 Zhan Wang1†
Zhan Wang1† Zenan Wang1†Shu Li2†
Zenan Wang1†Shu Li2† Binghao Li1Lingling Sun1
Binghao Li1Lingling Sun1 Hengyuan Li1Peng Lin1Shengdong Wang1
Hengyuan Li1Peng Lin1Shengdong Wang1 Wangsiyuan Teng1Xingzhi Zhou1
Wangsiyuan Teng1Xingzhi Zhou1 Zhaoming Ye1*
Zhaoming Ye1*
- 1Centre for Orthopaedic Research, Orthopedics Research Institute of Zhejiang University, Department of Orthopaedics, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
- 2Cancer Institute, Key Laboratory of Cancer Prevention and Intervention, Key Laboratory of Molecular Biology in Medical Sciences, National Ministry of Education, Department of Hematology, The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, China
γδ T cell-based immunotherapy for osteosarcoma (OS) has shown limited success thus far. DNA-demethylating agents not only induce tumor cell death but also have an immunomodulatory function. In this study, we have assessed the potential benefit of combining decitabine (DAC, a DNA demethylation drug) and γδ T cells for OS immunotherapy. DAC increased the expression of natural killer group 2D (NKG2D) ligands (NKG2DLs), including major histocompatibility complex class I-related chains B (MICB) and UL16-binding protein 1 (ULBP1), on the OS cell surface, making the cells more sensitive to recognition and destruction by cytotoxic γδ T cells. The upregulation of MICB and ULBP1 was due to promoter DNA demethylation. Importantly, the killing of OS cells by γδ T cells was partially reversed by blocking the NKG2D receptor, suggesting that the γδ T cell-mediated cytolysis of DAC-pretreated OS cells was mainly dependent on the NKG2D–NKG2DL axis. The in vivo results were consistent with the in vitro results. In summary, DAC could upregulate MICB and ULBP1 expression in OS cells, and combination treatment involving γδ T cell immunotherapy and DAC could be used to enhance the cytotoxic killing of OS cells by γδ T cells.
Introduction
Osteosarcoma (OS), which is the most common malignant primary bone tumor, predominantly affects children and young adults (1). The current standard treatment for OS includes preoperative neoadjuvant chemotherapy, surgical resection of the entire tumor, and postoperative chemotherapy (2). These treatment options have improved the 5-year survival of patients with localized OS to approximately 70%. However, the overall survival rate is ≤20% for patients with recurrence or distant metastases (3, 4). Moreover, since the 1980s, research on treatment for OS has fallen into stagnation (5). Therefore, there is an urgent need for a novel effective treatment for OS patients.
Although the combination of immunotherapy and other therapies is promising, T cell-based immunotherapy also presents challenges to those developing effective treatments for OS (6). In tumor microenvironments, γδ T cells act as an important innate immune cell population, mediating cytotoxic effects against a broad variety of neoplasms (7). Unlike αβ T cells, γδ T cells can distinguish tumor cells from normal cells and then kill them directly, without conventional major histocompatibility complex (MHC) restrictions (8). The antitumor effect of γδ T cells is induced by the interaction of γδ T cell receptors (TCRs) with tumor antigens. Additionally, other costimulatory receptors, such as the natural killer (NK) group 2D (NKG2D) receptor, expressed on γδ T cells can recognize NKG2D ligands (NKG2DLs), which can also induce the antitumor function of γδ T cells. These NKG2DLs on tumor cells include MHC class I-related chains A and B (MICA and MICB) and proteins in the UL16-binding protein family (ULBP1-6, mainly ULPBP1–3) (9). Clinical trials evaluating the use of γδ T cell immunotherapy for malignancies have confirmed its feasibility and acceptable safety profile (10–12). However, the effectiveness of γδ T cell-based immunotherapy for tumors is decreased because of its potential suppressive role in tumor microenvironments (13, 14).
Accumulating evidence has strongly indicated that γδ T cell immunotherapy for tumors could be combined with other therapies to improve clinical outcomes (6, 12, 14, 15). Hypomethylating agents such as 5-aza-2’-deoxycytidine [decitabine (DAC)] have been shown to have anticancer activity and additional immunomodulatory functions (16). Recently, several studies have reported that DAC-mediated hypomethylation restores NKG2DL expression in tumors and might, therefore, provide a clinically useful method for sensitizing tumors to γδ T cells (17, 18). However, the role of DAC in potentiating the antitumor effect of adoptive γδ T cell transfer remains unclear. We, therefore, investigated whether DAC could enhance the susceptibility of human OS cells to Vγ9Vδ2 T-cell-mediated cytotoxicity via the NKG2DL–NKG2D axis.
Materials and Methods
Cell Culture and Treatments
HOS and U2OS human OS cell lines were obtained from the Cell Collection of the Chinese Academy of Science (Shanghai, China). The HOS cells were cultured in Dulbecco’s Modified Eagle Medium (Gibco, Rockville, MD, USA) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) and 1% penicillin (100 U/mL). The U2OS cells were cultured in Roswell Park Memorial Institute 1640 medium (Gibco) supplemented with 10% fetal bovine serum and 1% penicillin (100 U/mL). All cells were cultured at 37°C in the humid atmosphere of a 5% CO2 incubator. Cells were counted and transferred to plates or dishes 1 day prior to treatment, and the medium was then replaced with medium containing DAC (Sigma-Aldrich, St. Louis, MO, USA) at different concentrations. New medium containing DAC was added every day, and the cells were then harvested and counted for further experiments (19).
In Vitro Expansion of Vγ9Vδ2 T Cells From Peripheral Blood Mononuclear Cells (PBMCs)
Peripheral blood samples were obtained from three healthy donors and the PBMCs were separated by density gradient centrifugation. The PBMCs were then seeded into 24-well culture plates. The detailed procedure for expanding human Vγ9Vδ2 T cells in vitro was described in our previous study (20). Negative magnetic-activated cell sorting (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) was used for purifying γδ T cells. Phenotypic analysis was performed by flow cytometry using anti-Vδ2-fluorescein isothiocyanate (cat. no. 331406, clone B6) and anti-CD3-allophycocyanin (cat. no. 300312, clone HIT3a) from BioLegend (San Diego, CA, USA).
The use of PBMCs was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine. Written informed consent was obtained from the three blood donors.
Cytotoxicity Assay
An MTS assay was performed to analyze the cytotoxic effects of γδ T cells on OS cells in vitro (19). OS cells (HOS and U2OS cells) were plated in triplicate onto 96-well flat-bottomed plates at 2 × 103, 5 × 103, and 1 × 104 cells/well for 72, 48, and 24 h of DAC pretreatment, respectively. γδ T cells were then added at the indicated effector/target (E:T) ratios and cocultured with OS cells for 4 h at 37°C in the humid atmosphere of a 5% CO2 incubator. Subsequently, all wells were gently washed with phosphate-buffered saline (PBS) twice to remove the γδ T cells. The percentage of viable cells was detected using an MTS assay (Promega, Madison, WI, USA). The absorbance was measured using a microplate reader at 490 nm.
To determine the cytotoxic pathways involved in γδ T cell-mediated cytolysis of OS cells, blocking experiments were carried out. In these experiments, γδ T cells were incubated with 10 µg/mL (saturating concentrations) of anti-human NKG2D (clone 149810; R&D Systems, Minneapolis, MN, USA), anti-pan-γδ TCR (clone B1; BD Biosciences, Franklin Lakes, NJ, USA), anti-human Fas ligand (FasL; clone NOK-2; BD Biosciences), or antitumor necrosis factor-related apoptosis-inducing ligand (Trail; clone RIK-2; BD Biosciences) for 30 min. The cells were then cocultured with OS cells to determine the cytotoxic pathways.
Enzyme-Linked Immunosorbent Assay (ELISA)
γδ T cells were cocultured with untreated or DAC-pretreated OS cells in triplicate at an E:T ratio of 5:1. After 4 h, the supernatants were collected to assess the secretion of interferon (IFN)-γ using a human IFN-γ ELISA kit (Dakewe Biotech, Shenzhen, China).
Reverse Transcription-Polymerase Chain Reaction
RNA was isolated using RNAiso Plus (TaKaRa Bio, Kusatsu, Japan) and subsequently transcribed into cDNA using a PrimeScript RT Reagent Kit (TaKaRa Bio). The mRNA was analyzed using a StepOnePlus Real-time PCR System (Applied Biosystems, Foster City, CA, USA) and SYBR Green (TaKaRa Bio). All primers used are listed in Table S2 in Supplementary Material. The amplification fold change was calculated using the 2−ΔΔCt method.
Flow Cytometry
For the analysis of apoptosis, target cells (γδ T cells) were stained with annexin V-phycoerythrin (PE) and 7-amino-actinomycin D (7-AAD) using a commercially available kit (BD Biosciences) according to the manufacturer’s instructions (21). The flow cytometry analysis of the target cells was performed on a FACSCanto flow cytometer (BD Biosciences).
The target cells were incubated with anti-human MICA, MICB, ULBP1, ULBP 2/5/6, or ULBP3 PE and its isotype control (R&D Systems) at 4°C. After 30 min, the cells were washed and analyzed using the FACSCanto flow cytometer. The mean fluorescence intensity (MFI) ratio was defined as the MFI of the specific staining relative to the MFI of the appropriate isotype control staining. Data analysis was performed using FlowJo vX software (TreeStar).
Bisulfite Sequencing
HOS cells were treated with 5 µM DAC or PBS for 3 days. Bisulfite treatment was then used to extract genomic DNA from these cells. The MICB promoter was amplified with the following primers: 5′-TTGTTGTTTAGGTTGGAGTGTAGT-3′ (forward) and 5′-TATAATCCCAACACTTTAAAAAATC-3′ (reverse). The ULBP1 promoter was amplified with the following primers: 5′-TGTTTTTAGTGGAGAGGTAAAAAA-3′ (forward) and 5′-AAAAAACTTACATAAAAATACTCAATAAC-3′ (reverse). Each amplified product was cloned into a pUC18 T-vector and subsequently sent for DNA sequencing (Sangon Biotech, Shanghai, China) (22, 23).
Western Blot Analysis
Osteosarcoma tumor tissues from mice were cut into pieces and lysed in radio immunoprecipitation assay lysis buffer to solubilize the proteins. The concentrations of protein extracts were determined using a bicinchoninic acid assay protein assay (Pierce, Rockford, IL, USA). An equal amount of protein was separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. The membranes were blocked using 5% nonfat milk and incubated with antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1,000; Abcam, Cambridge, UK), MICB (1:1,000; Abcam), or ULBP1 (1:1,000; Abcam) overnight at 4°C. A horseradish peroxidase-conjugated secondary antibody was added for 1 h at room temperature. Immunoblots were assessed using enhanced chemiluminescence (Millipore, Burlington, MA, USA) and recorded using a Bio-Rad XRS chemiluminescence detection system (Bio-Rad, Hercules, CA, USA).
In Vivo Experiments
Healthy 4-week-old female BALB/c-nu mice (which are immunodeficient) were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). HOS cells (1 × 106 in 20 µL PBS) were engrafted by injecting them through the tibial plateau in the primary spongiosa of the left tibia.
The mice were randomly assigned to four treatment groups to confirm the efficacy of γδ T cells + DAC treatment, with each group containing three mice. Group 1 consisted of mice treated with PBS and served as the control, group 2 consisted of mice treated with γδ T cells and PBS, group 3 consisted of mice treated with DAC and PBS, and group 4 consisted of mice treated with both DAC and γδ T cells. The concentration of DAC was 1 µg/g of body weight, and it was dissolved in a final volume of 100 µL PBS (19). DAC and PBS were administered by intraperitoneal injection, whereas the in vitro-generated γδ T cells (5 × 107 in 100 µL PBS) were administered by intravenous injection. The specific treatment plan is shown in Figure S1 in Supplementary Material. All mice were sacrificed at day 19 after treatment. The tumors were collected for measurement and weighing. The tumor volume (V) was estimated using the formula V = a × b2/2, where a is the maximum diameter of the tumor and b is the minimum diameter. The tumors were harvested for western blotting.
Statistical Analysis
The data are presented as the mean ± SD. Differences between groups were estimated using the χ2 test or Student’s t-test. Statistical analysis was performed using Microsoft Excel 2016 (Microsoft, Redmond, WA, USA) and SPSS statistical software for Windows, version 21.0 (SPSS, Chicago, IL, USA). All tests of significance were two-tailed tests and significance was defined as P < 0.05.
Results
Effect of DAC on Apoptosis and Phenotype of Human γδ T Cells
To assess the possible toxic effect of DAC when treating human γδ T cells, we performed cell apoptosis assays and analyzed the results using flow cytometry with annexin V-PE/7-AAD. DAC was added to γδ T cell cultures at 0, 5, 10, and 20 µM for 3 days. The media containing DAC were replenished every day. When DAC was used at 5 µM, no appreciable increase in the apoptosis of γδ T cells was observed (P > 0.05; Figure 1). Significantly increased apoptosis was detected when using ≥10 μM DAC. Subsequently, the DAC-induced expression of TCR and NKG2D in human γδ T cells was investigated using flow cytometry. Treatment with 5 µM DAC did not alter the percentage of γδ TCR+ and NKG2D+ γδ T cells (Figure S2 in Supplementary Material).
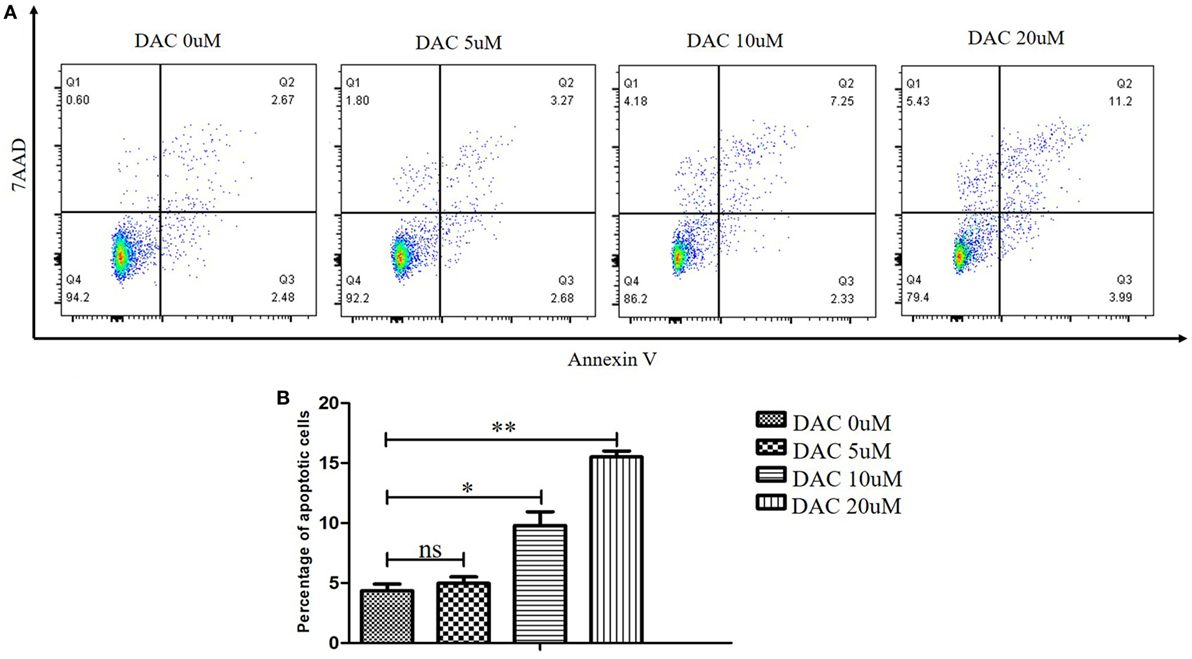
Figure 1. Effect of decitabine (DAC) on apoptosis in human γδ cells. DAC was added to γδ cell cultures at final concentrations ranging from 0 to 20 µM, or phosphate-buffered saline as vehicle control for 3 days. Media containing DAC was replenished every day. (A) Cell apoptosis was analyzed by flow cytometry with annexin V-PE/7-amino-actinomycin D (7-AAD). (B) The apoptotic rate was shown for three separate experiments. Error bars show SD among the replicates (*P < 0.05; **P < 0.01).
Cytotoxic Effects of γδ T Cells on DAC-Pretreated HOS and U2OS Cell Lines
To evaluate whether DAC treatment increased the susceptibility of OS cells to γδ T cell-mediated cytotoxicity, we performed MTS assays to evaluate the cytotoxic effects of γδ T cells on DAC-pretreated OS cell lines. As shown in Figure 2, treatment with γδ T cells or DAC alone slightly inhibited the proliferation of HOS and U2OS cells. However, γδ T cells in combination with DAC pretreatment for 24, 48, and 72 h exhibited significantly increased efficacy, indicating that DAC enhanced the susceptibility of OS cells to γδ T cell-mediated cytotoxicity.
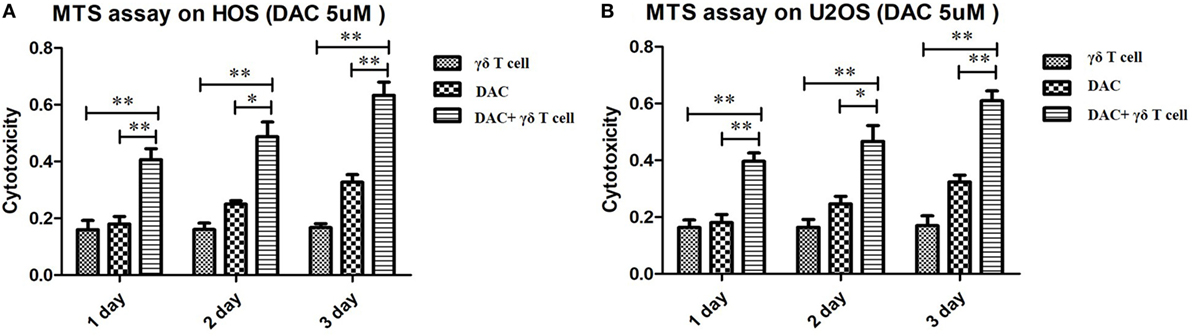
Figure 2. Cytotoxic assay using MTS method to evaluate the cytotoxic effects of γδ T cells on osteosarcoma (OS) cells pretreated with decitabine (DAC) (5 µM). (A) MTS assay on HOS cells. Treatment with γδ T cells or DAC alone slightly inhibited the growth of HOS cells. However, γδ T cells showed more efficiency in combination with DAC pretreatment for 1, 2, and 3 days. (B) MTS assay on U2OS cells. Similar to the results of HOS cells, DAC treatment can significantly enhance γδ T cells immunotherapy against OS cells. For (A) and (B), samples were run in triplicates. Error bars show standard deviation among the replicates. (γδ T cell: tumor cell = 5:1; *P < 0.05; **P < 0.01).
Upregulation of MICB and ULBP1 in HOS and U2OS Cell Lines After DAC Pretreatment
Previous studies have reported the role of the NKG2DL–NKG2D axis in the lysis of tumor cells by NK and γδ T cells (9, 24). Moreover, DAC treatment significantly increased NKG2DL expression in patients with acute myeloid leukemia (17). Whether DAC also increases NKG2DL expression in OS cells has not been previously explored. We assessed the cell-surface expression of NKG2DL in OS cells by PCR and flow cytometry. We found moderate to high levels of expression, with fivefold to >10-fold increases in the expression of the NKG2DLs MICB and ULBP1 in DAC-pretreated OS cells compared with PBS-pretreated OS cells. The expression of other NKG2DLs did not change significantly with DAC pretreatment (Figures 3A,B). Furthermore, the PCR results also showed that the expression levels of MICB and ULBP1 were significantly increased (P < 0.001) in DAC-pretreated OS cells compared with PBS-pretreated OS cells (Figure 3C).
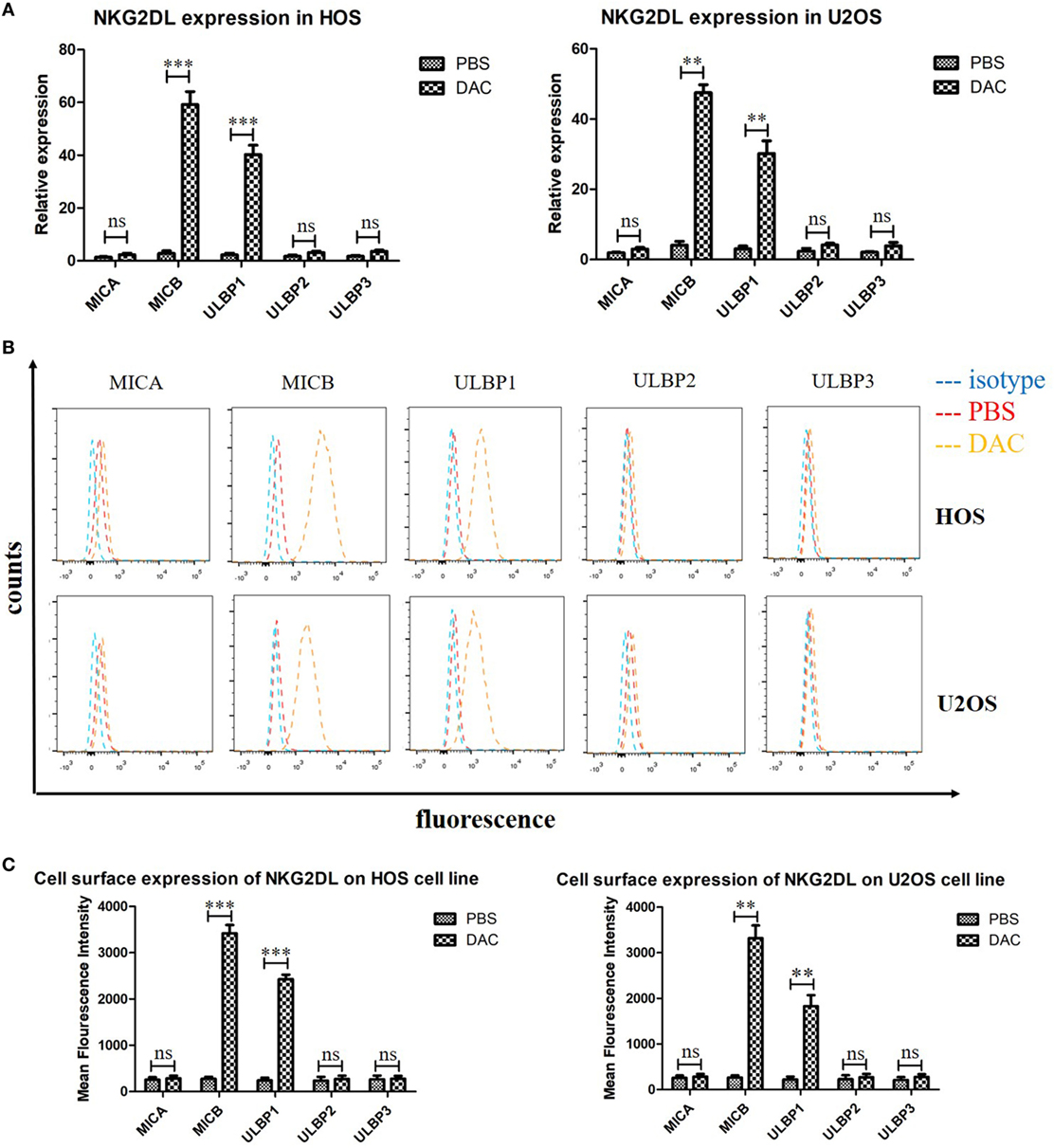
Figure 3. Increased expression of MICB and UL16-binding protein 1 (ULBP1) on osteosarcoma (OS) cells after 5 µM decitabine (DAC) treatment for 3 days. (A) NKG2DLs expression of HOS and U2OS cell lines were analyzed by reverse transcription-polymerase chain reaction. Expression levels are relative to GAPDH. MICB and ULBP1 expression was significantly increased after DAC treatment. While the rest NKG2DLs represented no obvious enhanced expression after DAC treatment. (B,C) Cell-surface expression of NKG2DL in OS cells was measured by flow cytometry and quantified by mean fluorescence intensity. The results were in line with PCR results. For (A,C), samples were run in triplicates. Error bars show standard deviation among the replicates (ns refers to no significance. ***P < 0.001).
DAC-Induced Demethylation at MICB and ULBP1 Promoter Sites
Decitabine treatment can activate methylated genes by promoter demethylation and the promotion of gene expression (25). Bisulfite sequencing revealed the high methylation levels of the MICB and ULBP1 promoters in untreated HOS cells, which were demethylated following DAC treatment (Figure 4). These results suggested that the upregulation of MICB and ULBP1 induced by DAC was associated with DNA demethylation.

Figure 4. Decitabine (DAC) induces DNA demethylation at the MICB and UL16-binding protein 1 (ULBP1) promoter region. Cells were treated with 5 µM DAC for 3 days, and the methylation state of MICB and ULBP1 promoter was checked by bisulfite sequencing. Each circle represents a single CpG dinucleotide on the strand. Five clones from each cell line were analyzed. The open and filled circles represent the unmethylated and methylated CpG islands, respectively.
Recognition and Targeting of DAC-Pretreated OS Cells by γδ T Cells via NKG2D–NKG2DL Interactions In Vitro
As DAC upregulated NKG2DL expression in OS cells, we speculated that γδ T cells recognize and target DAC-pretreated OS cells via NKG2D–NKG2DL interactions. To explore the molecular mechanisms involved in the interaction between γδ T cells and DAC-pretreated OS cells, a blocking experiment was performed to examine the effect of surface molecules on γδ T cell cytotoxicity. NKG2D blocking significantly reduced the cytotoxicity of γδ T cells against the DAC-pretreated OS cells (P < 0.01; Figure 5A). However, as shown in Figure 5A, NKG2D blocking had a weak effect on the cytotoxicity of γδ T cells against untreated OS cells.
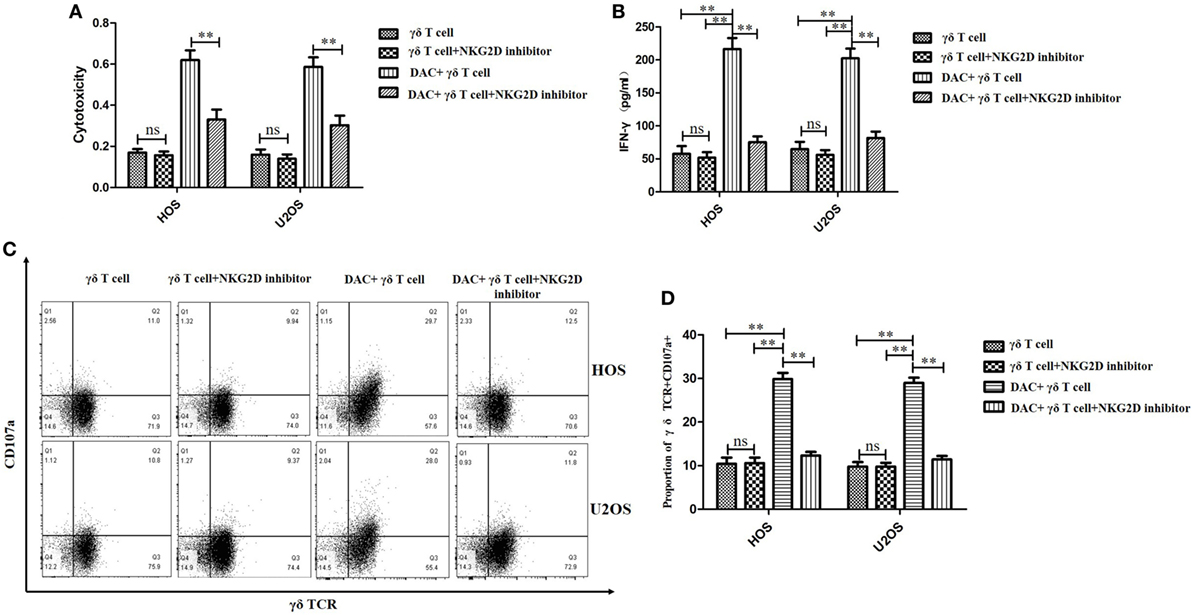
Figure 5. Decitabine (DAC) enhanced susceptibility of osteosarcoma (OS) cells to γδ T cell-mediated cytotoxicity via NKG2DL–natural killer group 2D (NKG2D) axis. Expanded γδ T cells were cocultured with OS cells for 4 h at an effector/target ratio of 5:1. (A) γδ T cells’ cytotoxicity on OS cells pretreated with DAC (5 µM) or phosphate-buffered saline for 3 days in the presence or absence of anti-NKG2D blocking antibodies (10 µg/mL) was determined by MTT assays. (B) Supernatants of (A) were harvested and assayed for interferon (IFN)-γ using a IFN-γ enzyme-linked immunosorbent assay kit. (C,D) Cell-surface expression of CD107a on γδ T cells was measured by flow cytometry and quantified by percent of γδ TCR+ CD107a+ T cells. For (A,B,D), samples were run in triplicates. Error bars show standard deviation among the replicates (γδ T cell: tumor cell = 5:1; ns refers to no significance; **P < 0.01).
Natural killer group 2D blocking significantly decreased the secretion of IFN-γ by γδ T cells when the OS cells were pretreated with DAC, but it had little effect on untreated OS cells (P < 0.01; Figure 5B). Moreover, the expression of CD107a was significantly increased during the γδ T cell killing of DAC-pretreated OS cells, and this expression was inhibited by blocking NKG2D. However, no obvious decrease in CD107a expression was observed during the γδ T cell killing of untreated OS cells after NKG2D blocking (P < 0.01; Figures 5C,D). Additionally, the cytotoxicity of the DAC + γδ T cells + NKG2D blocker group was significantly higher than that of the γδ T cells + NKG2D blocker group (P < 0.05). However, regarding IFN-γ secretion and CD107a expression, no significance was observed between these two groups. Although the TCR signaling pathway played an important role in γδ T cell-mediated cytotoxicity against untreated OS cells, for DAC-pretreated OS cells, the NKG2D–NKG2DL axis was the most important pathway. Approximately half of the killing of DAC-pretreated OS cells mediated by γδ T cells involved the NKG2D–NKG2DL axis (Figure 6). Taken together, these results suggested that the γδ T cell-mediated cytotoxic effects against DAC-pretreated OS cells were accomplished mainly through the NKG2D–NKG2DL pathway.
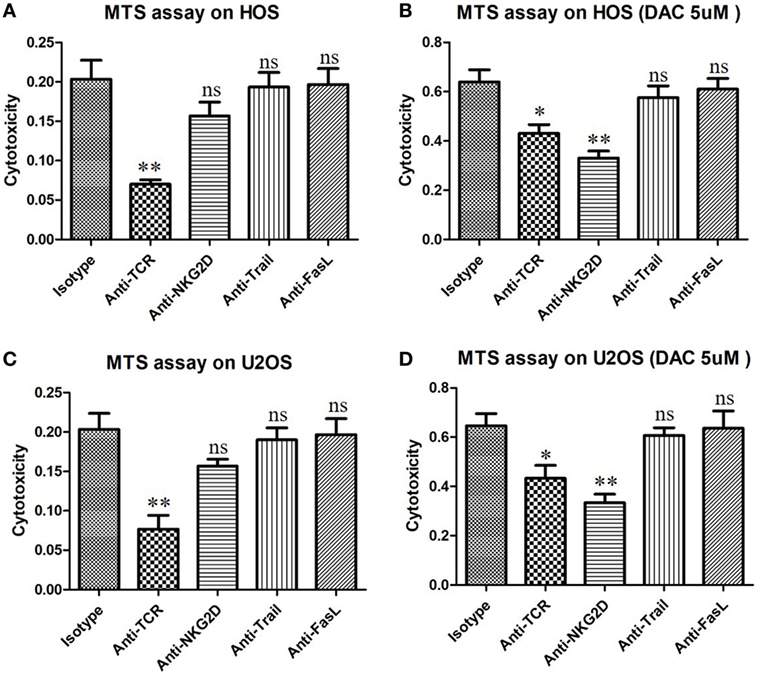
Figure 6. γδ T cells kill DAC-sensitized osteosarcoma (OS) cells mainly through the natural killer group 2D (NKG2D)–NKG2DL pathway. OS cells were pretreated with phosphate-buffered saline or decitabine (DAC) (5 µM) for 3 days. Then, γδ T cells were cocultured with DAC-pretreated OS cells for 4 h at an effector/target ratio of 5:1. Cytotoxicity of γδ T cells was determined using an MTS assay. γδ T cells were preincubated for 30 min with 10 µg/mL anti-human NKG2D, anti-pan-γδ T cell receptor (TCR), anti-Trail, or anti-FasL mAbs. Normally, TCR signal pathway was the key factor that can trigger the anti-OS activity of γδ T cells (A,B). However, for DAC-pretreated OS cells, NKG2D–NKG2DL axis was the most important pathway by which γδ T cells killing about half of them (C,D). Error bars show standard deviation among the replicates (*P < 0.05 vs. isotype control; **P < 0.01 vs. isotype control; ns, no significance vs. isotype control. mAb, monoclonal antibody).
Antitumor Effect of Combining Adoptive γδ T Cells With DAC Treatment In Vivo
To confirm the effects of γδ T cells + DAC treatment in vivo, we created an OS model by injecting HOS cells into nude mice through the tibial plateau in the primary spongiosa of the left tibia. Mice in the control group were treated with PBS. As shown in Figure 7A, all mice in the control group showed progressive tumor growth. The mean tumor volume in the groups treated with DAC or γδ T cells alone was slightly less than that in the control group, whereas there was a substantial decrease in the tumor volume in mice receiving γδ T cells + DAC treatment compared to the control group (P < 0.01; Figure 7B). In addition, the tumor weight of the group receiving combined treatment was significantly reduced compared with that of the control or single-treatment groups (P < 0.05; Figure 7C). Additionally, western blotting showed that the MICB and ULBP1 proteins in OS tumor tissues increased after DAC treatment (Figure 7D). These results indicated that γδ T cells + DAC treatment markedly inhibited the growth of OS cells in vivo.
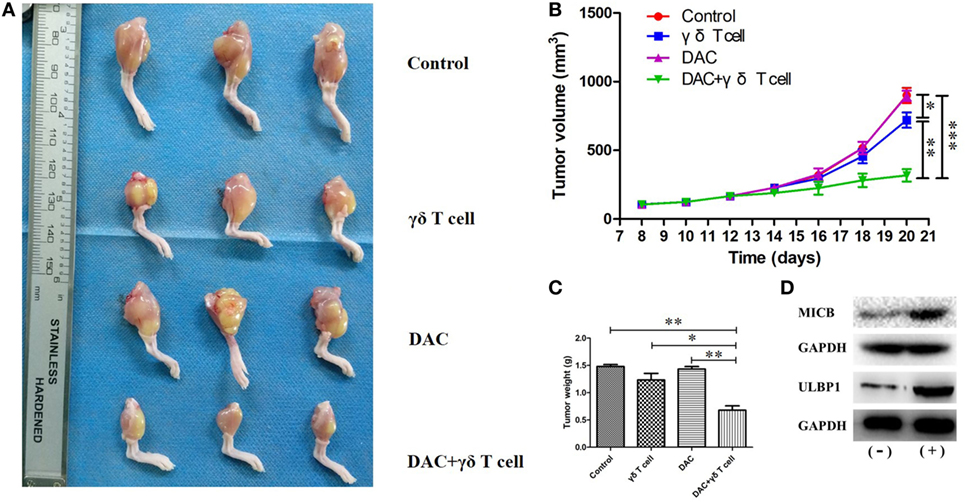
Figure 7. Mice receiving both γδ T cells and decitabine (DAC) showed significantly reduced tumor growth. (A) Mice from groups treated with γδ T cells + DAC showed less tumor burden than those mice untreated (control) or treated with DAC or γδ T cells alone. (B) Tumor volumes are significantly smaller in the group treated with γδ T cells + DAC compared with the control or DAC or γδ T cells group. (C) The group treated with γδ T cells + DAC showed significantly reduced tumor weight compared with the control or single-treatment group. Error bars show SD among the replicates. (D) Western blot confirmed that DAC treatment increased expression of MICB and UL16-binding protein 1 (ULBP1) in vivo. (−): without DAC treatment; (+): DAC treatment (ns refers to no significance; *P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
As a subpopulation of lymphocytes involved in the human innate immune system, γδ T cells can combat tumors and microbial infections. Unlike αβ T cells, γδ T cells can directly recognize and respond to antigens in an MHC-independent manner to effectively lyse tumor cells and microorganisms. Moreover, it is very difficult to achieve the required number of NK cells or αβ T cells in vitro to produce effective adoptive immunotherapy for tumors. In contrast, γδ T cells can be efficiently generated from PBMCs over a short period by in vitro culturing (26). Thus, γδ T cells have been suggested as a good candidate for cancer immunotherapy. Several clinical trials involving adoptive transfer of in vitro-expanded γδ T cells to control both hematological malignancies and solid tumors have been carried out, and they have shown promise regarding efficacy and safety (10–12, 27).
However, the efficacy of γδ T cell-based immunotherapy is limited because of its potential suppressive role in tumor microenvironments (13, 14). Regarding adoptive immunotherapy for treating tumors, human Vγ9Vδ2 T cells, which are the main γδ T cell population among PBMCs, are usually selected. However, Vδ2 T cells can exert immunosuppressive functions by producing suppressive cytokines, generating adenosine, upregulating negative costimulatory molecules, etc. They may also differentiate into cells with a regulatory phenotype similar to αβ Treg cells due to their high functional plasticity (28). Therefore, a better understanding of the functional role of Vδ2 T cells in tumor microenvironments will help to develop more effective antitumor immunotherapy.
Recently, the use of adoptive γδ T cell transfer in OS immunotherapy has become increasingly popular. In 2001, Kato et al. (29) documented the cytotoxicity of γδ T cells against OS cells, suggesting their potential use as anti-OS therapy. Preclinical studies have demonstrated the potential of using γδ T cells for OS treatment (30, 31). Although adoptive γδ T cell transfer alone has yielded promising clinical results, there is a strong rationale (based on preclinical research) for identifying the optimal combination treatment strategy to augment its efficacy (6, 32, 33).
Previous studies have shown that the cytotoxicity of γδ T cells against OS cells can be enhanced by other agents, such as trastuzumab (antihuman epidermal growth factor receptor 2 monoclonal antibody) and IFN-γ (20, 34). In addition, epigenetic modulators have recently been recognized as useful agents in cancer immunotherapy, especially γδ T cell-based immunotherapy (15). Hypomethylating agents, such as DAC, have been shown to have anticancer and immunomodulatory activities (16). Al-Romaih et al. (35) found that DAC-induced demethylation of growth arrest and DNA damage inducible alpha(GADD45A)and promoted apoptosis of OS cells. Zhang et al. (36) recently reported that DAC increased the levels of miR-370, an anti-oncogenic microRNA, resulting in the inhibition of OS cells. Regarding its mechanism of action in sarcoma immunotherapy, DAC may induce the upregulation of cancer/testis antigens, MHC molecules, and intracellular cell adhesion molecule-1 (ICAM-1) to promote immune recognition of sarcoma cells (37). Li et al. (19) reported that DAC treatment resulted in a CD8+ T cell-mediated anti-OS reaction via increasing expression of the MAGE-A family proteins and NY-ESO-1 in OS cells, both in vitro and in vivo, which has also been reported in other tumor cells (38, 39). A phase I trial combining DAC/dendritic cell vaccine was performed in two sarcoma patients, showing that this chemoimmunotherapeutic method was feasible and well tolerated, and providing a scientific basis for further studies (40).
Previous studies have focused on DAC treatment combined with adaptive immune cells but combinations involving innate immune cells are rare. Recent studies have reported that DAC-mediated hypomethylation restored NKG2DL expression in tumors and made tumor cells more sensitive to recognition and killing by NK cells (17, 18), indicating that the combined effect of DAC with the adoptive transfer of innate immune cells may be considerable. However, the combined effect of DAC with γδ T cells has not been previously explored. Thus, the purpose of our investigation was to demonstrate the effectiveness of this combination therapy for OS. DAC (5 µM) had no apparent cytotoxic effect on γδ T cells and did not alter the percentage of γδ TCR+ and NKG2D+ γδ T cells (Figure S2 in Supplementary Material). Cruz et al. (41) also found that DAC had minimal effects on the phenotype and function of T cells. In addition, Yang et al. (42) reported that treating tumor cells with DAC upregulated gene expression in a dose-dependent manner. Thus, in our study, 5 µM DAC was chosen for the subsequent experiments.
γδ T cells possess broad innate antitumor and anti-infective activities, which involve directly recognizing unprocessed phosphoantigens (PAgs) via cell-surface TCRs. In bacterial cells, PAgs are produced in the 1-deoxy-d-xylulose-5-phosphate pathway, whereas, in tumor cells, they are produced in the mevalonate pathway. Isopentenyl pyrophosphate (IPP) is one of the stress-induced PAgs in tumor cells, which can accumulate due to the inhibition of farnesyl pyrophosphate synthase (FPPS). FPPS is an enzyme that acts downstream of IPP in the mevalonate pathway. Moreover, inhibition of FPPS decreases the degree of prenylation, resulting in the accumulation of unprenylated Ras and Rap1A (43–45). Butyrophilin 3A (BTN3A, CD277) was recently identified as a critical mediator between PAgs and γδ T cells (46). BTN3A enhances γδ T cell-based immunotherapy for both hematological and solid tumors and has also been recognized as a beneficial prognostic marker in pancreatic ductal adenocarcinoma (47–49). However, precisely which tumor cell molecules are recognized by γδ TCRs remains unknown (46). In addition to the γδ TCR stimulus, the interactions involving NKG2D/NKG2DL, Trail/Trail-R, and Fas/FasL also activate the effector function of γδ T cells against tumors (26, 50).
Tumor cells express NKG2DL, which is a ligand of the NK cell-activating receptor NKG2D, on their cell surface. The interaction between NKG2D and NKG2DL triggers the cytolysis of tumor cells, such as OS cells (24). The NKG2D receptor is also expressed on the surface of γδ T cells, and γδ T cells have the same cytotoxic effect on tumor cells as NK cells. In the present study, we showed that γδ T cells exhibited significant efficacy when used in combination with DAC, suggesting that DAC may enhance the susceptibility of OS cells to γδ T cell-mediated cytotoxicity. Using PCR and flow cytometry, we then showed that DAC increased the expression of NKG2D ligands, primarily MICB and ULBP1, making OS cells more sensitive to recognition and destruction by cytotoxic γδ T cells. This effect of γδ T cells + DAC treatment may be unique to OS cells, because Zhang et al. (18) reported that DAC increased ULBP1 and ULBP3 expression in glioma cells and increased NK-mediated lysis of these cells in an NKG2D-dependent manner. In other words, NKG2DL expression may vary in different tumors after DAC treatment.
In the present study, DAC-induced demethylation of the promoter sites of MICB and ULBP1 in OS cells and promoted their expression, which was also reported by Tang et al. (23). Trail and FasL blocking had no obvious influence on the γδ T cell-mediated cytotoxic effect on OS cells regardless of DAC pretreatment (Figure 6). Although γδ T cell-mediated cytotoxicity against untreated OS cells occurred primarily in a TCR-dependent manner (Figures 6A,C), this mechanism was secondary (to a mechanism involving the NKG2D–NKG2DL axis) in DAC-pretreated OS cells (Figures 6B,D). Approximately half of the γδ T cell-mediated killing of the DAC-pretreated OS cells was reversed by blocking the NKG2D receptor. This suggested that the NKG2D–NKG2DL pathway played an important role during the γδ T cell-mediated cytolysis of DAC-pretreated OS cells. In the in vivo experiments in mice, no distinct side effects were observed. More importantly, we showed that combined γδ T cells + DAC treatment markedly inhibited the growth of OS, which provides a preclinical rationale for the use of DAC to sensitize OS to γδ T cell-mediated killing.
The present study has several limitations. First, the γδ T cells used in our study were all from healthy donors. Second, only HOS and U2OS cells were used in our study, and research with other human or murine OS cell lines and primary OS cells is required to verify the results of this study. Third, immunodeficient mice were used for the experiments with human OS cell lines, which limited the assessment of any additional effects of other adaptive immune cells (e.g., αβ T or B cells) in tumor microenvironments.
In summary, the results of the present study confirmed that DAC sensitizes OS cells to γδ T cell cytotoxicity. Furthermore, we demonstrated that the killing capacity of γδ T cells was potentiated by increasing the NKG2DL expression in DAC-pretreated OS cells. Adoptive transfer of γδ T cells combined with DAC treatment may represent a novel strategy for the treatment of OS and the findings of the present study provide evidence for the clinical application of this combination therapy.
Ethics Statement
This study was approved by the Human and Animal Research Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine. Written informed consent was obtained from all volunteers.
Author Contributions
ZY and ZhanW designed research; ZhanW, ZenanW, and SL performed research; ZhanW, BL, LS, PL, SW, WT, and XZ analyzed data; ZY, ZhanW ZenanW, and SL wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Orthopedics Research Institute of Zhejiang University for facility support and Jiangfeng Li for experimental technical support. This work was supported by the National Natural Science Foundation of China (Grant no. 81702118), Medical and Health Science and Technology Plan of Department of Health of Zhejiang Province (WKJ-ZJ-1821) and 2017 Zhejiang provincial medical and health science and technology project (Grant no. 2017KY392).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01239/full#supplementary-material.
Figure S1. Scheme of experimental groups and treatment procedure in vivo.
Figure S2. The human γδ T cells were identified by their expression of T cell receptor (TCR) and natural killer group 2D (NKG2D) after 5 µM decitabine (DAC) treatment for 1 day. The percentage of γδ TCR+ and NKG2D+ γδ T cells was analyzed by flow cytometry and no obvious change was observed between the two groups.
References
1. Damron TA, Ward WG, Stewart A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National Cancer Data Base Report. Clin Orthop Relat Res (2007) 459:40–7. doi:10.1097/BLO.0b013e318059b8c9
2. Harrison DJ, Geller DS, Gill JD, Lewis VO, Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther (2018) 18(1):39–50. doi:10.1080/14737140.2018.1413939
3. Kiany S, Huang G, Kleinerman ES. Effect of entinostat on NK cell-mediated cytotoxicity against osteosarcoma cells and osteosarcoma lung metastasis. Oncoimmunology (2017) 6(8):e1333214. doi:10.1080/2162402X.2017.1333214
4. Meyers PA, Schwartz CL, Krailo M, Kleinerman ES, Betcher D, Bernstein ML, et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. J Clin Oncol (2005) 23(9):2004–11. doi:10.1200/JCO.2005.06.031
5. Isakoff MS, Bielack SS, Meltzer P, Gorlick R. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol (2015) 33(27):3029–35. doi:10.1200/JCO.2014.59.4895
6. Wang Z, Li B, Ren Y, Ye Z. T-cell-based immunotherapy for osteosarcoma: challenges and opportunities. Front Immunol (2016) 7:353. doi:10.3389/fimmu.2016.00353
7. Chitadze G, Oberg HH, Wesch D, Kabelitz D. The ambiguous role of gammadelta T lymphocytes in antitumor immunity. Trends Immunol (2017) 38(9):668–78. doi:10.1016/j.it.2017.06.004
8. Tanaka Y, Morita CT, Tanaka Y, Nieves E, Brenner MB, Bloom BR. Natural and synthetic non-peptide antigens recognized by human gamma delta T cells. Nature (1995) 375(6527):155–8. doi:10.1038/375155a0
9. Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T. Activation of V gamma 9V delta 2 T cells by NKG2D. J Immunol (2005) 175(4):2144–51. doi:10.4049/jimmunol.175.4.2144
10. Aoki T, Matsushita H, Hoshikawa M, Hasegawa K, Kokudo N, Kakimi K. Adjuvant combination therapy with gemcitabine and autologous gammadelta T-cell transfer in patients with curatively resected pancreatic cancer. Cytotherapy (2017) 19(4):473–85. doi:10.1016/j.jcyt.2017.01.002
11. Kobayashi H, Tanaka Y, Yagi J, Minato N, Tanabe K. Phase I/II study of adoptive transfer of gammadelta T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol Immunother (2011) 60(8):1075–84. doi:10.1007/s00262-011-1021-7
12. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer (2011) 105(6):778–86. doi:10.1038/bjc.2011.293
13. Ye J, Ma C, Hsueh EC, Eickhoff CS, Zhang Y, Varvares MA, et al. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. J Immunol (2013) 190(5):2403–14. doi:10.4049/jimmunol.1202369
14. Paul S, Lal G. Regulatory and effector functions of gamma-delta (gammadelta) T cells and their therapeutic potential in adoptive cellular therapy for cancer. Int J Cancer (2016) 139(5):976–85. doi:10.1002/ijc.30109
15. Bhat J, Kabelitz D. gammadelta T cells and epigenetic drugs: a useful merger in cancer immunotherapy? Oncoimmunology (2015) 4(6):e1006088. doi:10.1080/2162402X.2015.1006088
16. Nie J, Liu L, Li X, Han W. Decitabine, a new star in epigenetic therapy: the clinical application and biological mechanism in solid tumors. Cancer Lett (2014) 354(1):12–20. doi:10.1016/j.canlet.2014.08.010
17. Vasu S, He S, Cheney C, Gopalakrishnan B, Mani R, Lozanski G, et al. Decitabine enhances anti-CD33 monoclonal antibody BI 836858-mediated natural killer ADCC against AML blasts. Blood (2016) 127(23):2879–89. doi:10.1182/blood-2015-11-680546
18. Zhang X, Rao A, Sette P, Deibert C, Pomerantz A, Kim WJ, et al. IDH mutant gliomas escape natural killer cell immune surveillance by downregulation of NKG2D ligand expression. Neuro Oncol (2016) 18(10):1402–12. doi:10.1093/neuonc/now061
19. Li B, Zhu X, Sun L, Yuan L, Zhang J, Li H, et al. Induction of a specific CD8+ T-cell response to cancer/testis antigens by demethylating pre-treatment against osteosarcoma. Oncotarget (2014) 5(21):10791–802. doi:10.18632/oncotarget.2505
20. Li Z, Xu Q, Peng H, Cheng R, Sun Z, Ye Z. IFN-gamma enhances HOS and U2OS cell lines susceptibility to gammadelta T cell-mediated killing through the Fas/Fas ligand pathway. Int Immunopharmacol (2011) 11(4):496–503. doi:10.1016/j.intimp.2011.01.001
21. Molhoek KR, McSkimming CC, Olson WC, Brautigan DL, Slingluff CL Jr. Apoptosis of CD4(+)CD25(high) T cells in response to sirolimus requires activation of T cell receptor and is modulated by IL-2. Cancer Immunol Immunother (2009) 58(6):867–76. doi:10.1007/s00262-008-0602-6
22. Li J, Xu X, Meng S, Liang Z, Wang X, Xu M, et al. MET/SMAD3/SNAIL circuit mediated by miR-323a-3p is involved in regulating epithelial-mesenchymal transition progression in bladder cancer. Cell Death Dis (2017) 8(8):e3010. doi:10.1038/cddis.2017.331
23. Tang KF, He CX, Zeng GL, Wu J, Song GB, Shi YS, et al. Induction of MHC class I-related chain B (MICB) by 5-aza-2’-deoxycytidine. Biochem Biophys Res Commun (2008) 370(4):578–83. doi:10.1016/j.bbrc.2008.03.131
24. Fernandez L, Valentin J, Zalacain M, Leung W, Patino-Garcia A, Perez-Martinez A. Activated and expanded natural killer cells target osteosarcoma tumor initiating cells in an NKG2D-NKG2DL dependent manner. Cancer Lett (2015) 368(1):54–63. doi:10.1016/j.canlet.2015.07.042
25. Seelan RS, Mukhopadhyay P, Pisano MM, Greene RM. Effects of 5-Aza-2’-deoxycytidine (decitabine) on gene expression. Drug Metab Rev (2018) 50:193–207. doi:10.1080/03602532.2018.1437446
26. Sun LL, Zhang ZL, Li YJ, Wang SD, Li HY, Li BH, et al. Zoledronic acid sensitizes rhabdomyosarcoma cells to cytolysis mediated by human gammadelta T cells. Oncol Lett (2017) 14(5):5597–604. doi:10.3892/ol.2017.6894
27. Kunzmann V, Smetak M, Kimmel B, Weigang-Koehler K, Goebeler M, Birkmann J, et al. Tumor-promoting versus tumor-antagonizing roles of gammadelta T cells in cancer immunotherapy: results from a prospective phase I/II trial. J Immunother (2012) 35(2):205–13. doi:10.1097/CJI.0b013e318245bb1e
28. Peters C, Kabelitz D, Wesch D. Regulatory functions of γδ T cells. Cell Mol Life Sci (2018) 75(12):2125–35. doi:10.1007/s00018-018-2788-x
29. Kato Y, Tanaka Y, Miyagawa F, Yamashita S, Minato N. Targeting of tumor cells for human gammadelta T cells by nonpeptide antigens. J Immunol (2001) 167(9):5092–8. doi:10.4049/jimmunol.167.9.5092
30. Muraro M, Mereuta OM, Carraro F, Madon E, Fagioli F. Osteosarcoma cell line growth inhibition by zoledronate-stimulated effector cells. Cell Immunol (2007) 249(2):63–72. doi:10.1016/j.cellimm.2007.11.005
31. Li Z, Tang J, Sun L, Ye Z. [Effect of zoledronate on the cytotoxicity of gammadelta T cells from PBMCs of osteosarcoma patients against osteosarcoma]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2013) 29(1):6–9.
32. Fisher JP, Heuijerjans J, Yan M, Gustafsson K, Anderson J. gammadelta T cells for cancer immunotherapy: a systematic review of clinical trials. Oncoimmunology (2014) 3(1):e27572. doi:10.4161/onci.27572
33. Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Front Immunol (2014) 5:636. doi:10.3389/fimmu.2014.00636
34. Liu M, Sun LL, Li YJ, Li HY, Zhang J, Li BH, et al. Trastuzumab enhanced the cytotoxicity of Vgamma9Vdelta2 T cells against zoledronate-sensitized osteosarcoma cells. Int Immunopharmacol (2015) 28(1):160–7. doi:10.1016/j.intimp.2015.06.002
35. Al-Romaih K, Sadikovic B, Yoshimoto M, Wang Y, Zielenska M, Squire JA. Decitabine-induced demethylation of 5′ CpG island in GADD45A leads to apoptosis in osteosarcoma cells. Neoplasia (2008) 10(5):471–80. doi:10.1593/neo.08174
36. Zhang W, Duan N, Zhang Q, Song T, Li Z, Zhang C, et al. DNA methylation mediated down-regulation of miR-370 regulates cell growth through activation of the Wnt/beta-Catenin signaling pathway in human osteosarcoma cells. Int J Biol Sci (2017) 13(5):561–73. doi:10.7150/ijbs.19032
37. Krishnadas DK, Bao L, Bai F, Chencheri SC, Lucas K. Decitabine facilitates immune recognition of sarcoma cells by upregulating CT antigens, MHC molecules, and ICAM-1. Tumour Biol (2014) 35(6):5753–62. doi:10.1007/s13277-014-1764-9
38. Griffiths EA, Srivastava P, Matsuzaki J, Brumberger Z, Wang ES, Kocent J, et al. NY-ESO-1 vaccination in combination with decitabine induces antigen-specific T-lymphocyte responses in patients with myelodysplastic syndrome. Clin Cancer Res (2018) 24(5):1019–29. doi:10.1158/1078-0432.CCR-17-1792
39. Leclercq S, Gueugnon F, Boutin B, Guillot F, Blanquart C, Rogel A, et al. A 5-aza-2’-deoxycytidine/valproate combination induces cytotoxic T-cell response against mesothelioma. Eur Respir J (2011) 38(5):1105–16. doi:10.1183/09031936.00081310
40. Krishnadas DK, Shusterman S, Bai F, Diller L, Sullivan JE, Cheerva AC, et al. A phase I trial combining decitabine/dendritic cell vaccine targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for children with relapsed or therapy-refractory neuroblastoma and sarcoma. Cancer Immunol Immunother (2015) 64(10):1251–60. doi:10.1007/s00262-015-1731-3
41. Cruz CR, Gerdemann U, Leen AM, Shafer JA, Ku S, Tzou B, et al. Improving T-cell therapy for relapsed EBV-negative Hodgkin lymphoma by targeting upregulated MAGE-A4. Clin Cancer Res (2011) 17(22):7058–66. doi:10.1158/1078-0432.CCR-11-1873
42. Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia (2014) 28(6):1280–8. doi:10.1038/leu.2013.355
43. Idrees AS, Sugie T, Inoue C, Murata-Hirai K, Okamura H, Morita CT, et al. Comparison of gammadelta T cell responses and farnesyl diphosphate synthase inhibition in tumor cells pretreated with zoledronic acid. Cancer Sci (2013) 104(5):536–42. doi:10.1111/cas.12124
44. Okamoto S, Kawamura K, Li Q, Yamanaka M, Yang S, Fukamachi T, et al. Zoledronic acid produces antitumor effects on mesothelioma through apoptosis and S-phase arrest in p53-independent and Ras prenylation-independent manners. J Thorac Oncol (2012) 7(5):873–82. doi:10.1097/JTO.0b013e31824c7d43
45. Gober HJ, Kistowska M, Angman L, Jeno P, Mori L, De Libero G. Human T cell receptor gammadelta cells recognize endogenous mevalonate metabolites in tumor cells. J Exp Med (2003) 197(2):163–8. doi:10.1084/jem.20021500
46. Gu S, Borowska MT, Boughter CT, Adams EJ. Butyrophilin3A proteins and Vγ9Vδ2 T cell activation. Semin Cell Dev Biol (2018). doi:10.1016/j.semcdb.2018.02.007
47. Benyamine A, Le Roy A, Mamessier E, Gertner-Dardenne J, Castanier C, Orlanducci F, et al. BTN3A molecules considerably improve Vgamma9Vdelta2T cells-based immunotherapy in acute myeloid leukemia. Oncoimmunology (2016) 5(10):e1146843. doi:10.1080/2162402X.2016.1146843
48. Zocchi MR, Costa D, Vene R. Zoledronate can induce colorectal cancer microenvironment expressing BTN3A1 to stimulate effector gammadelta T cells with antitumor activity. Oncoimmunology (2017) 6(3):e1278099.
49. Benyamine A, Loncle C, Foucher E, Blazquez JL, Castanier C, Chretien AS, et al. BTN3A is a prognosis marker and a promising target for Vgamma9Vdelta2 T cells based-immunotherapy in pancreatic ductal adenocarcinoma (PDAC). Oncoimmunology (2017) 7(1):e1372080. doi:10.1080/2162402X.2017.1372080
Keywords: decitabine, osteosarcoma, γδ T cell, MICB, UL16-binding protein 1
Citation: Wang Z, Wang Z, Li S, Li B, Sun L, Li H, Lin P, Wang S, Teng W, Zhou X and Ye Z (2018) Decitabine Enhances Vγ9Vδ2 T Cell-Mediated Cytotoxic Effects on Osteosarcoma Cells via the NKG2DL–NKG2D Axis. Front. Immunol. 9:1239. doi: 10.3389/fimmu.2018.01239
Received: 09 February 2018; Accepted: 17 May 2018;
Published: 01 June 2018
Edited by:
Fang-Ping Huang, University of Hong Kong, Hong KongReviewed by:
Amorette Barber, Longwood University, United StatesConstantinos Zambirinis, Memorial Sloan Kettering Cancer Center, United States
Copyright: © 2018 Wang, Wang, Li, Li, Sun, Li, Lin, Wang, Teng, Zhou and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhaoming Ye, eWV6aGFvbWluZ0B6anUuZWR1LmNu
†These authors have contributed equally to this work.