 Chen Chen1
Chen Chen1 Wei Qiu
Wei Qiu- 1Department of Neurology, The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 2Key Laboratory of the Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry, National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest of China, College of Life Sciences, Shaanxi Normal University, Xi’an, China
Multiple sclerosis (MS) is an immune-mediated demyelinating disease of the central nervous system. Growing evidence has proven that T helper 17 (Th17) cells are one of the regulators of neuroinflammation mechanisms in MS disease. Researchers have demonstrated that some microRNAs (miRNAs) are associated with disease activity and duration, even with different MS patterns. miRNAs regulate CD4+ T cells to differentiate toward various T cell subtypes including Th17 cells. In this review, we discuss the possible mechanisms of miRNAs in MS pathophysiology by regulating CD4+ T cell differentiation into Th17 cells, and potential miRNA targets for current disease-modifying treatments.
Introduction
Multiple sclerosis (MS) is an autoimmune disease characterized by chronic inflammatory demyelination in the central nervous system (CNS), which can result in cognitive decline and permanent disability among young adults. The etiology of MS has been widely studied, including virus infection, genetic predisposition, lack of vitamin D, occupational exposure, and toxins. It is accepted that MS is an inflammatory and neurodegenerative disease primarily driven by myelin-reactive CD4+ T helper 1 (Th1) cells, CD4+ T helper 17 (Th17) cells, CD8+ T cells, and B cells that target and damage the myelin sheath. CD4+ Th1 cells and CD4+ Th17 cells are the two subtypes of CD4+ T cells which have been intensively investigated in MS and its animal model, experimental autoimmune encephalomyelitis (EAE). In animal studies, adoptive transfer of myelin-specific Th1 cells into naïve recipient mice was sufficient to induce features of EAE (1). Th17 cells are a newly found player in the pathology of MS and EAE model. It has been shown that the proportion of Th17 cells in peripheral blood and interleukin (IL)-17 levels in serum were increased among MS patients (2). IL-17+-producing T cells were elevated in the active rather than in inactive areas of MS lesions, and significantly higher densities presented within acute lesions and active borders of chronic active lesions than in normal-appearing white matter (3). This suggested that Th17 cells and IL-17 involve in the MS pathogenesis.
MicroRNAs (miRNAs) are single-stranded, approximately 22 nt non-coding RNAs that are considered as key regulators in the complex network of gene expression at the posttranscriptional level. There are approximately 24,521 miRNA loci in 206 species, which produce 30,424 mature miRNAs including more than 2,500 mature miRNAs annotated in the miRBase database (v20, June 2013) (4). miRNAs are pluripotent components that participate in several biological processes including cell development and differentiation. Drosha and Dicer nucleases are the two major miRNA machinery enzymes that convert pri-miRNAs to mature miRNAs and control the production and function of mature miRNAs. Mature miRNAs loaded into the RNA-induced silencing complex directly target mRNAs, leading to target mRNA cleavage and lower protein expression through direct or indirect interference (5).
Studies have revealed that miRNAs may contribute to MS progression and responses to treatment (6). Th17 cells are characterized by expression of retinoic acid-related orphan receptor (ROR) γ and signal transducer and activator of transcription (STAT) 3. miRNAs play major roles in Th17 cell differentiation (7), particularly through the RORγ and STAT3 signal pathway. In this review, we discuss the relationships between miRNAs, Th17 cell differentiation, and MS, pointing out the pivotal roles of miRNA in the pathophysiology of MS, and miRNAs as potential targets for current disease-modifying treatments.
Th17 Cells in MS Pathogenesis
CCD4+ T cells mediate adaptive immunity to various pathogens and are critical for proper immune system homeostasis and host defense. CD4+ T cell-mediated autoimmunity has long been known as one of the most important aspects in MS pathogenesis. Th17 cells have been suggested as a new lineage of CD4+ T cells, which synthesize and secrete IL-17 and IL-22 to enhance cellular immune responses to autoimmune inflammation (8). Cytokines produced by Th17 cells are most likely to be critical pathological factors in autoimmune diseases, particularly MS. An over-exuberant response against self-antigens by Th17 cells induces several common autoimmune diseases (9) including MS. There is growing evidence from both animal models and human studies that Th17 cells and IL-17 play important roles in orchestrating MS progression. The frequency of Th17 cells is elevated in the blood and cerebrospinal fluid (CSF) of patients with MS and is higher during relapses (10). Th17 cell clones generated from the CSF and peripheral blood of MS patients expressed high levels of activation markers, adhesion molecules, and co-stimulatory molecules than Th1 clones (10). Activated Th17 cells migrate through the blood–brain barrier (BBB) into MS lesions. This migration is mediated by IL-17 and IL-22 that disrupt tight junction proteins in CNS endothelial cells (11). Th17 cells that are specific to myelin basic protein in active MS were associated with disease activity (12). After transmigration through the BBB, Th17 cells infiltrate at a high frequency into the acute MS lesions (13).
The Th17 cell lineage is characterized by expression of RORγ and STAT3, both of which are the basis for the cytokine profile, including IL-6, IL-12, IL-17, IL-22, and tumor necrosis factor (TNF), which mediate tissue inflammation. IL-17 is a cytokine can be secreted by multiple cells, such as activated T cells, natural killer cells, and neutrophils. IL-17-secreting CD4+ T cells (Th17 cells) are critical players in the pathology of EAE and MS. Similarly, most of the pathological functions of Th17 cells have been attributed to the secretion of cytokines, such as IL-17. IL-17 has many biological functions, such as recruiting both neutrophils and monocytes, regulating innate immunity, enhancing B cell functions, and regulating the release of pro-inflammatory cytokines including TNF and IL-1β (14). Furthermore, high levels of IL-17 exist in both serum and peripheral blood mononuclear cells (PBMCs) of MS patients (15). In vitro study, Th17 cells could be induced by two different conditions from naïve CD4+ T cells. One subset considered as non-pathogenic Th17 cells, was generated in the presence of TGF-β plus IL-6, which could abrogate Th17 cell-mediated pathology (16); the other subset was generated by IL-1β, IL-6, IL-23, and TGF-β, which was considered as pathogenic Th17 cells (17).
Dysregulated miRNAs in MS
MicroRNAs are an emerging group of promising biomarkers in various autoimmune diseases because of their small size and stable structure in body fluids. Studying the relationships between miRNAs and MS has been a hot topic in recent years. Growing evidence shows that miRNA expression profiles might facilitate identifying the different patterns of clinical progression of MS (18).
miRNA Profiling of Human Body Fluids
Many kinds of body fluids, such as blood, serum, plasma, CSF, and urine, can be a source to measure the expression level of miRNAs. The first study of circulating miRNA in plasma was performed by Siegel et al., revealing significant involvement of miRNAs in MS and suggesting that miRNAs may serve as potential prognostic and diagnostic biomarkers for MS (19). This study used microarray analysis to identify six plasma miRNAs, miR-614, miR-572, miR-648, miR-1826, miR-422a, and miR-22, which were significantly upregulated, and miR-1979 that was significantly downregulated in MS patients (19). miR-92a-1 was differentially expressed in relapsing–remitting MS (RRMS) versus secondary progressive MS (SPMS) and RRMS versus healthy controls (HCs). It was also associated with the expanded disability status scale and disease duration. The Let-7 family of miRNAs differentiated SPMS from HCs and RRMS from SPMS, miR-454 differentiated RRMS from SPMS, and miR-145 differentiated RRMS from HCs and RRMS from SPMS (19, 20). Other studies employed real-time RT-PCR and found higher expression of miR-155 in serum (21), and miR-141 and miR-200a in CD4+ T cells of MS patients in relapse than in remission (22). In addition, miR-141 and miR-200a may take part in promoting Th17 cell differentiation while inhibiting regulatory T (Treg) cells (22). miR-155 promotes T cell-driven inflammation by targeting heme oxygenase 1 (23). Using next-generation sequencing (NGS) and microarray analysis to test whole blood from MS patients, Keller et al. found that 16 miRNAs were downregulated and 22 miRNAs were upregulated in clinical isolation syndrome and RRMS. Five miRNAs were downregulated, and three miRNAs were upregulated as confirmed by microarray analysis. miR-16-2-3p was significantly upregulated, and miR-20a-5p and miR-7-1-3p were downregulated as measured by both methods (24). Compared with another study using microarray analysis, 26 miRNAs were downregulated, and 1 was upregulated in whole blood of MS patients. The downregulated group of miRNAs was found in all subtypes of MS. miR-17 and miR-20a, which were significantly under-expressed in MS, are regulators of genes involved in T cell activation (25). Sondergaard et al. investigated the expression of miRNAs in PBMCs as well as plasma and serum samples from RRMS patients by microarray analysis and identified miR-145, miR-660, and miR-939 as significantly and differentially distributed in plasma of RRMS patients compared with HCs (20).
To classify the possible function of deregulated miRNAs in target cells, many peripheral leukocyte subgroups have been isolated and examined. In a microarray analysis, 21 miRNAs had decreased expression, and 20 of them were shown to affect the expression of their target genes that are involved in the immune system (26). Studies using NGS to obtain miRNA expression profiles in a pilot cohort study of SPMS found that 97% of miRNA candidates were downregulated and 42 miRNAs were dysregulated in CD4+ T cells. Five miRNAs (miR-21-5p, miR-26b-5p, miR-29b-3p, miR-142-3p, and miR-155-5p) were significantly downregulated and confirmed by TaqMan assays, which targeted suppressor of cytokine signaling 6 that negatively regulates T cell activation (27). Another study using microarray analysis revealed increases of miR-128 and miR-27b in naïve CD4+ T cells and miR-340 in memory CD4+ T cells from patients with MS (28).
Compared with peripheral blood, CSF is more ideal to monitor CNS disease activity because of its close proximity to lesions, particularly the MS nidus. However, biomarkers in CSF are limited because a lumbar puncture is a traumatic procedure. Through global miRNA profiling, Haghikia et al. quantitatively confirmed that miR-922, miR-181c, and miR-633 in the CSF are differentially regulated in patients with MS (29) (Table 1). Another study demonstrated that miR-150 was elevated in MS and associated with markers of inflammation in CSF, such as the presence of oligoclonal bands, CSF cell counts, immunoglobulin G index, and candidate protein biomarkers C-X-C motif chemokine 13, matrix metallopeptidase 9, and osteopontin. This trend would be reversed after 12 months of treatment by natalizumab (30).
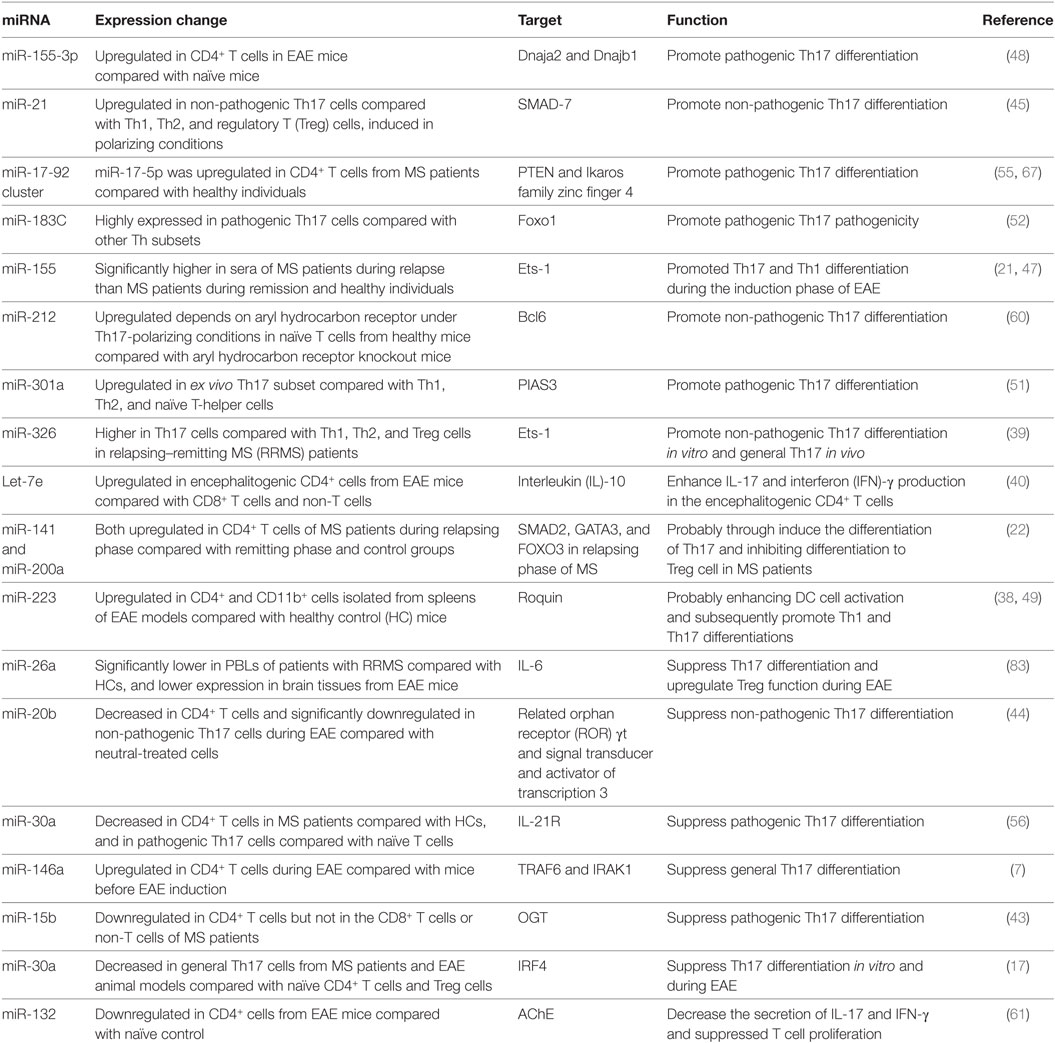
Table 1. MicroRNAs (miRNAs) involved in T helper 17 (Th17) cells development in multiple sclerosis (MS) and the experimental autoimmune encephalomyelitis (EAE) model.
miRNA Profiling of Lesions in Human and Animal Model
Brain-resident cells inside MS lesions may be more representative of immunological changes in MS patients. This type of study may provide important and new insights into pathological hallmarks and reveal potential targets for therapy. Junker et al. obtained miRNA profiles of active and inactive MS lesions using laser capture microdissection to isolate single cells for in vitro culture. As a result, 20 miRNAs were at least twice as abundant in active lesions and 22 miRNAs were at least twice as abundant in inactive lesions. miR-34a, miR-155, and miR-326, which were upregulated in active MS lesions, targeted the 3′-untranslated region of CD47 to reduce CD47 expression in brain-resident cells, particularly miR-155 (31). CD47, a ubiquitously expressed membrane glycoprotein, is abundantly expressed on phagocytic cells (32). The function of phagocytosis would be enhanced upon reduction of CD47 in macrophages. Lescher et al. analyzed miRNAs in human MS lesions together with myelin oligodendrocyte glycoprotein (MOG)35–55 peptide-induced EAE in C57/BL6 mice and MOG1–125 peptide-induced EAE in marmoset monkeys. The results demonstrated that the miRNA profiles of lesions in mice and marmoset monkeys were consistent with the miRNA profiles of active human MS lesions. miR-155, miR-326, miR-142-3p, miR-146a, miR-146b, and miR-142-5p were all significantly upregulated in active human MS lesions (33), and the miR-142-5p expression level was significantly increased in normal frontal white matter of MS patients, which was proven by upregulation of miR-142a-5p in the lumbar spinal cord at peak and post-peak phases of EAE, together with miR-142a-3p (34). However, miR-181a and miR-181-b levels in brain white matter from MS patients are downregulated (35).
Pathological and autopsy samples from patients are a valuable source that can reflect real pathological changes induced by a certain disease. In the case of MS, studies of brain tissue, circulating leukocytes, and fluids have shown altered expression of various miRNAs related to disease progression. miRNA biomarkers screened in specimens derived from MS lesions are more promising to represent the disease process, inflammatory cell motility, and/or therapeutic responses.
miRNAs Mediate Th17 Cell Differentiation in MS and the EAE Model
As mentioned earlier, various miRNAs are related to the complex biological networks of MS. Investigations of miRNAs have found upregulation of miR-29b (36), miR-141, miR-200a (22), miR-155 (37), miR-223 (38), miR-326 (39), let-7e (40), and miR-448 (41), and significant downregulation of miR-15a/16-1 (42) and miR-15b (43) in CD4+ T cells of MS patients and EAE models. miR-20b (44) was decreased, while miR-21 (45) and miR-590 (46) were increased significantly in Th17 cells compared with Th1, Th2, and inducible Treg cells. In vivo and/or in vitro studies had demonstrated that most of these miRNAs mediated Th17 cell differentiation. The mechanisms of miRNAs in Th17 cell differentiation are shown in Figure 1.
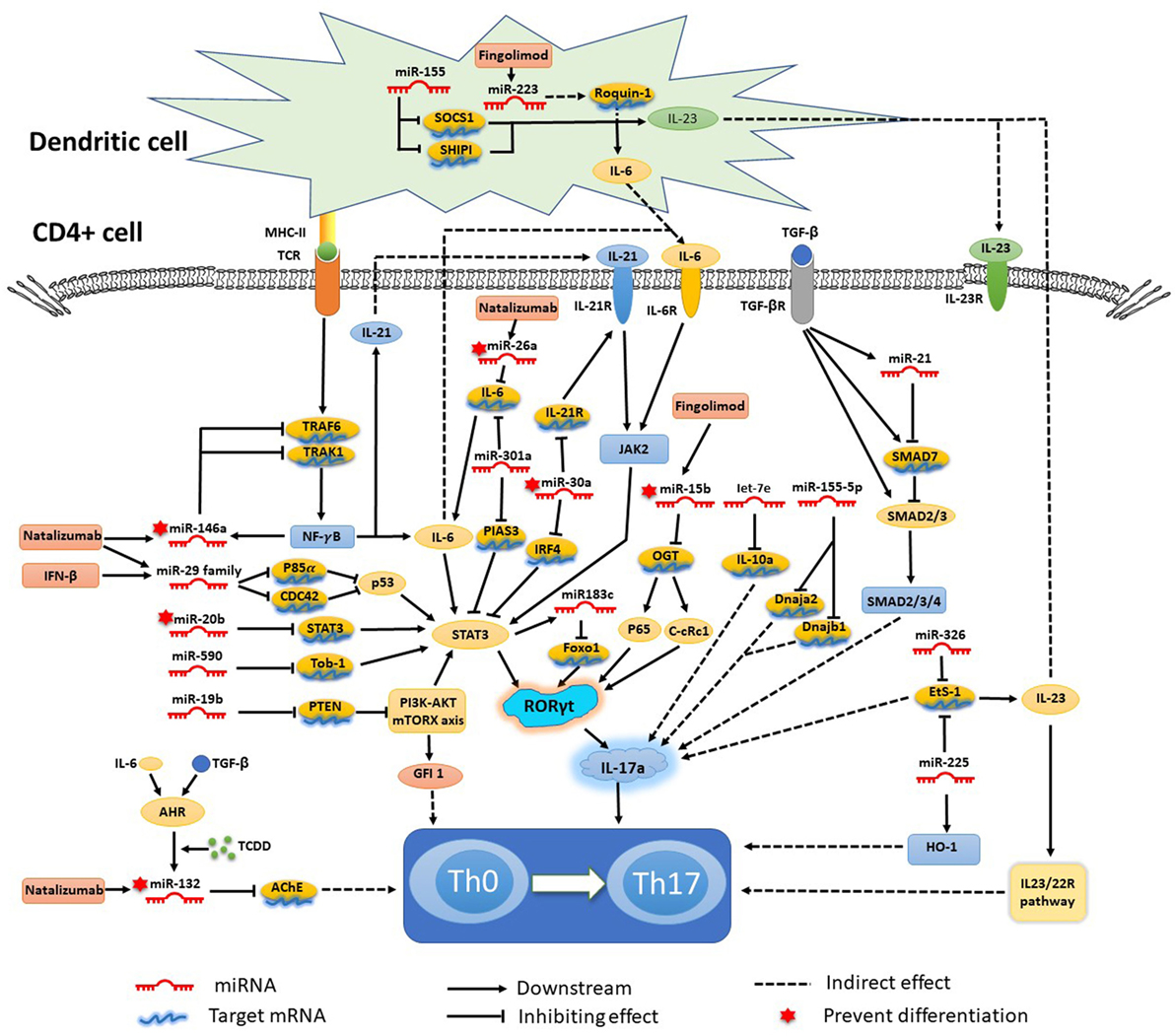
Figure 1. Mechanisms of specific microRNAs (miRNAs) in T helper 17 (Th17) cell differentiation and targets for current treatments. Most miRNAs promote Th17 cell differentiation and exacerbate multiple sclerosis and experimental autoimmune encephalomyelitis, while miRNA-15b, -20b, -26a, -30a, -132, and -146a show negative regulation of Th17 cell differentiation. The signal transducer and activator of transcription (STAT) 3-related orphan receptor (ROR) γt–interleukin (IL)-17 signaling pathway is downstream of most miRNAs.
Promotion in Th17 Cell Differentiation
Silencing or knockdown of miR-326, miR-155 (21), let-7e, and miR-21 attenuate EAE with fewer Th17 cells, while their overexpression leads to more inflammation in the CNS and severe EAE. By contrast, miR-20b shows opposing trends (44). Further study has indicated that miR-326 and miR-155 promote Th17 cell differentiation by translationally inhibiting Ets-1, a negative regulator of Th17 cell differentiation (39, 47). miR-155 and miR-223, which are confirmed to be upregulated in MS and EAE models, simultaneously promote Th17 and Th1 cell differentiation in EAE mice (21) with the requirement of optimal dendritic cell production of cytokines IL-1β, IL-6 and IL-23 (37, 38). miR-155 in Th17 cells can also cause autoimmune inflammation through the clinically relevant IL-23–IL-23R pathway (47). miR-155-3p and miR-155-5p are two key miRNAs produced by the miR-155 host gene. miR-155-3p promotes Th17 cell differentiation and autoimmune demyelination by suppressing two heat shock protein 40 genes, Dnaja2 and Dnajb1 (48). miR-223, a myeloid cell-specific miRNA, is specifically upregulated in spinal cords and lymphoid organs, and deficiency of miRNA-223 reduces Th17 cell infiltration into spinal cords by inhibiting dendritic cell activation (49). IL-10a, as a negative regulator of EAE by suppressing Th17 cells and promoting Th2 cells, is a selectively repressed target of let-7e (50). miR-21 is upregulated in non-pathogenic Th17 cells, which intrinsically promotes non-pathogenic Th17 cell differentiation and autoimmunity by targeting and inhibiting SMAD-7, a negative regulator of TGF-β signaling. Moreover, under-expression of miR-21 in CD4+ T cells leads to decreased SMAD2/3 activation and IL-2 suppression, resulting in reduced sensitivity to the effects of TGF-β in T cells (45). In vivo and in vitro studies of rodent EAE models showed that myelin antigen stimulation results in significant upregulation of miR-301a, miR-21, and miR-155 in CD4+ T cells (51). Using specific miRNA antagonists for in vitro modulation, the study revealed that miR-301a contributes to the development of the pathogenic Th17 subset by targeting PIAS3 mRNA through the IL-6/23–STAT3 pathway (51). miR-183C contains three important miRNAs (miR-183, miR-96, and miR-182), is a Dicer1-regulated miRNA and is significantly expressed in pathogenic Th17 cells. Overexpression of miR-96 specifically promotes pathological cytokine production in pathogenic Th17 cells, such as IL-17A, IL-17F, IL-22, and granulocyte-macrophage colony-stimulating factor, promoting pathological effects of Th17 cells and leading to a higher disease score in EAE models. Upregulation of miR-183C can be induced by IL-6 in Th17-polarized naïve T cells through IL-6-STAT3 signaling and suppressed by TGF-β (52). miR-448-upregulated miRNAs in CD4+ T cells (especially pathogenic Th17 cells) and CSF of MS patients were induced by IL-1β through the NF-κB pathway. The upregulation of miR-448 increases the expression levels of IL-17A and RORγt, favoring pathogenic Th17 cell differentiation through targeting protein tyrosine phosphatase non-receptor type 2 (41), as an anti-inflammatory player with the capacity to restrain the expression of pro-inflammatory mediators (53).
The miR-17-92 cluster, as a CD28 stimulation-dependent factor, is critical for Treg accumulation and functions during an autoimmune-mediated stress response (myelin-induced EAE models) (54). It also promotes Th17 cell differentiation and impairs induction of Treg cell differentiation. Lowing miR-17-92 expression results in anabatic EAE and failure of clinical remission (54). miR-17 and miR-19b are the two core components of the miR-17-92 cluster. miR-19b promotes non-pathogenic Th17 cell differentiation by repressing the expression of phosphatase and tensin homology (PTEN), a negative regulator of the PI3K–AKT–mTOR signaling pathway. miR-17 promotes pathogenic Th17 cell differentiation by inhibiting Ikaros family zinc finger 4 (IKZF4) (55).
Suppression of Th17 Cell Differentiation
miR-30a is downregulated in CD4+ T cells in MS patients and in pathogenic Th17 cells in EAE models. Overexpression of miR-30a inhibits pathogenic Th17 cell differentiation and reduces the severity of EAE by targeting mRNAs of IL-21 receptor and IRF4 (56). miR-30a also inhibits the proliferation and invasion of prostate cancer cells by targeting mRNA of sine oculis homeobox homolog 1 (57). miR-146a has been identified as a critical regulator that reduces inflammatory gene expression (58) with the opposing function to miR-155. Both of them are highly upregulated in human MS lesions. miR-146a controls Th17 cell differentiation by targeting TRAF6 and IRAK1, partially through modulation of the T cell autocrine IL-6/IL-21 pathway (7). Studies have also shown potential involvement of STAT3 and RORγt in miR-20b-induced non-pathogenic Th17 cell suppression in vitro, while the STAT3 gene has been reported as a candidate target of miR-20b (44). The orphan nuclear receptor RORγt has also been described as a key transcription factor that participates in CD4+ T cell differentiation toward the IL-17+ Th17 cell lineage. RORγt is also a potential target of NF-κB, especially c-Rel and p65, which are major factors in Th17 cell differentiation and autoimmune inflammatory disease. miR-15b is a downregulated miRNA in CD4+ T cells of MS patients and EAE models. miR-15b inhibits pathogenic Th17 cell differentiation by targeting its potential target, O-linked N-acetylglucosamine transferase (OGT), and suppresses RORγt through the NF-κB (c-Rel and p65) pathway (43). miR-132, which is a member of the miR-132/212 cluster, is highly expressed in the brain (59). The miR-132/212 cluster is upregulated by aryl hydrocarbon receptor (AHR) activation under Th17 cell-polarizing conditions and affects non-pathogenic Th17 cell differentiation (60). Activation of AHR by 2,3,7,8-tetrachlorodibenzo-pdioxin (TCCD) upregulates the expression of miR-132 in CD4+ cells, resulting in decreased IL-17 and interferon (IFN)-γ expression and suppressed T cell proliferation by targeting acetylcholinesterase (61).
miRNA Responses to Disease-Modifying Treatments
Investigations of miRNA response to clinical disease-modifying treatments are valuable. Several studies have focused on expression changes of miRNAs in MS and verified that miRNA expression correlates with the treatment response in MS.
Interferon-β
Interferon β-1b was the first disease-modifying drug recommended for MS treatment with long-term efficacy and good tolerability. IFN-β suppresses IL-23 production and increases IL-27 and IL-10 production by dendritic cells, lowers the ability to promote IL-17 expression by CD4+ T cells, and downregulates the expression of IL-17 and IL-10 by activated STAT3 and STAT1 (62).
Hecker et al. observed significant expression changes of miRNAs in PBMCs after 1 month of IFNβ-1b treatment. Three miRNAs (miR-29a-3p, miR-29c-3p, and miR-532-5p) were confirmed to be downregulated. In addition, the miR-29 family was associated with upregulated IFN-β-responsive genes (63). miR-29 induces apoptosis in a p53-dependent manner by directly targeting p85α and CDC42 that are negative regulators of p53 (64). p53 functions as an inflammation suppressor and is a crucial negative regulator of Th17 cell differentiation via the STAT3 signaling pathway. Another study also affirmed that the total expression change of miRNAs in PBMCs is markedly elevated after 3 and 6 months of IFN-β therapy compared with pre-treatment levels. miR-26a-5p, which is mainly expressed in neural tissues, has been identified as the most significantly upregulated miRNA in IFN-β-treated RRMS patients. However, the significant expression change was only found in IFNβ-responder RRMS patients after 3 months of treatment. The DLG4 gene is a potential target of miR-26a-5p, which encodes post-synaptic density protein 95 that plays a role in the signaling mechanisms of glutamate receptors (65).
Natalizumab
Natalizumab is a recombinant humanized immunoglobulin that blocks α4-integrin at the surface of activated T lymphocytes and other mononuclear leukocytes, preventing leukocytes from adhering to endothelial cells. It was notable that the frequency of Th17 cells increased in peripheral blood and IL-17 levels increased in serum of MS patients during natalizumab treatment and return to baseline after discontinuing natalizumab (66).
A recent study of miRNAs showed that miR-17 and miR-29 are upregulated in CD4+ T cells during relapse and downregulated after natalizumab treatment (67–69). miR-17 is a regulator of genes involved in T cell activation (25) and promotes Th17 cell differentiation by inhibiting IKZF4 (55). After 6 months of natalizumab therapy, miR-155 and miR-132 were upregulated in MS patients, whereas miR-146a and miR-26a were downregulated. Overexpression of miR-132 decreases IL-17 and IFN-γ expression and suppresses Th17 cell differentiation (61) by targeting acetylcholinesterase (70). Analyses of miRNAs in whole blood of MS patients also revealed significant expression changes. Let-7c and miR-125a-5p were decreased, while miR-642 was increased after 6 and 12 months of natalizumab therapy compared with the baseline. In addition, the first natalizumab infusion was sufficient to trigger the change in expression of these miRNAs. Furthermore, miR-320, miR-320b, and miR-629 were differentially and significantly expressed between progressive multifocal leukoencephalopathy and non-progressive multifocal leukoencephalopathy groups after 12 months of natalizumab therapy (71). In a longitudinal study on RRMS patients, miR-18a, miR-20b, miR-29a, and miR-103 in the blood were proved to be the most strongly upregulated miRNAs by natalizumab (72). EAE in miR-106a-363 (contain miR-20b)-deficient mice had an earlier onset of symptoms and a more severe disease course. Th17-related pro-inflammatory genes RORγt and STAT3 which were predicted as targets of miR-20b, were upregulated in the spinal cord tissue (72).
Fingolimod
Fingolimod is a sphingosine-1-phosphate analog that inhibits the egress of lymphocytes from lymphoid tissues and their recirculation, especially CCR7-expressing CD4+ and CD8+ T cells. Inhibited IL-17 and IFN expression has been found during fingolimod treatment. Moreover, the level of Th17 cells in peripheral blood fell dramatically at 1 month of treatment, but the percentage was increased among CD4+ T cells at 3 months until the end of follow-up (73).
A study of the transcriptome in circulating CD4+ T cells after 3 months of treatment identified 890 genes that were expressed differentially, 12 of which are precursors of mature miRNAs including miR-216b, miR-142, and miR-548c (74). miRNAs (miR-15b, miR-23a, and miR-223) in serum of MS patients showed slight reduction after fingolimod treatment (75), and a significant change was found after 6 months treatment (76). miR-15b suppresses Th17 differentiation by targeting OGT and suppresses RORγt through the NF-κB pathway (43). miR-223, a myeloid cell-specific miRNA, reduces Th17 cell infiltration into spinal cords by inhibiting dendritic cell activation (38, 49).
Hematopoietic Stem Cells Transplantation (HSCT)
In addition to the treatments demonstrated earlier, studies in recent decades found that HSCT could also have beneficial effects on MS. A recent study revealed that miR-16, miR-155, and miR-142-3p which were upregulated in CD4+ and CD8+ T cells, were significantly downregulated after autologous HSCT in MS patients. Meanwhile the expressions of these miRNAs returned to normal levels at 6 months and remained stable to the end of the follow-up (77). miR-16 and miR-142-3p had been reported in regulating Treg cells activity, while miR-155 was a positive Th17 cell differentiation regulator by inhibiting Ets-1, a negative regulator of Th17 cells differentiation (39, 47).
Advances in miRNA Techniques
We acknowledge different research groups may report different expression patterns of each miRNA even in the same disease stage and same tissues, that mainly due to different techniques for miRNA analysis. The RNA-seq and qRT-PCR are the two main methods for the expression pattern analysis for miRNAs. RNA-seq is good for large-scale analysis and occasionally used to identify novel ncRNAs including miRNAs. qRT-PCR was mostly used for the given miRNA analysis, and nowadays, several PCR-array kits were developed for large-scale known miRNA analysis in human, rat or mouse. Besides these two common methods, several new-developed methods were reviewed by Kalogianni et al. (78), such as hybridization chain reaction, target recycling, rolling circle amplification for signal enhancement, target amplification, and several sensing strategies without nucleic acid amplification. Taken together, in addition to the analysis methods, the absence of reproducibility and clear miRNA pattern identified in studies may be owing to differences in MS ethnic populations from different countries, disease status, ages, and genders.
To date, most studies are based on a single cell type, and the regulation of miRNAs among different cell types is still unknown. Recent studies indicated that extracellular vesicles including exosomes, microvesicles, and apoptotic bodies are bioactive vesicles working as miRNA carriers released by many living cells, of which, exosomes are smaller (30–100 nm) and originated from endosomal vesicles through secretion from intracellular luminal space, while microvesicles (100–1,000 nm, from activated or apoptotic eukaryotic cells) and apoptotic bodies (1–5 µm, from late stage of apoptotic cells) are formed by extensive plasma membrane budding. All these extracellular vesicles may serve as novel mediators for intracellular communication (79). Besides theses natural miRNA carriers, several man-made carriers were developed to transfer miRNAs into different cells or tissues to studying the function of miRNAs or for therapeutical uses. Because of the high molecular weight, low stability, negative charge, and high structural stiffness, it is difficult to transport miRNAs into the cytoplasm. Several methods were developed including liposomes, solid lipid nanoparticles, nanostructured lipid carriers, polymer-based nanoparticles, etc. (80). To address the limitations of polymeric and lipid-based nanoparticles, lipid–polymer hybrid nanoparticles have been developed (80). In addition, high-density lipoprotein was also used as miRNA carriers as it was used as endogenous vehicle for the transportation and metabolism of many different bioactive molecules including miRNAs (80). Furthermore, aptamer, peptide, antibody, and folate were used to combine with abovementioned methods for targeting miRNA delivery (80). Another important miRNAs delivering system is viral vectors. Now, there are several virus systems were developed for genes or ncRNAs (including miRNAs) delivery, such as lentivirus, retrovirus, adenovirus, adeno-associated virus, etc. Also, the abovementioned delivery system can be used to target or silence miRNAs expression, which is also very important for targeting endogenous miRNAs and identifying the role of miRNA or for the therapeutic uses. For miRNA silencing, one way is using oligonucleotides delivered by the abovementioned methods to inhibit miRNAs. Several chemical modification, such as antagomirs, 2′-MOEs, LNAs, and 2′-F/MOEs, were used to keep the stability of single-strand miRNA inhibitors (81). In addition, several small molecules were identified to inhibit miRNA function, such as polylysine and trypaflavine, which usually functioned as a inhibitor in miRNA-processing pathway (82). Another way to inhibit miRNA function is using viral vector to express multiple miRNA target mimics, which could bind endogenous miRNAs and left few miRNAs to bind their real targets.
Perspectives and Conclusion
In all, miRNAs have been shown to be key regulators in mediating CD4+ T cell differentiation toward Th17 cells, mostly through the STAT3 signaling pathway. Some miRNAs may be biomarkers and therapeutic targets in the diagnosis/prognosis and treatment of disease activity and progression. Modulating the expression of miRNAs by specific drugs might result in fewer Th17 cells or even inhibition of the functions of pathological Th17 cells, which would be a promising anti-inflammatory treatment for MS. Several methods have been developed to regulate the level of miRNAs in tissues or cells, which hold the opportunity for disease treatment by targeting the dysregulated miRNAs. Further study may focus on miRNAs involved in cell-to-cell communication, and a future challenge will be to characterize such communication between different cell types and the possible regulatory mechanisms of miRNAs in the whole immune system.
Author Contributions
CC reviewed the literature and was mainly responsible for writing the manuscript; YZ, JW, YY, and LP assisted in the conception of this review and figure preparation. WQ is the corresponding author and critically revised the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (grant number 81771300) and the Natural Science Foundation of Guangdong Province, China (grant number 2017A030313853).
References
1. Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, et al. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE). J Autoimmun (2001) 17:17–25. doi:10.1006/jaut.2001.0520
2. Li YF, Zhang SX, Ma XW, Xue YL, Gao C, Li XY. Levels of peripheral Th17 cells and serum Th17-related cytokines in patients with multiple sclerosis: a meta-analysis. Mult Scler Relat Disord (2017) 18:20–5. doi:10.1016/j.msard.2017.09.003
3. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol (2008) 172:146–55. doi:10.2353/ajpath.2008.070690
4. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res (2014) 42:D68–73. doi:10.1093/nar/gkt1181
5. Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, et al. TRBP recruits the Dicer complex to ago2 for microRNA processing and gene silencing. Nature (2005) 436:740–4. doi:10.1038/nature03868
6. Jagot F, Davoust N. Is it worth considering circulating microRNAs in multiple sclerosis? Front Immunol (2016) 7:129. doi:10.3389/fimmu.2016.00129
7. Li B, Wang X, Choi IY, Wang YC, Liu S, Pham AT, et al. miR-146a modulates autoreactive Th17 cell differentiation and regulates organ-specific autoimmunity. J Clin Invest (2017) 127:3702–16. doi:10.1172/JCI94012
8. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi:10.1016/j.immuni.2009.08.001
9. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine (2015) 74:5–17. doi:10.1016/j.cyto.2014.09.011
10. Brucklacher-Waldert V, Stuerner K, Kolster M, Wolthausen J, Tolosa E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain (2009) 132:3329–41. doi:10.1093/brain/awp289
11. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J (2010) 24:1023–34. doi:10.1096/fj.09-141978
12. Durelli L, Conti L, Clerico M, Boselli D, Contessa G, Ripellino P, et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann Neurol (2009) 65:499–509. doi:10.1002/ana.21652
13. Montes M, Zhang X, Berthelot L, Laplaud DA, Brouard S, Jin J, et al. Oligoclonal myelin-reactive T-cell infiltrates derived from multiple sclerosis lesions are enriched in Th17 cells. Clin Immunol (2009) 130:133–44. doi:10.1016/j.clim.2008.08.030
14. Xu X, Shao B, Wang R, Zhou S, Tang Z, Lu W, et al. Role of interleukin-17 in defense against pseudomonas aeruginosa infection in lungs. Int J Clin Exp Med (2014) 7:809–16.
15. Trenova AG, Slavov GS, Manova MG, Draganaova-Filipova MN, Mateva NG, Miteva LD, et al. Alterations in serum levels of IL-17 in contrast to TNF-alpha correspond to disease-modifying treatment in relapsing-remitting multiple sclerosis. Scand J Clin Lab Invest (2017) 77:283–8. doi:10.1080/00365513.2017.1303843
16. McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol (2007) 8:1390–7. doi:10.1038/ni1539
17. Zhao M, Sun D, Guan Y, Wang Z, Sang D, Liu M, et al. Disulfiram and diphenhydramine hydrochloride upregulate miR-30a to suppress IL-17-associated autoimmune inflammation. J Neurosci (2016) 36:9253–66. doi:10.1523/JNEUROSCI.4587-15.2016
18. Mancuso R, Hernis A, Agostini S, Rovaris M, Caputo D, Clerici M. MicroRNA-572 expression in multiple sclerosis patients with different patterns of clinical progression. J Transl Med (2015) 13:148. doi:10.1186/s12967-015-0504-2
19. Siegel SR, Mackenzie J, Chaplin G, Jablonski NG, Griffiths L. Circulating microRNAs involved in multiple sclerosis. Mol Biol Rep (2012) 39:6219–25. doi:10.1007/s11033-011-1441-7
20. Sondergaard HB, Hesse D, Krakauer M, Sorensen PS, Sellebjerg F. Differential microRNA expression in blood in multiple sclerosis. Mult Scler (2013) 19:1849–57. doi:10.1177/1352458513490542
21. Zhang J, Cheng Y, Cui W, Li M, Li B, Guo L. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol (2014) 266:56–63. doi:10.1016/j.jneuroim.2013.09.019
22. Naghavian R, Ghaedi K, Kiani-Esfahani A, Ganjalikhani-Hakemi M, Etemadifar M, Nasr-Esfahani MH. miR-141 and miR-200a, revelation of new possible players in modulation of Th17/Treg differentiation and pathogenesis of multiple sclerosis. PLoS One (2015) 10:e124555. doi:10.1371/journal.pone.0124555
23. Zhang J, Vandevenne P, Hamdi H, Van Puyvelde M, Zucchi A, Bettonville M, et al. Micro-RNA-155-mediated control of heme oxygenase 1 (HO-1) is required for restoring adaptively tolerant CD4+ T-cell function in rodents. Eur J Immunol (2015) 45:829–42. doi:10.1002/eji.201445066
24. Keller A, Leidinger P, Steinmeyer F, Stahler C, Franke A, Hemmrich-Stanisak G, et al. Comprehensive analysis of microRNA profiles in multiple sclerosis including next-generation sequencing. Mult Scler (2014) 20:295–303. doi:10.1177/1352458513496343
25. Cox MB, Cairns MJ, Gandhi KS, Carroll AP, Moscovis S, Stewart GJ, et al. MicroRNAs miR-17 and miR-20a inhibit T cell activation genes and are under-expressed in MS whole blood. PLoS One (2010) 5:e12132. doi:10.1371/journal.pone.0012132
26. Jernas M, Malmestrom C, Axelsson M, Nookaew I, Wadenvik H, Lycke J, et al. MicroRNA regulate immune pathways in T-cells in multiple sclerosis (MS). BMC Immunol (2013) 14:32. doi:10.1186/1471-2172-14-32
27. Sanders KA, Benton MC, Lea RA, Maltby VE, Agland S, Griffin N, et al. Next-generation sequencing reveals broad down-regulation of microRNAs in secondary progressive multiple sclerosis CD4+ T cells. Clin Epigenetics (2016) 8:87. doi:10.1186/s13148-016-0253-y
28. Guerau-de-Arellano M, Smith KM, Godlewski J, Liu Y, Winger R, Lawler SE, et al. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain (2011) 134:3578–89. doi:10.1093/brain/awr262
29. Haghikia A, Haghikia A, Hellwig K, Baraniskin A, Holzmann A, Decard BF, et al. Regulated microRNAs in the CSF of patients with multiple sclerosis: a case-control study. Neurology (2012) 79:2166–70. doi:10.1212/WNL.0b013e3182759621
30. Bergman P, Piket E, Khademi M, James T, Brundin L, Olsson T, et al. Circulating miR-150 in CSF is a novel candidate biomarker for multiple sclerosis. Neurol Neuroimmunol Neuroinflamm (2016) 3:e219. doi:10.1212/NXI.0000000000000219
31. Junker A, Krumbholz M, Eisele S, Mohan H, Augstein F, Bittner R, et al. MicroRNA profiling of multiple sclerosis lesions identifies modulators of the regulatory protein CD47. Brain (2009) 132:3342–52. doi:10.1093/brain/awp300
32. Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol (2001) 11:130–5. doi:10.1016/S0962-8924(00)01906-1
33. Lescher J, Paap F, Schultz V, Redenbach L, Scheidt U, Rosewich H, et al. MicroRNA regulation in experimental autoimmune encephalomyelitis in mice and marmosets resembles regulation in human multiple sclerosis lesions. J Neuroimmunol (2012) 246:27–33. doi:10.1016/j.jneuroim.2012.02.012
34. Talebi F, Ghorbani S, Chan WF, Boghozian R, Masoumi F, Ghasemi S, et al. MicroRNA-142 regulates inflammation and T cell differentiation in an animal model of multiple sclerosis. J Neuroinflammation (2017) 14:55. doi:10.1186/s12974-017-0832-7
35. Ghorbani S, Talebi F, Chan WF, Masoumi F, Vojgani M, Power C, et al. MicroRNA-181 variants regulate T cell phenotype in the context of autoimmune neuroinflammation. Front Immunol (2017) 8:758. doi:10.3389/fimmu.2017.00758
36. Smith KM, Guerau-de-Arellano M, Costinean S, Williams JL, Bottoni A, Mavrikis CG, et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J Immunol (2012) 189:1567–76. doi:10.4049/jimmunol.1103171
37. O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity (2010) 33:607–19. doi:10.1016/j.immuni.2010.09.009
38. Ifergan I, Chen S, Zhang B, Miller SD. Cutting edge: microRNA-223 regulates myeloid dendritic cell-driven Th17 responses in experimental autoimmune encephalomyelitis. J Immunol (2016) 196:1455–9. doi:10.4049/jimmunol.1501965
39. Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, et al. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol (2009) 10:1252–9. doi:10.1038/ni.1798
40. Guan H, Fan D, Mrelashvili D, Hao H, Singh NP, Singh UP, et al. MicroRNA let-7e is associated with the pathogenesis of experimental autoimmune encephalomyelitis. Eur J Immunol (2013) 43:104–14. doi:10.1002/eji.201242702
41. Wu R, He Q, Chen H, Xu M, Zhao N, Xiao Y, et al. MicroRNA-448 promotes multiple sclerosis development through induction of Th17 response through targeting protein tyrosine phosphatase non-receptor type 2 (PTPN2). Biochem Biophys Res Commun (2017) 486:759–66. doi:10.1016/j.bbrc.2017.03.115
42. Lorenzi JC, Brum DG, Zanette DL, de Paula ASA, Barbuzano FG, Dos SA, et al. miR-15a and 16-1 are downregulated in CD4+ T cells of multiple sclerosis relapsing patients. Int J Neurosci (2012) 122:466–71. doi:10.3109/00207454.2012.678444
43. Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao J, et al. MicroRNA-15b suppresses Th17 differentiation and is associated with pathogenesis of multiple sclerosis by targeting O-GlcNAc transferase. J Immunol (2017) 198:2626–39. doi:10.4049/jimmunol.1601727
44. Zhu E, Wang X, Zheng B, Wang Q, Hao J, Chen S, et al. miR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORgammat and STAT3. J Immunol (2014) 192:5599–609. doi:10.4049/jimmunol.1303488
45. Murugaiyan G, Da CA, Ajay AK, Joller N, Garo LP, Kumaradevan S, et al. MicroRNA-21 promotes Th17 differentiation and mediates experimental autoimmune encephalomyelitis. J Clin Invest (2015) 125:1069–80. doi:10.1172/JCI74347
46. Liu Q, Gao Q, Zhang Y, Li Z, Mei X. MicroRNA-590 promotes pathogenic Th17 cell differentiation through targeting Tob1 and is associated with multiple sclerosis. Biochem Biophys Res Commun (2017) 493:901–8. doi:10.1016/j.bbrc.2017.09.123
47. Hu R, Huffaker TB, Kagele DA, Runtsch MC, Bake E, Chaudhuri AA, et al. MicroRNA-155 confers encephalogenic potential to Th17 cells by promoting effector gene expression. J Immunol (2013) 190:5972–80. doi:10.4049/jimmunol.1300351
48. Mycko MP, Cichalewska M, Cwiklinska H, Selmaj KW. miR-155-3p drives the development of autoimmune demyelination by regulation of heat shock protein 40. J Neurosci (2015) 35:16504–15. doi:10.1523/JNEUROSCI.2830-15.2015
49. Satoorian T, Li B, Tang X, Xiao J, Xing W, Shi W, et al. MicroRNA223 promotes pathogenic T-cell development and autoimmune inflammation in central nervous system in mice. Immunology (2016) 148:326–38. doi:10.1111/imm.12611
50. Dai H, Ciric B, Zhang GX, Rostami A. Interleukin-10 plays a crucial role in suppression of experimental autoimmune encephalomyelitis by Bowman-Birk inhibitor. J Neuroimmunol (2012) 245:1–7. doi:10.1016/j.jneuroim.2012.01.005
51. Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci U S A (2012) 109:E1248–57. doi:10.1073/pnas.1114325109
52. Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, et al. The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression. Immunity (2016) 44:1284–98. doi:10.1016/j.immuni.2016.05.015
53. Scharl M, McCole DF, Weber A, Vavricka SR, Frei P, Kellermeier S, et al. Protein tyrosine phosphatase N2 regulates TNFalpha-induced signalling and cytokine secretion in human intestinal epithelial cells. Gut (2011) 60:189–97. doi:10.1136/gut.2010.216606
54. de Kouchkovsky D, Esensten JH, Rosenthal WL, Morar MM, Bluestone JA, Jeker LT. MicroRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J Immunol (2013) 191:1594–605. doi:10.4049/jimmunol.1203567
55. Liu SQ, Jiang S, Li C, Zhang B, Li QJ. miR-17-92 cluster targets phosphatase and tensin homology and Ikaros family zinc finger 4 to promote TH17-mediated inflammation. J Biol Chem (2014) 289:12446–56. doi:10.1074/jbc.M114.550723
56. Qu X, Zhou J, Wang T, Han J, Ma L, Yu H, et al. miR-30a inhibits Th17 differentiation and demyelination of EAE mice by targeting the IL-21R. Brain Behav Immun (2016) 57:193–9. doi:10.1016/j.bbi.2016.03.016
57. Zhu Q, Li H, Li Y, Jiang L. MicroRNA-30a functions as tumor suppressor and inhibits the proliferation and invasion of prostate cancer cells by down-regulation of SIX1. Hum Cell (2017) 30:290–9. doi:10.1007/s13577-017-0170-1
58. Saba R, Sorensen DL, Booth SA. MicroRNA-146a: a dominant, negative regulator of the innate immune response. Front Immunol (2014) 5:578. doi:10.3389/fimmu.2014.00578
59. Remenyi J, Hunter CJ, Cole C, Ando H, Impey S, Monk CE, et al. Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem J (2010) 428:281–91. doi:10.1042/BJ20100024
60. Nakahama T, Hanieh H, Nguyen NT, Chinen I, Ripley B, Millrine D, et al. Aryl hydrocarbon receptor-mediated induction of the microRNA-132/212 cluster promotes interleukin-17-producing T-helper cell differentiation. Proc Natl Acad Sci U S A (2013) 110:11964–9. doi:10.1073/pnas.1311087110
61. Hanieh H, Alzahrani A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: a new Ahr-based exploration. Eur J Immunol (2013) 43:2771–82. doi:10.1002/eji.201343486
62. Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic-Plese S. IFN-beta inhibits human Th17 cell differentiation. J Immunol (2009) 183:5418–27. doi:10.4049/jimmunol.0803227
63. Hecker M, Thamilarasan M, Koczan D, Schroder I, Flechtner K, Freiesleben S, et al. MicroRNA expression changes during interferon-beta treatment in the peripheral blood of multiple sclerosis patients. Int J Mol Sci (2013) 14:16087–110. doi:10.3390/ijms140816087
64. Park SY, Lee JH, Ha M, Nam JW, Kim VN. miR-29 miRNAs activate p53 by targeting p85 alpha and CDC42. Nat Struct Mol Biol (2009) 16:23–9. doi:10.1038/nsmb.1533
65. Cheng MC, Lu CL, Luu SU, Tsai HM, Hsu SH, Chen TT, et al. Genetic and functional analysis of the DLG4 gene encoding the post-synaptic density protein 95 in schizophrenia. PLoS One (2010) 5:e15107. doi:10.1371/journal.pone.0015107
66. Haas J, Schneider K, Schwarz A, Korporal-Kuhnke M, Faller S, von Glehn F, et al. Th17 cells: a prognostic marker for MS rebound after natalizumab cessation? Mult Scler (2017) 23:114–8. doi:10.1177/1352458516640609
67. Lindberg RL, Hoffmann F, Mehling M, Kuhle J, Kappos L. Altered expression of miR-17-5p in CD4+ lymphocytes of relapsing-remitting multiple sclerosis patients. Eur J Immunol (2010) 40:888–98. doi:10.1002/eji.200940032
68. Meira M, Sievers C, Hoffmann F, Derfuss T, Kuhle J, Kappos L, et al. miR-126: a novel route for natalizumab action? Mult Scler (2014) 20:1363–70. doi:10.1177/1352458514524998
69. Meira M, Sievers C, Hoffmann F, Rasenack M, Kuhle J, Derfuss T, et al. Unraveling natalizumab effects on deregulated miR-17 expression in CD4+ T cells of patients with relapsing-remitting multiple sclerosis. J Immunol Res (2014) 2014:897249. doi:10.1155/2014/897249
70. Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, et al. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity (2009) 31:965–73. doi:10.1016/j.immuni.2009.09.019
71. Munoz-Culla M, Irizar H, Castillo-Trivino T, Saenz-Cuesta M, Sepulveda L, Lopetegi I, et al. Blood miRNA expression pattern is a possible risk marker for natalizumab-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler (2014) 20:1851–9. doi:10.1177/1352458514534513
72. Ingwersen J, Menge T, Wingerath B, Kaya D, Graf J, Prozorovski T, et al. Natalizumab restores aberrant miRNA expression profile in multiple sclerosis and reveals a critical role for miR-20b. Ann Clin Transl Neurol (2015) 2:43–55. doi:10.1002/acn3.152
73. Teniente-Serra A, Hervas JV, Quirant-Sanchez B, Mansilla MJ, Grau-Lopez L, Ramo-Tello C, et al. Baseline differences in minor lymphocyte subpopulations may predict response to fingolimod in relapsing-remitting multiple sclerosis patients. CNS Neurosci Ther (2016) 22:584–92. doi:10.1111/cns.12548
74. Friess J, Hecker M, Roch L, Koczan D, Fitzner B, Angerer IC, et al. Fingolimod alters the transcriptome profile of circulating CD4+ cells in multiple sclerosis. Sci Rep (2017) 7:42087. doi:10.1038/srep42087
75. Fenoglio C, Ridolfi E, Cantoni C, De Riz M, Bonsi R, Serpente M, et al. Decreased circulating miRNA levels in patients with primary progressive multiple sclerosis. Mult Scler (2013) 19:1938–42. doi:10.1177/1352458513485654
76. Fenoglio C, De Riz M, Pietroboni AM, Calvi A, Serpente M, Cioffi SM, et al. Effect of fingolimod treatment on circulating miR-15b, miR23a and miR-223 levels in patients with multiple sclerosis. J Neuroimmunol (2016) 299:81–3. doi:10.1016/j.jneuroim.2016.08.017
77. Arruda LC, Lorenzi JC, Sousa AP, Zanette DL, Palma PV, Panepucci RA, et al. Autologous hematopoietic SCT normalizes miR-16, -155 and -142-3p expression in multiple sclerosis patients. Bone Marrow Transplant (2015) 50:380–9. doi:10.1038/bmt.2014.277
78. Kalogianni DP, Kalligosfyri PM, Kyriakou IK, Christopoulos TK. Advances in microRNA analysis. Anal Bioanal Chem (2018) 410:695–713. doi:10.1007/s00216-017-0632-z
79. Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci (2000) 113(Pt 19):3365–74.
80. Chen X, Mangala LS, Rodriguez-Aguayo C, Kong X, Lopez-Berestein G, Sood AK. RNA interference-based therapy and its delivery systems. Cancer Metastasis Rev (2018) 37:107–24. doi:10.1007/s10555-017-9717-6
81. Krutzfeldt J. Strategies to use microRNAs as therapeutic targets. Best Pract Res Clin Endocrinol Metab (2016) 30:551–61. doi:10.1016/j.beem.2016.07.004
82. Watashi K, Yeung ML, Starost MF, Hosmane RS, Jeang KT. Identification of small molecules that suppress microRNA function and reverse tumorigenesis. J Biol Chem (2010) 285:24707–16. doi:10.1074/jbc.M109.062976
Keywords: microRNA, T helper 17 cells, multiple sclerosis, treatment, experimental autoimmune encephalomyelitis
Citation: Chen C, Zhou Y, Wang J, Yan Y, Peng L and Qiu W (2018) Dysregulated MicroRNA Involvement in Multiple Sclerosis by Induction of T Helper 17 Cell Differentiation. Front. Immunol. 9:1256. doi: 10.3389/fimmu.2018.01256
Received: 16 February 2018; Accepted: 18 May 2018;
Published: 04 June 2018
Edited by:
Amy Lovett-Racke, The Ohio State University, United StatesReviewed by:
Takashi Yamamura, National Center of Neurology and Psychiatry, JapanVincent Van Pesch, Université Catholique de Louvain, Belgium
Copyright: © 2018 Chen, Zhou, Wang, Yan, Peng and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Qiu, cWl1d2VpMTIwQHZpcC4xNjMuY29t