ producing cells in an environment in which either high affinity ligands are available, or where IL-12 or CD40:CD40L co-stimulation are more easily established, either directly or due to activation of APCs.
Immunometabolic iNKT Cell Activation
The intracellular sphingolipid pool and subsequent CD1d ligand loading may be affected by TLR-activation or altered metabolism in the APC (65, 66). Several mechanisms were recently reported. For example, blocking glycolysis and increasing fatty acid oxidation (FAO) via AMPK provokes a CD1d-mediated iNKT cell cytokine response (65). AMPK is a nutrient-sensing kinase that is activated under low glucose conditions and blocks cellular glycolysis while promoting cell-sparing oxidative phosphorylation (67). Adiponectin, an adipokine produced by lean adipocytes that promotes insulin sensitivity, can directly activate AMPK (65, 68). Adiponectin overexpression in ob/ob obese mice protects against insulin resistance and AT inflammation (69), perhaps activating iNKT cells in a Th2-skewed manner through direct or indirect iNKT cell modulation. Conversely, TLR signaling leads to increased glycolysis, reduced FAO, and AMPK inhibition in the APC (70), but again leads to iNKT cell activation (58, 71). TLR-induced glycolysis is established via HIF-1α upregulation despite normoxic conditions, analogous to the Warburg effect (70). This pathway may be potentiated by mild hypoxia (72). In early obesity, relative hypoxia arises following adipocyte hypertrophy and hyperplasia and has been dubbed one of the initiating events in AT inflammation (1, 73). Indeed, hypoxia is also an important factor in cancer and in atherosclerosis (72, 74). iNKT cells are sensitive to HIF-1α activation and respond with a CD1d-mediated cytokine response (65). If and how the iNKT cell response is skewed toward an anti- or pro-inflammatory phenotype in these experiments, and whether different ligands are presented, remains to be determined. Notably, TLR4 signaling enhances atherosclerosis development in ApoE−/− mice in an iNKT cell-dependent manner (75). TLR4 signaling can be activated by LPS but also by excess free fatty acids, suggesting that nutrient overload mimics infection with regard to its downstream effects. Furthermore, during obesity, adipocytes produce the adipokine leptin to flag nutrient excess and diminish food intake. Leptin contributes to an iNKT cell response that results in anergy and PD-1 upregulation by directly triggering the leptin receptor expressed by iNKT cells (76, 77). Importantly, leptin-mediated iNKT cell activation still requires TCR triggering (77). These findings support the view that changing metabolic conditions determine the ligand pool and steer the iNKT cell response (Figure 2).
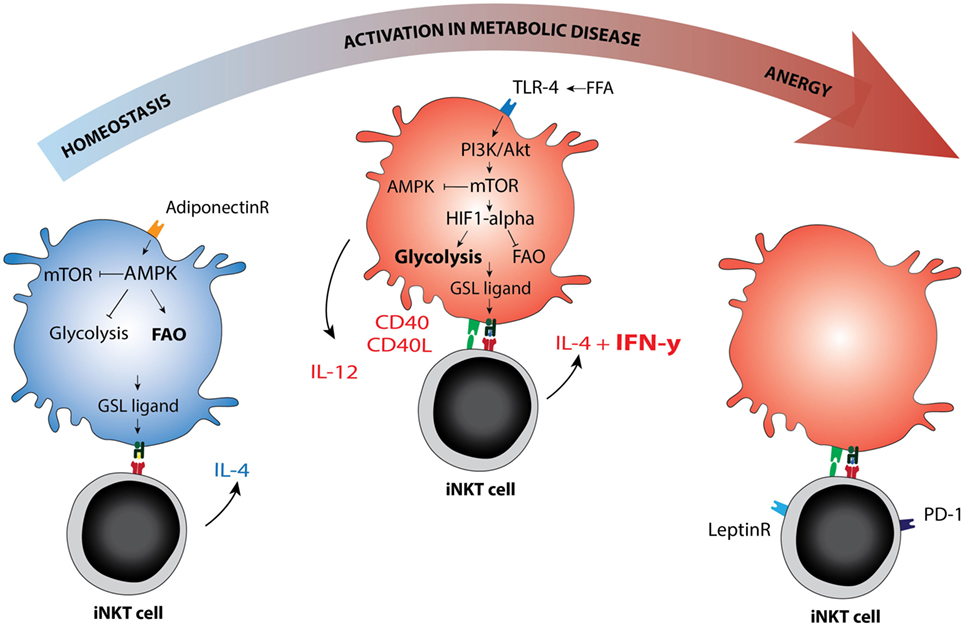
Figure 2. Immunometabolic activation of invariant natural killer T (iNKT) cells. Schematic representation of metabolic re-programming of iNKT cell function in immunometabolic disease. Metabolic changes in the antigen-presenting cell may affect the GSL ligand pool or alter the availability of co-stimulatory molecules. AMPK versus mTOR are depicted as the main metabolic regulators of the cellular metabolic program, they can each inhibit the other. AMPK is activated in low glucose conditions and can be activated by adiponectin, the adipokine secreted by lean adipocytes. AMPK activation drives fatty acid oxidation and is associated with cellular longevity. In lean fat- or homeostatic conditions, the iNKT cell response is mainly Th2 skewed (IL-4), but changes to Th1 in metabolic disease (IFN-γ). Whether AMPK or mTOR activation can be linked directly to Th1 or Th2 skewing of the iNKT cell response has not yet been studied to our knowledge. In dyslipidemic conditions, the hallmark of metabolic disease, FFA may activate mTOR via TLR4 signaling. The TLR4-driven glycolysis observed in metabolic disease is reminiscent of glycolysis observed under normoxic conditions in cancer (the Warburg effect). mTOR blocks AMPK and in that manner removes at least one of the brakes on CD40 upregulation and IL-12 production, the co-stimulatory requirements for a Th1 iNKT cell response. In late stage metabolic disease, iNKT cells have been described as anergic, while simultaneously upregulating PD-1. Blocking the leptin receptor on iNKT cells may reverse this anergic state. Abbreviations: GSL, glycosphingolipid; FFA, free fatty acids.
Besides the sphingolipid ligand pool, metabolic changes may also affect co-stimulatory molecules involved in iNKT activation, including CD40 and CD40L. For example, the amount of surface-expressed CD40 on macrophages and smooth muscle cells in human plaques correlates with the stage of atherosclerosis development (78). Possibly, ox-LDL signaling via LOX-1, a receptor for ox-LDL, is responsible for the CD40/CD40L upregulation (79). In addition to establishing a firm APC–iNKT cell interaction for Th1 skewing, CD40:CD40L signaling induces LDLR upregulation in human B-cells, enhancing iNKT cell activation (80). Low glucose conditions and AMPK activation causes lower baseline expression of CD40 by dendritic cells, decreased CD40 upregulation, and decreased IL-12 production upon LPS challenge (81). Moreover, AMPK inhibition leads to enhanced IL-12 production after LPS stimulation (81). IL-12 contributes to the formation of early atherosclerotic lesions in ApoE−/− mice and correlates positively with pulse wave velocity in healthy individuals, supporting a role for IL-12 in early atherogenesis in humans (82, 83).
Implications for Immunometabolic Disease
Immunometabolic diseases such as obesity, type 2 diabetes, and CVD are increasingly considered to be the downside of co-evolution of the immune system and metabolism (2). The growing body of immunometabolic diseases and the intricate web between metabolic dysregulation and inflammation emphasizes the need to understand metabolic programming of immune cells.
Current treatment for immunometabolic disease often targets dyslipidemia, with statins as mainstream therapy. However, we are learning now that the definition of dyslipidemia should extend far beyond cholesterol and triglyceride ratios, since ceramide and sphingolipid metabolism are closely involved in dyslipidemia and its consequences (17, 18, 39, 46). We are just starting to adequately link sphingolipid metabolism directly to metabolic disease and iNKT cell function, but the findings highlighted in this review indicate that the sphingolipid–iNKT cell axis holds promise for new treatment strategies. iNKT cells are unique in their restriction to lipid antigens and seem to possess all qualities required for immune-modulation. However, it is essential to unravel the underlying mechanism(s) directing the iNKT cell cytokine response, and to finally identify the endogenous lipid ligands involved. To this end, we may further explore the microbiome for extraction of potential lipid antigens, as iNKT cell–microbiome interaction has been firmly established (84–87). Additionally, we may further investigate naturally occurring iNKT cell subsets that skew toward Th1 or Th2 cytokine production, Th1/Th2 tissue distribution, plasticity, and the role of epigenetic memory (88, 89). Finally, co-stimulatory molecules, cytokines and adipokines involved in iNKT cell activation may be equally important in modulating iNKT cell function in immunometabolic disease.
Author Contributions
FV, MB, and HS wrote the manuscript. FV and HS created the figures. BL and EK provided critical evaluation and offered insightful suggestions to improve the content. All edited the manuscript and approved the final version for submission.
Conflict of Interest Statement
None of the authors have a competing financial interest in relation to the presented work. BL is employed by Nutricia Research and is leading a strategic alliance between University Medical Centre Utrecht/Wilhelmina Children’s Hospital and Nutricia Research, as indicated by the affiliations.
Funding
This work was supported by the Wilhelmina Children’s Hospital Research Fund, Dutch Topsector Life Sciences and Health TKI fund, and the Nutricia Research Fund. HS was supported by a Fellowship Clinical Research Talent of the University Medical Center Utrecht, and a VENI-NWO Innovational Research Incentive.
References
1. Lee YS, Wollam J, Olefsky JM. An integrated view of immunometabolism. Cell (2018) 172:22–40. doi:10.1016/j.cell.2017.12.025
2. Hotamisligil GS. Foundations of immunometabolism and implications for metabolic health and disease. Immunity (2017) 47:406–20. doi:10.1016/j.immuni.2017.08.009
3. Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res (2016) 57:2099–114. doi:10.1194/jlr.R066514
4. Cohen NR, Brennan PJ, Shay T, Watts GF, Brigl M, Kang J, et al. Shared and distinct transcriptional programs underlie the hybrid nature of iNKT cells. Nat Immunol (2013) 14:90–9. doi:10.1038/ni.2490
5. Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol (2013) 13:101–17. doi:10.1038/nri3369
6. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol (2010) 11:197–206. doi:10.1038/ni.1841
7. Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, et al. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity (2012) 37:574–87. doi:10.1016/j.immuni.2012.06.016
8. Lynch L, Michelet X, Zhang S, Brennan PJ, Moseman A, Lester C, et al. Regulatory iNKT cells lack expression of the transcription factor PLZF and control the homeostasis of T(reg) cells and macrophages in adipose tissue. Nat Immunol (2015) 16:85–96. doi:10.1038/ni.3047
9. Schipper HS, Rakhshandehroo M, van de Graaf SFJ, Venken K, Koppen A, Stienstra R, et al. Natural killer T cells in adipose tissue prevent insulin resistance. J Clin Invest (2012) 122:3343–54. doi:10.1172/JCI62739
10. Huh JY, Park J, Kim JI, Park YJ, Lee YK, Kim JB. Deletion of CD1d in adipocytes aggravates adipose tissue inflammation and insulin resistance in obesity. Diabetes (2017) 66:835–47. doi:10.2337/db16-1122
11. Wilson SB, Kent SC, Patton KT, Orban T, Jackson RA, Exley M, et al. Extreme Th1 bias of invariant Valpha24JalphaQ T cells in type 1 diabetes. Nature (1998) 391:177–81. doi:10.1038/34419
12. Kyriakakis E, Cavallari M, Andert J, Philippova M, Koella C, Bochkov V, et al. Invariant natural killer T cells: linking inflammation and neovascularization in human atherosclerosis. Eur J Immunol (2010) 40:3268–79. doi:10.1002/eji.201040619
13. Smith E, Croca S, Waddington KE, Sofat R, Griffin M, Nicolaides A, et al. Cross-talk between iNKT cells and monocytes triggers an atheroprotective immune response in SLE patients with asymptomatic plaque. Sci Immunol (2016) 1:1–14. doi:10.1126/sciimmunol.aah4081
14. Gorini F, Azzimonti L, Delfanti G, Scarfò L, Scielzo C, Bertilaccio MT, et al. Invariant NKT cells contribute to chronic lymphocytic leukemia surveillance and prognosis. Blood (2017) 129(26):3440–51. doi:10.1182/blood-2016-11-751065
15. Natori T, Morita M, Akimoto K, Koezuka Y. Agelasphins, novel antitumor and immunostimulatory cerebrosides from the marine sponge agelas-mauritianus. Tetrahedron (1994) 50:2771–84. doi:10.1016/S0040-4020(01)86991-X
16. Park WJ, Park JW. The effect of altered sphingolipid acyl chain length on various disease models. Biol Chem (2015) 396:693–705. doi:10.1515/hsz-2014-0310
17. Chaurasia B, Summers SA. Ceramides – lipotoxic inducers of metabolic disorders. Trends Endocrinol Metab (2015) 26:538–50. doi:10.1016/j.tem.2015.07.006
18. Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nat Rev Endocrinol (2017) 13:79–91. doi:10.1038/nrendo.2016.169
19. Brennan PJ, Tatituri RVV, Brigl M, Kim EY, Tuli AA, Sanderson JP, et al. Invariant natural killer T cells recognize lipid self antigen induced by microbial danger signals. Nat Immunol (2011) 12:1202–11. doi:10.1038/ni.2143
20. Brennan PJ, Tatituri RVV, Heiss C, Watts GFM, Hsu F-F, Veerapen N, et al. Activation of iNKT cells by a distinct constituent of the endogenous glucosylceramide fraction. Proc Natl Acad Sci U S A (2014) 111:13433–8. doi:10.1073/pnas.1415357111
21. Kain L, Costanzo A, Webb B, Holt M, Bendelac A, Savage PB, et al. Endogenous ligands of natural killer T cells are alpha-linked glycosylceramides. Mol Immunol (2015) 68:94–7. doi:10.1016/j.molimm.2015.06.009
22. Stax AM, Tuengel J, Girardi E, Kitano N, Allan LL, Liu V, et al. Autoreactivity to sulfatide by human invariant NKT cells. J Immunol (2017) 199:97–106. doi:10.4049/jimmunol.1601976
23. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. Activation CD1d-restricted and TCR-mediated of V. 14 NKT cells by glycosylceramides. Science (1997) 278:1626–9. doi:10.1126/science.278.5343.1626
24. McCarthy C, Shepherd D, Fleire S, Stronge VS, Koch M, Illarionov PA, et al. The length of lipids bound to human CD1d molecules modulates the affinity of NKT cell TCR and the threshold of NKT cell activation. J Exp Med (2007) 204:1131–44. doi:10.1084/jem.20062342
25. Guillaume J, Wang J, Janssens J, Remesh SG, Risseeuw MDP, Decruy T, et al. Galactosylsphingamides: new α-GalCer analogues to probe the F’-pocket of CD1d. Sci Rep (2017) 7:4276. doi:10.1038/s41598-017-04461-7
26. Im JS, Arora P, Bricard G, Molano A, Venkataswamy MM, Baine I, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity (2009) 30:888–98. doi:10.1016/j.immuni.2009.03.022
27. Sundararaj S, Zhang J, Krovi SH, Bedel R, Tuttle KD, Veerapen N, et al. Differing roles of CD1d2 and CD1d1 proteins in type I natural killer T cell development and function. Proc Natl Acad Sci U S A (2018) 115(6):E1204–13. doi:10.1073/pnas.1716669115
28. Chiu Y-H, Park S-H, Benlagha K, Forestier C, Jayawardena-Wolf J, Savage PB, et al. Multiple defects in antigen presentation and T cell development by mice expressing cytoplasmic tail-truncated CD1d. Nat Immunol (2002) 3:55–60. doi:10.1038/ni740
29. Roberts TJ, Sriram V, Spence PM, Gui M, Hayakawa K, Bacik I, et al. Recycling CD1d1 molecules present endogenous antigens processed in an endocytic compartment to NKT cells. J Immunol (2002) 168:5409–14. doi:10.4049/jimmunol.168.11.5409
30. Elewaut D, Lawton AP, Nagarajan NA, Maverakis E, Khurana A, Honing S, et al. The adaptor protein AP-3 is required for CD1d-mediated antigen presentation of glycosphingolipids and development of Valpha14i NKT cells. J Exp Med (2003) 198:1133–46. doi:10.1084/jem.20030143
31. Sugita M, Cao X, Watts GFM, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity (2002) 16:697–706. doi:10.1016/S1074-7613(02)00311-4
32. Chen X, Wang X, Keaton JM, Reddington F, Illarionov PA, Besra GS, et al. Distinct endosomal trafficking requirements for presentation of autoantigens and exogenous lipids by human CD1d molecules. J Immunol (2007) 178:6181–90. doi:10.4049/jimmunol.178.10.6181
33. Swift LL, Zhu M-Y, Kakkad B, Jovanovska A, Neely MD, Valyi-Nagy K, et al. Subcellular localization of microsomal triglyceride transfer protein. J Lipid Res (2003) 44:1841–9. doi:10.1194/jlr.M300276-JLR200
34. Van Kaer L, Parekh VV, Wu L. Invariant natural killer T cells as sensors and managers of inflammation. Trends Immunol (2013) 34:50–8. doi:10.1016/j.it.2012.08.009
35. van Eijkeren RJ, Krabbe O, Boes M, Schipper HS, Kalkhoven E. Endogenous lipid antigens for invariant natural killer T cells hold the reins in adipose tissue homeostasis. Immunology (2017) 153(2):179–89. doi:10.1111/imm.12839
36. Barral DC, Brenner MB. CD1 antigen presentation: how it works. Nat Rev Immunol (2007) 7:929–41. doi:10.1038/nri2191
37. Freigang S, Landais E, Zadorozhny V, Kain L, Yoshida K, Liu Y, et al. Scavenger receptors target glycolipids for natural killer T cell activation. J Clin Invest (2012) 122:3943–54. doi:10.1172/JCI62267
38. Angelo GD, Capasso S, Sticco L, Russo D. Glycosphingolipids: synthesis and functions. FEBS J (2013) 280:6338–53. doi:10.1111/febs.12559
39. Iqbal J, Walsh MT, Hammad SM, Hussain MM. Sphingolipids and lipoproteins in health and metabolic disorders. Trends Endocrinol Metab (2017) 28:506–18. doi:10.1016/j.tem.2017.03.005
40. Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, et al. Obesity-induced CerS6-dependent C16:0ceramide production promotes weight gain and glucose intolerance. Cell Metab (2014) 20:678–86. doi:10.1016/j.cmet.2014.08.002
41. Mesicek J, Lee H, Feldman T, Jiang X, Skobeleva A, Berdy-Shev EV, et al. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell Signal (2010) 22:1300–7. doi:10.1016/j.cellsig.2010.04.006
42. Patwardhan GA, Beverly LJ, Siskind LJ. Sphingolipids and mitochondrial apoptosis. J Bioenerg Biomembr (2016) 48:153–68. doi:10.1007/s10863-015-9602-3
43. Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature (2014) 508:406–10. doi:10.1038/nature13204
44. Laaksonen R, Ekroos K, Sysi-Aho M, Hilvo M, Vihervaara T, Kauhanen D, et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur Heart J (2016) 37:1967–76. doi:10.1093/eurheartj/ehw148
45. Edsfeldt A, Dunér P, Stahlman M, Mollet IG, Asciutto G, Grufman H, et al. Sphingolipids contribute to human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol (2016) 36:1132–40. doi:10.1161/ATVBAHA.116.305675
46. Sasset L, Zhang Y, Dunn TM, Di Lorenzo A. Sphingolipid de novo biosynthesis: a rheostat of cardiovascular homeostasis. Trends Endocrinol Metab (2016) 27:807–19. doi:10.1016/j.tem.2016.07.005
47. Chatterjee S, Bedja D, Mishra S, Amuzie C, Avolio A, Kass DA, et al. Inhibition of glycosphingolipid synthesis ameliorates atherosclerosis and arterial stiffness in apolipoprotein E-/-mice and rabbits fed a high-fat and -cholesterol diet. Circulation (2014) 129:2403–13. doi:10.1161/CIRCULATIONAHA.113.007559
48. Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol (2017) 19:175–91. doi:10.1038/nrm.2017.107
49. Abdul-Hammed M, Breiden B, Schwarzmann G, Sandhoff K. Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal β-glucocerebrosidase. J Lipid Res (2017) 58:563–77. doi:10.1194/jlr.M073510
50. Porubsky S, Speak AO, Salio M, Jennemann R, Bonrouhi M, Zafarulla R, et al. Globosides but not isoglobosides can impact the development of invariant NKT cells and their interaction with dendritic cells. J Immunol (2012) 189:3007–17. doi:10.4049/jimmunol.1201483
51. Zhou D, Mattner J, Cantu C III, Schrantz N, Yin N, Gao Y, et al. Lysosomal glycosphingolipid recognition by NKT cells. Science (2004) 306:1786–9. doi:10.1126/science.1103440
52. Schrantz N, Sagiv Y, Liu Y, Savage PB, Bendelac A, Teyton L. The Niemann-Pick type C2 protein loads isoglobotrihexosylceramide onto CD1d molecules and contributes to the thymic selection of NKT cells. J Exp Med (2007) 204:841–52. doi:10.1084/jem.20061562
53. Pereira C, Ribeiro H, Macedo M. From lysosomal storage diseases to NKT cell activation and back. Int J Mol Sci (2017) 18:502. doi:10.3390/ijms18030502
54. Torreno-Pina JA, Manzo C, Salio M, Aichinger MC, Oddone A, Lakadamyali M, et al. The actin cytoskeleton modulates the activation of iNKT cells by segregating CD1d nanoclusters on antigen-presenting cells. Proc Natl Acad Sci U S A (2016) 113(6):E772–81. doi:10.1073/pnas.1514530113
55. Park YK, Lee JW, Ko YG, Hong S, Park SH. Lipid rafts are required for efficient signal transduction by CD1d. Biochem Biophys Res Commun (2005) 327:1143–54. doi:10.1016/j.bbrc.2004.12.121
56. Lang GA, Maltsev SD, Besra GS, Lang ML. Presentation of α-galactosylceramide by murine CD1d to natural killer T cells is facilitated by plasma membrane glycolipid rafts. Immunology (2004) 112:386–96. doi:10.1111/j.1365-2567.2004.01896.x
57. Brigl M, Bry L, Kent SC, Gumperz JE, Brenner MB. Mechanism of CD1d-restricted natural killer T cell activation during microbial infection. Nat Immunol (2003) 4:1230–7. doi:10.1038/ni1002
58. Brigl M, Tatituri RVV, Watts GFM, Bhowruth V, Leadbetter EA, Barton N, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med (2011) 208:1163–77. doi:10.1084/jem.20102555
59. Mattner J, DeBord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature (2005) 434:525–9. doi:10.1038/nature03408
60. Nagarajan NA, Kronenberg M. Invariant NKT cells amplify the innate immune response to lipopolysaccharide. J Immunol (2007) 178:2706–13. doi:10.4049/jimmunol.178.5.2706
61. Oki S, Tomi C, Yamamura T, Miyake S. Preferential Th2 polarization by OCH is supported by incompetent NKT cell induction of CD40L and following production of inflammatory cytokines by bystander cells in vivo. Int Immunol (2005) 17:1619–29. doi:10.1093/intimm/dxh342
62. Kitamura H, Iwakabe K, Takashi Y, Nishimura S, Ohta A, Ohmi Y, et al. The natural killer T (NKT) cell ligand -galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med (1999) 189:1121–8. doi:10.1084/jem.189.7.1121
63. Liang P, Imamura M, Li X, Wu D, Fujio M, Guy RT, et al. Quantitative microarray analysis of intact glycolipid – CD1d interaction and correlation with cell-based cytokine production. J Am Chem Soc (2008) 130(37):12348–54. doi:10.1021/ja8012787
64. Tanaka S, Tsukada J, Suzuki W, Hayashi K, Tanigaki K, Tsuji M, et al. The interleukin-4 enhancer CNS-2 is regulated by notch signals and controls initial expression in NKT cells and memory-type CD4 T cells. Immunity (2006) 24:689–701. doi:10.1016/j.immuni.2006.04.009
65. Webb TJ, Carey GB, East JE, Sun W, Bollino DR, Kimball AS, et al. Alterations in cellular metabolism modulate CD1d-mediated NKT-cell responses. Pathog Dis (2016) 74:1–9. doi:10.1093/femspd/ftw055
66. Muindi K, Cernadas M, Watts GFM, Royle L, Neville DCA, Dwek RA, et al. Activation state and intracellular trafficking contribute to the repertoire of endogenous glycosphingolipids presented by CD1d. Proc Natl Acad Sci U S A (2010) 107:3052–7. doi:10.1073/pnas.0915056107
67. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol (2016) 17:618–25. doi:10.1038/ni.3466
68. Yamauchi T, Kamon J, Ito Y, TsuchLabela A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antLabeliabetic metabolic effects. Nature (2003) 423:762–9. doi:10.1038/nature01705
69. Kim J, Van De Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest (2007) 117:2621–37. doi:10.1172/JCI31021
70. McGettrick AF, O’Neill LAJ. How metabolism generates signals during innate immunity and inflammation. J Biol Chem (2013) 288:22893–8. doi:10.1074/jbc.R113.486464
71. Paget C, Mallevaey T, Speak AO, Torres D, Fontaine J, Sheehan KCF, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity (2007) 27:597–609. doi:10.1016/j.immuni.2007.08.017
72. Stienstra R, Netea-Maier RT, Riksen NP, Joosten LAB, Netea MG. Specific and complex reprogramming of cellular metabolism in myeloid cells during innate immune responses. Cell Metab (2017) 26:142–56. doi:10.1016/j.cmet.2017.06.001
73. Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell (2014) 157:1339–52. doi:10.1016/j.cell.2014.05.012
74. Dyck L, Lynch L. Cancer, obesity and immunometabolism – connecting the dots. Cancer Lett (2017) 417:11–20. doi:10.1016/j.canlet.2017.12.019
75. Andoh Y, Ogura H, Satoh M, Shimano K, Okuno H, Fujii S, et al. Natural killer T cells are required for lipopolysaccharide-mediated enhancement of atherosclerosis in apolipoprotein E-deficient mice. Immunobiology (2013) 218:561–9. doi:10.1016/j.imbio.2012.07.022
76. Favreau M, Menu E, Gaublomme D, Vanderkerken K, Faict S, Maes K, et al. Leptin receptor antagonism of iNKT cell function: a novel strategy to combat multiple myeloma. Leukemia (2017) 31:2678–85. doi:10.1038/leu.2017.146
77. Venken K, Seeuws S, Zabeau L, Jacques P, Decruy T, Coudenys J, et al. A bidirectional crosstalk between iNKT cells and adipocytes mediated by leptin modulates susceptibility for T cell mediated hepatitis. J Hepatol (2014) 60:175–82. doi:10.1016/j.jhep.2013.08.008
78. Bruemmer D, Riggers U, Holzmeister J, Grill M, Lippek F, Settmacher U, et al. Expression of CD40 in vascular smooth muscle cells and macrophages is associated with early development of human atherosclerotic lesions. Am J Cardiol (2001) 87(1):21–7. doi:10.1016/S0002-9149(00)01266-2
79. Li D, Liu L, Chen H, Sawamura T, Mehta JL. LOX-1, an oxidized LDL endothelial receptor, induces CD40/CD40L signaling in human coronary artery endothelial cells. Arterioscler Thromb Vasc Biol (2003) 23:816–21. doi:10.1161/01.ATV.0000066685.13434.FA
80. Allan LL, Hoefl K, Zheng DJ, Chung BK, Kozak FK, Tan R, et al. Apolipoprotein-mediated lipid antigen presentation in B cells provides a pathway for innate help by NKT cells. Blood (2009) 114:2411–6. doi:10.1182/blood-2009-04-211417
81. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, Deberardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood (2010) 115:4742–9. doi:10.1182/blood-2009-10-249540
82. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol (2011) 12:204–12. doi:10.1038/ni.2001
83. Yong K, Dogra G, Boudville N, Chan D, Adams L, Ching H, et al. Interleukin-12 is associated with arterial stiffness in healthy individuals. Am J Hypertens (2013) 26:159–62. doi:10.1093/ajh/hps032
84. Nieuwenhuis EES, Matsumoto T, Lindenbergh D, Willemsen R, Kaser A, Simons-oosterhuis Y, et al. Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J Clin Invest (2009) 119:1241–50. doi:10.1172/JCI36509
85. Olszak T, An D, Zeissig S, Vera M, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (2012) 489:489–94. doi:10.1126/science.1219328
86. Wei B, Wingender G, Fujiwara D, Chen DY, McPherson M, Brewer S, et al. Commensal microbiota and CD8+ T cells shape the formation of invariant NKT cells. J Immunol (2010) 184:1218–26. doi:10.4049/jimmunol.0902620
87. Wingender G, Stepniak D, Krebs P, Lin L, McBride S, Wei B, et al. Intestinal microbes affect phenotypes and functions of invariant natural killer T cells in mice. Gastroenterology (2012) 143:418–28. doi:10.1053/j.gastro.2012.04.017
88. Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-specific distribution of iNKT cells impacts their cytokine response. Immunity (2015) 43:566–78. doi:10.1016/j.immuni.2015.06.025
Keywords: immunometabolism, NKT, obesity, atherosclerosis, sphingolipid, AMPK, mTOR
Citation: Ververs FA, Kalkhoven E, van’t Land B, Boes M and Schipper HS (2018) Immunometabolic Activation of Invariant Natural Killer T Cells. Front. Immunol. 9:1192. doi: 10.3389/fimmu.2018.01192
Received: 31 March 2018; Accepted: 14 May 2018;
Published: 28 May 2018
Edited by:
Luc Van Kaer, Vanderbilt University, United StatesReviewed by:
Jae B. Kim, Seoul National University, South KoreaHiroshi Watarai, University of Tokyo, Japan
Copyright: © 2018 Ververs, Kalkhoven, van’t Land, Boes and Schipper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Henk S. Schipper, aC5zLnNjaGlwcGVyLTNAdW1jdXRyZWNodC5ubA==