 Sneha Sant1
Sneha Sant1 Ludivine Grzelak1,2
Ludivine Grzelak1,2 Zhongfang Wang1Angela Pizzolla1
Zhongfang Wang1Angela Pizzolla1 Marios Koutsakos1
Marios Koutsakos1 Jane Crowe3
Jane Crowe3 Thomas Loudovaris4
Thomas Loudovaris4 Stuart I. Mannering4
Stuart I. Mannering4 Glen P. Westall5Linda M. Wakim1
Glen P. Westall5Linda M. Wakim1 Jamie Rossjohn6,7,8Stephanie Gras6,7
Jamie Rossjohn6,7,8Stephanie Gras6,7 Michael Richards9
Michael Richards9 Jianqing Xu10
Jianqing Xu10 Paul G. Thomas11
Paul G. Thomas11 Liyen Loh1
Liyen Loh1 Thi H. O. Nguyen1*†
Thi H. O. Nguyen1*† Katherine Kedzierska1*†
Katherine Kedzierska1*†
- 1Department of Microbiology and Immunology, Peter Doherty Institute for Infection and Immunity, University of Melbourne, Melbourne, VIC, Australia
- 2École Normale Supérieure Paris-Saclay, Université Paris-Saclay, Cachan, France
- 3Deepdene Surgery, Deepdene, VIC, Australia
- 4Immunology and Diabetes Unit, St. Vincent’s Institute of Medical Research, Fitzroy, VIC, Australia
- 5Lung Transplant Unit, Alfred Hospital, Melbourne, VIC, Australia
- 6Infection and Immunity Program, Department of Biochemistry and Molecular Biology, Biomedicine Discovery Institute, Monash University, Clayton, VIC, Australia
- 7ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Clayton, VIC, Australia
- 8School of Medicine, Institute of Infection and Immunity, Cardiff University, Cardiff, United Kingdom
- 9Victorian Infectious Diseases Service, The Royal Melbourne Hospital, Peter Doherty Institute for Infection and Immunity, Parkville, VIC, Australia
- 10Shanghai Public Health Clinical Center, Institutes of Biomedical Sciences, Key Laboratory of Medical Molecular Virology of Ministry of Education/Health, Shanghai Medical College, Fudan University, Shanghai, China
- 11Department of Immunology, St Jude Children’s Research Hospital, Memphis, TN, United States
CD8+ T cells recognizing antigenic peptides derived from conserved internal viral proteins confer broad protection against distinct influenza viruses. As memory CD8+ T cells change throughout the human lifetime and across tissue compartments, we investigated how T cell receptor (TCR) composition and diversity relate to memory CD8+ T cells across anatomical sites and immunological phases of human life. We used ex vivo peptide-HLA tetramer magnetic enrichment, single-cell multiplex RT-PCR for both the TCR-alpha (TCRα) and TCR-beta (TCRβ) chains, and new TCRdist and grouping of lymphocyte interactions by paratope hotspots (GLIPH) algorithms to compare TCRs directed against the most prominent human influenza epitope, HLA-A*02:01-M158–66 (A2+M158). We dissected memory TCR repertoires directed toward A2+M158 CD8+ T cells within human tissues and compared them to human peripheral blood of young and elderly adults. Furthermore, we compared these memory CD8+ T cell repertoires to A2+M158 CD8+ TCRs during acute influenza disease in patients hospitalized with avian A/H7N9 virus. Our study provides the first ex vivo comparative analysis of paired antigen-specific TCR-α/β clonotypes across different tissues and peripheral blood across different age groups. We show that human A2+M158 CD8+ T cells can be readily detected in human lungs, spleens, and lymph nodes, and that tissue A2+M158 TCRαβ repertoires reflect A2+M158 TCRαβ clonotypes derived from peripheral blood in healthy adults and influenza-infected patients. A2+M158 TCRαβ repertoires displayed distinct features only in elderly adults, with large private TCRαβ clonotypes replacing the prominent and public TRBV19/TRAV27 TCRs. Our study provides novel findings on influenza-specific TCRαβ repertoires within human tissues, raises the question of how we can prevent the loss of optimal TCRαβ signatures with aging, and provides important insights into the rational design of T cell-mediated vaccines and immunotherapies.
Introduction
Influenza infections cause significant morbidity and mortality, with an estimated 3–5 million cases of severe illness and approximately 290,000–650,000 deaths worldwide annually1 (January 2018). In particular, young children, pregnant women, and elderly individuals are at high risk of severe influenza disease, often leading to high hospitalization rates. While current vaccination regimens, eliciting B cell and follicular T cell responses (1), are our most effective way of controlling annual influenza epidemics, the constantly changing nature of influenza viruses can easily evade preexisting host antibody responses (2). By contrast, killer CD8+ T cells are effective in inducing long-lived heterologous immunity toward different influenza strains (3, 4), thus leading to a rapid recovery of the host (5–7). CD8+ T cells eliminate influenza virus infection by recognizing conserved viral proteins presented by major histocompatibility complex (MHC) class I glycoproteins (8), thus protecting influenza-infected individuals against a diverse range of human-derived (i.e., H1N1 and H3N2), swine-derived (i.e., pandemic H1N1), and the more recently described deadly avian-derived (i.e., H5N1, H7N9, and H9N2) influenza viruses (9–11). Such recognition of peptide-MHC-I (p-MHC-I) complexes occurs via surface-expressed T cell receptors (TCRs) comprising of α and β chains. Following T cell activation, antigen-specific CD8+ T cells proliferate, migrate to the site of infection, and mount an immune response by killing virus-infected cells and producing anti-viral cytokines (12).
CD8+ T cells are highly specific in their interactions with p-MHC-I complexes (13, 14), and small changes within viral peptides can abrogate TCRαβ recognition, leading to viral escape from preexisting immunity (15–19). The vast array of different TCRs is generated by somatic recombination of variable (TRAV for TCRα chain and TRBV for TCRβ chain), diversity (within the β-chain only; Dβ), and joining (Jα and Jβ) gene segments in combination with varying lengths of the complementarity-determining region 3 (CDR3; CDR3α and CDR3β) loops. A set of different TCRαβ receptors, referred to as the TCRαβ repertoire, can recognize specific antigenic peptides in the context of MHC-I. TCRs exhibiting the same gene segment usage and amino acid (aa) sequences at CDR3α and CDR3β regions are defined as TCR clonotypes, and a range of TCR clonotypes displaying either near-identical or diverse features can recognize a given T cell epitope (20). Furthermore, TCR repertoires can be classified as “public,” when they express identical epitope-specific TCR sequences across multiple individuals (21–23), or “private,” when different individuals display distinct (non-overlapping) TCR clonotypes for the same p-MHC-I complex (21, 22, 24). The presence of “public” TCRαβ clonotypes is thought to have a selective evolutionary advantage, as these TCRs can be easily generated in different individuals (25–30).
As TCRs can greatly affect viral clearance, p-MHC-I avidity, and prevention from viral escape (31–33), it is important to understand TCR repertoire diversity and composition within influenza-specific CD8+ T cells directed at prominent human epitopes for designing a much-needed universal T cell-based influenza vaccine. To date, our knowledge of human influenza-specific CD8+ TCRs has been mainly focused on the HLA-A*02:01-restricted immunodominant M158–66 (A2+M158) epitope (7, 34–42), as HLA-A*02:01 is the most common HLA class I allele expressed at 1–54.5% (43) across different ethnic groups worldwide. For example, HLA-A*02:01 is expressed at 0.5–31% in Caucasians and 9–40% in native North Americans (42). Until recently (44), TCR analysis was mainly performed on bulk cells for a single TCRα or TCRβ chain, or following extensive CD8+ T cell cloning (45, 46). However, our recent studies using a single-cell multiplex RT-PCR approach (44) has showed, for the first time, direct ex vivo dissection of paired TCRαβ repertoires directed against the most prominent influenza A2+M158 epitope in healthy adults (36), elderly individuals (40), as well as in hospitalized influenza-infected elderly patients (7). In general, the A2+M158 TCRαβ repertoires were heavily biased toward TRBV19 and TRAV27, with public TRBV19/TRAV27-utilizing clonotypes being its main feature (34, 36), along with other less frequent diverse TCRs (37). Public TRBV19/TRAV27 motifs bearing CDR3α-GGSQGNL and CDR3β-SSIRSYEQ sequences are important for the optimal recognition of A2+M158 epitope, especially the CDR3β-IRS motif being key for the peg-notch mode of recognition (23, 36, 37). However, it is far from clear how the A2+M158 TCRαβ repertoire changes across different age groups, between different tissue compartments and following influenza virus infection. Especially, the composition of A2+M158 TCRαβ repertoire is largely unknown in human tissues, as previous studies have mainly focused on human peripheral blood (9, 10, 35, 36, 47, 48), with only one recent report from our group focusing on different subsets of influenza-specific CD8+ T cells in human lungs (41).
Here, we dissected A2+M158 TCRαβ repertoires across human tissues [lungs, spleen, and lymph node (LN)] and subsequently performed a comprehensive analysis of temporal dynamics of A2+M158 TCRs across different age groups (young/elderly), in the context of acute influenza infection (H7N9-infected hospitalized elderly patients), and across different anatomical locations. Using a combination of direct ex vivo tetramer enrichment, single-cell paired TCRαβ analyses and the newly established algorithm TCRdist (38) and grouping of lymphocyte interactions by paratope hotspots (GLIPH) (39), we compared and contrasted A2+M158 TCRαβ repertoires across the above-mentioned datasets. We show that human A2+M158 CD8+ T cells can be readily detected in human lungs, spleens, and LNs, and that the tissue A2+M158 TCRαβ repertoires reflect those from unmatched peripheral blood A2+M158 TCRαβ repertoires in healthy adults and hospitalized influenza-infected patients. Strikingly, elderly individuals displayed unique features within their A2+M158 TCRαβ repertoires, with largely expanded private TCRαβ clonotypes replacing, at least in part, the public TRBV19/TRAV27 TCRs. Our study provides novel findings on influenza-specific TCRαβ repertoires within human tissues and raises the question of how we can prevent the loss of optimal TCRαβ signatures with aging.
Materials and Methods
Human Peripheral Blood Samples
This study was approved by the University of Melbourne Human Ethics Committee (ID 1443389.3 and 0931311.5). Human experimental work was conducted according to the Declaration of Helsinki Principles and according to the Australian National Health and Medical Research Council (NHMRC) Code of Practice. All donors provided informed written consent for blood donation. Tissues from deceased organ donors were obtained following written informed consent from the next of kin. PMBCs were isolated from heparinized peripheral blood collected from healthy adult volunteers (AD, 22–59 years) at the University of Melbourne and elderly donors (ED, ≥60 years) recruited at Deepdene Medical Clinic (Deepdene, Australia), or from buffy packs of healthy adult donors (AD, 22–59 years) obtained from Australian Red Cross Blood Service (ARCBS). Peripheral blood samples from HLA-A*02:01+ H7N9-infected patients (A9, A10, and A79) were obtained from Shanghai Public Health Clinic Centre (SHAPHC) under the supervision of SHAPHC Ethics Committee (7). PBMCs were also obtained from one patient hospitalized at the Royal Melbourne Hospital in 2017 with seasonal-influenza virus infection and approved by the Monash Health Human Research Ethics Committee (HREC/15/MonH/64).
Human Tissue Samples
Human spleens, LNs (mesenteric and pancreatic), and lung tissue samples were obtained from deceased organ donors following family’s approval and approved by the Australian Red Cross Blood Service Ethics Committee (ID 2015#8). Spleen (SP) and mesenteric/pancreatic LN samples were procured from DonateLife Victoria. Lung (LG) tissue was obtained from the Alfred Hospital’s Lung Tissue Biobank (supported by the NHMRC Lung Fibrosis CRE). All tissue samples were stored and transported in cold PBS and processed within 18 h of procurement. Spleen and LNs were finely chopped and then enzymatically digested with type III Collagenase (1 mg/ml, Worthington, OH, USA) and DNase I (0.5 mg/ml, Roche, Basel, Switzerland) in RPMI media for 1 h at 37°C/5% CO2 (45 min for LN). Enzymatic digestion was halted by the addition of EDTA (0.01 mM) before cells were passed through a 70 µm sieve, washed, and then red blood cells were lysed with RBC lysis solution (0.168 M ammonium chloride, 0.01 mM EDTA, and 12 mM sodium bicarbonate in MilliQ water) before cryopreservation. Lung tissues were similarly processed, with the exception of 7 mg/ml type III Collagenase and 1 mg/ml DNase I digestion, as previously described (41).
Ex Vivo Detection of Influenza-Specific CD8+ T Cells
Between 2 × 107 and 5 × 107 of cells were thawed and washed in RPMI containing DNase I (1 mg/ml). Mouse splenic red pulp has been shown to contaminate magnetic column enrichments (49) and was also observed in our human spleen cell suspensions. Thus, to circumvent the contamination of our tetramer enrichments, spleen cells were thawed and passed through an LS column (Miltenyi Biotec, Bergisch Gladbach, Germany) before use. Blood and tissue cell preparations underwent tetramer-associated magnetic enrichment (TAME) to enrich for A2+M158-specific CD8+ T cells using the A2+M158-tetramer, essentially as previously described (40). Cell fractions (except for lung cells) were surface stained with anti-CD3-PECF594 (clone: UCHT1, BD, San Jose, CA, USA), anti-CD8-PerCpCy5.5 (clone: SK1, BD Pharmingen), anti-CD14-APCH7 (clone: MFP9, BD Pharmingen), anti-CD19-APCH7 (clone: SJ25C1, BD Pharmingen), anti-CD27-BV711 (clone: L128, BD Horizon), anti-CD45RA-FITC (clone: HI100, BD Pharmingen), and LIVE/DEAD™ Fixable Near-IR (Molecular Probes, Eugene, OR, USA). Lymphocytes isolated from lung tissue (41) and H7N9-infected patients (7) did not undergo TAME enrichment, instead, these samples were stained directly ex vivo with A2+M158-tetramer for 1 h at room temperature, washed and stained with surface markers, as previously described. Lung cells were surface stained as previously described (41) with anti-CD103-FITC (clone: Ber-ACT8, Biolegend), anti-CD45RO-PECy7 (clone: UCHL1, eBioscience), anti-CD69-BV421 (clone: FN50, Biolegend), anti-CD8-APCH7 (clone: SK1, Biolegend) and anti-CD3-APC (clone: OKT3, eBioscience). Splenic cells and PBMCs were also surface stained as above to compare the frequency of resident-T cells with lung, spleen and blood. Cells were resuspended in PBS containing 0.5% BSA/2 mM EDTA for single-cell sorting on a FACSAria III (BD Biosciences) or fixed with 1% PFA and acquired on an LSRFortessa II (BD Biosciences). FACS files were analyzed using FlowJo software (V10.2, Treestar, Ashland, OR, USA).
Single-Cell Paired TCRαβ Analysis
Following TAME or direct A2+M158-tetramer staining, single live CD3+CD8+tetramer+CD19−CD14− cells were sorted into pre-chilled 96-well twin.tec plates (Eppendorf, Hamburg, Germany) before performing multiplex-nested RT-PCR for paired TCRαβ analysis essentially as described (36, 40, 41, 44). TCRα and TCRβ sequences were parsed using the TCRdist algorithm (38). CDR3αβ pairs displaying the same aa sequences and V/J gene usage were defined as clonotypes. Heatmaps for CDR3α/β were made using Matplotlib (50). Circos plots were generated using Circos package (51).
Statistical Analysis
Statistical analysis was carried out using GraphPad prism software. Student’s t-test was performed for comparison between two groups. Statistical significance if present is described in figure legend.
TCRβ Cluster Analysis
CDR3β aa sequences obtained after parsing through TCRdist were used to construct clone network analysis using GLIPH (39). Based on the presence of unique motifs in a given dataset, GLIPH was used to calculate the probability of the occurrence of these unique motifs relative to their expected frequency in a naïve TCR dataset. The analysis returned a network of all CDR3β’s as nodes and global, local or singleton interactions as edges. Network clusters were generated using the clusterMaker plugin in Cytoscape V3.5.1.2
Datasets Used in this Study
Comprehensive analyses of A2+M158+CD8+ TCRαβ repertoire diversity was analyzed using paired TCRαβ datasets from donor spleens (SP234, SP235, SP156, and SP583), LNs (LN583), and lungs (LG9 and LG10) generated in this study, as well as from our previously published datasets from peripheral blood TCRαβ clonotypes found in adults (n = 3, AD9, AD16, and AD17) (36), elderly (n = 3, ED9, ED18, and ED31) (40), H7N9-infected patients (n = 3, A9, A10, and A79) (7), and one lung tissue [LG6 (41)]. Therefore, our total dataset (Table 1) comprised of 628 paired TCRαβ sequences that was parsed through TCRdist algorithm, analyzed using GLIPH, and visualized using Cytoscape to contribute to Figures 3–8. Sequence data from our study and the published studies have been made publicly available at VDJDb (52).
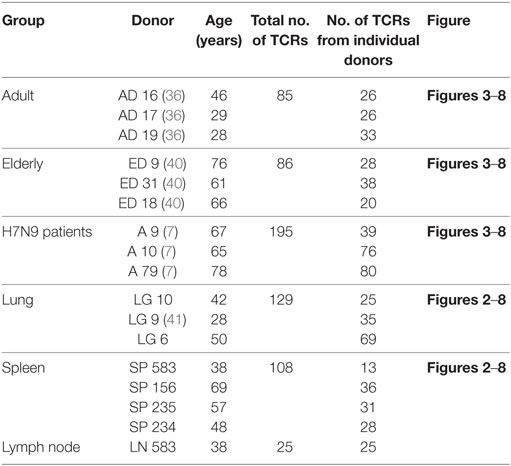
Table 1. Demographics of donors used for T cell receptor (TCR) analyses in this study.
Results
A2+M158+CD8+ T Cells Are Maintained Across Age, Tissues, and During Acute Influenza Infection
Effective CD8+ T cell responses are governed by several factors, including available numbers of naïve or memory CD8+ T cells against the pathogen, as well as the maintenance of antigen-experienced memory CD8+ T cells with aging and across tissue compartments (53–55). Using A2+M158-TAME (56, 57), A2+M158-specific CD8+ T cells can be readily detected in human peripheral blood of healthy adult and elderly individuals (Figure 1Ai) as well as human spleens (n = 3) and LNs (n = 1), and in unenriched human lungs (n = 3) (Figure 1B; Table 1). This clearly demonstrates that distribution of human influenza-specific A2+M158+CD8+ T cells across different anatomical sites is analogous to that of influenza-specific immunodominant DbPA224+CD8+ (24) and DbNP366+CD8+ (21) T cells in mice. Furthermore, effector memory A2+M158+CD8+ T cells can be detected directly ex vivo in hospitalized patients infected with influenza viruses via tetramer enrichment (Figure 1Aii). Thus, A2+M158+CD8+ T cells are prominent in HLA-A*02:01-expressing individuals of different ages across different tissue compartments, with comparable tetramer frequencies of total CD8+ T cells found between blood and tissues (Figure 1C).
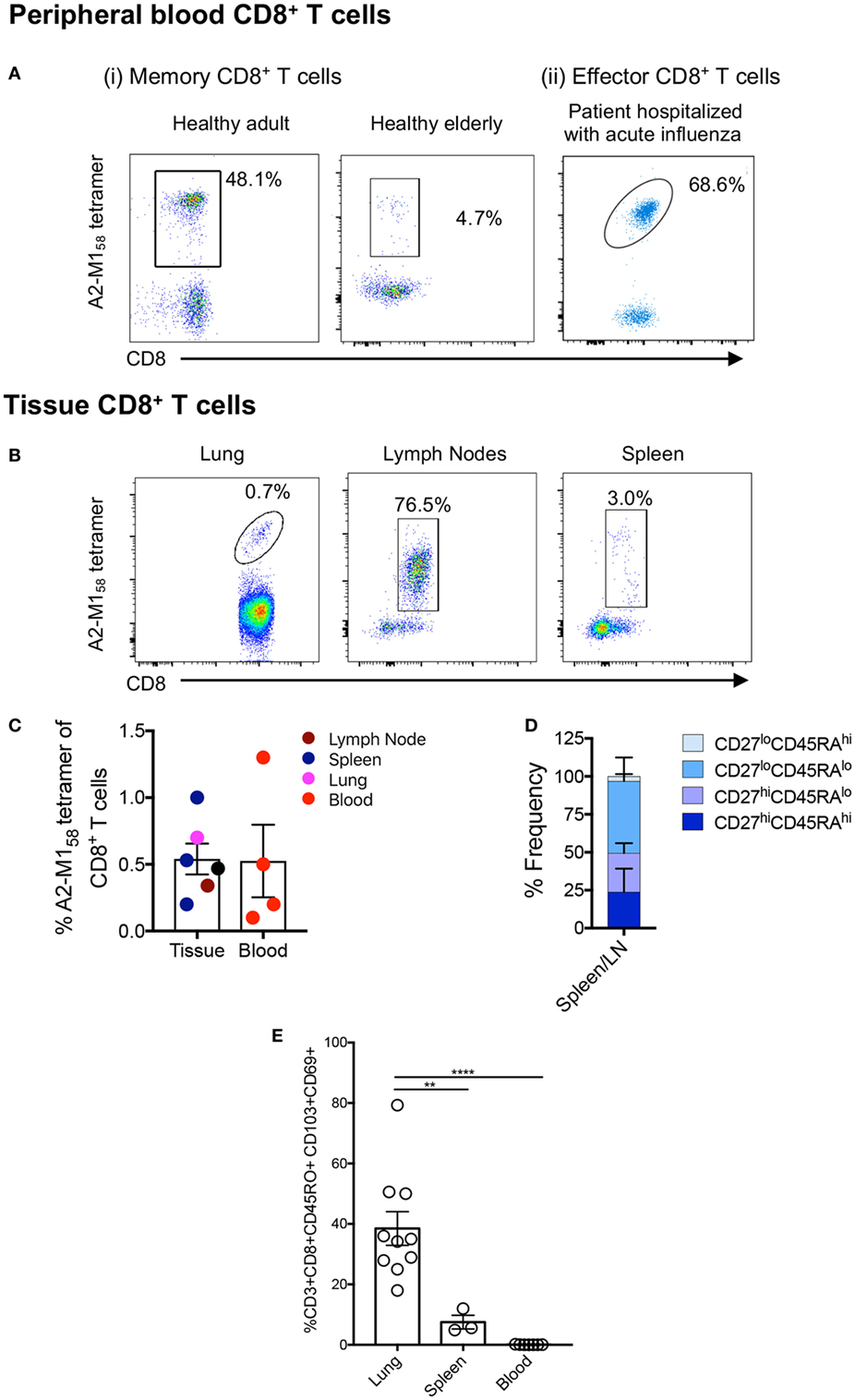
Figure 1. A2+M158-specific CD8+ T cells are prominent across age, tissue, and influenza infection. Representative FACS plots of A2+M158-specific memory CD8+ T cells in the blood of [(A) i] healthy adult and an elderly donor, [(A) ii] in the blood of influenza virus-infected hospitalized patient, and (B) in the lung, spleen, and lymph nodes (LNs) of deceased organ donors. Samples were enriched via tetramer-associated magnetic enrichment (TAME), except for the lung which was directly stained with tetramer. Frequencies of cells gated as viable CD14−CD19−CD3+CD8+ A2+M158-tetramer-positive events are shown. (C) Frequencies of A2/M158+CD8+ T cells in tissues (LNs n = 1 donor, spleen n = 4 donors, and lung n = 1 donor) and blood (n = 3 donors) for A2/M158+CD8+ T cells are shown. Except for the lung, which did not undergo TAME enrichment, A2+M158-tetramer+CD8+ T cell frequencies of total CD8+ T cells were calculated based on the total CD8+ T cell population. No statistical differences across distinct anatomical compartments were found. (D) CD45RA/CD27 memory profiles of A2+M158-specific CD8+ T cells within human tissues are shown. (E) Graph depicts the proportion of CD8+ TRMs (CD103+CD69+) among the total memory CD8+ T cell pool in the lung, spleen, and blood of donors (41). Dots represent individual donors (n = 3–10 donors, one-way ANOVA, Sidak’s multiple comparison).
We have previously characterized the memory phenotypes of A2+M158+CD8+ T cells from the blood of adult and elderly adult donors, which mainly expressed CD27hi, but not CD45RA (40), a marker highly expressed in naïve T cells. By contrast, A2+M158+CD8+ T cells in H7N9-infected patients displayed a more effector-like phenotype (CD38hiHLA-DRhiCD27loCD45RAlo) (7). In tissues, the A2+M158+CD8+ T cells were of a CD27hi/loCD45RAlo memory phenotype in spleens and LNs (Figure 1D), whereas in the lung, they were positive for the CD45RO+ memory marker and displayed a mixture of circulating (CD69−CD103− and CD69+CD103−) and tissue-resident (CD69+CD103+) memory phenotypes (41). Human spleens contained significantly less CD69+CD103+ tissue-resident memory (TRM) T cells compared to the lung, with relatively few TRMs observed in the blood (Figure 1E).
TCRαβ Repertoire of A2+M158+CD8+ T Cells in Human Tissues
To dissect A2+M158+ TCRαβ signatures within human tissues, we utilized tetramer staining together with single-cell sorting and multiplex RT-PCR approach (7, 36, 40, 41, 44) for spleens (n = 3), lungs (n = 4), and LN (n = 1), with spleens and LN following the TAME protocol and gating strategy, as shown in Figure S1 in Supplementary Material. Our data showed that the composition and distribution of A2+M158+ TCRαβ signatures within the human tissues (Table 2) was similar to that of TCRαβ clonotypes within peripheral blood of healthy individuals (36, 48). The vast majority of TCRαβ clonotypes in tissues utilized TRBV19 (87 ± 16%) and TRAV27 (36.8 ± 25%) (Table 2; Figures 2 and 3), similar to what has been observed in peripheral blood from healthy adults, with TRBV19 (100%) and TRAV27 (70 ± 21%) (36). The presence of the public TRBV19/TRAV27 TCRαβ was also observed across the tissues, with a mean of 13% (range 2–23%), detected in all the samples tested with the exception of one spleen (#234; Figures 2 and 3; Table 2), compared to a mean of 48% (15–67%) in healthy adults’ blood (36). Our data show, for the first time, that the distribution of human influenza-specific TCRαβ clonotypes in human tissues closely resembles TCRαβ repertoires in peripheral blood of healthy adults.
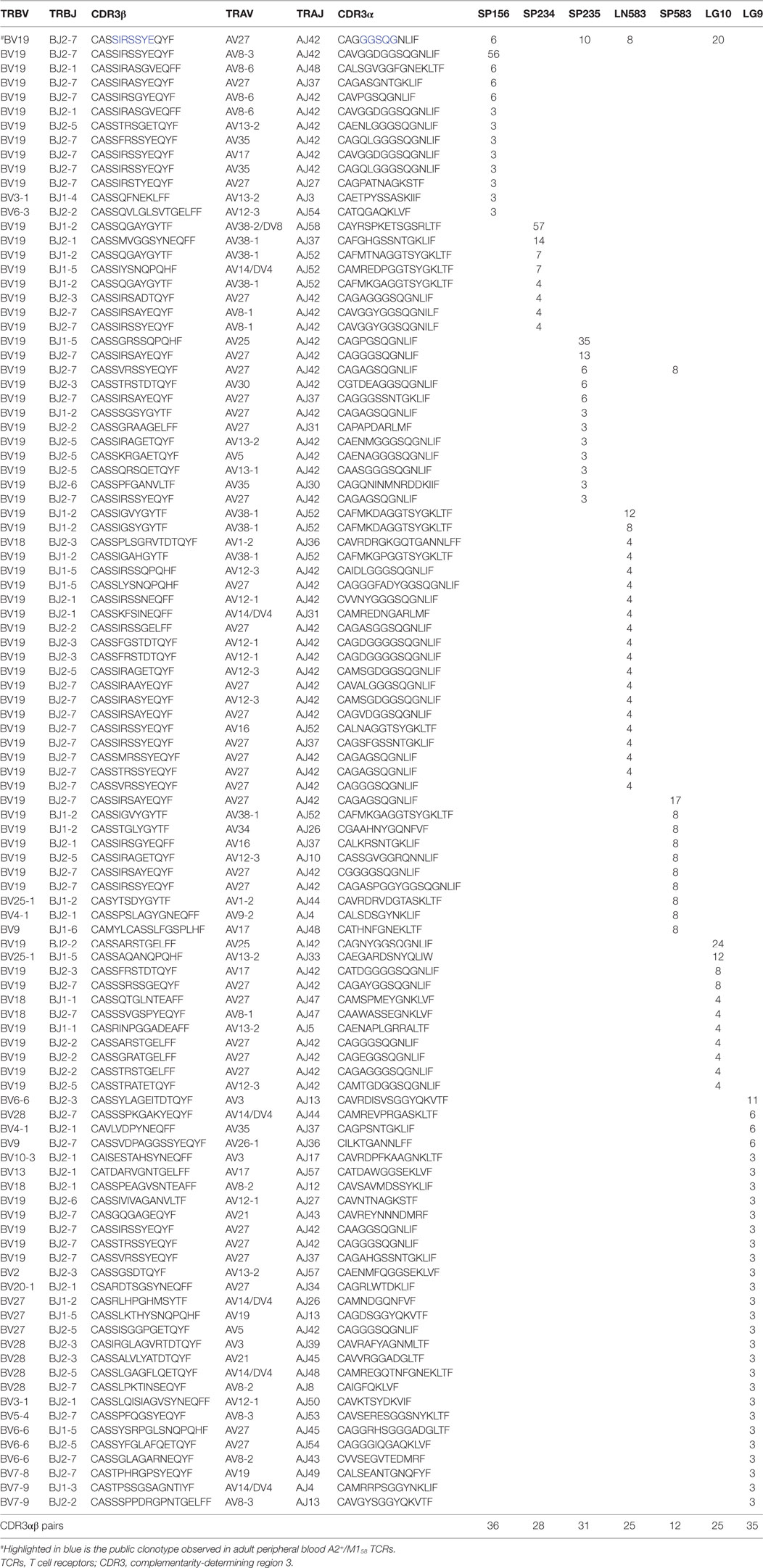
Table 2. Paired TRBV-TRBJ/TRAV-TRAJ clonotype frequencies of A2+M158 TCRs in human tissues.
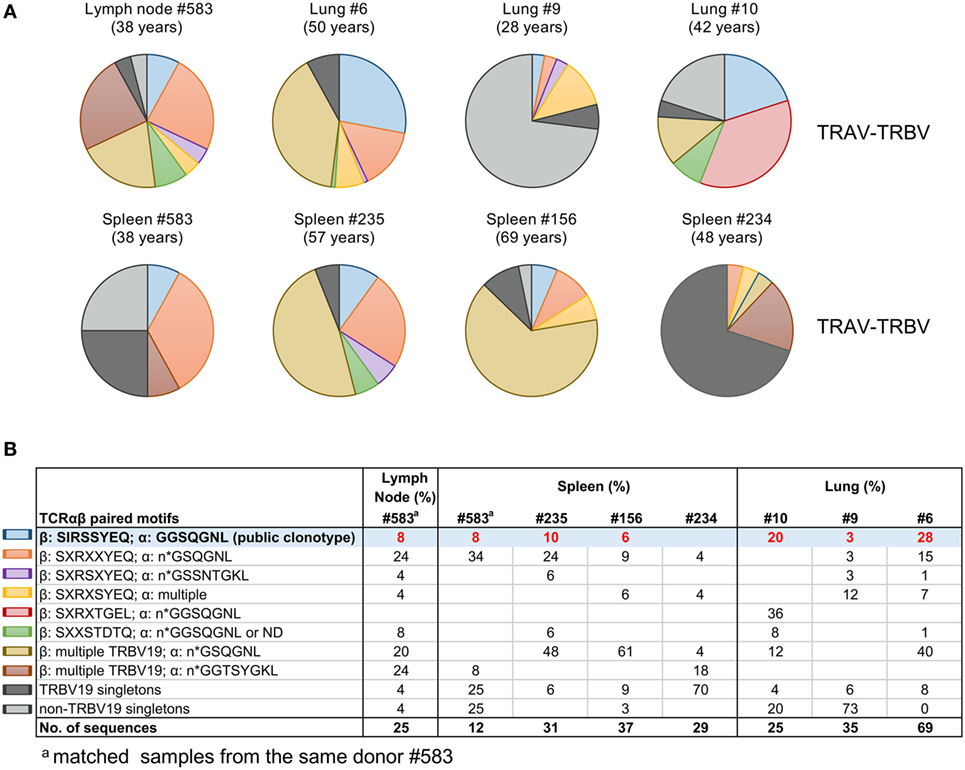
Figure 2. Tissue A2+M158-specific CD8+ T cell receptors (TCRs) display public clonotype signatures. (A) Pie-chart distributions and (B) table of paired TCRα/β clonotype repertoires of A2+M158-specific memory CD8+ T cells in human tissues. TRB-TRA clonotypes were grouped according to the presence of “public” [blue pie-slice in (A) and shaded in (B)] or public-like motifs (other colors). TRBV19 singletons are in dark gray and non-TRBV19 singletons in light gray. Singletons are defined as TCRβ chain clonotypes with variable TCRα chains. Matched samples from the same donor #583 are indicated.
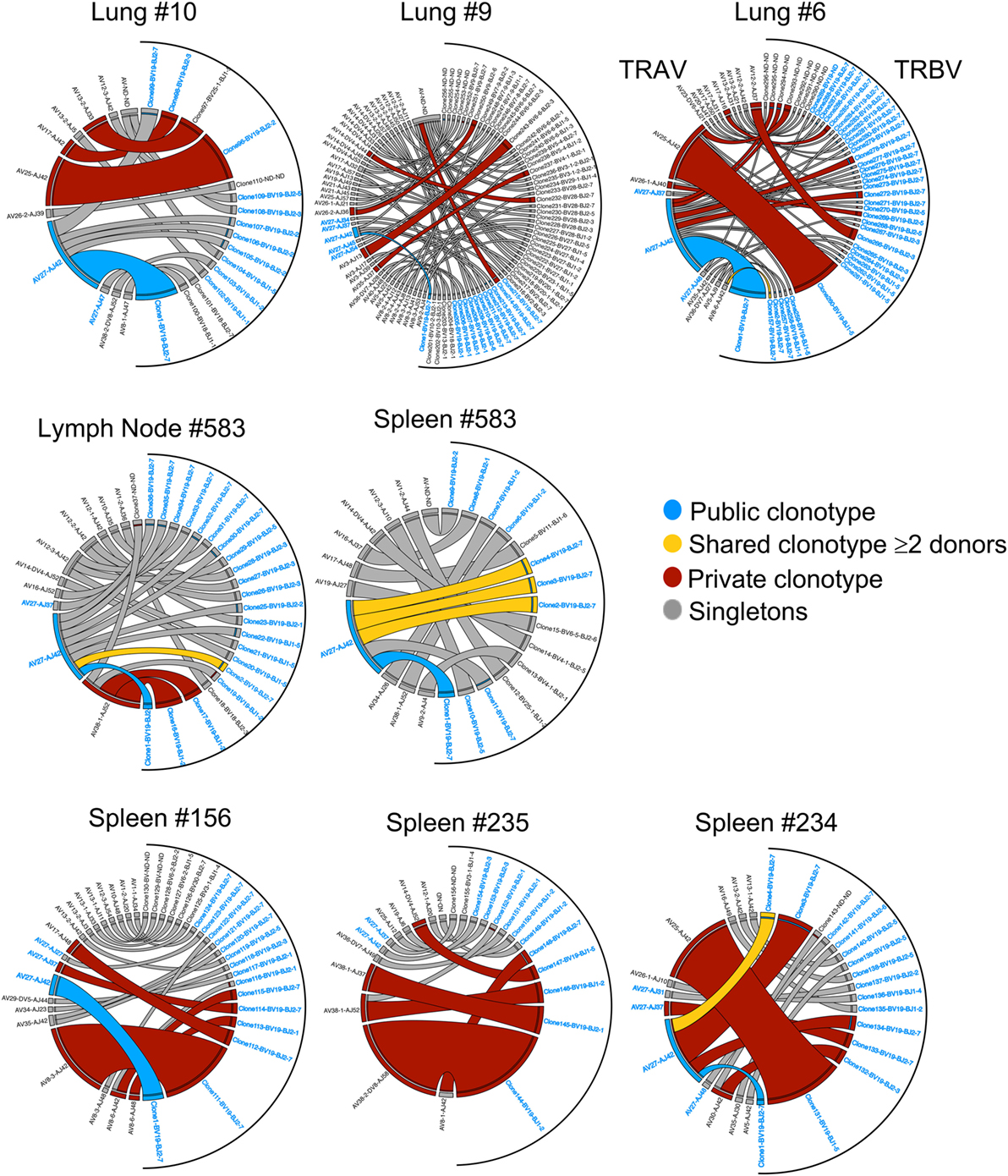
Figure 3. Clonotype frequency of tissue A2+M158-specific CD8+ T cell receptors (TCRs). Circos plots showing the distribution of TRA-TRB paired clonotypes across tissues (lung, spleen, and lymph node). Each segment defines individual clonotypes and width of segment correlates to frequency of the clonotype. Public clonotype is in blue segments, shared clonotypes between donors are in yellow, private or non-shared clonotypes observed more than once are in maroon, or only once as singletons in gray. For clonotypes having TRBV19/TRAV27 gene usage, the labels have been highlighted in blue. Circos plots were generated using Circos package (51).
We further performed comprehensive analyses of A2+M158+CD8+ TCRαβ repertoire diversity across human tissues using our newly generated dataset in comparison to our previously published datasets from peripheral blood TCRαβ clonotypes found in adults (n = 3) (36), elderly (n = 3) (40), H7N9-infected patients (n = 3) (7), and one lung tissue (LG6) (41). Our total dataset comprised of 628 paired TCRαβ sequences that was parsed through TCRdist algorithm (38). TCRdist was used to calculate a normalized Jensens–Shanon divergence to measure the gene segment usage by comparing the probability occurrence of our A2+M158+ epitope-specific TCRs against a background dataset of other epitope-specific TCRs (38). Gene segment pairing was represented as average-linkage dendrograms (Figure 4), where each clonotype presented a constant vertical height, and the gene segment within each clonotype was joined by curve segments to neighboring genes. Thickness of the segment indicated the number of unique clones sharing the same gene, with the TRBV segment almost completely comprising of TRBV19 (in red) across all peripheral blood adult/elderly donors and spleen and LN samples. With respect to background TCRs (38), the analysis of A2+M158+ TCR repertoire indicated 16-fold enrichments for TRBV19, 8- to 16-fold enrichments for TRAV27 gene usage, 2-fold enrichments for TRBJ2-7, and 4- to 8-fold enrichments for TRAJ42, with each arrow corresponding to a 2-fold increase or decrease (Figure 4). Overall, we found no striking differences for the most preferred A2+M158+CD8+ gene usage represented by TRBV19/TRBJ2-7/TRAV27/TRAJ42 (36). However, A2+M158+CD8+ TCRα-chain within healthy elderly, elderly H7N9-infected patients, spleen, and lung donors displayed increased variability within Vα and Jα gene usage. Thus, our data show that the TCRαβ diversity within A2+M158+CD8+ T cells results from a diverse gene usage at Vα, Jα, and Jβ locus.
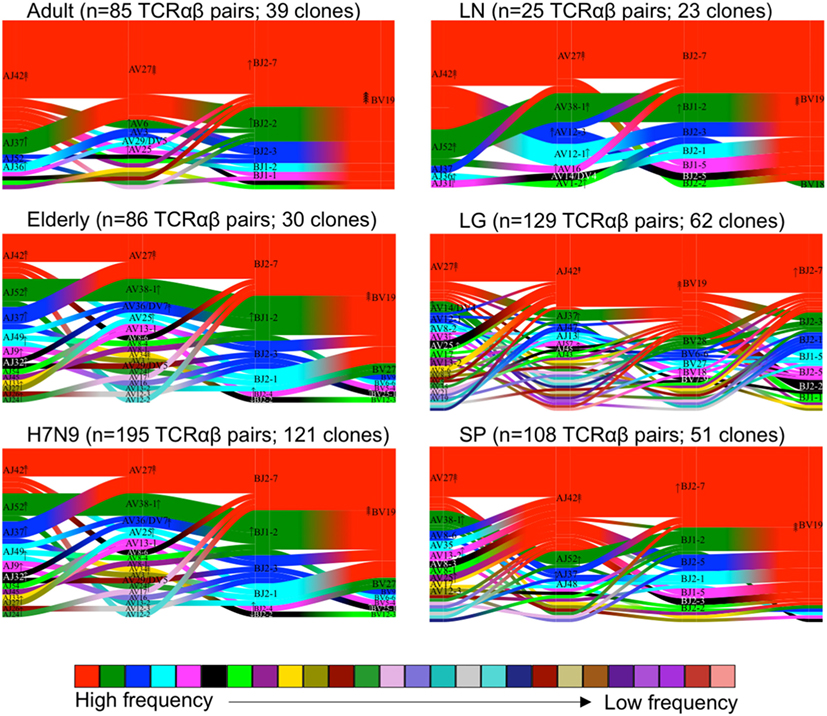
Figure 4. Maintenance of clonal diversity across age group and tissue compartment. Gene segment usage and pairing landscapes are shown for each group. Each clonotype is assigned the same vertical length irrespective of clonotype size. Each vertical stack reflects the V and J gene segment usage and pairing is shown by curved connecting lines. Genes are ranked in color by the frequency distribution with red being the highest frequency, followed by green, dark blue, aqua, magenta, black, and thereafter. Enrichment or depletion of gene usage is indicated by up or down arrows, respectively, where one arrowhead correlates to a two-fold increase or decrease.
Conserved CDR3α and CDR3β Length Usage in Human Tissues
As CDR3 loops play an important role in p-MHC-I contact (58), we next examined the CDR3 aa length within A2+M158+CD8+ TCRs in human tissues, taking into consideration the size of each clonotype to highlight the contribution of the expanded clonotypes. As shown by our previous data, A2+M158+CD8+ TCRβs within the peripheral blood of adults (36) and H7N9-infected patients (7) utilized mainly CDR3β lengths of eight aa, representing 92 ± 7 and 94 ± 3.5%, respectively. Such strong bias toward CDR3β lengths of eight aa was also found in human tissues (mean 66.3 ± 31%) (Figure 5A) but was greatly reduced in healthy elderly donors (40). CDR3α loops on the other hand encompassed a wide range of CDR3α lengths, displaying specific variations across different datasets (Figure 5B). Cumulative analysis across different groups showed that only ED#9 (66%) and SP#235 (67%) showed the preferred seven aa length as reported for adult (68 ± 30.5%) A2+M158+CD8+ TCRα chains, while the remaining donors from healthy elderly individuals, H7N9-infected elderly patients and tissues had longer CDR3α lengths (Figure 5B). Similar observations were reported by Gil et al. for A2+M158+CD8+ TCRα chains within elderly donors (34). Interestingly, CDR3α chains with seven aa in the elderly maintained a biased pairing with TRAV27 and TRAJ42 genes. Thus, our paired TCRαβ analyses highlight the plasticity of TRBV19 genes with a preferential CDR3β length of eight aa to pair with diverse TRAV regions with CDR3α’s across different lengths.
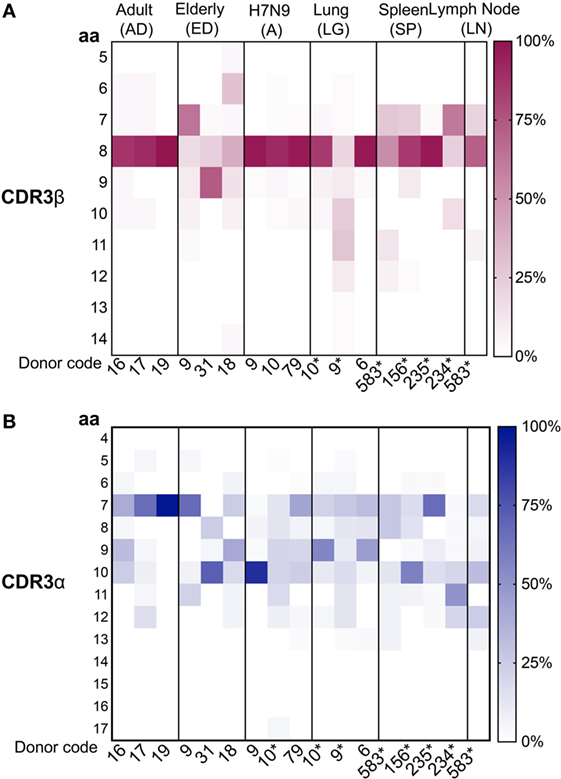
Figure 5. Conserved complementarity-determining region 3 (CDR3)β and diverse CDR3α amino acid (aa) length usage. Distribution of (A) CDR3β and (B) CDRα aa length usage across adult, elderly, lung, spleen, lymph node (LN) (n = 1), and H7N9 hospitalized patients (all groups have n = 3 unless specified). CDR3 lengths for donors marked by “*” were generated from this study. Data from unmarked donors were taken from our previous studies as described [adult (36), elderly (40), H7N9 hospitalized patients (7), and lung (41)]. Heatmaps were generated using Matplotlib package (50).
Large Dominant Private TCRαβ Clusters Dominate A2+M158+CD8+ T Cells in the Elderly Donors
To further understand the distribution of A2+M158+CD8+ TCRs based on the level of similarity within the CDR3 regions, TCRdist (38) computes the similarity-weighted hamming distance between given TCRs in a group and presents the two-dimensional kernel principal component analysis (a non-linear form of PCA) of TCR clusters reflecting the clonal expansions and gene usage. TCRdist also allows identification of top-scoring CDR3α and CDR3β motifs crucial in recognizing the p-MHC-I complex. TCRdist quantifies all these by comparing a given set of epitope-specific TCRs against a background set of publicly available non-epitope-specific TCR dataset. Using TCRdist, we observed that adult, H7N9-infected elderly and tissue samples exhibited TCR Vβ clusters dictated by large clonotypes with TRBV19 usage (Figure 6A, in red), while, the dispersion in TCR Vα clusters resulted in the presence of large clonotypes of non-TRAV27 usage (Figure 6A). Conversely, elderly TCR clusters highlighted the presence of large private clonotypes with decreased TRBV19/TRAV27 gene usage (Figure 6A, second row panels). However, despite such reduced frequency of TRBV19 and TRAV27 gene usage in elderly donors, analysis of the key “RS” motif in TRBV-CDR3β and the glycine-rich “GG” motif in TRAV27-CDR3α clearly demonstrates a preservation of those conserved CDR3 residues across age groups, tissues and following influenza virus infection (Figure 6B). The patterns observed in our TCR clusters were further complemented by our TCRlogo summary of CDR3 aa sequences and corresponding gene frequencies (Figure 7). As compared to the adult and H7N9-infected elderly (Figures 7A,C; higher proportion of red shades), elderly adult TCRs had lower probability scores (probability of the TCR being generated from germ-line TCRs; blue shades) (Figure 7B). Similarly, tissues also presented a large cluster with high probability scores (Figures 7D–F). Observations at a single chain level further showed that elderly A2+M158+CD8+ TCRs were composed of more diverse and rarer TCRs, as shown by high proportion of TCRs with low probability scores (Figure 7). However, elderly donors TCRs still presented “RS” motif containing TRBV19 (five distinct TCRs) pairing with “GG” containing diverse TRAV27 (five distinct TCRs). The representation of “RS” and “GG” motif was more pronounced in adult, H7N9 patients, and tissues (Figure 7). Therefore, our analyses highlight that despite large clonal expansions of private TCRαβ clonotypes found in the elderly individuals (36, 37), the key motifs are still present in the memory A2+M158+CD8+ T cell pool, but with much lower probability for the elderly individuals. Most striking is that these key motifs are still efficiently recruited following severe influenza virus infection in the elderly patients (7).
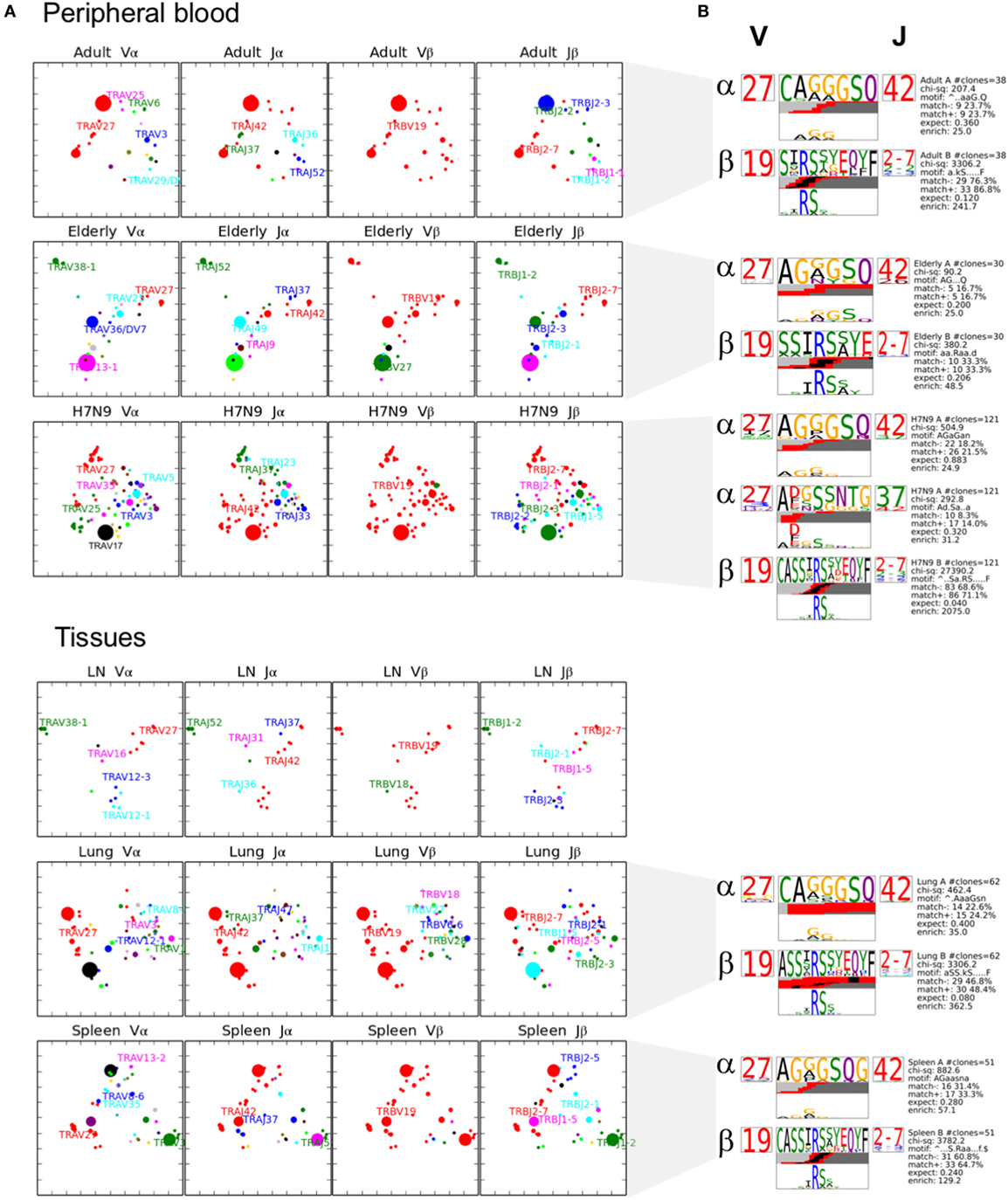
Figure 6. Private clonotypes dominate elderly T cell receptors (TCRs), while public TCRs are represented in blood and across different tissues. (A) 2D kernel principal component analysis (kPCA) projections for healthy adult, healthy elderly, elderly H7N9 hospitalized patients, lung, lymph node (LN) (n = 1), and spleen (all groups have n = 3 unless specified). Size of the clone correlates with clonotype size and color correlates to gene usage. Most prevalent gene usages are mentioned within the plots matching with clonotype color. Each row represents group and each column is the same 2D kPCA projection of the four gene segment usage (Vα, Jα, Vβ, and Jβ). (B) Using TCRlogo in TCRdist algorithm, top-scoring complementarity-determining region 3 (CDR3)β and CDR3α motifs are shown for each group, except for LN. V and J gene usage are indicated left and right of motifs, respectively, and bottom panel highlights the motif enriched by calculating against a background dataset of naïve non-A2+M158-specific CD8+ TCRs.
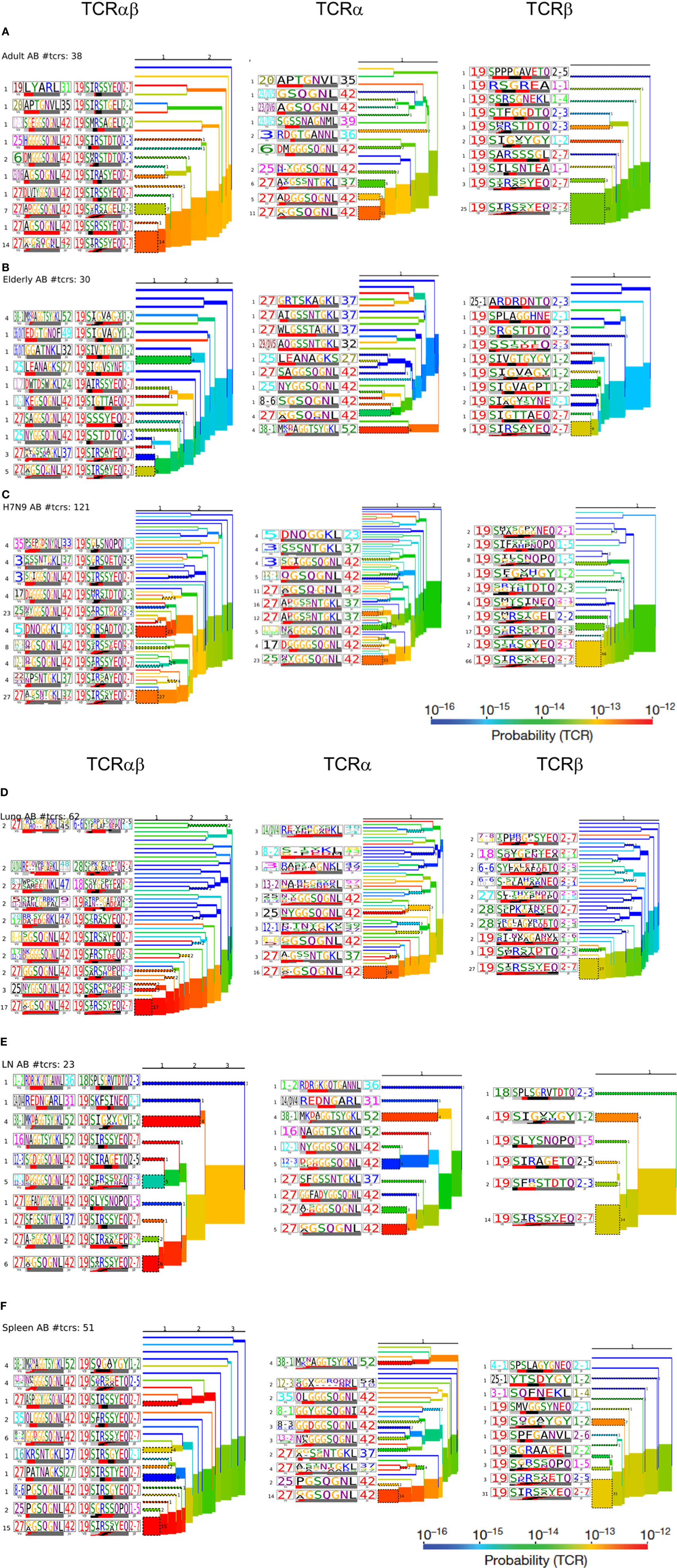
Figure 7. Hierarchal clustering of T cell receptors (TCRs) highlights diversity of aging A2+M158-specific CD8+ TCRs: TCRαβ, TCRα, and TCRβ clustering along with corresponding TCRlogos for (A) adult, (B) elderly adult, (C) elderly H7N9 patients, (D) lungs, (E) lymph nodes, and (F) spleen. Number on the branches and next to TCRlogos depicts number of TCRs contributing to the cluster. Color of the branches indicates the TCR probability generation scores.
A2+M158-Specific Public CDR3βs Form Highly Connected Similarity Networks
As the public (or near-public) TCRβ clonotypes are a key signature of the A2+M158+CD8+ TCRαβ repertoire with heavy bias for TRBV19, we further dissected in-depth CDR3β clusters across age groups, tissues, and following influenza virus infection. Using GLIPH, we focused on TCRβ chains by identifying enriched motifs independent of the V-J chain usage (39). We generated a network of all CDR3β regions defined as nodes connected by neighboring (by sequence similarity) CDR3β nodes, which have global or local interactions or no interactions at all (defined as singletons). We then visualized the A2+M158+ CDR3β network using the clusterMaker plugin of Cytoscape (59) across age groups, anatomical locations, and during acute influenza virus infection. Our network organization of CDR3β regions (628 sequences) across all the datasets demonstrated that the most abundant public CDR3β sequences comprising the “RS” motif formed a dense network with many interconnecting nodes within and between the different donor groups (Figure 8). For example, nodes representing shared TCRβ clonotypes between groups were “most-dense” and found mainly in the center of the cluster. Rarer private CDR3β sequences sharing no motifs formed singletons (disconnected nodes), while CDR3β clonotypes with low level of motif sharing clustered around the edges of the public network, highly evident for the large private clonotypes observed in our healthy elderly cohort.
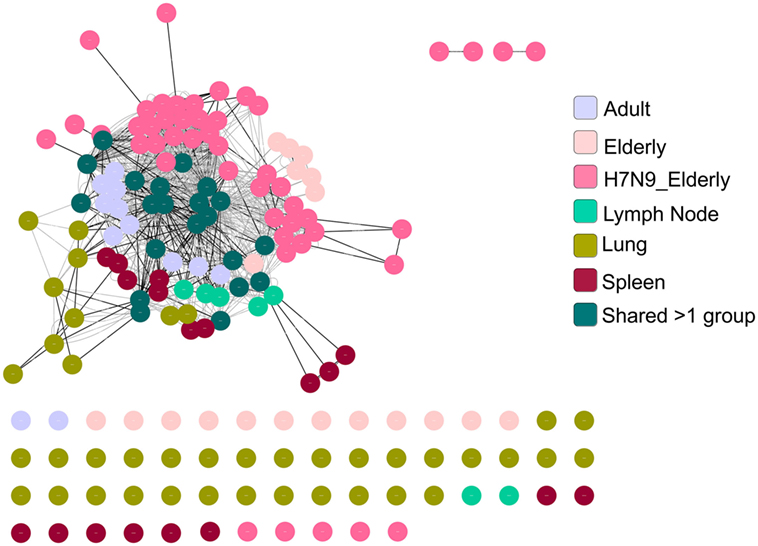
Figure 8. A2+M158-specific complementarity-determining region 3 (CDR3)βs forms a dense cluster comprising public T cell receptors (TCRs). TCR network organization of CDR3β from A2+M158-specific CD8+ TCR repertoire from adult, elderly, elderly H7N9 patients, spleen, lymph node, and lung to study the level of sequence similarity within and between donors. Black lines are global interactions (a pair of TCRs that share the same length CDR3 and differ by less than a certain number of amino acid) and the gray lines are local interactions. Node (dot) represent unique clonotype, edge is the global (black line) or local (gray line) interactions between the nodes. Colors of the node indicate the group to which the node belongs as shown in the legend.
Taken together, our analysis showed that aging had the greatest impact on the A2+M158+ memory CD8+ T cell repertoire, with a considerable loss of public TCRαβ signatures being paralleled with large clonal expansions of private TCRαβ clonotypes. TCRαβ analysis during acute H7N9 infection in elderly donors revealed that the immune system was capable of recruiting diverse TRBV19, rather than largely expanded private TCRs, in spite of considerably low frequency of public TCRs. Memory antigen-specific TCRs in tissues displayed the main features of the public TCRαβ clonotypes along with the presence of other diverse TCR repertoires, as summarized in Figure 9. Overall, this study establishes the framework for understanding antigen-specific TCRs across the human life span (young and old) and different anatomical locations, thus providing a clearer picture on the antigen-specific “TCRome,” with implications for the rational design of T cell-mediated vaccines and immunotherapies.
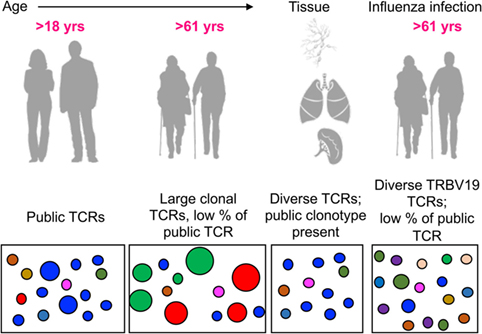
Figure 9. Modeling the dynamic changes of A2+M158-specific CD8+ T cell receptor (TCR) repertoires with age, tissue location, and during influenza infection. A conceptual figure summarizing evolution of A2+M158+ CD8+ TCR repertoires in young adults (with large public clonotypes depicted in blue), elderly adults (displaying loss of public clonotypes and presence of large private clonotypes in green and red), tissues (with public clonotypes along with diverse set of TCRs), and influenza-infected elderly adults (displaying diverse TRBV19s and public clonotypes).
Discussion
Preexisting memory CD8+ T cells can recognize internal conserved peptides derived from influenza viruses, irrespectively of the strain, thus mount effective immune responses toward newly emerging viruses (8–10, 60). This can be exemplified by a positive correlation between the presence of functional influenza-specific CD8+ T cells and rapid recovery in patients hospitalized with severe avian H7N9 virus (7, 10). Several factors can affect virus-specific CD8+ T cell responses, including numbers and quality of influenza-specific T cells, aging, disease, as well as the composition and diversity of TCRαβ repertoire against a given epitope, which has been well documented in mouse studies (54, 61, 62). With respect to influenza infection, human studies have mostly focused on antigen-specific CD8+ T cells residing in peripheral blood (11, 34, 42, 48). Our study presents the first data on influenza-specific A2+M158+CD8+ T cell repertoires across different human tissue compartments, including LNs, spleens, and lungs, and provides comprehensive analyses on temporal dynamics and maintenance of epitope-specific CD8+ T cells with aging, influenza virus infection, and across different tissue compartments.
We dissected A2+M158+CD8+ T cells across young and elderly age groups, during severe influenza infection and within human lung tissues from our published studies (7, 36, 40, 41), along with new TCRαβ data generated from additional tissues sampled across different anatomical sites. Using human A2+M158-tetramer magnetic enrichment or direct-staining (for lungs) for isolation of epitope-specific CD8+ T cells directly ex vivo, we observed the presence of A2+M158+CD8+ T cells in human spleen, mesenteric/pancreatic LNs, and in the lung. The frequency of these cells was comparable to memory or effector A2+M158+CD8+ T cells found in peripheral blood. The presence of A2+M158+CD8+ T cells in the human lung using tetramer staining was first reported very recently by our group, and for one donor (LG6), TCR analysis was previously performed showing a high degree of TCRαβ clonotype sharing between different tissue-resident T cell subsets (41). Here, we have performed further TCRαβ analysis on two additional A2+ lung donors as well as source different human tissues from deceased adults for our study. Hence, to the best of our knowledge, our study is the first to isolate and characterize influenza-specific CD8+ T cells from multiple different human tissues. Most strikingly, the presence of A2+M158+CD8+ T cells in LNs and secondary tissues supports the idea that human spleen and distal LNs serve as reservoirs of antigen-specific memory CD8+ T cells. This has been well established in mouse models of local and systemic virus challenge, showing that effector or memory CD8+ T cells can traffic to distal tissue during a secondary challenge, irrespectively of location of primary infection, and contribute to the overall immune response on secondary infection (54, 61, 63). Future in-depth studies should highlight the functional relevance of these antigen-specific CD8+ T cells in comparison to tissue-resident T cells residing at the site of primary infection (lungs).
Utilizing paired single-cell TCRαβ sequencing technology, we dissected TCRαβ repertoires of A2+M158+CD8+ T cells isolated from tissues and compared those to our published datasets of A2+M158-specific TCRαβ repertoires. We performed comparative analyses utilizing TCRdist allowing dissection of gene usage within Vα, Jα, Vβ, and Jβ segments. The A2+M158-specific TCRαβ repertoire is characterized by the dominant presence of the TRAV27/TRBV19 TCR signature presenting the public CDR3αβ motif “GGSQGNL”/“SIRSYEQ” (36). In support of our initial observations that the elderly adults had less of the public A2+M158 TCR signatures (40), our new analyses encompassing age, tissue compartment, and infection, showed that aging had the greatest impact on memory A2+M158+ CD8+ T cells repertoire, leading to a reduced frequency and lower probability of TCR clonotypes presenting the TRAV27/TRBV19 TCRαβ signatures. Moreover, the presence of TCRs with low generation probability corresponded to the high frequency of private clonotypes and a loss of public clonotypes, thus highly reflected aging-associated changes in TCR repertoires in elderly at a steady state. Importantly, these TRAV27/TRBV19 TCRs display the key motifs for A2+M158 epitope recognition, similar to those found in adults, tissues, and influenza-infected patients. Thus, any T cell vaccine development needs to consider strategies to maintain high frequency of the optimal TCRαβ signatures for efficient immunity against influenza infection in the elderly individuals, who are most vulnerable to severe influenza disease.
A striking observation was the variable aa lengths of the CDR3α loop across all the datasets, although longer CDR3α lengths were preferentially observed in the elderly donors, similar to our previous findings (34). Conversely, CDR3β length was consistently maintained at eight aa, with the exception of elderly memory A2+M158+TCRαβ repertoires (nine aa length preferred). The CDR3α length has been previously shown to affect the p-MHC-I affinity (37). A comparison between the JM22 TCR (public TRAV27/TRBV19 motif “AGSQGNL”/“SIRSYEQ”) with that of F6 TCR (TRAV27/TRBV19 motif “AIGSSNTGKL”/“SIRSYEQ”), both displaying identical CDR3β, showed that longer CDR3α loop reduced the affinity of F6, due to possible high entropic cost in stabilizing the CDR3α loop.
Using GLIPH, our TCRβ network analyses showed that public clonotype and “public-like” clonotypes formed dense clusters, while more private clonotypes formed either independent smaller clusters or remained disconnected. Our observation of a dense network within memory, young versus old, and across different tissue locations suggests the likelihood of structural constraints of diverse TCRs to accommodate recognition of the featureless A2+M158 topology. However, antigen recognition is mediated by interactions of both TCRα and TCRβ with the p-MHC-I complex, thus differences observed in the CDR3α length usage could hamper p-MHC-I recognition irrespective of the presence of the “best fit” TCRβ. Future studies are needed to correlate our findings with structural studies and functional characteristics of TCRs displaying restricted TCRβ chain pairing with diverse CDR3α chains.
In our study, we used a single-cell approach for TCRαβ analysis across different age groups and tissue compartments. This approach allowed us to gather data on the paired TCRα and TCRβ signatures obtained from single cells as well as dissect TCRαβ clonotypes in human samples with limited cell numbers. On the other hand, bulk sorted-A2+M158+CD8+ T cells would provide more depth (a higher number of TCR sequences) to the study, however, a major disadvantage of using a bulk TCR approach is its inability to obtain paired TCRαβ chains. Thus, single-cell sequencing techniques identifying paired TCRαβ sequences are a very powerful tool for understanding the contribution of both alpha and beta TCR chains to the overall TCR repertoire in health and disease.
Overall, our study provides comprehensive analyses of A2+M158-specific TCRαβ repertoires to understand temporal dynamics of TCRαβ repertoire selection and maintenance across young and aged donors, during human acute influenza infection, at primary site of infection (lungs), and at tissues distal to primary site of infection. Our analyses emphasize the remarkable clonal stability of the A2+M158-specific TCRαβ repertoire across different tissue locations and show the greatest impact of aging on influenza-specific TCRαβ repertoire. Future studies are needed to fully understand how aging impacts recruitment of influenza-specific TCRs in the elderly during acute influenza disease. Compilation of similar analyses, for other immunodominant CD8+ T cell epitopes across different HLA alleles, across human lifespan and tissues could greatly benefit any T cell-based vaccines and immunotherapies.
Ethics Statement
This study was approved by the University of Melbourne Human Ethics Committee (ID 1443389.3 and 0931311.5). Human experimental work was conducted according to the Declaration of Helsinki Principles and according to the Australian National Health and Medical Research Council Code of Practice. All donors provided informed written consent for blood donation. Tissues from deceased organ donors were obtained following written informed consent from the next of kin. PMBCs were isolated from heparinized peripheral blood collected from healthy adult volunteers (AD, 22–59 years) at the University of Melbourne and elderly donors (ED, 60 years) recruited at Deepdene Medical Clinic (Deepdene, Australia), or from buffy packs of healthy adult donors (AD, 22–59 years) obtained from Australian Red Cross Blood Service (ARCBS). Peripheral blood from HLA-A*02:01+ H7N9-infected patients (A9, A10, and A79) were obtained from Shanghai Public Health Clinic Centre (SHAPHC) under the supervision of SHAPHC Ethics Committee (7). PBMCs were also obtained from one patient hospitalized at the Royal Melbourne Hospital in 2017 with seasonal-influenza virus infection and approved by the Monash Health Human Research Ethics Committee (HREC/15/MonH/64).
Author Contributions
SS, TN, and KK designed the research. SS, TN, ZW, AP, LW, MK, and LG performed and/or analyzed the research. GW provided lung donor tissue. TL and SM provided spleen and lymph node donor tissues. JX and MR provided influenza patient samples. JC provided elderly adult donor samples. SS, LG, ZW, LL, AP, GW, MK, TN, LW, JC, JR, SG, PT, and KK contributed to manuscript preparation. SS, TN, and KK wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a past co-authorship with one of the authors KK.
Acknowledgments
We thank ImmunoID facility for FACS sorting. We thank Jeremy Chase Crawford for assistance with TCRdist algorithm. The Australian National Health and Medical Research Council (NHMRC) Program Grant (1071916) to KK supported this work. SS is a recipient of Victoria India Doctoral Scholarship and Melbourne International Fee Remission Scholarship, University of Melbourne. MK is a recipient of Melbourne International Research Scholarship and Melbourne International Fee Remission Scholarship. KK is an NHMRC Senior Research Level B Fellow. JR is supported by ARC Laureate fellowship. SG is a Monash Senior Research Fellow. SIM is supported by a JDRF Career Development Award (5-CDA-2014-210-A-N) and NHMRC project grant (GNT1123586).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01453/full#supplementary-material.
Figure S1. Single-cell sorting approach for A2+M158-specific CD8+ T cells from spleen/LN. Gating strategy for isolation of A2+M158-specific CD8+ T from spleen and lymph node post magnetic enrichment. Single lymphocytes were gated on CD3+ cells and excluded for LIVE/DEAD+/CD14+/CD19+ cells (dump channel) before gating on A2+M158-tetramer+CD8+ T cells. Frequencies shown are based on the parent gate, therefore, A2+M158-tetramer+CD8+ T cell frequencies were calculated based on the total CD8+ T cell population.
Footnotes
- ^http://www.who.int/mediacentre/factsheets/fs211/en/ (Accessed: January 31, 2018).
- ^http://www.cytoscape.org/.
References
1. Koutsakos M, Wheatley AK, Loh L, Clemens EB, Sant S, Nussing S, et al. Circulating TFH cells, serological memory, and tissue compartmentalization shape human influenza-specific B cell immunity. Sci Transl Med (2018) 10(428). doi:10.1126/scitranslmed.aan8405
2. Clemens EB, van de Sandt C, Wong SS, Wakim LM, Valkenburg SA. Harnessing the power of T cells: the promising hope for a universal influenza vaccine. Vaccines (Basel) (2018) 6(2). doi:10.3390/vaccines6020018
3. Yewdell JW, Bennink JR, Smith GL, Moss B. Influenza A virus nucleoprotein is a major target antigen for cross-reactive anti-influenza A virus cytotoxic T lymphocytes. Proc Natl Acad Sci U S A (1985) 82(6):1785–9. doi:10.1073/pnas.82.6.1785
4. Braciale TJ. Immunologic recognition of influenza virus-infected cells. II. Expression of influenza A matrix protein on the infected cell surface and its role in recognition by cross-reactive cytotoxic T cells. J Exp Med (1977) 146(3):673–89. doi:10.1084/jem.146.3.673
5. Nguyen Thi. H. O, Koutsakos M, Grant EJ, Doherty PC, Kedzierska K. Towards future T cell-mediated influenza vaccines. Infect Dis Transl Med (2016) 2(1):20–9. doi:10.11979/idtm.201601004
6. Nussing S, Sant S, Koutsakos M, Subbarao K, Nguyen THO, Kedzierska K. Innate and adaptive T cells in influenza disease. Front Med (2018) 12(1):34–47. doi:10.1007/s11684-017-0606-8
7. Wang Z, Zhu L, Nguyen THO, Wan Y, Sant S, Quinones-Parra SM, et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat Commun (2018) 9(1):824. doi:10.1038/s41467-018-03243-7
8. McMichael AJ, Gotch FM, Noble GR, Beare PA. Cytotoxic T-cell immunity to influenza. N Engl J Med (1983) 309(1):13–7. doi:10.1056/NEJM198307073090103
9. Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med (2013) 19(10):1305–12. doi:10.1038/nm.3350
10. Wang Z, Wan Y, Qiu C, Quinones-Parra S, Zhu Z, Loh L, et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8(+) T cells. Nat Commun (2015) 6:6833. doi:10.1038/ncomms7833
11. Kreijtz JH, de Mutsert G, van Baalen CA, Fouchier RA, Osterhaus AD, Rimmelzwaan GF. Cross-recognition of avian H5N1 influenza virus by human cytotoxic T-lymphocyte populations directed to human influenza A virus. J Virol (2008) 82(11):5161–6. doi:10.1128/JVI.02694-07
12. Moskophidis D, Assmann-Wischer U, Simon MM, Lehmann-Grube F. The immune response of the mouse to lymphocytic choriomeningitis virus. V. High numbers of cytolytic T lymphocytes are generated in the spleen during acute infection. Eur J Immunol (1987) 17(7):937–42. doi:10.1002/eji.1830170707
13. Gras S, Burrows SR, Turner SJ, Sewell AK, McCluskey J, Rossjohn J. A structural voyage toward an understanding of the MHC-I-restricted immune response: lessons learned and much to be learned. Immunol Rev (2012) 250(1):61–81. doi:10.1111/j.1600-065X.2012.01159.x
14. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol (2014) 33:169–200. doi:10.1146/annurev-immunol-032414-112334
15. Berkhoff EG, Boon AC, Nieuwkoop NJ, Fouchier RA, Sintnicolaas K, Osterhaus AD, et al. A mutation in the HLA-B*2705-restricted NP383-391 epitope affects the human influenza A virus-specific cytotoxic T-lymphocyte response in vitro. J Virol (2004) 78(10):5216–22. doi:10.1128/JVI.78.10.5216-5222.2004
16. Gras S, Kedzierski L, Valkenburg SA, Laurie K, Liu YC, Denholm JT, et al. Cross-reactive CD8+ T-cell immunity between the pandemic H1N1-2009 and H1N1-1918 influenza A viruses. Proc Natl Acad Sci U S A (2010) 107(28):12599–604. doi:10.1073/pnas.1007270107
17. Valkenburg SA, Quinones-Parra S, Gras S, Komadina N, McVernon J, Wang Z, et al. Acute emergence and reversion of influenza A virus quasispecies within CD8+ T cell antigenic peptides. Nat Commun (2013) 4:2663. doi:10.1038/ncomms3663
18. Quinones-Parra S, Loh L, Brown LE, Kedzierska K, Valkenburg SA. Universal immunity to influenza must outwit immune evasion. Front Microbiol (2014) 5:285. doi:10.3389/fmicb.2014.00285
19. Kedzierska K, Guillonneau C, Gras S, Hatton LA, Webby R, Purcell AW, et al. Complete modification of TCR specificity and repertoire selection does not perturb a CD8+ T cell immunodominance hierarchy. Proc Natl Acad Sci U S A (2008) 105(49):19408–13. doi:10.1073/pnas.0810274105
20. Turner SJ, Kedzierska K, La Gruta NL, Webby R, Doherty PC. Characterization of CD8+ T cell repertoire diversity and persistence in the influenza A virus model of localized, transient infection. Semin Immunol (2004) 16(3):179–84. doi:10.1016/j.smim.2004.02.005
21. Kedzierska K, Turner SJ, Doherty PC. Conserved T cell receptor usage in primary and recall responses to an immunodominant influenza virus nucleoprotein epitope. Proc Natl Acad Sci U S A (2004) 101(14):4942–7. doi:10.1073/pnas.0401279101
22. Cibotti R, Cabaniols JP, Pannetier C, Delarbre C, Vergnon I, Kanellopoulos JM, et al. Public and private V beta T cell receptor repertoires against hen egg white lysozyme (HEL) in nontransgenic versus HEL transgenic mice. J Exp Med (1994) 180(3):861–72. doi:10.1084/jem.180.3.861
23. Stewart-Jones GB, McMichael AJ, Bell JI, Stuart DI, Jones EY. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol (2003) 4(7):657–63. doi:10.1038/ni942
24. Turner SJ, Diaz G, Cross R, Doherty PC. Analysis of clonotype distribution and persistence for an influenza virus-specific CD8+ T cell response. Immunity (2003) 18(4):549–59. doi:10.1016/S1074-7613(03)00087-6
25. Yassai M, Bosenko D, Unruh M, Zacharias G, Reed E, Demos W, et al. Naive T cell repertoire skewing in HLA-A2 individuals by a specialized rearrangement mechanism results in public memory clonotypes. J Immunol (2011) 186(5):2970–7. doi:10.4049/jimmunol.1002764
26. Venturi V, Kedzierska K, Price DA, Doherty PC, Douek DC, Turner SJ, et al. Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination. Proc Natl Acad Sci U S A (2006) 103(49):18691–6. doi:10.1073/pnas.0608907103
27. Garcia KC, Adams JJ, Feng D, Ely LK. The molecular basis of TCR germline bias for MHC is surprisingly simple. Nat Immunol (2009) 10(2):143–7. doi:10.1038/ni.f.219
28. Fazilleau N, Cabaniols JP, Lemaitre F, Motta I, Kourilsky P, Kanellopoulos JM. Valpha and Vbeta public repertoires are highly conserved in terminal deoxynucleotidyl transferase-deficient mice. J Immunol (2005) 174(1):345–55. doi:10.4049/jimmunol.174.1.345
29. Kedzierska K, Thomas PG, Venturi V, Davenport MP, Doherty PC, Turner SJ, et al. Terminal deoxynucleotidyltransferase is required for the establishment of private virus-specific CD8+ TCR repertoires and facilitates optimal CTL responses. J Immunol (2008) 181(4):2556–62. doi:10.4049/jimmunol.181.4.2556
30. Turner SJ, Doherty PC, McCluskey J, Rossjohn J. Structural determinants of T-cell receptor bias in immunity. Nat Rev Immunol (2006) 6(12):883–94. doi:10.1038/nri1977
31. Messaoudi I, Guevara Patino JA, Dyall R, LeMaoult J, Nikolich-Zugich J. Direct link between mhc polymorphism, T cell avidity, and diversity in immune defense. Science (2002) 298(5599):1797–800. doi:10.1126/science.1076064
32. Venturi V, Price DA, Douek DC, Davenport MP. The molecular basis for public T-cell responses? Nat Rev Immunol (2008) 8(3):231–8. doi:10.1038/nri2260
33. Culshaw A, Ladell K, Gras S, McLaren JE, Miners KL, Farenc C, et al. Germline bias dictates cross-serotype reactivity in a common dengue-virus-specific CD8(+) T cell response. Nat Immunol (2017) 18(11):1228–37. doi:10.1038/ni.3850
34. Gil A, Yassai MB, Naumov YN, Selin LK. Narrowing of human influenza A virus-specific T cell receptor alpha and beta repertoires with increasing age. J Virol (2015) 89(8):4102–16. doi:10.1128/JVI.03020-14
35. Neller MA, Ladell K, McLaren JE, Matthews KK, Gostick E, Pentier JM, et al. Naive CD8(+) T-cell precursors display structured TCR repertoires and composite antigen-driven selection dynamics. Immunol Cell Biol (2015) 93(7):625–33. doi:10.1038/icb.2015.17
36. Valkenburg SA, Josephs TM, Clemens EB, Grant EJ, Nguyen TH, Wang GC, et al. Molecular basis for universal HLA-A*0201-restricted CD8+ T-cell immunity against influenza viruses. Proc Natl Acad Sci U S A (2016) 113(16):4440–5. doi:10.1073/pnas.1603106113
37. Chen G, Yang X, Ko A, Sun X, Gao M, Zhang Y, et al. Sequence and structural analyses reveal distinct and highly diverse human CD8(+) TCR repertoires to immunodominant viral antigens. Cell Rep (2017) 19(3):569–83. doi:10.1016/j.celrep.2017.03.072
38. Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature (2017) 547(7661):89–93. doi:10.1038/nature22383
39. Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nature (2017) 547(7661):94–8. doi:10.1038/nature22976
40. Nguyen THO, Sant S, Bird NL, Grant EJ, Clemens EB, Koutsakos M, et al. Perturbed CD8(+) T cell immunity across universal influenza epitopes in the elderly. J Leukoc Biol (2018) 103(2):321–39. doi:10.1189/jlb.5MA0517-207R
41. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest (2018) 128(2):721–33. doi:10.1172/JCI96957
42. Quinones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci U S A (2014) 111(3):1049–54. doi:10.1073/pnas.1322229111
43. Middleton D, Menchaca L, Rood H, Komerofsky R. New allele frequency database: http://www.allelefrequencies.net/. Tissue Antigens (2003) 61(5):403–7. doi:10.1034/j.1399-0039.2003.00062.x
44. Wang GC, Dash P, McCullers JA, Doherty PC, Thomas PG. T cell receptor alphabeta diversity inversely correlates with pathogen-specific antibody levels in human cytomegalovirus infection. Sci Transl Med (2012) 4(128):128ra42. doi:10.1126/scitranslmed.3003647
45. Bousso P, Casrouge A, Altman JD, Haury M, Kanellopoulos J, Abastado JP, et al. Individual variations in the murine T cell response to a specific peptide reflect variability in naive repertoires. Immunity (1998) 9(2):169–78. doi:10.1016/S1074-7613(00)80599-3
46. Wang C, Sanders CM, Yang Q, Schroeder HW Jr, Wang E, Babrzadeh F, et al. High throughput sequencing reveals a complex pattern of dynamic interrelationships among human T cell subsets. Proc Natl Acad Sci U S A (2010) 107(4):1518–23. doi:10.1073/pnas.0913939107
47. van de Sandt CE, Hillaire ML, Geelhoed-Mieras MM, Osterhaus AD, Fouchier RA, Rimmelzwaan GF. Human influenza A virus-specific CD8+ T-cell response is long-lived. J Infect Dis (2015) 212(1):81–5. doi:10.1093/infdis/jiv018
48. Grant EJ, Josephs TM, Valkenburg SA, Wooldridge L, Hellard M, Rossjohn J, et al. Lack of heterologous cross-reactivity toward HLA-A*02:01 restricted viral epitopes is underpinned by distinct alphabetaT cell receptor signatures. J Biol Chem (2016) 291(47):24335–51. doi:10.1074/jbc.M116.753988
49. Franken L, Klein M, Spasova M, Elsukova A, Wiedwald U, Welz M, et al. Splenic red pulp macrophages are intrinsically superparamagnetic and contaminate magnetic cell isolates. Sci Rep (2015) 5:12940. doi:10.1038/srep12940
50. Hunter JD. Matplotlib: A 2D graphics environment. Comput Sci Eng (2007) 9(3):90–5. doi:10.1109/MCSE.2007.55
51. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res (2009) 19(9):1639–45. doi:10.1101/gr.092759.109
52. Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, et al. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res (2018) 46(D1):D419–27. doi:10.1093/nar/gkx760
53. Gordon CL, Miron M, Thome JJ, Matsuoka N, Weiner J, Rak MA, et al. Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J Exp Med (2017) 214(3):651–67. doi:10.1084/jem.20160758
54. Masopust D, Vezys V, Usherwood EJ, Cauley LS, Olson S, Marzo AL, et al. Activated primary and memory CD8 T cells migrate to nonlymphoid tissues regardless of site of activation or tissue of origin. J Immunol (2004) 172(8):4875–82. doi:10.4049/jimmunol.172.8.4875
55. Jameson SC, Masopust D. Understanding subset diversity in T cell memory. Immunity (2018) 48(2):214–26. doi:10.1016/j.immuni.2018.02.010
56. Moon JJ, Chu HH, Pepper M, McSorley SJ, Jameson SC, Kedl RM, et al. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity (2007) 27(2):203–13. doi:10.1016/j.immuni.2007.07.007
57. Nguyen TH, Tan AC, Xiang SD, Goubier A, Harland KL, Clemens EB, et al. Understanding CD8+ T-cell responses toward the native and alternate HLA-A*02:01-restricted WT1 epitope. Clin Transl Immunology (2017) 6(3):e134. doi:10.1038/cti.2017.4
58. Miles JJ, McCluskey J, Rossjohn J, Gras S. Understanding the complexity and malleability of T-cell recognition. Immunol Cell Biol (2015) 93(5):433–41. doi:10.1038/icb.2014.112
59. Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc (2007) 2(10):2366–82. doi:10.1038/nprot.2007.324
60. Fiore AE, Uyeki TM, Broder K, Finelli L, Euler GL, Singleton JA, et al. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm Rep (2010) 59(RR–8):1–62.
61. Marshall DR, Turner SJ, Belz GT, Wingo S, Andreansky S, Sangster MY, et al. Measuring the diaspora for virus-specific CD8+ T cells. Proc Natl Acad Sci U S A (2001) 98(11):6313–8. doi:10.1073/pnas.101132698
62. Cukalac T, Chadderton J, Zeng W, Cullen JG, Kan WT, Doherty PC, et al. The influenza virus-specific CTL immunodominance hierarchy in mice is determined by the relative frequency of high-avidity T cells. J Immunol (2014) 192(9):4061–8. doi:10.4049/jimmunol.1301403
Keywords: T cell receptor repertoire, influenza A virus, human T lymphocytes, CD8+ T cells, aging, tissues
Citation: Sant S, Grzelak L, Wang Z, Pizzolla A, Koutsakos M, Crowe J, Loudovaris T, Mannering SI, Westall GP, Wakim LM, Rossjohn J, Gras S, Richards M, Xu J, Thomas PG, Loh L, Nguyen THO and Kedzierska K (2018) Single-Cell Approach to Influenza-Specific CD8+ T Cell Receptor Repertoires Across Different Age Groups, Tissues, and Following Influenza Virus Infection. Front. Immunol. 9:1453. doi: 10.3389/fimmu.2018.01453
Received: 18 April 2018; Accepted: 12 June 2018;
Published: 27 June 2018
Edited by:
Fabio Luciani, University of New South Wales, AustraliaReviewed by:
Christoph Wülfing, University of Bristol, United KingdomKristin Ladell, Cardiff University, United Kingdom
Victor Appay, INSERM, France
Copyright: © 2018 Sant, Grzelak, Wang, Pizzolla, Koutsakos, Crowe, Loudovaris, Mannering, Westall, Wakim, Rossjohn, Gras, Richards, Xu, Thomas, Loh, Nguyen and Kedzierska. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thi H. O. Nguyen, dGhvLm5ndXllbkB1bmltZWxiLmVkdS5hdQ==;
Katherine Kedzierska, a2tlZHpAdW5pbWVsYi5lZHUuYXU=
†These authors have contributed equally to this work.