 Luisa J. Ströh
Luisa J. Ströh Thomas Krey
Thomas Krey- Institute of Virology, Hannover Medical School, Hannover, Germany
Numerous antibodies have been described that potently neutralize a broad range of hepatitis C virus (HCV) isolates and the majority of these antibodies target the binding site for the cellular receptor CD81 within the major HCV glycoprotein E2. A detailed understanding of the major antigenic determinants is crucial for the design of an efficient vaccine that elicits high levels of such antibodies. In the past 6 years, structural studies have shed additional light on the way the host’s humoral immune system recognizes neutralization epitopes within the HCV glycoproteins. One of the most striking findings from these studies is that the same segments of the E2 polypeptide chain induce antibodies targeting distinct antigen conformations. This was demonstrated by several crystal structures of identical polypeptide segments bound to different antibodies, highlighting an unanticipated intrinsic structural flexibility that allows binding of antibodies with distinct paratope shapes following an “induced-fit” mechanism. This unprecedented flexibility extends to the entire binding site for the cellular receptor CD81, underlining the importance of dynamic analyses to understand (1) the interplay between HCV and the humoral immune system and (2) the relevance of this structural flexibility for virus entry. This review summarizes the current understanding how neutralizing antibodies target structurally flexible epitopes. We focus on differences and common features of the reported structures and discuss the implications of the observed structural flexibility for the viral replication cycle, the full scope of the interplay between the virus and the host immune system and—most importantly—informed vaccine design.
Introduction
Approximately 71 million people worldwide are chronically infected with hepatitis C virus (HCV), which is one of the major causes of liver cirrhosis, liver failure, and hepatocellular carcinoma (1). Small-molecule drugs targeting HCV proteins termed direct-acting antivirals achieve cure rates of >95% (2), but high treatment costs, lack of awareness about hepatitis C, the emergence of multi-drug resistant viruses, and the need to protect patients from re-infection indicate that a prophylactic vaccine is still urgently required. In most viral infections, neutralizing antibodies (nAbs) are in the first line of defense of the adaptive immune response. For HCV, rapid induction of nAbs along with a broadly reactive T-cell response correlates with spontaneous clearance during acute infection and several studies highlighted the role of humoral immunity for the control both in the acute and chronic phase of infection (3, 4).
The two glycoproteins E1 and E2 of HCV are the major targets for nAbs. In particular, the receptor-binding glycoprotein E2 contains major antigenic determinants of HCV, mostly overlapping with binding sites for cellular receptors, including scavenger receptor class B type 1 (SR-B1) (5), the low-density lipoprotein receptor (LDLr) (6), and the tetraspanin CD81 (7). In addition to an extensive disulfide bridge network involving 8 and 18 conserved cysteines in E1 and E2, respectively, both proteins are heavily glycosylated in their N-terminal ectodomains (8, 9). Glycans are important for protein folding and affect epitope presentation and/or accessibility (10). The C-terminal transmembrane domains of E1 and E2 are anchored in the lipid envelope and interact to form an E1E2 heterodimer in HCV particles that are associated with lipoproteins and therefore also termed “lipo-viro particles” (11). Moreover, an E1 trimer observed at the surface of cell culture-derived HCV (HCVcc) and pseudoparticles suggested the presence of E1E2 heterodimers assembled as heterohexameric complexes (12, 13). However, due to the lack of structural information, many features of the architecture and glycoprotein arrangement at the surface of infectious HCV particles remain elusive.
E2 contains four hypervariable regions (HVR) termed HVR1 (residues 384–410 in the prototype H77 sequence), HVR2 (residues 460–485) (14, 15), HVR3 (residues 431–466) (16), and the intergenotypic variable region (igVR, residues 570–580) (17). The fact that the HVR1 interacts with SR-B1 and LDLr during virus entry (5, 6) would per se render this segment an interesting target for nAbs. Indeed, the first described HCV neutralization epitope is localized in HVR1 (18). However, nAbs targeting the HVR1 tend to be mostly strain specific, making the HVR1 less interesting for vaccine design (19). Although viruses lacking the HVR1 infect chimpanzees (20) they are more susceptible to neutralization by patient sera and other human mAbs (21–24), indicating that the HVR1 masks neutralization epitopes and serves as an “immune decoy,” recombinant glycoproteins lacking the HVR1 are not superior vaccine antigens (25). In addition, the binding of poorly neutralizing Abs to HVR1 can block the binding of broadly neutralizing Abs (bnAbs) to adjacent, conserved regions on E2 (26). These observed antagonistic effects suggest that the induction of anti-HVR1 Abs can interfere with a protective humoral response against HCV infection. By contrast, both HVR2 and the igVR seem neither to be direct targets for nAbs nor be directly involved in receptor binding. Nevertheless, similar to HVR1, both regions were found to modulate the accessibility of the CD81-binding site and the presentation of neutralizing epitopes on the E2 ectodomain (17, 27).
Neutralization Epitopes
On the quest to develop a safe and efficient B-cell vaccine, numerous neutralization epitopes within the HCV glycoproteins have been mapped using a variety of approaches. Peptide scanning approaches using overlapping peptide libraries or random peptide display libraries have revealed a number of linear epitopes, but such an approach is not suitable to identify residues that contribute to conformation-sensitive epitopes (27). Another powerful approach is alanine scanning, probing panels of protein variants with distinct amino acid substitutions for binding to the Abs of interest (28–33). However, amino acid substitution frequently results in protein misfolding and thereby in false contact residues in case of conformation-sensitive epitopes—as illustrated for the bnAb AR3C, where the crystal structure revealed different contact residues than expected from previous alanine scanning (34). This pitfall is often alleviated by the use of non-competing conformational Abs to probe overall protein conformation and cross-competition analysis using a panel of well-characterized nAbs. In vitro studies of antibody escape can provide or confirm information about key epitopes (35–43). The gold standard to identify neutralization epitopes still remains the structural analysis of the immune complex, however, HCV glycoproteins are difficult to crystallize and only one neutralization epitope has been structurally characterized in complex with the E2 ectodomain to date (34). The combination of peptide and alanine scanning together with Ab cross-competition studies have yielded different nomenclature systems to describe and cluster epitopes on E2 to date such as antigenic domain A–E (44), antigenic region 1–5 (45), and epitope I–III (46). Of note, extensive overlap exists between these three systems of epitope nomenclature (47).
E1 is less immunogenic but two regions targeted by nAbs have been identified: residues 192–202 (in the prototype H77 sequence), which are recognized by the weakly nAb H-111 (48) and residues 313–324, which interact with the cross-reactive nAbs IGH-526 and IGH-505 (49, 50).
E2 Structure and Conformational Flexibility
The two crystal structures of E2 core fragments, one in complex with the non-nAb 2A12 and the other with the bnAb AR3C, show that E2 features a central immunoglobulin (Ig)-like β-sandwich with two adjacent layers, one in front and one at the back (34, 51). Several regions of the protein are found in loop configurations or are disordered suggesting a high flexibility in parts of the structure (34). The igVR forms a disulfide-constrained loop within a flexible region spanning residues 567–596 but HVR1 and HVR2 are not included in the expression construct (51) or are only partially resolved in the electron density (34). Both structures are highly similar in the overall fold but the disulfide bond connectivity differs, suggesting that E2 features an enhanced plasticity compared to other viral glycoproteins, allowing for rather drastic local structural changes without affecting the overall fold of E2 (52). Of note, free thiol groups within the viral glycoproteins are required for virus entry (53), indicating a functional role of the observed plasticity.
To date, no detailed structural information on the CD81–E2 interaction is available but different techniques including alanine scanning mutagenesis, negative stain electron microscopy, nAb competition experiments, and in silico docking have been applied to map the CD81-binding site on E2 (34, 54, 55). Critical contact residues include highly conserved residues W420, Y527, W529, G530, D535 (54), and the G436WLAGLF motif (55, 56) most of which are located within (1) a conserved N-terminal region (aa412–423), (2) a front layer region (aa428–446), and (3) an adjacent loop named CD81-binding loop (aa518–542). The majority of α-E2 bnAbs identified to date compete with CD81 for binding to E2. Hence, it is not surprising that their epitopes overlap with one or more of these three regions corresponding to three antigenic regions named epitope I, II, and III (46) (Figure 1). Interestingly, not all Abs targeting one of these three epitopes neutralize HCV infection, in spite of similar contact residues (57–59). For non-nAbs directed against epitope II interference with neutralization by nAbs targeting epitope I was proposed (60), but also cooperativity effects between nAbs directed against epitope I and nAbs targeting epitope II have been reported (29).
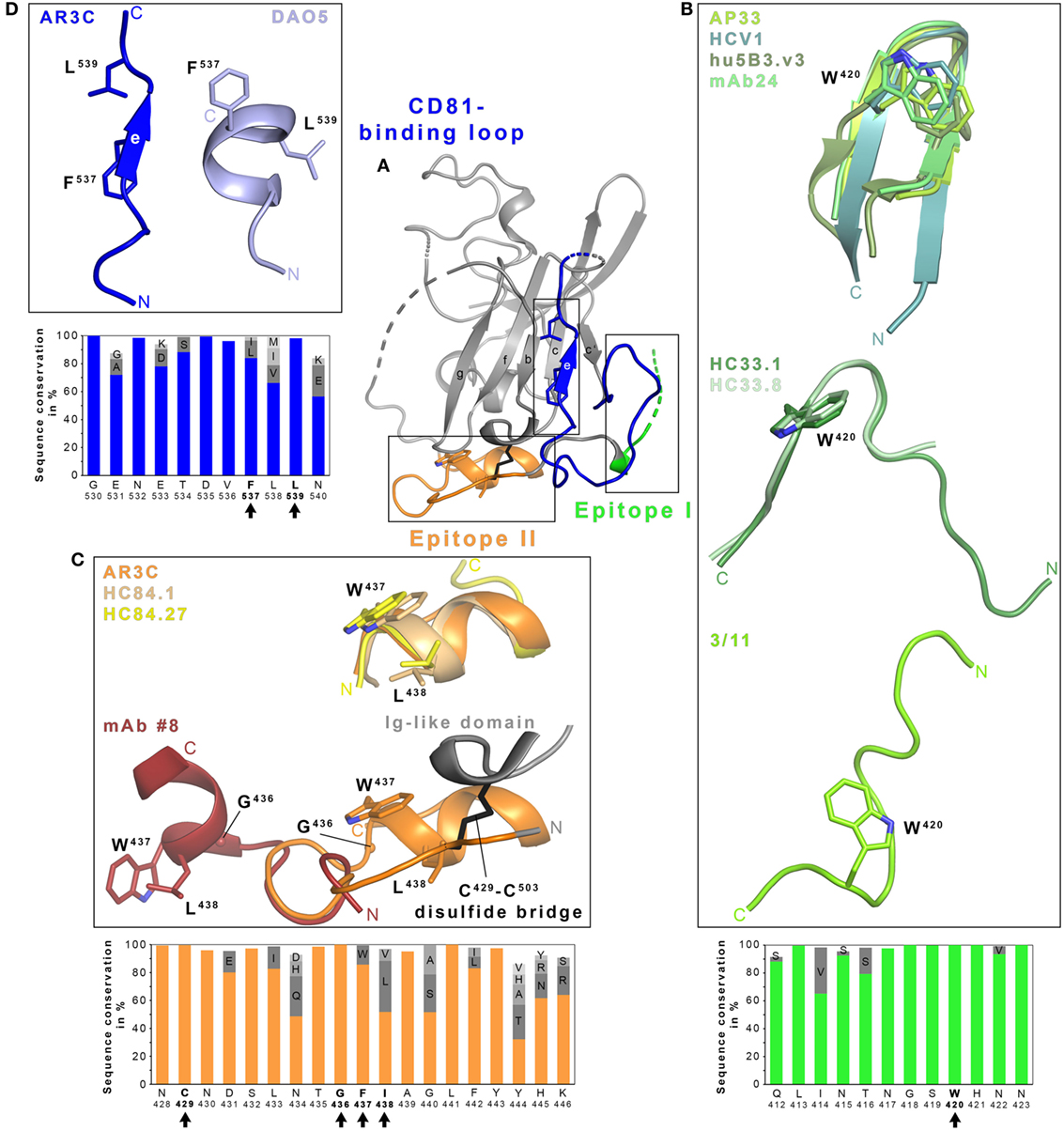
Figure 1. Structural flexibility of the hepatitis C virus (HCV) E2 glycoprotein. (A) Cartoon representation of the E2 ectodomain crystallized in complex with AR3C Fab (PDB 4MWF). The composite CD81-binding site consisting of epitope I (aa412–423; green), epitope II (aa428–446; orange), and the CD81-binding loop (aa518–542; blue) is highlighted in color and sidechains of selected residues are displayed as sticks. (B–D) Close-up views of the three antigenic sites mentioned above. (B) Epitope I is disordered in the context of the E2 structure but a synthetic epitope peptide folds as β-hairpin in complex with neutralizing antibodies (nAbs) AP33 (PDB 4GAJ), HCV1 (PDB 4DGV), hu5B3.v3 (PDB 4HS8), and mAb24 (PDB 5VXR) (upper panel). By contrast, the same peptide adopts two distinct extended conformations in complex with nAbs HC33.1 (PDB 4XVJ) and HC33.8 (PDB 5FGC) (middle panel) or 3/11 (PDB 4WHY; bottom panel), respectively. The peptide in the nAb HC33.4 complex (PDB 5FGB) adopts an amino acid backbone conformation identical to the one in complex with nAb HC33.8 and is not shown for simplicity. (C) Superposition of the epitope II peptide structure in complex with HC84.1 and HC84.27 Fabs (PDB 4JZN and 4JZO, respectively) onto the E2 ectodomain structure (upper panel) reveals a conserved 1.5-turn α-helix (aa437–442) with an extended C-terminal segment containing aa443–446. Superposition of the N-terminal loop of epitope II (aa430–434) from the peptide structure in complex with mAb #8 (PDB 4HZL) onto its counterparts in the E2 structure suggests that the short α-helix flips out to expose residues W437 and L438 for mAb #8 binding (bottom panel). (D) Residues 532–540 of the CD81-binding loop were observed in an extended conformation in the context of the E2 ectodomain structure (A) and in a helical conformation in the DAO5 Fab–E2 peptide complex structure (PDB 5NPJ) suggesting thereby a putative open and closed conformation of the immunoglobulin-like domain. Amino acid sequence conservation of the respective antigenic site was calculated across the six HCV genotypes for 481 isolate sequences (100 sequences each for genotypes 1, 2, 3, 6, and 70, 9 and 2 sequences for genotypes 4, 5, and 7, respectively) obtained and analyzed from the ViPR database (http://www.viprbrc.org) and is shown below each close-up view. Residues with side chains shown as sticks are highlighted by black arrows.
Within the last years a number of crystallographic studies have revealed molecular details of how Abs interact with these three epitopes, illustrating a great structural heterogeneity in particular within the epitope I (26, 41, 61–66), but also epitope II (59, 67–69), and more recently epitope III comprising the CD81-binding loop and parts of the core Ig-like domain (58). Of note, all three segments are largely conserved in sequence across all HCV genotypes and subtypes (Figure 1).
In the E2 core–AR3C Fab complex structure, epitope I is mostly disordered but synthetic peptides mimicking this epitope were complexed and crystallized with Fabs from bnAbs isolated from immunized rodents or from HCV-infected individuals. Human nAb HCV1, mouse nAbs AP33 and mAb24, and humanized and affinity-matured nAbs MRCT10.v362 and hu5B3.v3 (derived from AP33 and mu5B3, respectively) bind such epitope I peptides in a very similar β-hairpin conformation (Figure 1B) (41, 62, 63, 65, 66). However, the superposition of the linear epitope in complex with AP33 and HCV1 Fab reveal a 22° difference in the binding angle highlighting that both Abs engage the epitope on E2 from different directions (62). nAb paratopes are similar in shape and surface charge but interactions with E2 are realized by different nAb residues resulting in small conformational differences within a conserved β-hairpin conformation. In both cases, residues L413, N415, G418, and W420 of E2 are deeply buried in Fab-peptide interface.
By contrast, the same peptide is recognized in an extended conformation in a deep cleft between heavy and light chains of the Fab from the rat nAb 3/11 (Figure 1B) (61). E2 residues N415, W420, and H421 are especially critical for the 3/11-antigen interaction in line with epitope mapping by alanine scanning mutagenesis (32, 61). A third conformation of epitope I is recognized by a group of human mAbs named HC33 that were isolated from HCV-infected blood donors (Figure 1B) (64). In complex with the HC33.1 Fab, residues I414 and N415 form an anti-parallel β-sheet with strand F of the heavy chain variable region Ig domain and the remaining part of the peptide is recognized in an extended coil conformation. This interaction mode results in a turn (residues T416–S419) superimposing with the turn observed in the β-hairpin conformation in complex with HCV1 and AP33 Fabs. Residues L413, G418, and W420 constitute key anchors for the interaction and are deeply buried in the HC33.1-E2 interface (29, 64). An adaptive mutation N417S is associated with a shift of an N-linked glycosylation site from N417 to N415 that abolishes neutralization by nAbs HCV1, AP33, and mAb24. Residue N415 is buried by HCV1, AP33, and mAb24 but it is solvent-exposed in the HC33.1 Fab-peptide complex structure and allowing for glycosylation at N415 (29, 41, 64). Mutations N417S and N415D enhance the sensitivity to HC33.1 neutralization but also to neutralization by other human nAbs targeting different epitopes, suggesting that this region has a global impact on the conformation of HCV glycoproteins (41).
In the complex structures of the related HC33.4 and HC33.8 Fabs, a similar extended conformation is observed for E2 aa418–423 and aa415–423, respectively (26) (Figure 1B), but N-terminal residues aa412–414 are disordered. Although the HVR1-residue K408 was identified by alanine scanning mutagenesis to be part of the HC33.8- and HC33.4- but not of the HC33.1-epitope, no structural evidence for further epitope–paratope interactions beyond epitope I was observed (26). In summary, epitope I adopts at least three distinct conformations and greatly differs in its nAb interactions depending on the individual nAb. However, in all cases, the hydrophobic interaction networks involves W420, which is strictly conserved across HCV genotypes (Figure 1B) and serves also as a critical residue for CD81 binding (54). A recent electron microscopy study demonstrated that the HCV1 Fab binds soluble E2 from different angles of approach thereby further highlighting the conformational flexibility in epitope I (70).
At the surface of HCV particles, the epitope is either present in different conformations or readily converts between them (i.e., with a minimal kinetic barrier for conversion) and individual nAbs bind the epitope with their particular conformational selectivity. Indeed, the dose-dependent neutralization of nAbs 3/11 and AP33 suggests that the different conformations are in a dynamic equilibrium and can be converted in either direction (61). Interestingly, in silico predictions of the peptide alone propose a β-hairpin similar to the one observed in complex with Fabs from HCV1, AP33, and mAb24 (64). Together with the fact that the β-hairpin was observed in the majority of Fab complex structures, this suggests that the β-hairpin represents a preferred, but extremely unstable conformation on the HCV particle that can be readily converted into different conformations following an “induced-fit” binding mode to the antibody. This is further supported by the reported differences in neutralization potency of nAbs targeting the three described epitope I conformations (39, 71). The observation that nAbs targeting this segment usually have a broad neutralization activity suggests that genotype-specific sequence variations do not dictate the predominant epitope I conformation, although neutralization efficiency may be modulated by these sequence variations (61). The observed structural flexibility could explain the limited immunogenicity to the epitope I observed in HCV-infected patients (72).
Similarly, Fab-peptide structures provided molecular insights into recognition of epitope II. Structural information is available for epitope II in complex with different Abs—potent human nAbs on the one hand and weakly and non-nAbs derived from immunization with synthetic peptides on the other hand. When recognized by potent nAbs HC84.1, HC84.27, and the affinity-maturated nAb HC84.26.5D, an E2 peptide comprising aa434–446 forms a short α-helix spanning residues W437–F442 with an extended conformation on the C-terminal side comprising residues 443–446 (68, 69). This short α-helix can also be found in the AR3C Fab–E2 complex structure (34). Two other crystal structures of murine Fabs from the non-nAb #12 and the weakly neutralizing mAb #8 reveal an epitope that is located few amino acids upstream, but also includes the short α-helix (59, 67). Of note, residues W437 and L438 crucial for binding of mAb #12 and #8 are not accessible in the AR3C Fab–E2 complex, suggesting that a conformational change exposing these two residues is required to allow E2 binding. In line with this observation, superposition of the respective peptide structures using the N-terminus, which should be anchored to the Ig-like domain via a disulfide bridge (C429–C503), reveals that the C-terminal α-helix has to flip out to allow for mAb #8 binding (Figure 1C). This flexibility has been attributed to the strictly conserved G436 constituting a hinge between N- and C-terminus of the polypeptide chain, thereby resulting in an open and a closed state of E2 that implies the different presentation of epitope II (59, 70). Potent nAbs HC84.1, HC84.26.5D, and HC84.27 recognize the closed state similar to AR3C, indicating that this represents the preferred state of E2 in the viral particle and the open state targeted by weakly or non-nAbs is less frequently observed on virus particles. However, minor differences in the spatial arrangement of the C-terminal part of epitope II (aa443–446) (69) suggests that additional local structural changes may also occur in the closed conformation (59, 69).
A detailed functional and structural analysis of the non-nAb DAO5 provided a glimpse onto conformational changes in the CD81-binding loop (epitope III) and the adjacent part of the Ig-like domain (58) (Figure 1D). In the AR3C–E2 complex structure, the CD81-binding loop is stabilized by the Fab and the side chains of residues F537 and L539 (located on β-strand E) are buried inside the hydrophobic core of the Ig-like domain resembling a hypothetical closed conformation (34). In the absence of stabilizing Fab interactions, residues 524–535 are disordered and F537 is solvent-exposed (51). The crystal structure of non-nAb DAO5 Fab in complex with the E2 peptide aa532–540 reveals a helical conformation in which residues F537 and L539 are buried in the Fab interface, suggesting that on E2 they need to be solvent exposed to allow for interaction with DAO5 in a putative open conformation. A high sequence conservation within this region suggests that the observed conformational flexibility in the Ig-like domain is an intrinsic feature of E2. Indeed, both conformations are present simultaneously on infectious particles; hence, it is tempting to speculate that the open conformation recognized by non-Ab DAO5 acts as an immunological decoy that distracts the humoral immune system from the relevant CD81-binding conformation (58).
In addition to the static crystal structures, representing snapshots of an apparently highly dynamic protein, solution-based studies such as hydrogen deuterium exchange mass spectrometry (HDXMS) and limited proteolysis help to characterize flexible regions in E2 (51, 70). HDXMS detects the deuterium incorporation into the backbone amides when proteins are exposed to deuterated solvent. The exchange rate depends on the conformational flexibility and accessibility of individual residues to the solvent (73). As expected, HDXMS data confirmed the high structural flexibility in the E2 front layer including the composite CD81-binding site overlapping with epitopes of most bnAbs (70). Moreover, HVR1, HVR2, and igVR are highly flexible and heterogeneous in presented conformations in addition to being hypervariable in sequence (70). Interestingly, despite its unique conformational flexibility E2 has a high thermal stability when compared to proteins from thermophilic organisms or other viral envelope proteins such as HIV-1 env or influenza hemagglutinin presumably also due to its dense disulfide bridge network (70).
Conclusion and Future Perspectives
The conformational flexibility within HCV E2 extends to the entire composite CD81-binding site, which overlaps most of the conserved neutralization epitopes present in E2. This finding raises the question how such a conformational flexibility emerges during virus evolution? Which functional importance does this flexibility have—or in other words—which selective advantage does this flexibility provide for the virus?
One possible explanation could be that the observed conformations represent different stages during virus entry, where a number of changes in environmental conditions (e.g., receptor binding, endosomal acidification, or a putative conformational change to fuse viral and endosomal membrane) may require different glycoprotein conformations. However, all nAbs mentioned above targeting epitope I block CD81 binding, suggesting that the different epitope I conformations can be adopted upstream of receptor binding. To date, the epitope I conformation in complex with CD81 remains elusive, but a conformationally flexible surface could be required for receptor binding. It is estimated that ~30% of protein–protein interactions include disordered protein regions (74) and the region within the large extracellular loop of CD81 thought to interact with E2 was described to display marked conformational fluctuations (75, 76). Therefore, a conformationally flexible surface on the glycoprotein may be favorable to establish a highly specific receptor interaction via an ordered interface following an induced fit binding mechanism. Structural studies on E2 in complex with CD81 will be required to further address this hypothesis.
Another possible explanation could be that the observed conformational flexibility is required for a putative dynamic rearrangement at the virus surface during infection of the host cell, resulting in exposure of the conserved receptor-binding region in E2—similar to the structural dynamics or “virus breathing” described for the related flaviviruses [reviewed in Ref. (77)]. Such an “opening” rearrangement would be in line with the observed time- and temperature-modulated exposure of neutralization epitopes on HCV virions (78).
A third possible explanation could be that this flexibility constitutes a viral mechanism to efficiently evade from nAbs. In general, the stability of peptides has been reported to directly correlate with their capacity to induce a humoral immune response (79), suggesting that conformational flexibility implies a modest immunogenicity. In line with this finding, immunization with a synthetic HCV epitope I peptide did not elicit bnAbs, likely due to its intrinsic structural flexibility (80) and several studies reported that even a cyclic variant of epitope I does not elicit high titers of Abs neutralizing HCV infection (80, 81).
This has important implications for vaccine design, suggesting that— although many subunit vaccine candidates based on HCV glycoproteins are currently under development—an unmodified form of the latter is limited in its capacity to elicit nAbs. Structure-guided stabilization of neutralization epitopes within E2 toward the conformation targeted by nAbs can potentially improve its immunogenic properties. Alternatively, a recent innovative approach termed epitope-focused vaccine design (82, 83) facilitates the design of epitope-specific immunogens to elicit nAbs where conventional vaccines failed to raise an immune response. For this purpose, a structurally characterized neutralization epitope is grafted onto an unrelated protein scaffold containing a segment with an identical backbone conformation. A successful example of this strategy is the development of an epitope scaffold presenting a single neutralization epitope of the human respiratory syncytial virus F protein and its neutralization potency can potentially be further augmented by the incorporation of further neutralization epitopes (82). Epitope-focused design has also been applied to HCV neutralization epitopes (80, 84) albeit with limited success. However, more recently an anti-idiotypic Ab, which also functions by mimicking a neutralization epitope on an unrelated protein (in this case an antibody), was demonstrated to robustly induce HCVcc-nAbs (85), suggesting that epitope-focused immunogens represent a viable strategy to develop a safe and efficient B cell vaccine and elicit a protective nAb response.
Author Contributions
LS, KN, and TK participated in the design and coordination of the manuscript. All authors wrote, read, and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors [TK].
Acknowledgments
We apologize to all our colleagues who have contributed to this area of research but whose work could not be cited or discussed here due to space limitations. We thank Thomas Pietschmann and Alexander W. Tarr for critical reading of the manuscript.
Funding
This work was supported by funding to TK by the Deutsche Forschungsgemeinschaft within project B10 of the Collaborative Research Centre SFB900 “Chronic Infections: Microbial Persistence and Its Control” as well as an individual DFG research grant (KR2880/3-1).
References
2. Naggie S, Muir AJ. Oral combination therapies for hepatitis C virus infection: successes, challenges, and unmet needs. Annu Rev Med (2017) 68:345–58. doi:10.1146/annurev-med-052915-015720
3. Swadling L, Klenerman P, Barnes E. Ever closer to a prophylactic vaccine for HCV. Expert Opin Biol Ther (2013) 13(8):1109–24. doi:10.1517/14712598.2013.791277
4. Ball JK, Tarr AW, McKeating JA. The past, present and future of neutralizing antibodies for hepatitis C virus. Antiviral Res (2014) 105:100–11. doi:10.1016/j.antiviral.2014.02.013
5. Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J (2002) 21(19):5017–25. doi:10.1093/emboj/cdf529
6. Prentoe J, Serre SB, Ramirez S, Nicosia A, Gottwein JM, Bukh J. Hypervariable region 1 deletion and required adaptive envelope mutations confer decreased dependency on scavenger receptor class B type I and low-density lipoprotein receptor for hepatitis C virus. J Virol (2014) 88(3):1725–39. doi:10.1128/JVI.02017-13
7. Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, et al. Binding of hepatitis C virus to CD81. Science (1998) 282(5390):938–41. doi:10.1126/science.282.5390.938
8. Meunier JC, Fournillier A, Choukhi A, Cahour A, Cocquerel L, Dubuisson J, et al. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J Gen Virol (1999) 80(Pt 4):887–96. doi:10.1099/0022-1317-80-4-887
9. Goffard A, Dubuisson J. Glycosylation of hepatitis C virus envelope proteins. Biochimie (2003) 85(3–4):295–301. doi:10.1016/S0300-9084(03)00004-X
10. Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, et al. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol (2010) 84(22):11905–15. doi:10.1128/JVI.01548-10
11. Piver E, Boyer A, Gaillard J, Bull A, Beaumont E, Roingeard P, et al. Ultrastructural organisation of HCV from the bloodstream of infected patients revealed by electron microscopy after specific immunocapture. Gut (2017) 66(8):1487–95. doi:10.1136/gutjnl-2016-311726
12. Falson P, Bartosch B, Alsaleh K, Tews BA, Loquet A, Ciczora Y, et al. Hepatitis C virus envelope glycoprotein E1 forms trimers at the surface of the virion. J Virol (2015) 89(20):10333–46. doi:10.1128/JVI.00991-15
13. Freedman H, Logan MR, Hockman D, Koehler Leman J, Law JL, Houghton M. Computational prediction of the heterodimeric and higher-order structure of gpE1/gpE2 envelope glycoproteins encoded by hepatitis C virus. J Virol (2017) 91(8). doi:10.1128/JVI.02309-16
14. Kato N, Ootsuyama Y, Ohkoshi S, Nakazawa T, Sekiya H, Hijikata M, et al. Characterization of hypervariable regions in the putative envelope protein of hepatitis C virus. Biochem Biophys Res Commun (1992) 189(1):119–27. doi:10.1016/0006-291X(92)91533-V
15. Weiner AJ, Brauer MJ, Rosenblatt J, Richman KH, Tung J, Crawford K, et al. Variable and hypervariable domains are found in the regions of HCV corresponding to the flavivirus envelope and NS1 proteins and the pestivirus envelope glycoproteins. Virology (1991) 180(2):842–8. doi:10.1016/0042-6822(91)90104-J
16. Troesch M, Meunier I, Lapierre P, Lapointe N, Alvarez F, Boucher M, et al. Study of a novel hypervariable region in hepatitis C virus (HCV) E2 envelope glycoprotein. Virology (2006) 352(2):357–67. doi:10.1016/j.virol.2006.05.015
17. McCaffrey K, Boo I, Poumbourios P, Drummer HE. Expression and characterization of a minimal hepatitis C virus glycoprotein E2 core domain that retains CD81 binding. J Virol (2007) 81(17):9584–90. doi:10.1128/JVI.02782-06
18. Farci P, Shimoda A, Wong D, Cabezon T, De Gioannis D, Strazzera A, et al. Prevention of hepatitis C virus infection in chimpanzees by hyperimmune serum against the hypervariable region 1 of the envelope 2 protein. Proc Natl Acad Sci U S A (1996) 93(26):15394–9. doi:10.1073/pnas.93.26.15394
19. Vieyres G, Dubuisson J, Patel AH. Characterization of antibody-mediated neutralization directed against the hypervariable region 1 of hepatitis C virus E2 glycoprotein. J Gen Virol (2011) 92(Pt 3):494–506. doi:10.1099/vir.0.028092-0
20. Forns X, Thimme R, Govindarajan S, Emerson SU, Purcell RH, Chisari FV, et al. Hepatitis C virus lacking the hypervariable region 1 of the second envelope protein is infectious and causes acute resolving or persistent infection in chimpanzees. Proc Natl Acad Sci U S A (2000) 97(24):13318–23. doi:10.1073/pnas.230453597
21. Bartosch B, Verney G, Dreux M, Donot P, Morice Y, Penin F, et al. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J Virol (2005) 79(13):8217–29. doi:10.1128/JVI.79.13.8217-8229.2005
22. Bankwitz D, Steinmann E, Bitzegeio J, Ciesek S, Friesland M, Herrmann E, et al. Hepatitis C virus hypervariable region 1 modulates receptor interactions, conceals the CD81 binding site, and protects conserved neutralizing epitopes. J Virol (2010) 84(11):5751–63. doi:10.1128/JVI.02200-09
23. Prentoe J, Jensen TB, Meuleman P, Serre SB, Scheel TK, Leroux-Roels G, et al. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J Virol (2011) 85(5):2224–34. doi:10.1128/JVI.01594-10
24. Prentoe J, Velazquez-Moctezuma R, Foung SK, Law M, Bukh J. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology (2016) 64(6):1881–92. doi:10.1002/hep.28705
25. Law JLM, Logan M, Wong J, Kundu J, Hockman D, Landi A, et al. Role of the E2 hypervariable region (HVR1) in the immunogenicity of a recombinant HCV vaccine. J Virol (2018) 92:e02141-17. doi:10.1128/JVI.02141-17
26. Keck ZY, Girard-Blanc C, Wang W, Lau P, Zuiani A, Rey FA, et al. Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. J Virol (2016) 90(6):3112–22. doi:10.1128/JVI.02458-15
27. Alhammad Y, Gu J, Boo I, Harrison D, McCaffrey K, Vietheer PT, et al. Monoclonal antibodies directed toward the hepatitis C virus glycoprotein E2 detect antigenic differences modulated by the N-terminal hypervariable region 1 (HVR1), HVR2, and intergenotypic variable region. J Virol (2015) 89(24):12245–61. doi:10.1128/JVI.02070-15
28. Johansson DX, Voisset C, Tarr AW, Aung M, Ball JK, Dubuisson J, et al. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc Natl Acad Sci U S A (2007) 104(41):16269–74. doi:10.1073/pnas.0705522104
29. Keck Z, Wang W, Wang Y, Lau P, Carlsen TH, Prentoe J, et al. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J Virol (2013) 87(1):37–51. doi:10.1128/JVI.01941-12
30. Gopal R, Jackson K, Tzarum N, Kong L, Ettenger A, Guest J, et al. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog (2017) 13(12):e1006735. doi:10.1371/journal.ppat.1006735
31. Pierce BG, Keck ZY, Lau P, Fauvelle C, Gowthaman R, Baumert TF, et al. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc Natl Acad Sci U S A (2016) 113(45): E6946–54. doi:10.1073/pnas.1614942113
32. Tarr AW, Owsianka AM, Timms JM, McClure CP, Brown RJ, Hickling TP, et al. Characterization of the hepatitis C virus E2 epitope defined by the broadly neutralizing monoclonal antibody AP33. Hepatology (2006) 43(3):592–601. doi:10.1002/hep.21088
33. Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med (2008) 14(1):25–7. doi:10.1038/nm1698
34. Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science (2013) 342(6162):1090–4. doi:10.1126/science.1243876
35. Keck ZY, Saha A, Xia J, Wang Y, Lau P, Krey T, et al. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol (2011) 85(20):10451–63. doi:10.1128/JVI.05259-11
36. Gal-Tanamy M, Keck ZY, Yi M, McKeating JA, Patel AH, Foung SK, et al. In vitro selection of a neutralization-resistant hepatitis C virus escape mutant. Proc Natl Acad Sci U S A (2008) 105(49):19450–5. doi:10.1073/pnas.0809879105
37. Bailey JR, Wasilewski LN, Snider AE, El-Diwany R, Osburn WO, Keck Z, et al. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J Clin Invest (2015) 125(1):437–47. doi:10.1172/JCI78794
38. Chung RT, Gordon FD, Curry MP, Schiano TD, Emre S, Corey K, et al. Human monoclonal antibody MBL-HCV1 delays HCV viral rebound following liver transplantation: a randomized controlled study. Am J Transplant (2013) 13(4):1047–54. doi:10.1111/ajt.12083
39. Keck ZY, Angus AG, Wang W, Lau P, Wang Y, Gatherer D, et al. Non-random escape pathways from a broadly neutralizing human monoclonal antibody map to a highly conserved region on the hepatitis C virus E2 glycoprotein encompassing amino acids 412-423. PLoS Pathog (2014) 10(8):e1004297. doi:10.1371/journal.ppat.1004297
40. Keck ZY, Li SH, Xia J, von Hahn T, Balfe P, McKeating JA, et al. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol (2009) 83(12):6149–60. doi:10.1128/JVI.00248-09
41. Gu J, Hardy J, Boo I, Vietheer P, McCaffrey K, Alhammad Y, et al. Escape of hepatitis C virus from epitope I neutralization increases sensitivity of other neutralization epitopes. J Virol (2018) 92(9):e02066-17. doi:10.1128/JVI.02066-17
42. Keck ZY, Olson O, Gal-Tanamy M, Xia J, Patel AH, Dreux M, et al. A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. J Virol (2008) 82(12):6067–72. doi:10.1128/JVI.00252-08
43. Velazquez-Moctezuma R, Law M, Bukh J, Prentoe J. Applying antibody-sensitive hypervariable region 1-deleted hepatitis C virus to the study of escape pathways of neutralizing human monoclonal antibody AR5A. PLoS Pathog (2017) 13(2):e1006214. doi:10.1371/journal.ppat.1006214
44. Fuerst TR, Pierce BG, Keck Z-Y, Foung SKH. Designing a B cell-based vaccine against a highly variable hepatitis C virus. Front Microbiol (2018) 8:2692. doi:10.3389/fmicb.2017.02692
45. Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, et al. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A (2012) 109(16):6205–10. doi:10.1073/pnas.1114927109
46. Drummer HE. Challenges to the development of vaccines to hepatitis C virus that elicit neutralizing antibodies. Front Microbiol (2014) 5:329. doi:10.3389/fmicb.2014.00329
47. Fauvelle C, Colpitts CC, Keck ZY, Pierce BG, Foung SK, Baumert TF. Hepatitis C virus vaccine candidates inducing protective neutralizing antibodies. Expert Rev Vaccines (2016) 15(12):1535–44. doi:10.1080/14760584.2016.1194759
48. Keck ZY, Sung VM, Perkins S, Rowe J, Paul S, Liang TJ, et al. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J Virol (2004) 78(13):7257–63. doi:10.1128/JVI.78.13.7257-7263.2004
49. Kong L, Kadam RU, Giang E, Ruwona TB, Nieusma T, Culhane JC, et al. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314-324 in complex with antibody IGH526. J Mol Biol (2015) 427(16):2617–28. doi:10.1016/j.jmb.2015.06.012
50. Meunier JC, Russell RS, Goossens V, Priem S, Walter H, Depla E, et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol (2008) 82(2):966–73. doi:10.1128/JVI.01872-07
51. Khan AG, Whidby J, Miller MT, Scarborough H, Zatorski AV, Cygan A, et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature (2014) 509(7500):381–4. doi:10.1038/nature13117
52. Castelli M, Clementi N, Sautto GA, Pfaff J, Kahle KM, Barnes T, et al. HCV E2 core structures and mAbs: something is still missing. Drug Discov Today (2014) 19(12):1964–70. doi:10.1016/j.drudis.2014.08.011
53. Fraser J, Boo I, Poumbourios P, Drummer HE. Hepatitis C virus (HCV) envelope glycoproteins E1 and E2 contain reduced cysteine residues essential for virus entry. J Biol Chem (2011) 286(37):31984–92. doi:10.1074/jbc.M111.269605
54. Owsianka AM, Timms JM, Tarr AW, Brown RJ, Hickling TP, Szwejk A, et al. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol (2006) 80(17):8695–704. doi:10.1128/JVI.00271-06
55. Harman C, Zhong L, Ma L, Liu P, Deng L, Zhao Z, et al. A view of the E2-CD81 interface at the binding site of a neutralizing antibody against hepatitis C virus. J Virol (2015) 89(1):492–501. doi:10.1128/JVI.01661-14
56. Drummer HE, Boo I, Maerz AL, Poumbourios P. A conserved Gly436-Trp-Leu-Ala-Gly-Leu-Phe-Tyr motif in hepatitis C virus glycoprotein E2 is a determinant of CD81 binding and viral entry. J Virol (2006) 80(16):7844–53. doi:10.1128/JVI.00029-06
57. Duan H, Kachko A, Zhong L, Struble E, Pandey S, Yan H, et al. Amino acid residue-specific neutralization and nonneutralization of hepatitis C virus by monoclonal antibodies to the E2 protein. J Virol (2012) 86(23):12686–94. doi:10.1128/JVI.00994-12
58. Vasiliauskaite I, Owsianka A, England P, Khan AG, Cole S, Bankwitz D, et al. Conformational flexibility in the immunoglobulin-like domain of the hepatitis C virus glycoprotein E2. MBio (2017) 8(3):e00382–17. doi:10.1128/mBio.00382-17
59. Deng L, Ma L, Virata-Theimer ML, Zhong L, Yan H, Zhao Z, et al. Discrete conformations of epitope II on the hepatitis C virus E2 protein for antibody-mediated neutralization and nonneutralization. Proc Natl Acad Sci U S A (2014) 111(29):10690–5. doi:10.1073/pnas.1411317111
60. Zhang P, Zhong L, Struble EB, Watanabe H, Kachko A, Mihalik K, et al. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc Natl Acad Sci U S A (2009) 106(18):7537–41. doi:10.1073/pnas.0902749106
61. Meola A, Tarr AW, England P, Meredith LW, McClure CP, Foung SK, et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J Virol (2015) 89(4):2170–81. doi:10.1128/JVI.02190-14
62. Kong L, Giang E, Nieusma T, Robbins JB, Deller MC, Stanfield RL, et al. Structure of hepatitis C virus envelope glycoprotein E2 antigenic site 412 to 423 in complex with antibody AP33. J Virol (2012) 86(23):13085–8. doi:10.1128/JVI.01939-12
63. Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A (2012) 109(24):9499–504. doi:10.1073/pnas.1202924109
64. Li Y, Pierce BG, Wang Q, Keck ZY, Fuerst TR, Foung SK, et al. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J Biol Chem (2015) 290(16):10117–25. doi:10.1074/jbc.M115.643528
65. Potter JA, Owsianka AM, Jeffery N, Matthews DJ, Keck ZY, Lau P, et al. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J Virol (2012) 86(23):12923–32. doi:10.1128/JVI.02052-12
66. Pantua H, Diao J, Ultsch M, Hazen M, Mathieu M, McCutcheon K, et al. Glycan shifting on hepatitis C virus (HCV) E2 glycoprotein is a mechanism for escape from broadly neutralizing antibodies. J Mol Biol (2013) 425(11):1899–914. doi:10.1016/j.jmb.2013.02.025
67. Deng L, Zhong L, Struble E, Duan H, Ma L, Harman C, et al. Structural evidence for a bifurcated mode of action in the antibody-mediated neutralization of hepatitis C virus. Proc Natl Acad Sci U S A (2013) 110(18):7418–22. doi:10.1073/pnas.1305306110
68. Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog (2013) 9(5):e1003364. doi:10.1371/journal.ppat.1003364
69. Keck Z-Y, Wang Y, Lau P, Lund G, Rangarajan S, Fauvelle C, et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology (2016) 64(6):1922–33. doi:10.1002/hep.28850
70. Kong L, Lee DE, Kadam RU, Liu T, Giang E, Nieusma T, et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc Natl Acad Sci U S A (2016) 113(45):12768–73. doi:10.1073/pnas.1609780113
71. Desombere I, Fafi-Kremer S, Van Houtte F, Pessaux P, Farhoudi A, Heydmann L, et al. Monoclonal anti-envelope antibody AP33 protects humanized mice against a patient-derived hepatitis C virus challenge. Hepatology (2016) 63(4):1120–34. doi:10.1002/hep.28428
72. Tarr AW, Owsianka AM, Jayaraj D, Brown RJ, Hickling TP, Irving WL, et al. Determination of the human antibody response to the epitope defined by the hepatitis C virus-neutralizing monoclonal antibody AP33. J Gen Virol (2007) 88(Pt 11):2991–3001. doi:10.1099/vir.0.83065-0
73. Konermann L, Pan J, Liu YH. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev (2011) 40(3):1224–34. doi:10.1039/c0cs00113a
74. Petsalaki E, Russell RB. Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol (2008) 19(4):344–50. doi:10.1016/j.copbio.2008.06.004
75. Cunha ES, Sfriso P, Rojas AL, Roversi P, Hospital A, Orozco M, et al. Mechanism of structural tuning of the hepatitis C virus human cellular receptor CD81 large extracellular loop. Structure (2017) 25(1):53–65. doi:10.1016/j.str.2016.11.003
76. Kitadokoro K, Ponassi M, Galli G, Petracca R, Falugi F, Grandi G, et al. Subunit association and conformational flexibility in the head subdomain of human CD81 large extracellular loop. Biol Chem (2002) 383(9):1447–52. doi:10.1515/BC.2002.164
77. Kuhn RJ, Dowd KA, Beth Post C, Pierson TC. Shake, rattle, and roll: impact of the dynamics of flavivirus particles on their interactions with the host. Virology (2015) 479-480:508–17. doi:10.1016/j.virol.2015.03.025
78. Sabo MC, Luca VC, Ray SC, Bukh J, Fremont DH, Diamond MS. Hepatitis C virus epitope exposure and neutralization by antibodies is affected by time and temperature. Virology (2012) 422(2):174–84. doi:10.1016/j.virol.2011.10.023
79. Camacho CJ, Katsumata Y, Ascherman DP. Structural and thermodynamic approach to peptide immunogenicity. PLoS Comput Biol (2008) 4(11):e1000231. doi:10.1371/journal.pcbi.1000231
80. Pierce BG, Boucher EN, Piepenbrink KH, Ejemel M, Rapp CA, Thomas WD Jr, et al. Structure-based design of hepatitis C virus vaccines that elicit neutralizing antibody responses to a conserved epitope. J Virol (2017) 91(20):e01032-17. doi:10.1128/JVI.01032-17
81. Sandomenico A, Leonardi A, Berisio R, Sanguigno L, Foca G, Foca A, et al. Generation and characterization of monoclonal antibodies against a cyclic variant of hepatitis C virus E2 epitope 412-422. J Virol (2016) 90(7):3745–59. doi:10.1128/JVI.02397-15
82. Correia BE, Bates JT, Loomis RJ, Baneyx G, Carrico C, Jardine JG, et al. Proof of principle for epitope-focused vaccine design. Nature (2014) 507(7491):201–6. doi:10.1038/nature12966
83. Burton DR. Scaffolding to build a rational vaccine design strategy. Proc Natl Acad Sci U S A (2010) 107(42):17859–60. doi:10.1073/pnas.1012923107
84. He L, Cheng Y, Kong L, Azadnia P, Giang E, Kim J, et al. Approaching rational epitope vaccine design for hepatitis C virus with meta-server and multivalent scaffolding. Sci Rep (2015) 5:12501. doi:10.1038/srep12501
Keywords: hepatitis C virus, glycoprotein E2, neutralizing antibodies, conformational flexibility, immunoglobulin-like domain, CD81-binding site, vaccine design
Citation: Ströh LJ, Nagarathinam K and Krey T (2018) Conformational Flexibility in the CD81-Binding Site of the Hepatitis C Virus Glycoprotein E2. Front. Immunol. 9:1396. doi: 10.3389/fimmu.2018.01396
Received: 05 April 2018; Accepted: 05 June 2018;
Published: 18 June 2018
Edited by:
Steven Foung, Stanford University, United StatesReviewed by:
Heidi Drummer, Burnet Institute, AustraliaJannick Prentoe, University of Copenhagen, Denmark
Copyright: © 2018 Ströh, Nagarathinam and Krey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Krey, a3JleS50aG9tYXNAbWgtaGFubm92ZXIuZGU=