 Michael Bauer1,2*
Michael Bauer1,2* Sina M. Coldewey1,2,3
Sina M. Coldewey1,2,3 Margit Leitner2Bettina Löffler2,4Sebastian Weis1,2,5
Margit Leitner2Bettina Löffler2,4Sebastian Weis1,2,5 Reinhard Wetzker1
Reinhard Wetzker1
- 1Department of Anesthesiology and Intensive Care Medicine, Jena University Hospital, Jena, Germany
- 2Center for Sepsis Control and Care (CSCC), Jena University Hospital, Jena, Germany
- 3Septomics Research Center, Jena University Hospital, Jena, Germany
- 4Institute of Medical Microbiology, Jena University Hospital, Jena, Germany
- 5Center for Infectious Disease and Infection Control, Jena University Hospital, Jena, Germany
Development of organ dysfunction discriminates sepsis from uncomplicated infection. The paradigm shift implicated by the new sepsis-3 definition holds that initial impairment of any organ can pave the way for multiple organ dysfunction and death. Moreover, the role of the systemic inflammatory response, central element in previous sepsis definitions, has been questioned. Most strikingly, a so far largely underestimated defense mechanism of the host, i.e., “disease tolerance,” which aims at maintaining host vitality without reducing pathogen load, has gained increasing attention. Here, we summarize evidence that a dysregulation of critical cellular signaling events, also in non-immune cells, might provide a conceptual framework for sepsis-induced dysfunction of parenchymal organs in the absence of significant cell death. We suggest that key signaling mediators, such as phosphoinositide 3-kinase, mechanistic target of rapamycin, and AMP-activated protein kinase, control the balance of damage and repair processes and thus determine the fate of affected organs and ultimately the host. Therapeutic targeting of these multifunctional signaling mediators requires cell-, tissue-, or organ-specific approaches. These novel strategies might allow stopping the domino-like damage to further organ systems and offer alternatives beyond the currently available strictly supportive therapeutic options.
Introduction
The advent of the new sepsis-3 definition, published in 2016 prompted a reappraisal of organ dysfunction as the hallmark of sepsis. Sepsis is now defined as “life-threatening organ dysfunction caused by a dysregulated host response to infection” (1). With this definition, the conceptual focus shifted from exclusive attention to the host inflammatory response, i.e., Systemic Inflammatory Response Syndrome (2, 3), to the multifactorial tissue damage occurring during the progression of infection to sepsis. In the current conceptual framework, sepsis can be construed as a pathogen-induced imbalance of host damage and repair processes that trigger failure of either resistance or “disease tolerance” mechanisms (4). In these processes, metabolic adaptation is of outstanding significance (1, 5–7).
Certain critical mediators, most notably related to phosphoinositide 3-kinase (PI3K), mechanistic target of rapamycin (mTOR), and AMP-activated protein kinase (AMPK) signaling, have dual functions in metabolism and defense signaling. In this “perspective,” we summarize evidence that substantial changes in these signaling pathways during sepsis occur not only in the immune system but also in parenchymal cells. To target (parenchymal) cellular metabolic and signaling dysfunctions will open new avenues for therapeutic interventions.
Septic Shock, Sepsis-Related Dysoxia, and Energy Crisis
Severe metabolic dysregulation and circulatory failure are central to the definition of septic shock (1, 3) and sepsis mortality rises dramatically when there is cardiovascular impairment (8, 9). Microvascular thrombosis and low blood pressure act together with the sepsis-induced endothelial capillary leakage syndrome. These factors can aggravate and perpetuate organ damage through impaired oxygen availability (10). In addition, impaired mitochondrial function and biogenesis as well as presumably mitophagy have been identified as common features in non-survivors of sepsis (11). These findings support our hypothesis that impaired cellular metabolic processes occur early and widespread across the organism during sepsis development. We assume that the resulting “metabolic crisis” in multiple organs is accompanied by an increasing “misfiring” of signaling mediators, further deteriorating metabolic functions. Predominantly key players of anabolic processes, such as PI3K or mTOR, or of cellular maintenance, including AMPK, get out of control (12, 13). These cellular metabolic dysfunctions also impact detoxification mechanisms, the failure of which perpetuates metabolic deterioration (see Figure 1). Accordingly, “septic shock” is now defined as a “subset of sepsis in which underlying circulatory and cellular/metabolic abnormalities are profound enough to substantially increase mortality” (1). Thus, the current concept of (multi-) organ dysfunction focuses on the deleterious impact of already failed organ functions, most notably cardiovascular dysfunction, as a perpetuator of damage. Supposedly, interfering with the discussed signaling events will allow intervening at a much earlier stage.
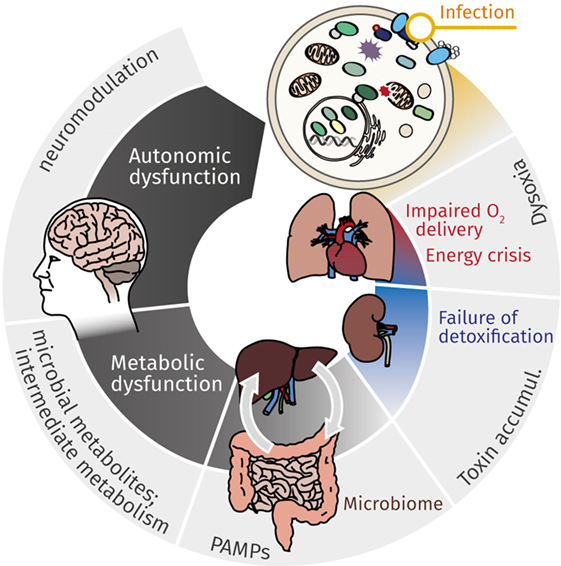
Figure 1. Mutual effects of cellular energy metabolism and organ dysfunction cause continuous deterioration during sepsis. While cardiovascular impairment leads to reduced O2 delivery, metabolic dysfunction, most markedly mitochondrial dysfunction, contributes to impaired cellular energy supply. This energy crisis contributes to failure of detoxification, which in turn affects primarily kidney and liver. Accumulating toxins, gut microbiome with (antibiotic-induced) dysbiosis, and microbial metabolites (along the gut–liver axis, via organ–organ cross-talk) aggravate and perpetuate deteriorating organ function. The resulting metabolic catastrophe ultimately results in neurologic manifestations and autonomic dysfunction, which again contributes to continuing the downward spiral of metabolic derangement and organ dysfunction.
End Organ Failure—Insights from Systems Medicine
A paradigmatic example of sepsis-related organ failure is excretory dysfunction of the liver. Sepsis accounts for approximately 20% of admissions for jaundice, a rate that is only surpassed by malignant compression of the bile duct (14). Whereas the traditional view of liver failure would imply (pericentral) necrosis or apoptosis of parenchymal cells, this has been excluded as the predominant mechanism in septic shock by the pioneering work of Hotchkiss et al. (15) revealing that severe organ dysfunction occurs despite remarkably well preserved tissue structure. A systems biology approach analyzing the hepatocyte response to infection found reprogramming of metabolic functions in parallel with severity-dependent disruption of phase I and II biotransformation and excretory failure (16, 17). This characteristic phenotype of excretory failure critically depends on PI3Kγ signaling as a triggering event. This important role of PI3Kγ is also supported by interventions into upstream localized G protein-coupled receptors. As such, the long known role of C5aR in sepsis (18, 19), which signals through PI3Kγ, might reflect the central importance of the PI3K-Akt-mTOR axis. Taken together, these findings indicate that liver dysfunction does not result from a loss of viable parenchyma but from cellular dysfunctions that are potentially reversible and thus amenable to therapeutic intervention. Whereas inhibition of PI3Kγ could be a strategy to counter liver failure, the same signaling molecule might confer protection in other organs, e.g., in the heart (20). This highlights the need to achieve organ and cell-specific targeting of these processes in multi-organ dysfunction (21, 22).
Altered Signaling in Sequential Stress Events and by Comorbidities
Liver dysfunction also strongly challenges overall metabolic and immunological homeostasis in the critically ill and frequently promotes progression to multi-organ failure. Prolonged hepatic dysfunction interferes with the adaptive immune system (23). Accordingly, pre-existing liver disease is a risk factor for the progression of bacterial infections to sepsis with increased odds ratios for hospitalization, ICU admission, and death (24). Similarly, patients with septic complications in the presence of chronic liver disease, most notably cirrhosis, have a poor prognosis due to development of acute-on-chronic liver failure (25). Altered signaling in chronic liver disease is closely connected to signaling processes mediating liver dysfunction during sepsis-triggered hepatic and extrahepatic organ failure (26, 27). In both conditions, metabolic master regulators, such as mTOR and AMPK, are in the center of the regulatory cascade. These and other key regulators, like the classical NFκB signaling pathway (28), probably govern the processes promoting damage and functional recovery. The underlying mechanisms might be amenable to therapy (1, 6, 7). Shock and cell death can further aggravate organ dysfunction, but in a small proportion of patients only (29).
Imbalance Between Damage and Repair Processes as a Unifying Concept of Organ Failure
Organisms can counter infections via two distinct strategies: resistance and disease tolerance (4, 30–32). While resistance mechanisms and immunopathology have been in the focus of sepsis research for the past decades, disease tolerance and resilience remained largely underrated in animal and human biology (4).
Resistance mechanisms aim at reducing pathogen burden by pathogen destruction and elimination. The term disease tolerance summarizes a diversity of mechanisms that enable an organism to cope with a given stressor without eliminating it. In the context of infections and sepsis, these mechanisms provide tissue damage control and repair to sustain host tissue integrity and organ function even without reducing pathogen burden (32, 33). Disease tolerance can thus result in a relative increase in host fitness in the presence of a disease burden that would otherwise greatly reduce vitality (34). A disadvantage of these mechanisms is that inadequate tolerance can result in persistent infections and possibly relapses. Disease tolerance undoubtedly has a genetic basis but, as yet, this aspect has scarcely been investigated. Studies in Drosophila suggest that components other than the immune system are crucial for tolerance and endurance of infections (35).
Although we are far from knowing all factors involved, disease outcome seems to strongly depend on a fine balance between resistance and disease tolerance mechanisms that are further influenced by environmental factors [see, e.g., Ref. (36)].
Therapeutic Strategies Derived from Stress Biology
That tolerance to infections is not exclusively linked to the immune system was further substantiated by the finding that anthracyclines, long known as chemotherapeutic agents, attenuated lung injury in a rodent model of sepsis (37). The protective effect can presumably be attributed to the induction of damage repair pathways via DNA damage responses (37). The underlying principle, systemic induction of maintenance and repair processes by stressors, such as cytotoxic agents or radiation, seems to be a general phenomenon, as demonstrated in C. elegans (38), among other organisms. These results indicate that tolerance can be pharmaceutically enhanced and that the principle of hormesis—a general fitness benefit of low doses of a stressor—is applicable to counter infections and sepsis (39).
Over the past century, however, the predominant medical interventions against infectious diseases have involved targeting resistance mechanisms, e.g., via vaccination, and direct reduction of pathogen load via antimicrobial drugs (40). But this strategy comes at the expense of drug resistance against many classes of antibiotics (41). Moreover, whereas this strategy has proven to be very successful against a broad range of infectious diseases, it has often failed in the treatment of sepsis (42). Futile antibiotic therapy in sepsis is a common phenomenon, even if the pathogen is tested sensitive to the given drug. This underlines the need for host-directed supportive therapies. In this connection, disease tolerance mechanisms emerge as therapeutic targets in the treatment of infectious diseases and offer fundamentally novel concepts in the treatment of sepsis (4, 32, 34, 43). Exploiting disease tolerance to counter sepsis also seems promising in view of increasing antibiotic resistance, as it does not generate selective pressure and cannot be opposed by antimicrobial resistance development. Thus, tolerance strategies are considered more stable (30, 44). Moreover, as tissue damage triggered by an excessive immune response is suggested to be the predominant cause of multi-organ failure, which results in poor disease outcome, damage control and repair mechanisms are promising targets for new therapeutic interventions. And, indeed, stress responsive genes that prevent the deleterious effects of damage-associated molecular patterns, such as free heme/iron (7, 45), DNA damage (37), or oxidative damage (46) have been shown in experimental systems to protect against bacterial infections without reducing host pathogen burden. In these tolerance settings, stabilizing organ functions has priority and pathogen eradication might be insufficient. However, unrestricted tolerance carries the risk of creating persistent reservoirs of pathogens in the host population (44, 47–49). To reduce this risk, therapeutically used tolerance responses have to be strictly controlled. Timing and extent of tolerance induction seem of eminent importance. Pathogen persistence and dissemination might be prevented by suppressing tolerance mediators or by provoking specific immune responses. Finally, pathogen persistence mechanisms might be affected directly to clear an infection and re-achieve balanced tolerance and resistance.
Intracellular “Wiring” of Metabolism and Host Defense Mechanisms
Immunity and cellular metabolism are intricately connected, as the proteins PI3K, Akt, mTOR, HIF1alpha, and PKC are not only well described mediators of resistance responses but also drive energy-demanding anabolic processes, including glucose storage, protein synthesis, and proliferation. In contrast, disease tolerance is closely connected to cellular maintenance reactions, including the unfolded protein response and autophagy. Both processes are predominantly controlled by the signaling mediator AMPK. The interplay of mTOR and AMPK can, thus, serve as paradigm for the critical balance between resistance and disease tolerance. Apparently, both defense mechanisms become dysregulated during sepsis. Whereas excessive activation of resistance responses can lead to immunopathology, inappropriate disease tolerance might entail fulminant infection or long-term pathogen persistence. The interpretation of sepsis as collapsing tolerance and resistance responses obtains increasing support by recent experimental data, in which mTOR and AMPK, as their master regulators, play central roles: For example, mortality of septic mice could be drastically decreased after treatment with the specific mTOR inhibitor rapamycin (50) and the mTOR–HIF1alpha pathway is required for metabolic activation of trained monocytes (51). On the other hand, AICAR and metformin as agonists of AMPK have been shown to prevent sepsis in mice with similar efficacy (52, 53). Moreover, while AMPK suppresses resistance responses controlled by mTOR (54, 55), mTOR blocks disease tolerance and maintenance responses mediated by AMPK (56). Reduced mortality of septic mice by treatment with either mTOR inhibitors or AMPK activating agents could thus be interpreted as either directly or indirectly suppressing excessive resistance reactions promoted by mTOR. However, recent studies report opposed effects. Under certain conditions, stimulation of AMPK by metformin significantly increases the mortality of septic mice (57), indicating that the effects are context- and dose-dependent. Besides this paradigmatic antagonism of AMPK and mTOR-related pathways, Akt and forkhead box O (FOXO) have been recently found to mediate infection-induced cachexia, as indicated by Drosophila FOXO mutants surviving infection longer than wt conspecifics (58). As outlined in Figure 2, we assume that dysregulated mTOR-, AMPK-, and related signaling pathways are the molecular basis underlying infection-triggered organ dysfunction. The intricate connection of metabolism, resistance responses, and disease tolerance suggests a mechanism for controlled resource allocation to either immunity or repair processes. Aggravating factors, such as comorbidities or environmental stresses, contribute to pushing disease tolerance and resistance out of this delicate equilibrium.
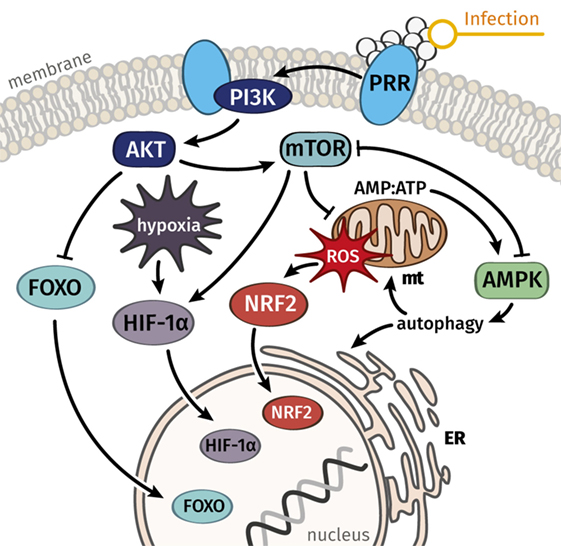
Figure 2. Key signal mediators connect stress responses to metabolism and can serve to explain a balance between damage and repair processes in immune cells and the majority of parenchymal cells. We hypothesize that dysregulated signaling and metabolic functions, which govern resistance responses and disease tolerance, are the common underlying cause of septic organ failure. Whereas AMPK is considered a key mediator of disease tolerance, mTOR and connected pathways regulate resistance responses and the metabolic reprogramming connected to immune activation. Abbreviations: AMPK, AMP-activated protein kinase; ER, endoplasmic reticulum, FOXO, forkhead box O; HIF-1α, hypoxia-inducible factor-1α; mt, mitochondrion; mTOR, mechanistic target of rapamycin; NRF2, nuclear factor erythroid 2-related factor 2; PI3K, phosphoinositide 3-kinase; PRR, pattern recognition receptor; ROS, reactive oxygen species.
Outlook—Purposeful Manipulation of Signals Mediating Damage and Repair
The recognition of the close interrelation between metabolism and host defense mechanisms opens new perspectives to sepsis treatment. Most notably, stimulating disease tolerance during sepsis might be a strategy to support patients in surviving septic shock. The fundamental concept of the hormetic nature of mild-to-moderate stress has been observed as a highly conserved phenomenon across kingdoms. Applying such principles, e.g., induction of the DNA-damage response with low doses of anthracyclines, might add to the therapeutic options in treating septic organ failure. However, timing and targeting of such interventions will be challenging. Manipulation of the relevant signals is likely to produce cell- and tissue-specific (side) effects. Hence, before signaling mediators can be used therapeutically, it will be necessary to overcome the significant hurdles posed by these multifunctional signaling molecules. Nanoparticle-based targeted drug-delivery (21, 22, 59) might help to specifically tackle this problem.
The manipulation of key signaling pathways can be expected to significantly add to armamentarium of sepsis therapy, in particular, in the light of increasing antibiotic resistance.
Author Contributions
MB, SC, ML, SW, BL, and RW wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
MB declares that he is co-founder and share-holder of a company aiming to develop PI3K inhibitors for target-specific treatment of organ dysfunction. RW holds patents related to the therapeutic use of PI3K inhibitors. All other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the Federal Ministry of Education and Research (BMBF), Germany, FKZ 01EO1502.
References
1. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA (2016) 315:801–10. doi:10.1001/jama.2016.0287
2. Bone RC. Toward an epidemiology and natural history of SIRS (systemic inflammatory response syndrome). JAMA (1992) 268:3452–5. doi:10.1001/jama.268.24.3452
3. Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Intensive Care Med (2003) 29:530–8. doi:10.1007/s00134-003-1662-x
4. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science (2012) 335:936–41. doi:10.1126/science.1214935
5. Weber M, Lambeck S, Ding N, Henken S, Kohl M, Deigner HP, et al. Hepatic induction of cholesterol biosynthesis reflects a remote adaptive response to pneumococcal pneumonia. FASEB J (2012) 26:2424–36. doi:10.1096/fj.11-191957
6. Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence (2014) 5:66–72. doi:10.4161/viru.26907
7. Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, et al. Metabolic adaptation establishes disease tolerance to sepsis. Cell (2017) 169:1263–1275.e14. doi:10.1016/j.cell.2017.05.031
8. Merx MW, Weber C. Sepsis and the heart. Circulation (2007) 116:793–802. doi:10.1161/CIRCULATIONAHA.106.678359
9. Thomas-Rueddel DO, Poidinger B, Weiss M, Bach F, Dey K, Häberle H, et al. Hyperlactatemia is an independent predictor of mortality and denotes distinct subtypes of severe sepsis and septic shock. J Crit Care (2015) 30:439.e1–6. doi:10.1016/j.jcrc.2014.10.027
10. Opal SM, van der Poll T. Endothelial barrier dysfunction in septic shock. J Intern Med (2015) 277:277–93. doi:10.1111/joim.12331
11. Carré JE, Orban J-C, Re L, Felsmann K, Iffert W, Bauer M, et al. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med (2010) 182:745–51. doi:10.1164/rccm.201003-0326OC
12. Reiling JH, Sabatini DM. Stress and mTORture signaling. Oncogene (2006) 25:6373–83. doi:10.1038/sj.onc.1209889
13. Cheng SC, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol (2016) 17:406–13. doi:10.1038/ni.3398
14. Whitehead MW, Hainsworth I, Kingham JGC. The causes of obvious jaundice in South West Wales: perceptions versus reality. Gut (2001) 48:409–13. doi:10.1136/gut.48.3.409
15. Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, et al. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med (1999) 27:1230–51. doi:10.1097/00003246-199907000-00002
16. Recknagel P, Gonnert FA, Westermann M, Lambeck S, Lupp A, Rudiger A, et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med (2012) 9:e1001338. doi:10.1371/journal.pmed.1001338
17. Bauer M, Press AT, Trauner M. The liver in sepsis: patterns of response and injury. Curr Opin Crit Care (2013) 19:123–7. doi:10.1097/MCC.0b013e32835eba6d
18. Riedemann NC, Guo RF, Ward PA. Novel strategies for the treatment of sepsis. Nat Med (2003) 9:517–24. doi:10.1038/nm0503-517
19. Wrann CD, Tabriz NA, Barkhausen T, Klos A, Van Griensven M, Pape HC, et al. The phosphatidylinositol 3-kinase signaling pathway exerts protective effects during sepsis by controlling C5a-mediated activation of innate immune functions. J Immunol (2007) 178:5940–8. doi:10.4049/jimmunol.178.9.5940
20. Coldewey SM, Benetti E, Collino M, Pfeilschifter J, Sponholz C, Bauer M, et al. Elevation of serum sphingosine-1-phosphate attenuates impaired cardiac function in experimental sepsis. Sci Rep (2016) 6:27594. doi:10.1038/srep27594
21. Press AT, Traeger A, Pietsch C, Mosig A, Wagner M, Clemens MG, et al. Cell type-specific delivery of short interfering RNAs by dye-functionalised theranostic nanoparticles. Nat Commun (2014) 5:5565. doi:10.1038/ncomms6565
22. Press AT, Ramoji A, Vd Lühe M, Rinkenauer AC, Hoff J, Butans M, et al. Cargo–carrier interactions significantly contribute to micellar conformation and biodistribution. NPG Asia Mater (2017) 9:e444. doi:10.1038/am.2017.161
23. Taylor NJ, Nishtala A, Manakkat Vijay GK, Abeles RD, Auzinger G, Bernal W, et al. Circulating neutrophil dysfunction in acute liver failure. Hepatology (2013) 57:1142–52. doi:10.1002/hep.26102
24. van Hoek AJ, Andrews N, Waight PA, Stowe J, Gates P, George R, et al. The effect of underlying clinical conditions on the risk of developing invasive pneumococcal disease in England. J Infect (2012) 65:17–24. doi:10.1016/j.jinf.2012.02.017
25. Bruns T, Zimmermann HW, Stallmach A. Risk factors and outcome of bacterial infections in cirrhosis. World J Gastroenterol (2014) 20:2542–54. doi:10.3748/wjg.v20.i10.2542
26. Horn P, Metzing UB, Steidl R, Romeike B, Rauchfuss F, Sponholz C, et al. Chemerin in peritoneal sepsis and its associations with glucose metabolism and prognosis: a translational cross-sectional study. Crit Care (2016) 20:39. doi:10.1186/s13054-016-1209-5
27. von Loeffelholz C, Docke S, Lock JF, Lieske S, Horn P, Kriebel J, et al. Increased lipogenesis in spite of upregulated hepatic 5’AMP-activated protein kinase in human non-alcoholic fatty liver. Hepatol Res (2017) 47:890–901. doi:10.1111/hepr.12825
28. Liu SF, Malik AB. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol (2006) 290:L622–45. doi:10.1152/ajplung.00477.2005
29. Kortgen A, Paxian M, Werth M, Recknagel P, Rauchfuss F, Lupp A, et al. Prospective assessment of hepatic function and mechanisms of dysfunction in the critically ill. Shock (2009) 32:358–65. doi:10.1097/SHK.0b013e31819d8204
30. Råberg L, Sim D, Read AF. Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science (2007) 318:812–4. doi:10.1126/science.1148526
31. Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol (2008) 8:889–95. doi:10.1038/nri2432
32. Soares MP, Gozzelino R, Weis S. Tissue damage control in disease tolerance. Trends Immunol (2014) 35:483–94. doi:10.1016/j.it.2014.08.001
33. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell (2015) 160:816–27. doi:10.1016/j.cell.2015.02.010
34. Ayres JS, Schneider DS. Tolerance of infections. Annu Rev Immunol (2012) 30:271–94. doi:10.1146/annurev-immunol-020711-075030
35. Ayres JS, Freitag N, Schneider DS. Identification of Drosophila mutants altering defense of and endurance to Listeria monocytogenes infection. Genetics (2008) 178:1807–15. doi:10.1534/genetics.107.083782
36. Howick VM, Lazzaro BP. Genotype and diet shape resistance and tolerance across distinct phases of bacterial infection. BMC Evol Biol (2014) 14:56. doi:10.1186/1471-2148-14-56
37. Figueiredo N, Chora A, Raquel H, Pejanovic N, Pereira P, Hartleben B, et al. Anthracyclines induce DNA damage response-mediated protection against severe sepsis. Immunity (2013) 39:874–84. doi:10.1016/j.immuni.2013.08.039
38. Ermolaeva MA, Segref A, Dakhovnik A, Ou HL, Schneider JI, Utermohlen O, et al. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature (2013) 501:416–20. doi:10.1038/nature12452
39. Weis S, Rubio I, Ludwig K, Weigel C, Jentho E. Hormesis and defense of infectious disease. Int J Mol Sci (2017) 18:1273. doi:10.3390/ijms18061273
40. Fauci AS, Morens DM. The perpetual challenge of infectious diseases. N Engl J Med (2012) 366:454–61. doi:10.1056/NEJMra1108296
41. Aminov R. History of antimicrobial drug discovery: major classes and health impact. Biochem Pharmacol (2017) 133:4–19. doi:10.1016/j.bcp.2016.10.001
42. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence (2014) 5:4–11. doi:10.4161/viru.27372
43. Medzhitov R. Septic shock: on the importance of being tolerant. Immunity (2013) 39:799–800. doi:10.1016/j.immuni.2013.10.012
44. Schafer JF. Tolerance to plant disease. Annu Rev Phytopathol (1971) 9:235–52. doi:10.1146/annurev.py.09.090171.001315
45. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med (2010) 2:51ra71. doi:10.1126/scitranslmed.3001118
46. Sahoo M, Del Barrio L, Miller MA, Re F. Neutrophil elastase causes tissue damage that decreases host tolerance to lung infection with Burkholderia species. PLoS Pathog (2014) 10:e1004327. doi:10.1371/journal.ppat.1004327
47. Miller MR, White A, Boots M. The evolution of parasites in response to tolerance in their hosts: the good, the bad, and apparent commensalism. Evolution (2006) 60:945–56. doi:10.1554/05-654.1
48. Kahl BC, Becker K, Loffler B. Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clin Microbiol Rev (2016) 29:401–27. doi:10.1128/CMR.00069-15
49. Wood TK. Combatting bacterial persister cells. Biotechnol Bioeng (2016) 113:476–83. doi:10.1002/bit.25721
50. Lee PS, Wilhelmson ASK, Hubner AP, Reynolds SB, Gallacchi DA, Chiou TT, et al. mTORC1-S6K activation by endotoxin contributes to cytokine up-regulation and early lethality in animals. PLoS One (2010) 5:e14399. doi:10.1371/journal.pone.0014399
51. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1 alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi:10.1126/science.1250684
52. Escobar DA, Botero-Quintero AM, Kautza BC, Luciano J, Loughran P, Darwiche S, et al. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J Surg Res (2015) 194:262–72. doi:10.1016/j.jss.2014.10.009
53. Mulchandani N, Yang WL, Khan MM, Zhang F, Marambaud P, Nicastro J, et al. Stimulation of brain AMP-activated protein kinase attenuates inflammation and acute lung injury in sepsis. Mol Med (2015) 21:637–44. doi:10.2119/molmed.2015.00179
54. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi:10.1016/j.molcel.2008.03.003
55. Rutherford C, Speirs C, Williams JJL, Ewart MA, Mancini SJ, Hawley SA, et al. Phosphorylation of Janus kinase 1 (JAK1) by AMP-activated protein kinase (AMPK) links energy sensing to anti-inflammatory signaling. Sci Signal (2016) 9:10. doi:10.1126/scisignal.aaf8566
56. Hardie DG. AMP-activated protein kinase: a key regulator of energy balance with many roles in human disease. J Intern Med (2014) 276:543–59. doi:10.1111/joim.12268
57. Zha QB, Wei HX, Li CG, Liang YD, Xu LH, Bai WJ, et al. ATP-induced inflammasome activation and pyroptosis is regulated by AMP-activated protein kinase in macrophages. Front Immunol (2016) 7:597. doi:10.3389/fimmu.2016.00597
58. Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. Akt and foxo dysregulation contribute to infection-induced wasting in Drosophila. Curr Biol (2006) 16:1977–85. doi:10.1016/j.cub.2006.08.052
Keywords: sepsis, organ dysfunction, metabolic adaptation, disease tolerance, signaling
Citation: Bauer M, Coldewey SM, Leitner M, Löffler B, Weis S and Wetzker R (2018) Deterioration of Organ Function As a Hallmark in Sepsis: The Cellular Perspective. Front. Immunol. 9:1460. doi: 10.3389/fimmu.2018.01460
Received: 18 December 2017; Accepted: 12 June 2018;
Published: 26 June 2018
Edited by:
Tobias Schuerholz, Universitätsmedizin Rostock, GermanyReviewed by:
Dror Mevorach, Hadassah Medical Center, IsraelMarkus Bosmann, Boston University, United States
Hugo Caire Castro-Faria-Neto, Fundação Oswaldo Cruz (Fiocruz), Brazil
Copyright: © 2018 Bauer, Coldewey, Leitner, Löffler, Weis and Wetzker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael Bauer, bWljaGFlbC5iYXVlckBtZWQudW5pLWplbmEuZGU=