 Xueyi Li1
Xueyi Li1 Yanfang Yu1,2
Yanfang Yu1,2 Boris Gorshkov1Stephen Haigh1Zsuzsanna Bordan1
Boris Gorshkov1Stephen Haigh1Zsuzsanna Bordan1 Daniel Weintraub1Radu Daniel Rudic3
Daniel Weintraub1Radu Daniel Rudic3 Trinad Chakraborty4
Trinad Chakraborty4 Scott A. Barman3Alexander D. Verin1Yunchao Su3
Scott A. Barman3Alexander D. Verin1Yunchao Su3 Rudolf Lucas1,3David W. Stepp3
Rudolf Lucas1,3David W. Stepp3 Feng Chen1,2*
Feng Chen1,2* David J. R. Fulton1,3*
David J. R. Fulton1,3*
- 1Vascular Biology Center, Medical College of Georgia at Augusta University, Augusta, Georgia
- 2Department of Forensic Medicine, Nanjing Medical University, Nanjing, Jiangsu, China
- 3Department of Pharmacology and Toxicology, Medical College of Georgia at Augusta University, Augusta, GA, United States
- 4Institute for Medical Microbiology, Justus-Liebig University Giessen, Giessen, Germany
Pneumonia is a leading cause of death in children and the elderly worldwide, accounting for 15% of all deaths of children under 5 years old. Streptococcus pneumoniae is a common and aggressive cause of pneumonia and can also contribute to meningitis and sepsis. Despite the widespread use of antibiotics, mortality rates for pneumonia remain unacceptably high in part due to the release of bacterial toxins. Pneumolysin (PLY) is a cholesterol-dependent toxin that is produced by Streptococcus, and it is both necessary and sufficient for the development of the extensive pulmonary permeability edema that underlies acute lung injury. The mechanisms by which PLY disrupts the pulmonary endothelial barrier are not fully understood. Previously, we found that reactive oxygen species (ROS) contribute to the barrier destructive effects of PLY and identified an unexpected but potent role of Hsp70 in suppressing ROS production. The ability of Hsp70 to influence PLY-induced barrier dysfunction is not yet described, and the goal of the current study was to identify whether Hsp70 upregulation is an effective strategy to protect the lung microvascular endothelial barrier from G+ bacterial toxins. Overexpression of Hsp70 via adenovirus-mediated gene transfer attenuated PLY-induced increases in permeability in human lung microvascular endothelial cells (HLMVEC) with no evidence of cytotoxicity. To adopt a more translational approach, we employed a pharmacological approach using geranylgeranylacetone (GGA) to acutely upregulate endogenous Hsp70 expression. Following acute treatment (6 h) with GGA, HLMVECs exposed to PLY displayed improved cell viability and enhanced endothelial barrier function as measured by both Electric Cell-substrate Impedance Sensing (ECIS) and transwell permeability assays compared to control treated cells. PLY promoted increased mitochondrial ROS, decreased mitochondrial oxygen consumption, and increased caspase 3 cleavage and cell death, which were collectively improved in cells pretreated with GGA. In mice, IP pretreatment with GGA 24 h prior to IT administration of PLY resulted in significantly less Evans Blue Dye extravasation compared to vehicle, indicating preserved endothelial barrier integrity and suggesting that the acute upregulation of Hsp70 may be an effective therapeutic approach in the treatment of lung injury associated with pneumonia.
Introduction
Pneumonia is a pulmonary infection that can affect people of all ages, but is most severe in the elderly, children, and the immunocompromised. Infection and subsequent inflammation compromise the function of lung endothelial and epithelial barriers resulting in alveolar flooding, impaired gas exchange, and eventually lung consolidation, which collectively underlie the development of acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS) (1). Multiple pathogens can promote pneumonia, including bacteria, fungi, and viruses. Bacteria, in particular Gram positive (G+) bacteria, are the most common cause (2, 3). Currently there are no effective pharmacological approaches for ALI/ARDS and patients treated with anti-inflammatory steroids treatment remain at significant risk of mortality (4).
A hallmark feature of ALI/ARDS is dysfunction of the pulmonary microvascular endothelial barrier resulting in an imbalance of Starling’s forces and the passage of excess fluid into the alveoli (1). The mechanisms underlying the loss of the microvascular barrier function are complicated and involve diverse mechanisms in multiple cell types which can vary depending on the causative agent. A major cause of ALI is G+ bacteria including Streptococcus pneumoniae which accounts for up to half of all community-acquired pneumonia (CAP) cases in the US and CAP is the most frequent cause of ARDS (3). Greater than 500,000 yearly cases of pneumonia and 25,000 pneumococcal-related deaths are reported in the US alone, resulting in a health-care burden that exceeds $5 billion dollars (5). The first-line treatment for Streptococcus pneumonia is antibiotic therapy. However, the onset of ARDS is resistant to antibiotics and paradoxically, bacteriolytic antibiotics can exacerbate lung injury (6). One likely reason for this is the G+ toxin, pneumolysin (PLY), which is produced in Streptococcus and released by autolysis and in greater amounts in the presence of antibiotics that compromise the bacterial cell wall (7). PLY is a 53-kDa intracellular protein which belongs to the cholesterol-dependent cytolysin family (8). Upon binding to cholesterol molecules on the cell membrane of target cells, PLY induces the macromolecular assembly of ring shaped pores that promote calcium influx and alter intracellular signaling (9, 10). Subsequent to these changes, G+-toxins robustly increase the intracellular production of reactive oxygen species (ROS) (6, 7, 11–13). Elevated ROS have been shown to have important roles in regulating a number of physiological and pathophysiological events, including cell apoptosis, survival, proliferation and migration, cell metabolism, DNA damage, inflammation, and disruption of the endothelial barrier (14). The major sources of ROS in endothelial cells are the NADPH oxidases (NOX enzymes), uncoupled eNOS, and the mitochondria. G+-toxins have been reported to activate PKC and alter eNOS fidelity to disrupt the balance of nitric oxide and superoxide (13), activate NADPH oxidase (7), and increase mitochondria-derived ROS (mtROS) (15). There are also significant interactions between these ROS generating systems, where mitochondrial ROS can activate NOX enzymes and vice versa and increased ROS can lead to eNOS uncoupling and ROS production. Mitochondrial DNA (mtDNA) is highly sensitive to ROS and loss of mtDNA integrity can result in mitochondrial dysfunction, ATP deprivation, and cell apoptosis (16–19). NADPH-derived ROS have been shown to promote oxidative damage of mitochondrial proteins in particularly, complex I and complex II, which results in increased mitoROS production (20). Increased mitoROS can activate NADPH oxidase promoting a feed-forward relationship (20, 21). NOX2 has been identified as a potential target for mitochondrial superoxide production in endothelial cells (21, 22) and increased mtROS, secondary to a partial deficiency of mitochondrial superoxide dismutase, can trigger a cytosolic oxidative burst (21). On the other hand, inhibitors that reduce mtROS can also attenuate cytosolic ROS (21–23). ROS can deplete BH4 and alter the S-glutathionylation of eNOS, compromising NO formation and increasing ROS production (24, 25).
Mitochondria are increasingly recognized for their contributions to inflammation, and mtROS is a key factor mediating this process in endothelial cells in response to both physiological and pathophysiological stressors (16, 19). Along with the activation of NOX enzymes, mtROS also promote the activation of endothelial cells and increase proinflammatory cytokines (26) in a manner synergistic with cytosolic ROS. Antioxidants targeted to the mitochondria can reduce endothelial inflammation in hypertension animal models (27). The inflammatory process during pneumonia is complex, and different in the young versus elderly patients. In aged patients, inflammation may initiate at slowly in the early phase compared to young patients, but is more robust and enduring in the later stages (28). This deregulated inflammatory response is a risk factor for death in elderly patients, and broad anti-inflammatory strategies can result in compromised elimination of pathogens.
Heat-shock proteins (Hsp) are intracellular “chaperones” that guide the behavior (folding, function, and fate) of newly synthesized proteins and also associate stably with many signaling molecules (29). They are critical to the maintenance of cellular homeostasis under both physiological and stressed conditions (30). The major chaperones are Hsp90 and Hsp70 which together with co-chaperones mediate protein folding, complex assembly, intracellular transport, and also degradation (31, 32). Previously, we and others have shown that Hsp90 binds specifically to Nox proteins to regulate enzyme stability and ROS production (33, 34). We also found that Hsp90 inhibitors can robustly upregulate Hsp70 expression and that Hsp70 alone can potently suppress NOX-derived ROS production in human pulmonary arterial endothelial cells (35).
Hsp70 has also been shown to be important for cell survival (36) and regulating mitoROS and mtDNA integrity (37), but whether it can provide protection from PLY-induced endothelial injury and loss of endothelial barrier function by suppressing ROS is not yet known. Therefore, the goals of this study were to assess the importance of Hsp70 in protecting the pulmonary microvascular endothelium from PLY, to identify the underlying mechanisms, and to advance the possibility of targeting Hsp70 as a promising therapy for Streptococcus-induced pneumonia.
Materials and Methods
Cells and Reagents
Human lung microvascular endothelial cells (HLMVECs) were obtained from Lonza. Cells were cultured with 5% CO2 at 37°C using EBM-2 MV medium supplemented with EGM-2 MV that was purchased from Lonza. All of the in vitro experiments were performed using passage 3 to passage 5 HLMVECs. PLY was a gift from Dr. Trinad Chakraborty (Institute for Medical Microbiology, Justus-Liebig University, Giessen, Germany) PLY was purified from a recombinant Listeria innocua 6a strain expressing LPS-free PLY. Geranylgeranylacetone (GGA, Sigma) was prepared in DMSO. Tempol, LPS, Glucose, pyruvate, and l-glutamine were obtained from Millipore Sigma. Oligomycin, FCCP, and antimycin were provided in the Seahorse XF Cell Mito Stress Test Kit from Agilent. FITC dextran (Fluorescein isothiocyanate–dextran 4000 and Fluorescein isothiocyanate–dextran 70000) were obtained from Sigma. Antibodies for western blotting included Hsp70 from BD Bioscience, cleaved caspase 3, NF-κB, and GAPDH were from Cell Signaling and Hsp90 from BD Bioscience. Nox1 antibody from Sigma. GFP and Hsp70 adenoviruses were generated in house using established methodologies (38, 39).
Animals
8- to 10-weeks-old male C57BL6 mice, weighing 19–21 g were obtained from Harlan and were kept at the animal facilities at Augusta University. All animal studies conformed to National Institutes of Health guidelines. The experimental procedure was approved by the Augusta University Institutional Animal Care and Use Committee.
Assessment of Pulmonary Vascular Barrier Function In Vivo
Mice were pretreated with 500 mg/kg GGA (in ethanol) administered IP 24 h prior to toxin instillation. Mice were anesthetized with IP ketamine (150 mg/kg) and acetylpromazine (15 mg/kg), the trachea was exposed and PLY (60 ng) instilled IT for 6 h via a 20-gauge catheter. Evans blue dye (EBD)/albumin mixture (30 mg/kg in saline; 0.5% EBD conjugated to 4% BSA, Fraction V; Sigma-Aldrich, St. Louis, MO, USA) was injected into the tail vein, 2 h prior the conclusion of the experiment, in order to assess vascular leak. The lungs were homogenized, incubated with formamide (18 h at + 60°C), and centrifuged at 5,000 × g for 30 min. The optical density of the supernatant was determined spectrophotometrically at 620–750 nm. The concentration of extravasated EBD in the lungs was calculated by using a standard curve (micrograms of EBD per gram of wet lung tissue), as described previously (40).
Western Blotting
Cells were washed three times with HBSS before lysing with Laemmli sample buffer. Lysed cells were briefly sonicated to ensure protein extraction, proteins size fractionated by SDS PAGE and transferred to nitrocellulose membranes. Membranes were incubated overnight at 4°C with antibodies diluted to the manufacturer’s specifications. Secondary IgG antibodies (Invitrogen) conjugated with horseradish peroxidase were used to detect antigen–antibody complexes.
Cell Viability Assay
Cell viability was assessed using the Muse Cell Analyzer (Millipore Sigma). HLMVECs were seeded at a density of 6 × 105 in 6-well plates with complete EBM-2 MV culture medium with or without 30 µM GGA or DMSO for 6 h. PLY was added to dishes 4 h before the viability test. The percentage of live HLMVECs was determined using the Muse cell count and viability kit (Millipore Sigma). In brief, 50 µl of suspended HLMVECs was mixed with 450 µl of count and viability reagent, gently mixed, and injected into the Muse Cell Analyzer. Statistical analysis was performed based on three independent experiments. p Value was compared to the cell treated with GGA plus PLY group.
Endothelial Cell Permeability Assays
Transwell Cell Permeability Assay
Sub confluent HLMVECs were split into 24 transwell inserts with a 0.4-µm pore sized filter. Following seeding, cells were maintained in complete EBM-2 MV medium and then treated for overnight with or without 30 µM GGA. Prior to exposure to PLY, complete medium was removed, and HLMVECs were washed carefully with HBSS three times before transitioning to FITC-dextran containing serum-free medium. Fresh FITC-conjugated dextran was prepared using serum-free medium and 50 ng/ml PLY was added to the endothelial cells. The lower compartment was filled with 1.5 ml of serum-free medium without FITC-dextran. At each time point, 50 µl of lower chamber medium was transferred to a 96-well plate for quantitation. The amount of FITC-dextran permeating through the HLMVEC monolayer into the lower chamber was measured using plate reader with excitation wave length 488 nm and emission wave length 520 nm. Three independent experiments were performed and the data are shown as mean ± SEM.
Transendothelial Electrical Resistance Measurements
Transendothelial electrical resistance was measured in HLMVEC using electric cell-substrate impedance sensing equipment (ECIS). HLMVECs were split into ECIS array chambers (8W10E) at a density of 1,000 cells per well according to the manufacturer’s instructions. On the following day, complete medium was removed, and cells were washed with HBSS for three times. The cells were then cultured in serum-free medium and exposed to PLY to initiate changes in barrier function. Normalized resistance (Ohms) representing HLMVEC barrier integrity was recorded for up to 3 h. Data were aggregated as the mean normalized resistance of eight individual wells.
MitoSOX Assay
Human lung microvascular endothelial cells were transferred into glass-bottomed dishes and cultured in complete EBM-2 MV medium and pretreated with or without 30 µM GGA. On the following day, cells were washed in HBSS, the culture medium changed to serum-free EBM-2. MitoSOX Red and MitoTracker Green were diluted with pre-warmed appropriate medium to a final concentration of 5 µM and 250 nM, respectively. Medium containing MitoSOX Red and MitoTracker Green was applied to the HLMVECs, stimulated with PLY and cells were incubated in 37°C for 15 min prior to observation using a Zeiss 780 inverted confocal microscope. MitoSOX Red mitochondrial superoxide indicator signal was measured at excitation/emission wavelength of 510/580 nm, and MitoTracker Green was detected using an excitation/emission wavelength of 488/516 nm. Wells without dyes were tested for MitoSOX Red and MitoTracker Green signals, respectively, as background signals. Nuclear blue was used to stain the nucleus. Equivalent experiments were performed using a fluorescent plate reader (POLARstar OMEGA) for quantification. After subtracting the background from the signal recorded in the presence of each dye, the ratio of MitoSOX Red signal over MitoTracker Green was used to determine the amount of mitochondrial ROS. The data are presented as fold change over control HLMVEC cells.
Mitochondrial Stress Assay
Mitochondrial stress in HLMVEC was assessed using the Seahorse XF96 analyzer. Low passage number HLMVECs were cultured in the XF96 well plate at a density of 7.0 × 103 cell per well in complete EBM-2 MV medium with or without GGA (30 µM) overnight. The sensor cartridge of a XF96 seahorse plate was hydrated overnight at 37°C in a non CO2 incubator. The following day, fresh seahorse assay medium was prepared with the addition of glucose (10 mM), pyruvate (1.0 mM), and l-glutamine (2 mM). The pH of the medium was adjusted to 7.4 using NaOH. Cell confluency was assessed under light microscopy, and the plate was incubated in a non CO2 incubator for 1 h. After calibration of the sensor cartridge, the XF96 plate was placed into the seahorse instrument and OCR measured as pmole per minute per cell. Analysis of mitochondrial function was made using changes in OCR in the presence of oligomycin (inhibits mitochondrial ATP synthase), and phenylhydrazone (FCCP, mitochondrial membrane potential) which were injected at final concentrations of 1.0 µM each. Lastly, antimycin A was injected at a final concentration of 0.5 µM to inhibit both complex I and III. After each injection, the Seahorse instrument measured OCR for three times at five time points. The average of three measurements was used for data analysis.
Statistical Analysis
Data are presented as means ± SEM. Single comparisons were made using a Student’s t-test and multiple comparisons were made using one way ANOVA with an appropriate post hoc test (Tukey). p < 0.05 was considered a statistically significant difference.
Results
Hsp70 Protects HLMVECs From PLY-Induced Barrier Destruction
Pneumolysin has been well documented to disrupt barrier function in HLMVEC and increase endothelial permeability (12, 13, 41, 42). To determine the effect of Hsp70 on barrier function, HLMVECs were transduced with adenoviruses encoding Hsp70 (pAd Hsp70-GFP, 60MOI) or GFP (Control, 60MOI) overnight and then transferred to an ECIS plate. HLMVECs transduced with the Hsp70 adenovirus were significantly protected from PLY-induced barrier dysfunction as compared to control cells (Figure 1A). The protective effect of Hsp70 on barrier function was then confirmed using a distinct approach, the transwell permeability assay. HLMVECs transduced with pAd Hsp70-GFP or pAd-GFP were seeded into the upper compartments of transwell plates. FITC-dextran (70 kDa) was added to the medium in the upper compartments of test wells, the background wells remained without dextran and all were treated with PLY. FITC-dextran flow through was measured in the lower compartments using a fluorescence capable plate reader (BMG POLARstar Omega). Consistent with the ECIS assay, HLMVEC expressing Hsp70 were protected from PLY-induced barrier destruction (Figure 1B, left panel). Expression of transgenes was confirmed using fluorescent microscopy and western blot (Figure 1B, right panel).
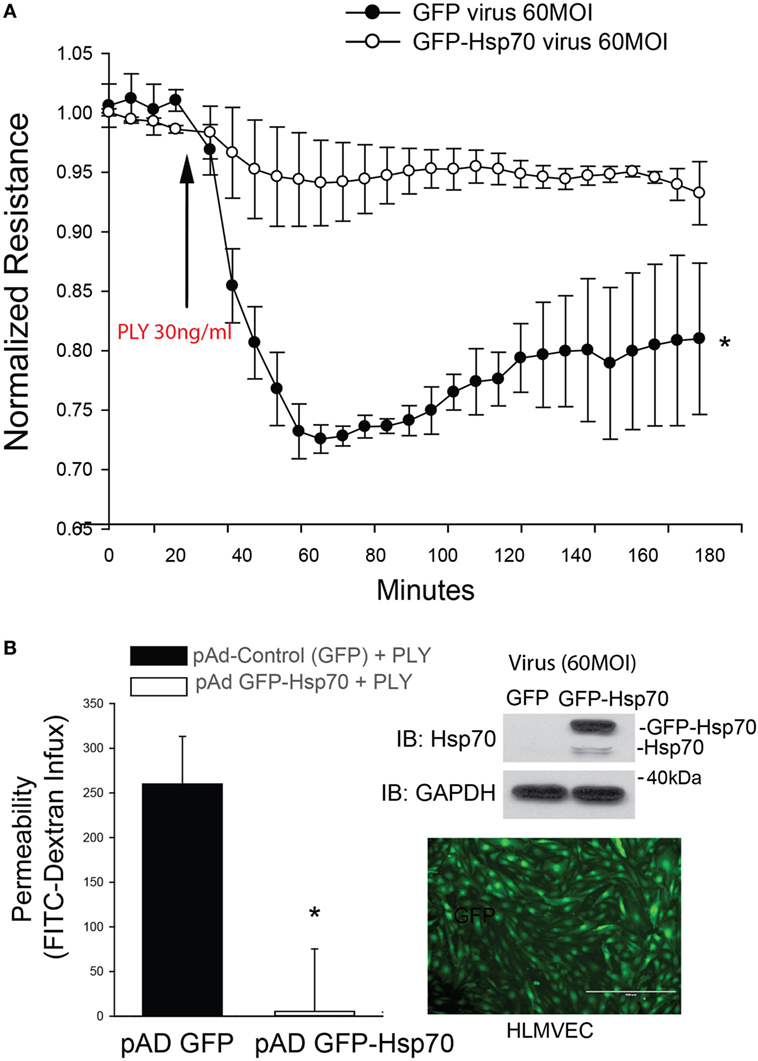
Figure 1. Hsp70 provides robust protection against pneumolysin (PLY)-induced EC barrier disruption. In panel (A) human lung microvascular endothelial cells were transduced with pAD-GFP (Control) or pAD-GFP-Hsp70 at 60MOI and 48 h later, cells were assessed for barrier function using transendothelial resistance as monitored by electric cell-substrate impedance sensing (ECIS) in the presence and absence of PLY (30 ng/ml). In panel (B) (left) HLMEC were grown in transwells and similarly transduced with GFP or GFP-Hsp70 and the flux of FITC-dextran (70 kDa) into the bottom chamber determined using a fluorescent plate reader. On the right (top panel) relative expression of the GFP-Hsp70 transgene relative to endogenous and (bottom panel) image showing the expression pattern of GFP in transduced cells. Data are shown as mean ± SEM (n = 3 wells for each treatment). *p < 0.05 versus control.
The Pharmacological Inducer of Hsp70 Expression, GGA Protects HLMVECs From PLY-Induced Increases in Permeability
Geranylgeranylacetone (GGA) pharmacologically induces the upregulation of Hsp70 via actions on HSF1 to increase HSP70 gene transcription (43). GGA exhibits low cellular toxicity and has been widely used in Japan for antiulcer therapy (44). HLMVECs treated with GGA overnight exhibited a dose-dependent increase in baseline microvascular barrier electrical resistance (Figure 2A). In contrast, PLY induced an acute and pronounced decrease in HLMVEC resistance, reflecting severe barrier disruption. However, in cells cultured with GGA, there was a dose-dependent protective effect from PLY-induced hyperpermeability (Figure 2B). Gram negative (G−) bacterial toxins can also be a significant cause of pneumonia and compromised endothelial barrier function. To assess whether Hsp70 protects against the G− bacterial toxin, LPS, HLMVEC were cultured in ECIS arrays and treated overnight with GGA (30 µM) or vehicle (DMSO). In control cells, exposure to LPS (1 µg/ml) resulted in a more gradual loss of barrier function that peaked at approximately 15 h. In HLMVEC pretreated with GGA, there was a significantly reduced ability of LPS to disrupt endothelial barrier function (Figure 2C).
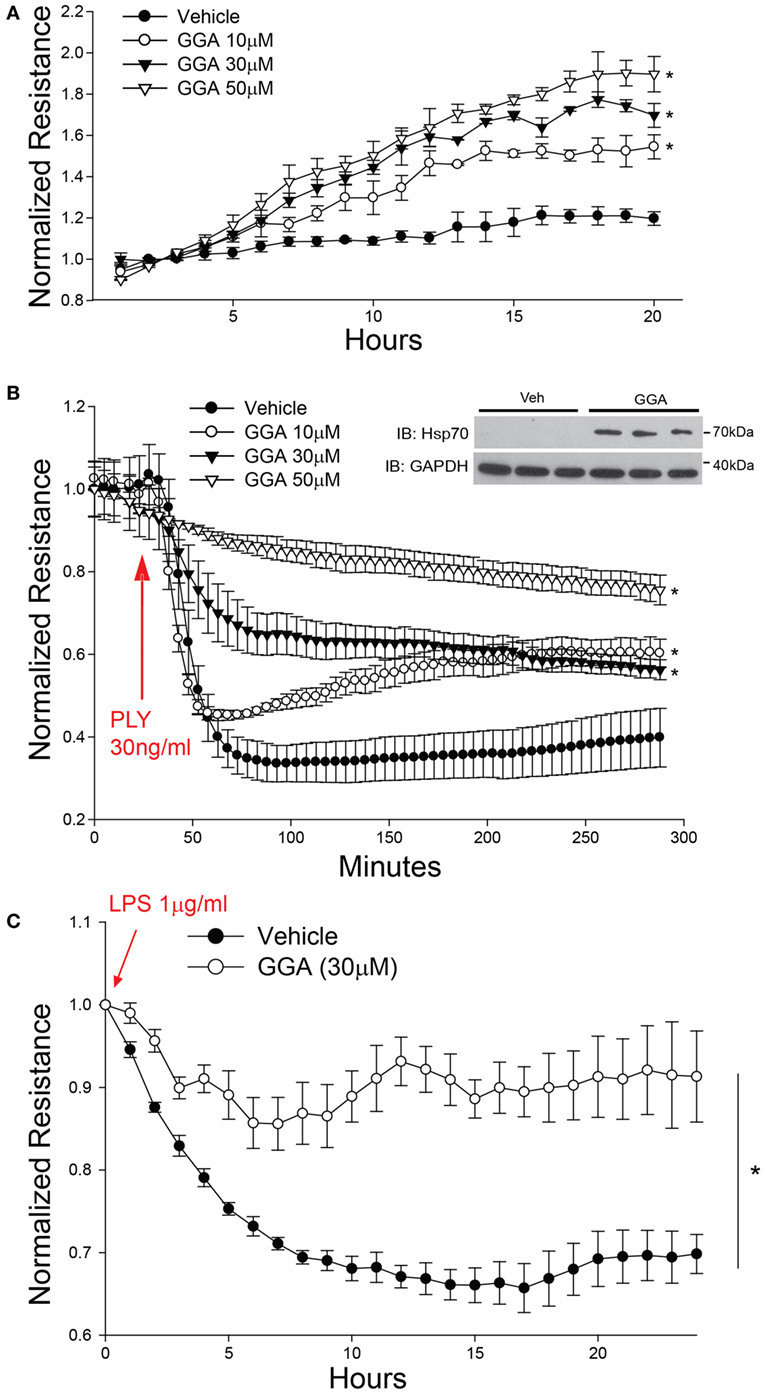
Figure 2. Acute pharmacological upregulation of Hsp70 protects against pneumolysin (PLY) and LPS-induced EC barrier disruption in vitro. In panel (A) human lung microvascular endothelial cells (HLMVEC) were plated in electric cell-substrate impedance sensing (ECIS) arrays, treated with the indicated concentrations of geranylgeranylacetone (GGA) and changes in barrier strength determined by ECIS over time. Data shown as mean ± SED (n = 4 wells for each treatment), *p < 0.05. In panel (B) HLMVEC were treated with the indicated concentrations of GGA overnight and then were treated with or without PLY (30 ng/ml) and barrier function determined by ECIS. Data are shown as mean ± SEM (n = 4 wells for each treatment). *p < 0.05 versus vehicle. In panel (C) HLMVEC were pretreated overnight with GGA (30 µM) and then exposed to LPS (1 µg/ml). Data shown as mean ± SEM (n = 4 wells for each treatment). *p < 0.05 versus vehicle (DMSO).
PLY-induced EC dysfunction has been associated with its ability to upregulate ROS production (13). To determine the importance of ROS in mediating the loss of barrier function, we employed the superoxide inhibitor, tempol (TEM) in transwell assays. Consistent with previous findings, HLMVECs that were pretreated with GGA for overnight showed a robust decrease in dextran permeability in response to PLY (Figures 3A–B). Although transcellular dextran flux cannot be completely excluded, the decreased passage of both the 4- and 70-kDa FITC-dextran into the lower compartment reflects impaired endothelial monolayer integrity. Inhibition of superoxide with tempol also provided acute protection against PLY-induced barrier disruption (Figures 3A–B). GGA induced upregulation of Hsp70 provided more protection than that afforded by TEM.
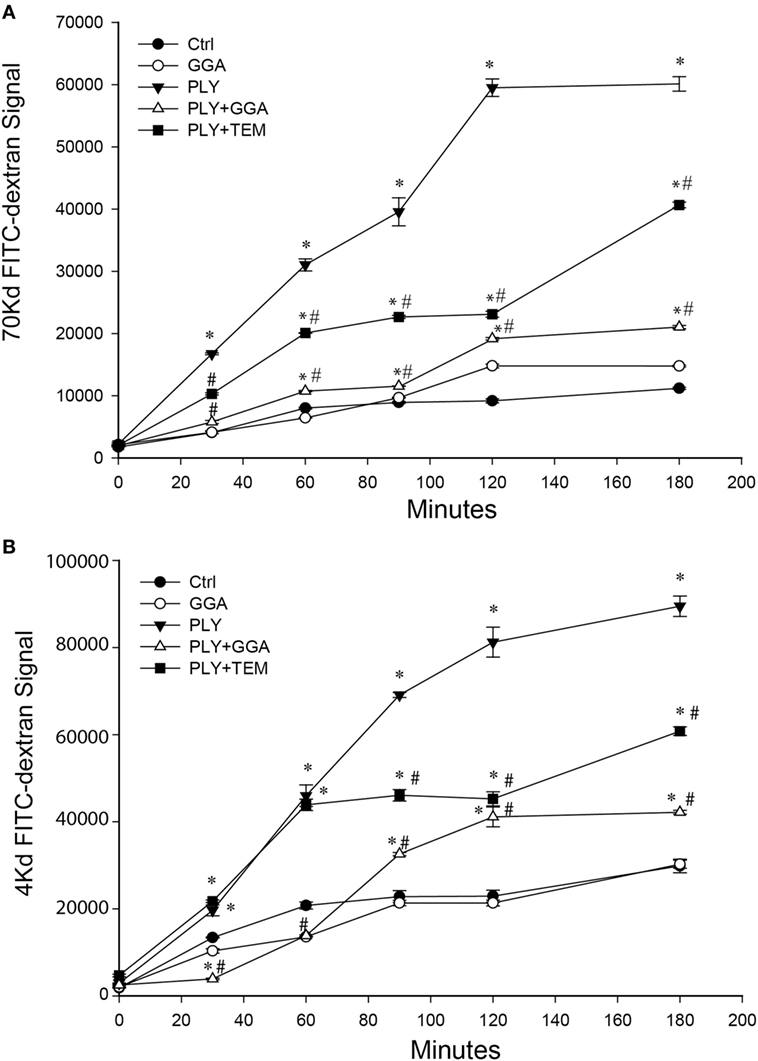
Figure 3. Geranylgeranylacetone (GGA) and TEMPOL protect Human lung microvascular endothelial cell (HLMVEC) from pneumolysin (PLY)-induced barrier disruption. In panel (A) 70-kDa FITC-dextran permeability of HLMVEC pretreated with vehicle, GGA (30 µM) or tempol (TEM, 100 μM) then stimulated with PLY (50 ng/ml). *p < 0.05 versus control, #p < 0.05 versus PLY. Data shown as mean ± SEM (n = 3 wells for each treatment). (B) 4-kDa FITC-dextran permeability of HLMVEC pretreated with GGA (30 µM) or tempol (100 μM) then stimulated with PLY (50 ng/ml). *p < 0.05 versus control, #p < 0.05 versus PLY. Data shown as mean ± SEM (n = 3 wells for each treatment).
GGA Protects HLMVECs From PLY Caused Mitochondrial Damage
Mitochondria are a major source of ROS production in endothelia cells. Given that Hsp70 and superoxide scavengers can protect HLMVECs from PLY-induced increases in permeability, we next investigated whether the barrier protection afforded by Hsp70 might be mediated through changes in mitochondrial ROS production. HLMVECs were plated on glass bottom dishes, pretreated with 30 µM GGA for 24 h and then loaded with MitoSOX Red and MitoTracker Green. Serum-free medium containing 50 ng/ml PLY or vehicle was added prior to measurement of ROS. Using MitoTracker Green to label mitochondria, we were able to identify that mitochondrial numbers were similar among groups, while red florescence, which represented the ROS produced in the mitochondrial compartment, was significantly increased in HLMVEC stimulated with PLY (Figures 4A,B). In cells that were pretreated with GGA, PLY-failed to stimulate mitochondrial ROS production and cells pretreated with Tempol also demonstrated an attenuation of PLY-stimulated mitochondrial ROS production.
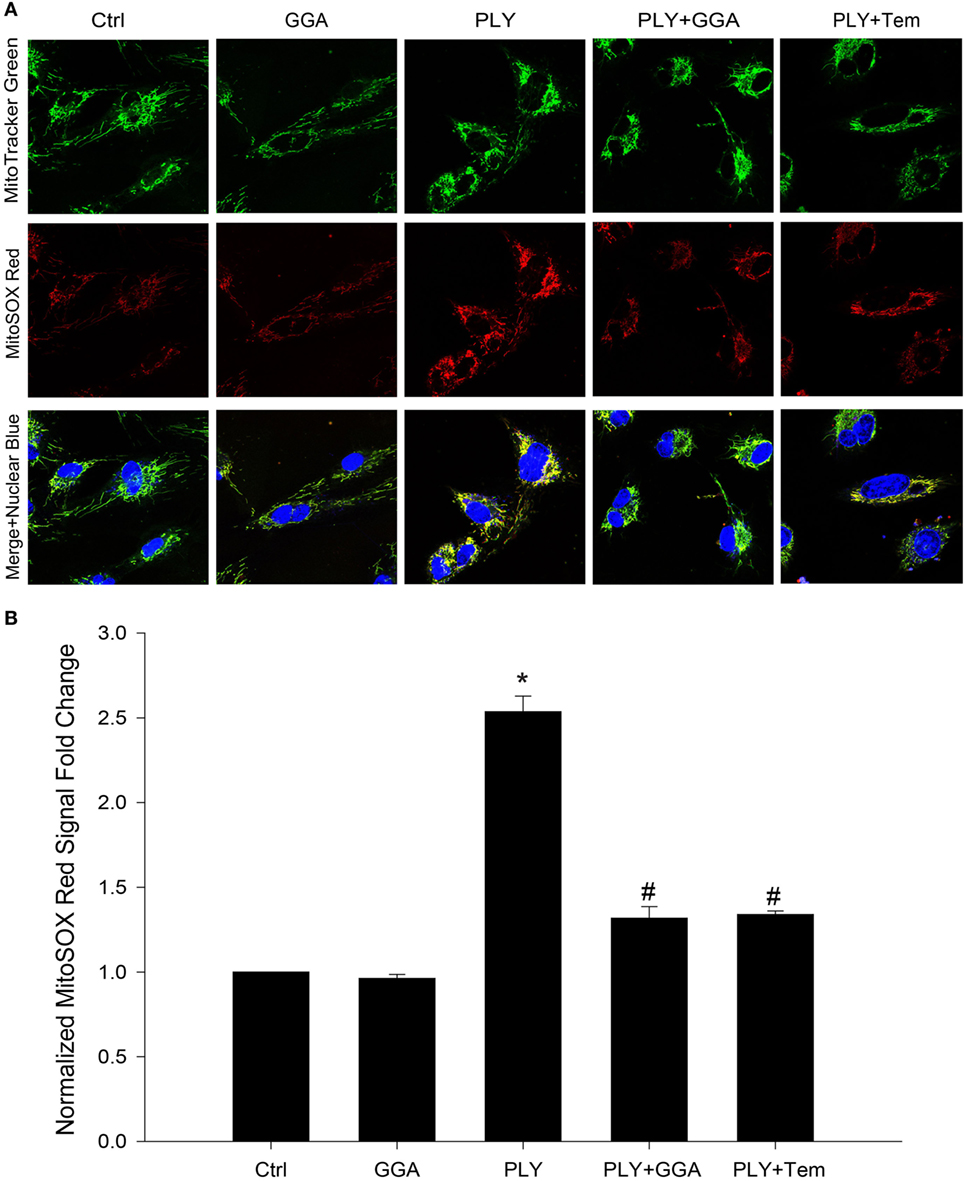
Figure 4. Geranylgeranylacetone (GGA) protects against pneumolysin (PLY)-induced mitochondrial reactive oxygen species (ROS) production. In panel (A) representative confocal images of MitoSox red and MitoTracker green stained human lung microvascular endothelial cell reporting the degree of mitochondria localized ROS. In panel (B) analysis of fluorescent signal from MitoSox red normalized to MitoTracker green (n = 4 wells for each treatment). Data shown as mean ± SEM *p < 0.05 versus control, #p < 0.05 versus PLY.
We next assessed whether PLY alters mitochondrial function using the Seahorse extracellular flux analyzer. HLMVECs were pretreated with or without GGA to induce Hsp70 expression and then challenged with or without PLY. Basal rates of mitochondrial respiration were similar among groups (Figures 5A,B). The addition of oligomycin which inhibits ATP synthase (complex V) reduced oxygen consumption in all groups, but less so in cells that were treated with PLY (Figures 5A,B bottom left). This reflects a diminished ability of cells to use oxidative phosphorylation to generate ATP and the preservation of this ability in cells pretreated with GGA. The addition of the protonophore, FCCP [Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone] collapses the proton gradient, disrupting the mitochondrial membrane potential, and drives maximal oxygen consumption (complex IV). Maximal respiration rates were decreased in HLMVEC exposed to PLY compared to control and significantly preserved in cells pretreated with GGA (Figure 5B, bottom right). Collectively, these data indicate that PLY compromises mitochondrial respiration and that upregulation of Hsp70 protects against the PLY-induced loss of function.
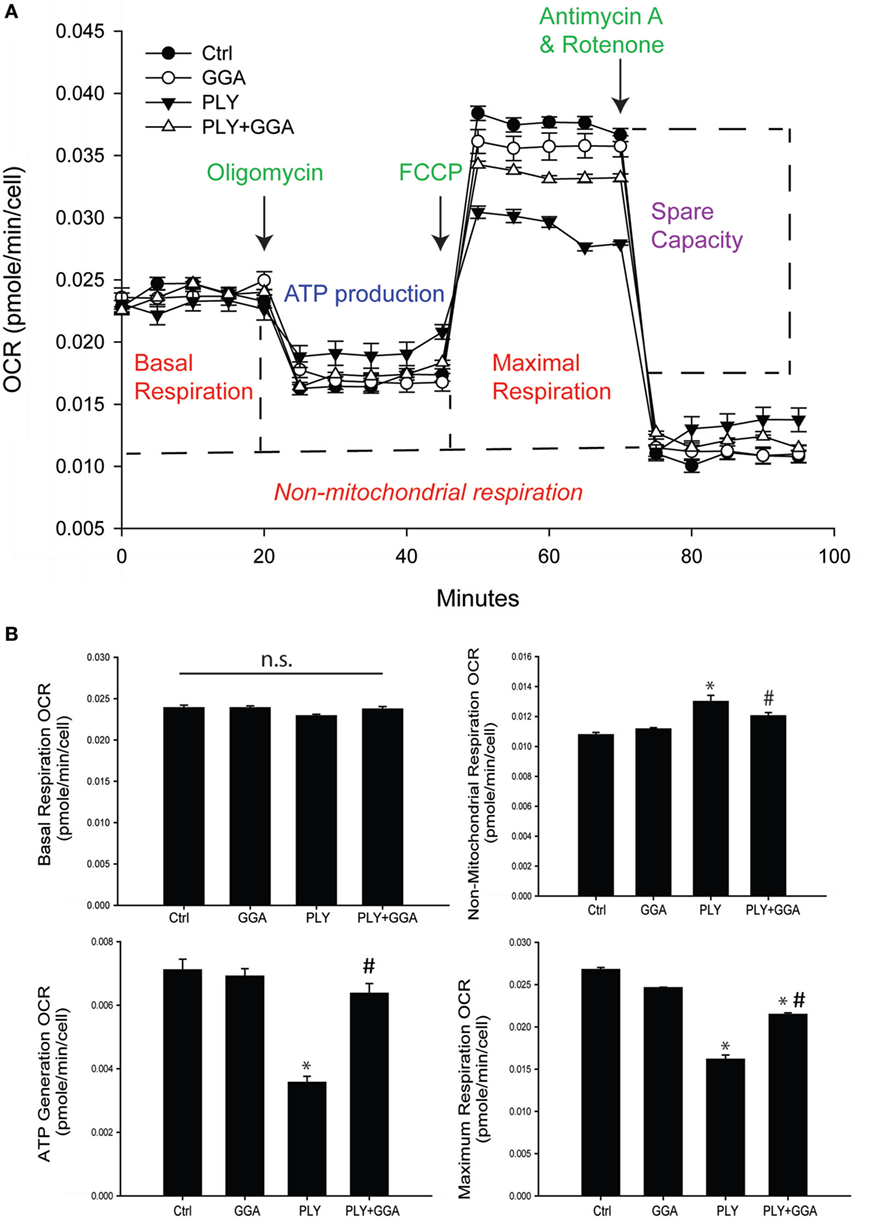
Figure 5. Geranylgeranylacetone (GGA) protects human lung microvascular endothelial cells from pneumolysin (PLY)-induced mitochondrial dysfunction. (A) XF96 seahorse Mito stress assay profile with arrows showing the time of injections of oligomycin, carbonyl cyanide p-trifluoromethoxy-phenylhydrazone (FCCP), and antimycin A. Data are represented as mean ± SEM (n = 5 for each treatment). (B) Mitochondria function data were generated using the XF96 seahorse Mito stress assay. Basal respiration OCR (OCR before adding oligomycin-OCR and after adding antimycin A). Non-mitochondrial respiration OCR (stressed OCR after adding antimycin A). ATP generation OCR (basal respiration OCR after adding oligomycin). Maximum respiration OCR (stressed OCR after adding FCCP-stressed OCR after adding antimycin A). Data are shown as mean ± SEM. *p < 0.05 versus control, #p < 0.05 versus PLY.
GGA Protects HLMVECs From PLY-Induced Cell Death
Hsp70 is an established cytoprotective factor that can reduce cell death (45, 46). We next assessed whether Hsp70 upregulation decreases cell death in response to PLY. HLMVECs were challenged with PLY and cell viability assessed by Muse flow cytometer. PLY (50 ng/ml) resulted in a significant decrease in cell viability (Figure 6A), which was significantly higher in cells pretreated with GGA (30 µM). To assess whether GGA impacts PLY-induced apoptosis, we evaluated the expression levels of cleaved caspase 3, a commonly used marker of apoptosis in HLMVEC. PLY increased the expression of cleaved caspase 3, an effect that was decreased in cells pretreated with GGA. Neither PLY nor GGA treatment altered the expression levels of total caspase 3 or the loading control, Hsp90 (Figure 6B). Apoptosis can be triggered by both cytosolic ROS and mtROS. MitoSOX assays revealed less mtROS and decreased NOX1 protein expression in HLMVEC treated with GGA (Figure 6C). Given that mitochondrial, and mtROS can both increase inflammatory signaling, we assessed phosphorylated-NF-κB p65 (Ser536) levels in HLMVEC treated with 50 ng/ml PLY with or without GGA. NF-κB p65 phosphorylation was decreased with GGA treatment (Figure 6D).
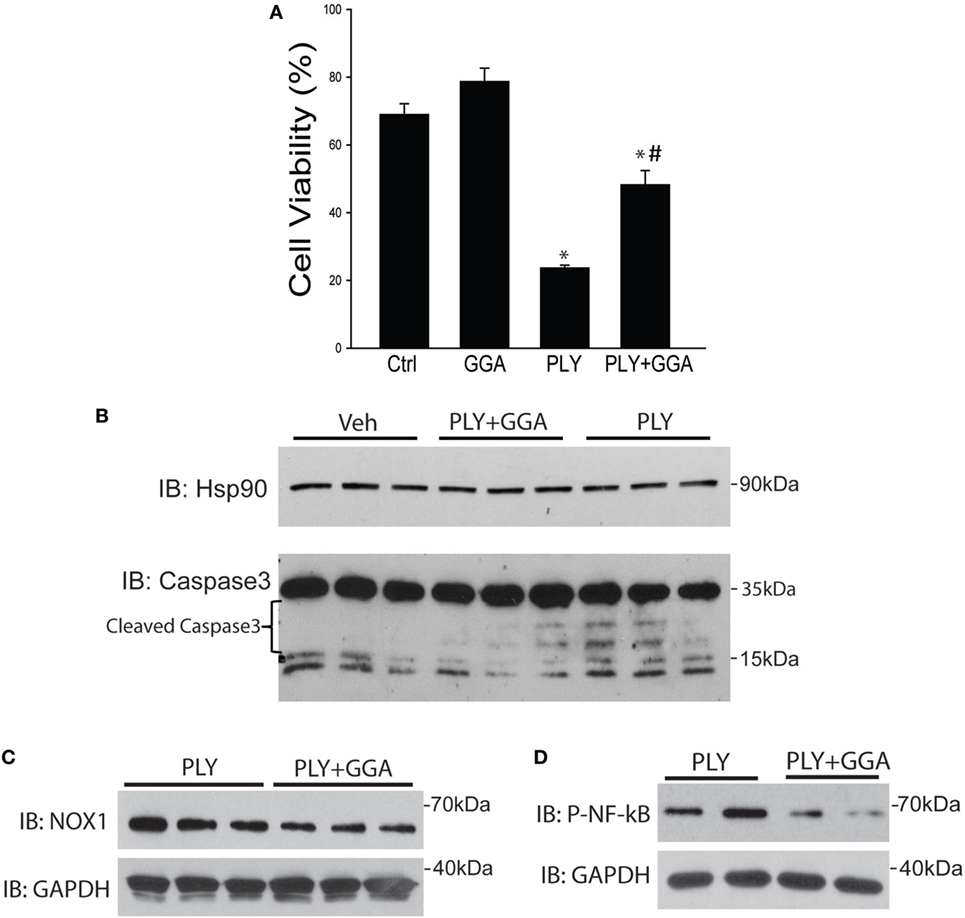
Figure 6. Geranylgeranylacetone (GGA) protects against pneumolysin (PLY)-induced Human lung microvascular endothelial cells (HLMVEC) apoptosis. In panel (A) HLMVEC were pretreated 6 h with GGA (30 µM) and then challenged with PLY (50 ng/ml) for 4 h and cell viability determined (MUSE flow cytometry). Data are shown as mean ± SEM, *p < 0.05 versus control, #p < 0.05 versus PLY. In panel (B) HLMVEC were challenged pretreated with vehicle or GGA (30 µM, 6 h) and then challenged with PLY (50 ng/ml) for 4 h. Cells were lysed and the level of cleaved caspase 3 was determined by western blot (n = 3). (C) HLMVECs were pretreated with vehicle or GGA overnight and then challenged with PLY (50 ng/ml) for 4 h. Cells were lysed and NOX1 levels determined by western blot. (D) HLMVECs were pretreated with vehicle or GGA (30 µM, 6 h) and then challenged with PLY (50 ng/ml) for 4 h. Cells were lysed and levels of phosphorylated (P-) NF-κB p65 were determined by western blot.
GGA Protects Mice From PLY-Induced ALI
To assess the translational relevance of Hsp70 upregulation in a model of ALI, mice were administered GGA (500 mg/kg IP) or vehicle overnight and then challenged with IT PLY (60 ng). Endothelial barrier integrity was assessed by EBD extravasation. A single dose of GGA significantly increased Hsp70 protein expression in lung tissues relative to the loading control, GADPH (Figure 7A). Neither vehicle nor GGA alone influenced baseline levels of EBD in lung tissue, however, in vehicle treated mice, PLY administration evoked a significant increase in pulmonary vascular permeability and extravascular leak of EBD. Pretreatment with GGA significantly blunted the ability of PLY to induce EBD extravasation into the lungs consistent with greater preservation of the pulmonary endothelial barrier (Figure 7B).
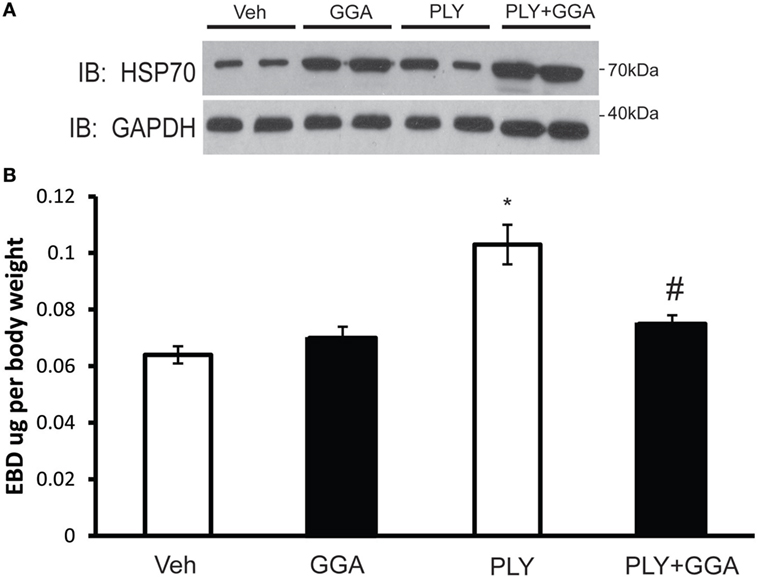
Figure 7. Geranylgeranylacetone (GGA) provides protection against pneumolysin (PLY)-induced vascular leak in vivo. Mice were administered vehicle (10% ethanol, IP) or GGA (500 ng/kg, IP) and, 24 h later, were challenged with IT PLY 60 ng/mouse. Evans blue dye-albumin (EBD, 30 mg/kg-2 h) was injected IV via the tail vein 2 h before the administration of PLY to assess pulmonary vascular leak (n = 5–6). After the mice were sacrificed, lung tissue was lysed and the levels of Hsp70 and GAPDH were determined by Western blot (A), and then the levels of EBD in lung tissue were determined spectrophotometrically at 620–750 nm (B). Data are shown as mean ± SEM, *p < 0.05 versus control, #p < 0.05 versus PLY.
Discussion
Herein, we show that genetic and pharmacological upregulation of Hsp70 provides robust protection from bacterial toxin-induced destruction of the endothelial barrier both in vitro and in vivo. Increased Hsp70 expression, achieved through adenovirus-mediated gene transfer or activation of HSF with GGA, protected the integrity of the pulmonary microvascular endothelial barrier following challenge with both G+ and G− bacterial toxins. The G+-toxin PLY promoted increased mitochondrial ROS, decreased mitochondrial function, increased caspase C cleavage, and increased cell death, effects that were mitigated by GGA treatment. In mice, GGA upregulated pulmonary Hsp70 expression and provided significant protection from PLY-induced pulmonary microvascular permeability. Collectively, these results suggest that Hsp70 can protect against bacterial toxin-induced increases in mitochondrial ROS which contribute to the loss of pulmonary endothelial barrier integrity.
Pneumolysin and other cholesterol-dependent pore-forming cytolysins from G+-bacteria such as listeriolysin O have been shown to form plasma membrane pores that stimulate calcium entry and promote disruption of the endothelial barrier (12, 13, 41, 42, 47). Multiple mechanisms have been proposed, including activation of PKC, disruption of NO signaling, arginase induction, and inhibition of ENaC. A connecting theme between these mechanisms is the ability of G+ toxins to increase ROS, but the sources of superoxide appear to be multifactorial and are incompletely defined (13). PLY has been shown to induce cell death and inflammation in the lung (48) and, in conjunction with elevated ROS it has been shown to promote cell death and apoptosis in multiple cell types including cardiomyocytes (49) epithelial cells (49–51), neurons (15, 52) and cerebral endothelial cells (53), and human umbilical vein endothelial cells (49). The ability of PLY to induce cell death in lung microvascular endothelial cells has not previously been described. Using MitoSOX red, we observed that PLY-stimulated ROS in the mitochondria of HLMVEC. An increase in ROS occurred alongside compromised mitochondrial function and increased cell death. Whether the increase in mitochondrial ROS contributes to PLY-induced cell death is not yet established.
Hsp70 belongs to large family (>13 members) of related 70 kDa Hsp that are both ubiquitous and highly conserved (54). Hsp70 family members share a similar structural organization composed of three basic domains: an N-terminal domain that encodes a highly conserved ATP-binding site, the M or middle domain binds which binds to numerous substrates, and the C-terminal domain facilitates protein folding and the binding of co-chaperones. Some of the Hsp70 genes are constitutively expressed and others are inducible in response to various stressors including increased heat (55), ROS (56), osmolarity (57), and toxins (58) to name a select few. The most abundant Hsp70s are HspA1A (Hsp70-1) and HspA1B (Hsp70-2). To simplify, we will use Hsp70 when referring to HspA1A. Hsp70 is found in the cytoplasm, nucleus, mitochondria, cell membranes, and in the extracellular space (59). Hsp70 influences many aspects of cellular function including binding to nascent proteins, preventing protein aggregation, facilitating protein folding and stability, and regulating protein activity. The upregulation of Hsp70 in response to cell stress provides a survival advantage (46, 60). Hsp70 has been shown to influence cell survival via multiple mechanisms (45, 60, 61), which may differ based on the cell type and stimulus. We found that upregulation of Hsp70 provided protection against PLY-induced ROS, mitochondrial dysfunction and ultimately caspase 3 cleavage and cell death. Cell death is an important mechanism underlying ALI (62) and the ability of Hsp70 to provide cell survival may underlie is ability to preserve barrier integrity. While our studies focused primarily on the role of G+ toxins, we also showed that Hsp70 upregulation can protect against the loss of barrier function induced by LPS. These data are consistent with other studies in which upregulation of a distinct Hsp70 family member, HspA12B, protects against the loss of barrier function in HUVECs (63). Hsp70 has also been shown to protect against hyperoxia-stimulated loss of endothelial barrier function (39).
Another important finding of our study was the ability of Hsp70 to suppress the production of ROS. Previously, we have shown that upregulation of Hsp70 inhibits the activity of the NADPH oxidases which are the major sources of cellular ROS (35, 64). In this study, we show that Hsp70 suppressed NOX1 expression which is consistent with our previous findings and protects against PLY-stimulated mitochondria ROS. How Hsp70 reduces mitochondrial ROS production in HLMVEC is not yet fully understood and evidence in the literature in other cell types suggest that multiple mechanisms are involved. PLY opens pores in the plasma membrane leading to a pronounced calcium influx that can impair mitochondrial coupling and promote superoxide formation. Within the inner membrane of mitochondria is an ATP sensitive potassium channel (mitoKATP) that can be activated by ATP deprivation and calcium overload. Studies have shown that GGA and Hsp70 reduce mitoROS in cardiomyocytes and protect from ischemia reperfusion injury. The protective effect of increased Hsp70 expression is abolished with the mitoKATP inhibitor, 5-hydroxydecanoate, which does not alter Hsp70 expression. These data suggest that Hsp70 reduces mitoROS accumulation by facilitating the opening of the mitoKATP channel. NADPH derived ROS have been shown to promote oxidative damage of mitochondrial proteins in particularly, complex I and complex II, which results in increased mitoROS production (20). Increased mitoROS can activate NADPH oxidase locking both enzymes into a feed forward relationship (20–22). The upregulation of Hsp70 in HLMVEC would be expected to break this vicious cycle between mtROS and NADPH oxidase through actions on both pathways. Hsp70 has also been shown to attenuate oxidative phosphorylation (65) which may reduce mitoROS generation. Hsp70 can also impair mitochondrial proteostasis (66) which may compromise local antioxidant (SOD2) pathways or improve the function of enzymes in the electron transport chain. Hsp70 has also been shown to inhibit NADPH oxidase activity (35) which can secondarily impact mitoROS. Finally, increased expression of Hsp70 has been shown to upregulate Akt-eNOS activity and the resulting increase in NO could indirectly suppress mitochondrial ROS by quenching superoxide (67).
Others have shown that Hsp70 can protect against cell death by mechanisms upstream of the mitochondria and by suppressing the ability of ROS (H2O2) to induce mitochondrial dysfunction (68, 69). There is little Hsp70 (1A1) in the mitochondria of normal cells; however, tumor cells have significantly increased amounts which provide a survival advantage. Depletion of Hsp70 in tumor cells disrupts mitochondria function and increases cell death (70). There is also a constitutive mitochondrial isoform of Hsp70 (HspA9 or motalin) which has important roles in protein translocation and although it is not induced by stress, it has also been shown to be important for cell survival (71). In our study, we upregulated Hsp70 using GGA and using adenoviral mediated gene transfer of a specific Hsp70 (HspA1A). How Hsp70 traffics to the mitochondria is not completely understood. Hsp70 can interact with lipids and in particular cardiolipin which is localized to the mitochondria (72). In addition to actions on Nox enzymes and the mitochondria, Hsp70 upregulation is also associated with reduced inflammation and the upregulation of antioxidant pathways (73). Inhibition of mitochondrial complexes I, II, and III has been shown to protect cardiomyocytes from hypoxia induced injury by reducing p38 phosphorylation as well as inflammatory signaling (74). Interestingly, blockade of ROS diffusion in the mitochondria using anion inhibitors also protects cardiomyocytes (74), which suggests an important role of mtROS in the initiation of inflammation during conditions of cellular stress. As the pulmonary inflammatory response can be very different in young versus elderly patients (28), future studies may want to compare the protective effects of Hsp70 in both young and aged animal models of PLY-induced ALI. Previously, we and others have shown that increased intracellular calcium is important in mediating the loss of endothelial barrier function in response to PLY (13, 75). Whether Hsp70 upregulation alters calcium-dynamics in HLMVEC is not yet known. Recent studies have revealed that PLY can activate TLR4 (76, 77) which is also the primary target of LPS. However, the kinetics of the loss of barrier function induced by PLY and LPS are quite different suggesting different mechanisms and furthermore, Hsp70 has been shown to both support and stimulate TLR4 signaling (78).
Hsp90 inhibitors have been shown to protect the endothelial barrier from G− bacterial toxins and are potent anti-inflammatory agents (79, 80). However, Hsp90 inhibitors also upregulate Hsp70 and the prosurvival actions of Hsp70 limit the effectiveness of these agents in anticancer strategies (81). In acute settings, whether the effectiveness of Hsp90 inhibitors relates to their ability to upregulate Hsp70 remains to be determined. Hsp70 has been shown to be released into the extracellular space through a yet to be identified mechanism. Increased circulating Hsp70 levels reflect heightened inflammation and poor outcome and autoantibodies against Hsp70 have been observed in a number of diseases (59). Although, upregulation of Hsp70 may promote cell survival and potentially increase extracellular Hsp70, the negative effects of long-term upregulation appear to be minor. GGA is approved for use as an antiulcer medication in Japan and appears to have low toxicity. Any potential risks are expected to be lower in the short-term treatment of ALI.
In conclusion, our study reveals that Hsp70 upregulation is a rapid and potent modality to protect the pulmonary endothelial barrier from the G+ bacterial toxin, PLY. Attractive features of this approach include the ability to rapidly upregulate Hsp70 with pharmacological agents and a broad spectrum of protection against both G+ toxins, G− toxins and hyperoxia. Hsp70 also targets multiple pathways including mitochondrial function, ROS production, and cell death which are key mechanisms that underlie the loss of endothelial barrier integrity. Upregulation of Hsp70 may be of high clinical significance in the management of ARDS/ALI-related pulmonary barrier dysfunction.
Ethics Statement
All animal studies conformed to National Institutes of Health guidelines. The experimental procedure was approved by the Augusta University Institutional Animal Care and Use Committee.
Author Contributions
XL: conceptual ideas, performed experiments, and wrote paper. YY and BG: performed experiments. SH and ZB: critical reading and performed experiments. DW, RR, SB, AV, and YS: critical reading. TC: provided reagents. RL: conceptual ideas, reagents, and critical reading. DS and DF: conceptual ideas and critical reading. FC: conceptual ideas, experiments, and critical reading.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by NIH grants P01 HL101902-01A1 (AV and DF), R01HL124773 (SB and DF), R01HL125926 (DS and DF), extramural Success Award from the Vice President for Research at Augusta University (to RL), ADA grant #1-16-IBS-196 (to AV, RL, and DF), as well as by SFB grant TR-84 “Innate Immunity of the Lung” from the German Research Foundation (DFG) (to TC). RL is a Mercator Fellow of the DFG.
References
1. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest (2012) 122:2731–40. doi:10.1172/JCI60331
2. O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet (2009) 374:893–902. doi:10.1016/S0140-6736(09)61204-6
3. Dreyfuss D, Ricard JD. Acute lung injury and bacterial infection. Clin Chest Med (2005) 26:105–12. doi:10.1016/j.ccm.2004.10.014
4. Matthay MA, McAuley DF, Ware LB. Clinical trials in acute respiratory distress syndrome: challenges and opportunities. Lancet Respir Med (2017) 5:524–34. doi:10.1016/S2213-2600(17)30188-1
5. Weycker D, Strutton D, Edelsberg J, Sato R, Jackson LA. Clinical and economic burden of pneumococcal disease in older US adults. Vaccine (2010) 28:4955–60. doi:10.1016/j.vaccine.2010.05.030
6. Lucas R, Czikora I, Sridhar S, Zemskov E, Gorshkov B, Siddaramappa U, et al. Mini-review: novel therapeutic strategies to blunt actions of pneumolysin in the lungs. Toxins (Basel) (2013) 5:1244–60. doi:10.3390/toxins5071244
7. Martner A, Dahlgren C, Paton JC, Wold AE. Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect Immun (2008) 76:4079–87. doi:10.1128/IAI.01747-07
8. Gilbert RJ. Cholesterol-dependent cytolysins. Adv Exp Med Biol (2010) 677:56–66. doi:10.1007/978-1-4419-6327-7_5
9. Tilley SJ, Orlova EV, Gilbert RJ, Andrew PW, Saibil HR. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell (2005) 121:247–56. doi:10.1016/j.cell.2005.02.033
10. Gilbert RJ, Jimenez JL, Chen S, Andrew PW, Saibil HR. Structural basis of pore formation by cholesterol-binding toxins. Int J Med Microbiol (2000) 290:389–94. doi:10.1016/S1438-4221(00)80049-1
11. Kwon IS, Kim J, Rhee DK, Kim BO, Pyo S. Pneumolysin induces cellular senescence by increasing ROS production and activation of MAPK/NF-kappaB signal pathway in glial cells. Toxicon (2017) 129:100–12. doi:10.1016/j.toxicon.2017.02.017
12. Chen F, Wang Y, Rafikov R, Haigh S, Zhi WB, Kumar S, et al. RhoA S-nitrosylation as a regulatory mechanism influencing endothelial barrier function in response to G(+)-bacterial toxins. Biochem Pharmacol (2017) 127:34–45. doi:10.1016/j.bcp.2016.12.014
13. Chen F, Kumar S, Yu Y, Aggarwal S, Gross C, Wang Y, et al. PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+-toxins. PLoS One (2014) 9:e99823. doi:10.1371/journal.pone.0099823
14. Di A, Mehta D, Malik AB. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium (2016) 60:163–71. doi:10.1016/j.ceca.2016.02.002
15. Braun JS, Hoffmann O, Schickhaus M, Freyer D, Dagand E, Bermpohl D, et al. Pneumolysin causes neuronal cell death through mitochondrial damage. Infect Immun (2007) 75:4245–54. doi:10.1128/IAI.00031-07
16. Mikhed Y, Daiber A, Steven S. Mitochondrial oxidative stress, mitochondrial DNA damage and their role in age-related vascular dysfunction. Int J Mol Sci (2015) 16:15918–53. doi:10.3390/ijms160715918
17. Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res (2009) 37:2539–48. doi:10.1093/nar/gkp100
18. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta (2009) 1787:1395–401. doi:10.1016/j.bbabio.2009.06.009
19. Caja S, Enriquez JA. Mitochondria in endothelial cells: sensors and integrators of environmental cues. Redox Biol (2017) 12:821–7. doi:10.1016/j.redox.2017.04.021
20. Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med (2011) 51:1289–301. doi:10.1016/j.freeradbiomed.2011.06.033
21. Kroller-Schon S, Steven S, Kossmann S, Scholz A, Daub S, Oelze M, et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal (2014) 20:247–66. doi:10.1089/ars.2012.4953
22. Nazarewicz RR, Dikalova AE, Bikineyeva A, Dikalov SI. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am J Physiol Heart Circ Physiol (2013) 305:H1131–40. doi:10.1152/ajpheart.00063.2013
23. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res (2008) 102:488–96. doi:10.1161/CIRCRESAHA.107.162800
24. Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature (2010) 468:1115–8. doi:10.1038/nature09599
25. Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A (1998) 95:9220–5. doi:10.1073/pnas.95.16.9220
26. Li X, Fang P, Yang WY, Chan K, Lavallee M, Xu K, et al. Mitochondrial ROS, uncoupled from ATP synthesis, determine endothelial activation for both physiological recruitment of patrolling cells and pathological recruitment of inflammatory cells. Can J Physiol Pharmacol (2017) 95:247–52. doi:10.1139/cjpp-2016-0515
27. Dikalov SI, Ungvari Z. Role of mitochondrial oxidative stress in hypertension. Am J Physiol Heart Circ Physiol (2013) 305:H1417–27. doi:10.1152/ajpheart.00089.2013
28. Boyd AR, Orihuela CJ. Dysregulated inflammation as a risk factor for pneumonia in the elderly. Aging Dis (2011) 2:487–500.
29. Nollen EA, Morimoto RI. Chaperoning signaling pathways: molecular chaperones as stress-sensing ’heat shock’ proteins. J Cell Sci (2002) 115:2809–16.
30. Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell (1998) 94:471–80. doi:10.1016/S0092-8674(00)81588-3
31. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) (2003) 228:111–33. doi:10.1177/153537020322800201
32. Sharma K, Vabulas RM, Macek B, Pinkert S, Cox J, Mann M, et al. Quantitative proteomics reveals that Hsp90 inhibition preferentially targets kinases and the DNA damage response. Mol Cell Proteomics (2012) 11:M111.014654. doi:10.1074/mcp.M111.014654
33. Chen F, Pandey D, Chadli A, Catravas JD, Chen T, Fulton DJ. Hsp90 regulates NADPH oxidase activity and is necessary for superoxide but not hydrogen peroxide production. Antioxid Redox Signal (2011) 14:2107–19. doi:10.1089/ars.2010.3669
34. Madrigal-Matute J, Fernandez-Garcia CE, Gomez-Guerrero C, Lopez-Franco O, Munoz-Garcia B, Egido J, et al. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc Res (2012) 95:116–23. doi:10.1093/cvr/cvs158
35. Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, et al. Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production. Arterioscler Thromb Vasc Biol (2012) 32:2989–99. doi:10.1161/ATVBAHA.112.300361
36. Jolly C, Morimoto RI. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J Natl Cancer Inst (2000) 92:1564–72. doi:10.1093/jnci/92.19.1564
37. Tyc J, Klingbeil MM, Lukes J. Mitochondrial heat shock protein machinery hsp70/hsp40 is indispensable for proper mitochondrial DNA maintenance and replication. MBio (2015) 6(1):e02425-14. doi:10.1128/mBio.02425-14
38. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature (1999) 399:597–601. doi:10.1038/21218
39. Kondrikov D, Fulton D, Dong Z, Su Y. Heat shock protein 70 prevents hyperoxia-induced disruption of lung endothelial barrier via caspase-dependent and AIF-dependent pathways. PLoS One (2015) 10:e0129343. doi:10.1371/journal.pone.0129343
40. Gonzales JN, Gorshkov B, Varn MN, Zemskova MA, Zemskov EA, Sridhar S, et al. Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol (2014) 306:L497–507. doi:10.1152/ajplung.00086.2013
41. Lucas R, Yang G, Gorshkov BA, Zemskov EA, Sridhar S, Umapathy NS, et al. Protein kinase C-alpha and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am J Respir Cell Mol Biol (2012) 47:445–53. doi:10.1165/rcmb.2011-0332OC
42. Xiong C, Yang G, Kumar S, Aggarwal S, Leustik M, Snead C, et al. The lectin-like domain of TNF protects from listeriolysin-induced hyperpermeability in human pulmonary microvascular endothelial cells – a crucial role for protein kinase C-alpha inhibition. Vascul Pharmacol (2010) 52:207–13. doi:10.1016/j.vph.2009.12.010
43. Otaka M, Yamamoto S, Ogasawara K, Takaoka Y, Noguchi S, Miyazaki T, et al. The induction mechanism of the molecular chaperone HSP70 in the gastric mucosa by geranylgeranylacetone (HSP-inducer). Biochem Biophys Res Commun (2007) 353:399–404. doi:10.1016/j.bbrc.2006.12.031
44. Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured Guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology (1996) 111:345–57. doi:10.1053/gast.1996.v111.pm8690199
45. Li CY, Lee JS, Ko YG, Kim JI, Seo JS. Heat shock protein 70 inhibits apoptosis downstream of cytochrome c release and upstream of caspase-3 activation. J Biol Chem (2000) 275:25665–71. doi:10.1074/jbc.M906383199
46. Riabowol KT, Mizzen LA, Welch WJ. Heat shock is lethal to fibroblasts microinjected with antibodies against hsp70. Science (1988) 242:433–6. doi:10.1126/science.3175665
47. Witzenrath M, Gutbier B, Hocke AC, Schmeck B, Hippenstiel S, Berger K, et al. Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit Care Med (2006) 34:1947–54. doi:10.1097/01.CCM.0000220496.48295.A9
48. Garcia-Suarez Mdel M, Florez N, Astudillo A, Vazquez F, Villaverde R, Fabrizio K, et al. The role of pneumolysin in mediating lung damage in a lethal pneumococcal pneumonia murine model. Respir Res (2007) 8:3. doi:10.1186/1465-9921-8-3
49. Zhou A, Wang H, Lan K, Zhang X, Xu W, Yin Y, et al. Apoptosis induced by pneumolysin in human endothelial cells involves mitogen-activated protein kinase phosphorylation. Int J Mol Med (2012) 29:1025–30. doi:10.3892/ijmm.2012.946
50. Feldman C, Anderson R, Cockeran R, Mitchell T, Cole P, Wilson R. The effects of pneumolysin and hydrogen peroxide, alone and in combination, on human ciliated epithelium in vitro. Respir Med (2002) 96:580–5. doi:10.1053/rmed.2002.1316
51. Li P, Shi J, He Q, Hu Q, Wang YY, Zhang LJ, et al. Streptococcus pneumoniae induces autophagy through the inhibition of the PI3K-I/Akt/mTOR pathway and ROS hypergeneration in A549 cells. PLoS One (2015) 10:e0122753. doi:10.1371/journal.pone.0122753
52. Braun JS, Sublett JE, Freyer D, Mitchell TJ, Cleveland JL, Tuomanen EI, et al. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J Clin Invest (2002) 109:19–27. doi:10.1172/JCI12035
53. Bermpohl D, Halle A, Freyer D, Dagand E, Braun JS, Bechmann I, et al. Bacterial programmed cell death of cerebral endothelial cells involves dual death pathways. J Clin Invest (2005) 115:1607–15. doi:10.1172/JCI23223
54. Brocchieri L, Conway de Macario E, Macario AJ. hsp70 genes in the human genome: conservation and differentiation patterns predict a wide array of overlapping and specialized functions. BMC Evol Biol (2008) 8:19. doi:10.1186/1471-2148-8-19
55. Tissieres A, Mitchell HK, Tracy UM. Protein synthesis in salivary glands of Drosophila melanogaster: relation to chromosome puffs. J Mol Biol (1974) 84:389–98. doi:10.1016/0022-2836(74)90447-1
56. Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler Thromb Vasc Biol (2001) 21:321–6. doi:10.1161/01.ATV.21.3.321
57. Shim EH, Kim JI, Bang ES, Heo JS, Lee JS, Kim EY, et al. Targeted disruption of hsp70.1 sensitizes to osmotic stress. EMBO Rep (2002) 3:857–61. doi:10.1093/embo-reports/kvf175
58. Koller M, Hensler T, Konig B, Prevost G, Alouf J, Konig W. Induction of heat-shock proteins by bacterial toxins, lipid mediators and cytokines in human leukocytes. Zentralbl Bakteriol (1993) 278:365–76. doi:10.1016/S0934-8840(11)80853-4
59. Radons J. The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones (2016) 21:379–404. doi:10.1007/s12192-016-0676-6
60. Jaattela M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases. EMBO J (1998) 17:6124–34. doi:10.1093/emboj/17.21.6124
61. Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, et al. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med (2004) 200:425–35. doi:10.1084/jem.20040531
62. Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, et al. Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest (2017) 127:4124–35. doi:10.1172/JCI94495
63. Kang Q, Chen Y, Zhang X, Yu G, Wan X, Wang J, et al. Heat shock protein A12B protects against sepsis-induced impairment in vascular endothelial permeability. J Surg Res (2016) 202:87–94. doi:10.1016/j.jss.2015.12.034
64. Chen F, Haigh S, Yu Y, Benson T, Wang Y, Li X, et al. Nox5 stability and superoxide production is regulated by C-terminal binding of Hsp90 and CO-chaperones. Free Radic Biol Med (2015) 89:793–805. doi:10.1016/j.freeradbiomed.2015.09.019
65. Wang L, Schumann U, Liu Y, Prokopchuk O, Steinacker JM. Heat shock protein 70 (Hsp70) inhibits oxidative phosphorylation and compensates ATP balance through enhanced glycolytic activity. J Appl Physiol (2012) 113:1669–76. doi:10.1152/japplphysiol.00658.2012
66. Leu JI, Barnoud T, Zhang G, Tian T, Wei Z, Herlyn M, et al. Inhibition of stress-inducible HSP70 impairs mitochondrial proteostasis and function. Oncotarget (2017) 8:45656–69. doi:10.18632/oncotarget.17321
67. Zhou C, Bai J, Jiang C, Ye L, Pan Y, Zhang H. Geranylgeranylacetone attenuates myocardium ischemic/reperfusion injury through HSP70 and Akt/GSK-3beta/eNOS pathway. Am J Transl Res (2017) 9:386–95.
68. Chong KY, Lai CC, Lille S, Chang C, Su CY. Stable overexpression of the constitutive form of heat shock protein 70 confers oxidative protection. J Mol Cell Cardiol (1998) 30:599–608. doi:10.1006/jmcc.1997.0623
69. Polla BS, Kantengwa S, Francois D, Salvioli S, Franceschi C, Marsac C, et al. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proc Natl Acad Sci U S A (1996) 93:6458–63. doi:10.1073/pnas.93.13.6458
70. Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett (2007) 581:3702–10. doi:10.1016/j.febslet.2007.05.039
71. Deocaris CC, Kaul SC, Wadhwa R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones (2006) 11:116–28. doi:10.1379/CSC-144R.1
72. McCallister C, Kdeiss B, Nikolaidis N. HspA1A, a 70-kDa heat shock protein, differentially interacts with anionic lipids. Biochem Biophys Res Commun (2015) 467:835–40. doi:10.1016/j.bbrc.2015.10.057
73. Hirota K, Nakamura H, Arai T, Ishii H, Bai J, Itoh T, et al. Geranylgeranylacetone enhances expression of thioredoxin and suppresses ethanol-induced cytotoxicity in cultured hepatocytes. Biochem Biophys Res Commun (2000) 275:825–30. doi:10.1006/bbrc.2000.3392
74. Kulisz A, Chen N, Chandel NS, Shao Z, Schumacker PT. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol (2002) 282:L1324–9. doi:10.1152/ajplung.00326.2001
75. Wippel C, Fortsch C, Hupp S, Maier E, Benz R, Ma J, et al. Extracellular calcium reduction strongly increases the lytic capacity of pneumolysin from Streptococcus pneumoniae in brain tissue. J Infect Dis (2011) 204:930–6. doi:10.1093/infdis/jir434
76. Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, et al. Recognition of pneumolysin by toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A (2003) 100:1966–71. doi:10.1073/pnas.0435928100
77. Dessing MC, Hirst RA, de Vos AF, van der Poll T. Role of toll-like receptors 2 and 4 in pulmonary inflammation and injury induced by pneumolysin in mice. PLoS One (2009) 4:e7993. doi:10.1371/journal.pone.0007993
78. Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem (2002) 277:15028–34. doi:10.1074/jbc.M200497200
79. Joshi AD, Dimitropoulou C, Thangjam G, Snead C, Feldman S, Barabutis N, et al. Heat shock protein 90 inhibitors prevent LPS-induced endothelial barrier dysfunction by disrupting RhoA signaling. Am J Respir Cell Mol Biol (2014) 50:170–9. doi:10.1165/rcmb.2012-0496OC
80. Chatterjee A, Snead C, Yetik-Anacak G, Antonova G, Zeng J, Catravas JD. Heat shock protein 90 inhibitors attenuate LPS-induced endothelial hyperpermeability. Am J Physiol Lung Cell Mol Physiol (2008) 294:L755–63. doi:10.1152/ajplung.00350.2007
Keywords: pneumolysin, endothelial barrier, reactive oxygen species, mitochondria, Hsp70
Citation: Li X, Yu Y, Gorshkov B, Haigh S, Bordan Z, Weintraub D, Rudic RD, Chakraborty T, Barman SA, Verin AD, Su Y, Lucas R, Stepp DW, Chen F and Fulton DJR (2018) Hsp70 Suppresses Mitochondrial Reactive Oxygen Species and Preserves Pulmonary Microvascular Barrier Integrity Following Exposure to Bacterial Toxins. Front. Immunol. 9:1309. doi: 10.3389/fimmu.2018.01309
Received: 02 March 2018; Accepted: 25 May 2018;
Published: 12 June 2018
Edited by:
Christoph Thiemermann, Queen Mary University of London, United KingdomReviewed by:
Andrey V. Kozlov, Institute for Experimental and Clinical Traumatology (LBG), AustriaAndrew C. B. Cato, Karlsruher Institut für Technologie, Germany
Copyright: © 2018 Li, Yu, Gorshkov, Haigh, Bordan, Weintraub, Rudic, Chakraborty, Barman, Verin, Su, Lucas, Stepp, Chen and Fulton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Chen, ZmNoZW5AbmptdS5lZHUuY24=;
David J. R. Fulton, ZGZ1bHRvbkBhdWd1c3RhLmVkdQ==