 Yunben Yang1,2†
Yunben Yang1,2† Chunjing Xu1,3†
Chunjing Xu1,3† Dang Wu1,4†
Dang Wu1,4† Zhen Wang1,3
Zhen Wang1,3 Pin Wu1,5
Pin Wu1,5 Lili Li1,2
Lili Li1,2 Jian Huang1,3*Fuming Qiu1,2*
Jian Huang1,3*Fuming Qiu1,2*
- 1Cancer Institute (Key Laboratory of Cancer Prevention and Intervention, National Ministry of Education), The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Medical Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Surgical Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
- 4Department of Radiation Oncology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
- 5Department of Thoracic Surgery, The Second Affiliated Hospital, Zhejiang University School of Medicine, Zhejiang University, Hangzhou, China
Increasing evidence suggests that intestinal microbiota dysbiosis and chronic inflammation contribute to colorectal cancer (CRC) development. γδ T cells represent a major innate immune cell population in the intestinal epithelium that is involved in the maintenance of gut homeostasis, inflammation regulation, and carcinogenesis. The important contributions of γδ T cells are (i) to perform a protective role in the context of barrier damage and pathogenic microorganism translocation; (ii) to exert either pro- or anti-inflammatory effects at different inflammatory stages; and (iii) to boost the crosstalk between immune cells and tumor microenvironment, inducing a cascade of suppressive immune responses. Understanding the crucial role of γδ T cells would enable us to manipulate these cells during the CRC sequence and improve the efficacy of tumor therapy.
Introduction
Colorectal cancer (CRC) is the third most-common cancer in the world with high mortality (1–3). Evidence suggests that CRC development is linked to the change of environmental factors including dietary behavior, obesity, and infection, which further lead to the alteration of microbial composition (4, 5). The majority of the microbiota resides on the surface of the intestine and is indispensable for physiologic homeostasis; microbial dysbiosis has been linked to chronic inflammation and cancer development (6–8). Metagenomic analysis of specimens (mucosal tissues or fecal samples) from patients with CRC and from healthy controls shows dramatic differences in the microbial community structure (9–11). Moreover, compared to those colonized with healthy microbiota, transfer of cancer-associated microbiota into germ-free mice significantly increases the tumor burden, indicating that altered microbiota exacerbate CRC formation (12–15). From a mechanistic point of view, microbiota can generate potential oncometabolites that reshape the polarization of immune cells, thus contributing to aberrant inflammatory processes and loss of prevention of inappropriate immune responses (16–18). For example, reduced abundance of butyrate-producing bacteria, in patients with CRC, is associated with impaired barrier function and regulatory T cell generation (19). Dysbiosis-driven chronic inflammation evolves as a characteristic that fosters hallmarks of cancer, and triggers uncontrolled immune responses, resulting in loss of homeostasis and consequently in a vicious cycle contributing to tumorigenesis (16, 20). Altered commensal bacteria, chronic inflammation, and CRC development do not exist independently; they influence each other in reciprocal causation. Alterations of microbiota can occur prior to the initial macroscopic observation of colonic tumor formation, and bacterial translocation across the barrier may cause enhanced chronic inflammation (13); the recruitment of immune cells and changes in cancer cell metabolism drive the microbial structure remodeling (21). Innate immune cells act as significant mediators in these processes by participating in bidirectional regulation of inflammatory responses (22).
γδ T cells, a unique population of innate-like T lymphocytes, recognize the superstructure of antigen without the requirement of major histocompatibility complex (MHC) molecules (22, 23). In humans, the two major subsets of γδ T cells, comprising of Vδ1+ and Vγ9Vδ2 T cells, are identified by their Vδ chain usage. Vδ1+ (associated with various Vγ elements) T cells are the initial γδ T cells derived from thymus, which ultimately reside in colonic epithelial tissues and make up the majority of intraepithelial lymphocytes (IELs). Normal development and antimicrobial capacity acquisition of Vδ1+ T cells rely on each cell receiving proper instructions from the gut microbiota (24, 25). During microbial dysbiosis, resident Vδ1+ T cells are indispensable for triggering early protective inflammatory responses, which are essential for wound healing (22, 24, 26). Once the inflammatory response persists, Vδ1+ T cells subtly provide the foundation for angiogenesis, survival signal pathway activation, and myeloid-derived suppressor cell (MDSC) recruitment, thereby generating a chronic pre-cancerous inflammatory environment (27–29). In such cancer or stressed cells, isopentenyl pyrophosphate (IPP) gradually accumulates and boosts the activation of Vγ9Vδ2 T cells, which only take up a small fraction of lymphocytes in peripheral blood (PB), but harbor an antineoplastic effect (30, 31). However, the local tumor microenvironment (TME) has a profound influence on these recruited Vγ9Vδ2 T cells, which are gradually polarized into cancer-promoting phenotypes (e.g., Th17-like cells) (32). Taken together, tumor progression is facilitated by continuous presence of inflammatory cells and cytokines in the TME (21, 33).
This review highlights the plasticity of γδ T cells in different environments, thereby linking microbiota, inflammation, and CRC.
Induction of γδ T Cell Activation and Polarization by Microbiota
γδ IELs, the major population of IELs, are localized in the intestinal surface, establishing the most intimate communication with microbiota (34, 35). In fact, commensal bacteria are necessary for the γδ IELs to release protective factors following mucosal damage, through both myeloid differentiation primary response 88 (MYD88)-dependent [e.g., keratinocyte-derived chemokine and chemokine (C-X-C motif) ligand 9 (CXCL9)] and MYD88-independent (e.g., macrophage inflammatory protein 2α, RegIIIγ) pathways (36, 37). Compared to that in wild-type mice, expression of antimicrobial cytokines is significantly reduced in germ-free mice, which may, in turn, be restored by transplanting microbiota harvested from conventionalized mice (37). Microbial effects on γδ IELs can be mediated by epithelial cells (ECs) (38–40). In a recent study, Hoytema van Konijnenburg et al. indicated that antimicrobial responses and movement dynamics of γδ IELs were impaired in mice with MYD88-deficient ECs. Interestingly, EC-mediated microbial sensing also controls metabolic switch of γδ IELs in response to infection (38). Thus, a cellular network of microbiota, ECs, and γδ IELs, in which microbiota-stimulated ECs regulate motility and energy metabolism of γδ IELs, is essential for the maintenance of homeostasis (Figure 1).
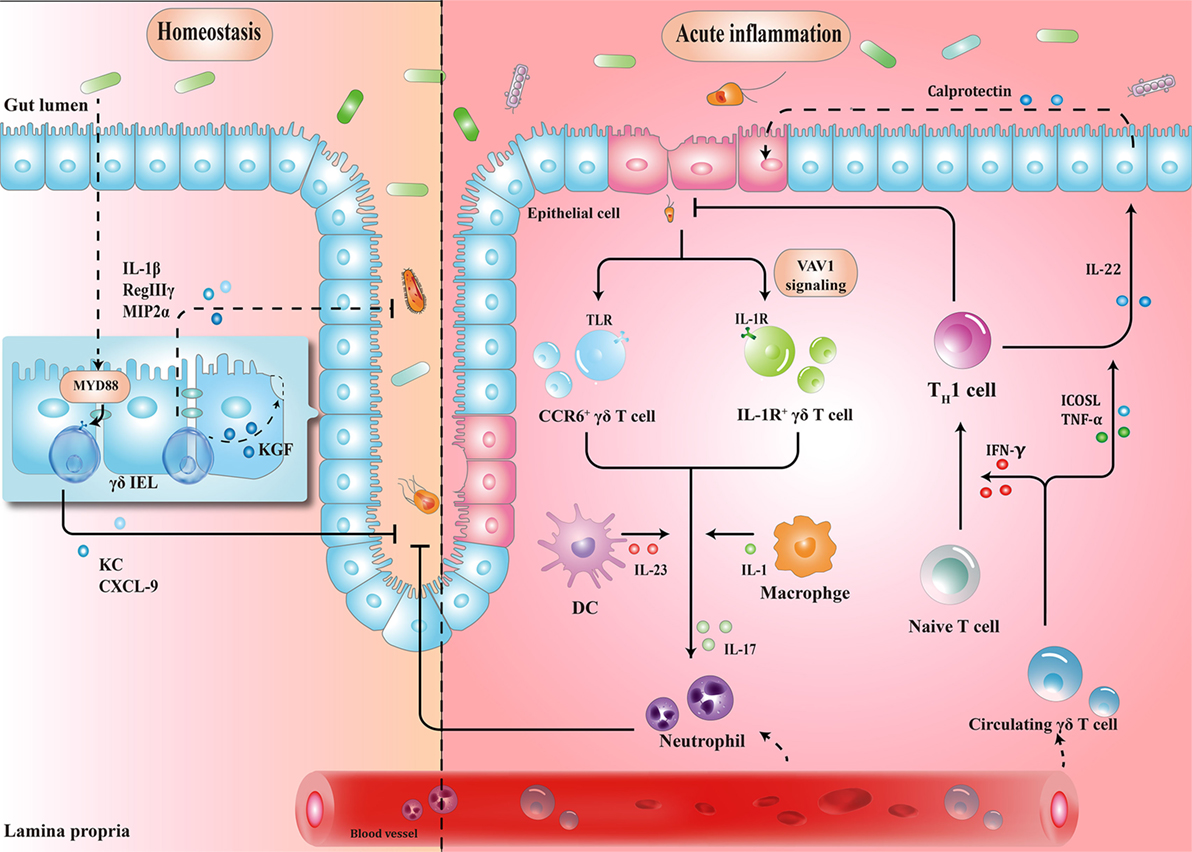
Figure 1. Schematic overview of the protective roles of γδ T cells in homeostasis maintenance and immune surveillance. (1) Physiologically, the crosstalk between microbiota, epithelial cells (ECs), and γδ T cells enhances barrier stabilization. (2) During acute inflammation, neutrophils are stimulated by IL-17 from γδ T cells, and recruited to eliminate pathogens. Meanwhile, microbe-activated circulating γδ T cells promote cytotoxic responses with Th1-committed αβ T cells and potentiate the release of calprotectin in an inducible T-cell co-stimulator ligand (ICOSL)/tumor necrosis factor α (TNF-α)-dependent manner. IEL, intraepithelial lymphocyte; MYD88, myeloid differentiation primary response 88; KC, keratinocyte-derived chemokine; CXCL, chemokine (C-X-C motif) ligand; MIP2α, macrophage inflammatory protein 2α; CCR, C-C motif chemokine receptor; TLR, toll-like receptor; IFN-γ, interferon γ; IL-17, interleukin-17.
When barrier damage results in microfloral translocation, γδ T cells are indispensable for triggering early acute inflammatory responses against the penetration of microbiota across the impaired intestinal mucosa (34). For example, TCRγδ-deficient mice are more susceptible to harmful microbial cues and show early spread of pathogens to other organs (37). Importantly, a compelling feature seen in γδ T cells is their ability to secrete interleukin-17 (IL-17) in early pathogen invasion, which seems to benefit the host initially (41–43). Mechanistically, it is probable that γδ T cells recognize pathogen-associated molecular patterns through a series of toll-like receptors (TLRs), indicating the direct effect of bacterial products to drive the expansion of γδ T cells (44). For example, in response to pathogenic bacteria, C-C motif chemokine receptor 6 (CCR6)+ γδ T cells (predominately Vγ2) produce IL-17 through TLR1 and TLR2, which in turn form a positive feedback loop to recruit immune cells, thus launching the inflammatory response to eliminate pathogens before Th17 cells get activated (43). As such, commensal bacteria-induced IL-1R1+ γδ T cells provide the main source of IL-17 via the guanine nucleotide exchange factor VAV1 (45). Microbial metabolites or antigens may also exert indirect effects on γδ T cells. In response to altered micro-environmental cues, dendritic cells (DCs) can communicate with IL-17-producing γδ T (γδT17) cells via cell-to-cell contact or different cytokines, revealing a crosstalk between the immune system and microbiota (Figure 1) (46).
Circulating Vγ9Vδ2 T cells strategically migrate from blood to the infected intestine (47, 48). On one hand, γδ T cells promote Th1-committed αβ T cell responses via interferon γ (IFN-γ) production and enhance the secretion of inflammatory cytokines including tumor necrosis factor α (TNF-α) (49, 50). On the other hand, γδ T cells, employed as antigen-presenting cells, potentiate the secretion of IL-22 by CD4+ cells and release of calprotectin in an inducible T-cell co-stimulator ligand (ICOSL)/TNF-α-dependent manner (Figure 1) (51).
These studies suggest that dysbiosis can induce γδ T cell activation to trigger early protective inflammatory responses.
Participation of γδ T Cells in Chronic Inflammation
γδ T cells are continuously activated by sustained exposure to bacterial metabolites, thereby leading to the exhaustion of protective γδ T cell subtypes and activation of chronic inflammation (5, 18). In this regard, one representative example is inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC) (52–54). Although γδ T cells (especially Vδ1+) are reported to be significantly increased in the inflamed mucosa of patients with UC, the role of γδ T cells in human CD remains a matter of debate [reviewed in Ref. (55)], suggesting the discrepancy of immunological background between UC and CD (55, 56). Early studies, focused on the change in proportion of γδ T cells in CD (either in PB or tissue samples), reported diverse results (57–60), probably due to the limitation of small sample size and different γδ T cell subpopulations. For example, a recent study compared γδ T cell subsets in PB from 40 patients having CD with that from healthy controls, and the reduction extent of γδ T cell subsets was addressed, especially for CD8+ γδ T cells (61). The authors showed that deficiency of this γδ T cell subset could affect the immune responses to pathogens in patients with CD (62). Lately, Kadivar et al. found decreased levels of CD8αβ+ γδ T cells (predominately Vδ1+) in inflamed mucosa, associated with worse disease activity, whereas increased proportion of CD8αβ+ γδ T cells was observed in anti-TNF-α-treated patients with CD (63). Based on these results, γδ T cells cannot be simply recognized as one homogeneous population; however, further studies would be required to define the functions of different γδ T cell subtypes for interpreting the pathology of human IBD.
The protective role of γδ T cells in exerting wound-healing responses has been suggested in murine colitis model. γδ T cells preserve homeostasis by removal of impaired ECs; secretion of growth factors to promote epithelial regeneration; regulation of αβ T cell functions to limit excessive inflammatory response; and enhancement of granulocyte infiltration (64–66). Importantly, one layer of these processes is associated with protective IL-17 production (28, 41, 67); Lee et al. showed that γδ T cells were the major producers of protective IL-17 in the retinoid-related orphan receptor γt-dependent and IL-23-independent manner (68). Secretion of IL-17 could activate Act-1 (a key adaptor protein of IL-17 receptor), which attenuated inflammation and immobilized the localization of occluding (a tight junction protein) to prevent excessive intestinal permeability (Figure 2) (68). Another crucial anti-inflammatory pathway of γδ T cells is linked to the recruitment of MDSCs (29). Sun et al. observed that TCRγδ-deficient mice were more susceptible to dextran sulfate sodium-induced colitis, with a reduced expression of IL-18 and CXCL5 relative to wild-type mice, which was important for the subsequent Gr-1+CD11b+ suppressor cell infiltration (29). Moreover, mice reconstituted with γδT17 cells (mainly Vγ6) had ameliorated intestinal inflammation and were associated with increased frequency of Gr-1+CD11b+ cells, whereas mice with IFN-γ producing γδ T cells had no significant difference, indicating a protective role of γδT17 cells (but not IFN-γ producing γδ T cells) to a certain extent (29).
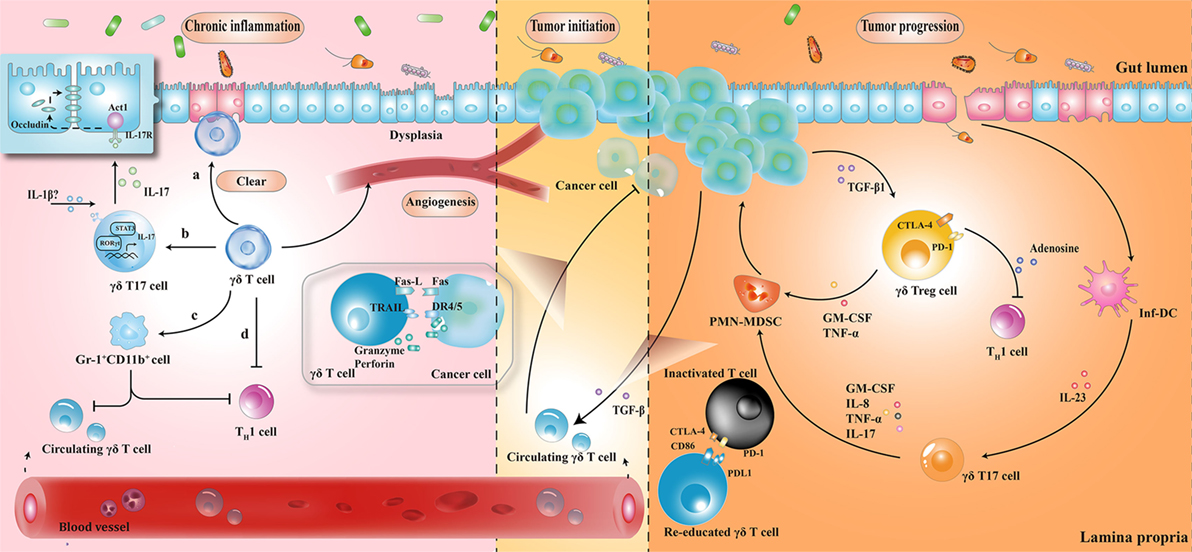
Figure 2. The roles played by γδ T cells at different stages of colorectal cancer development. (1) During chronic inflammation, γδ T cells can limit excessive inflammatory response and maintain homeostasis by (a) removal of impaired epithelial cells; (b) secretion of protective IL-17; (c) enhancement of Gr-1+CD11b+ suppressor cell infiltration; and (d) regulation of αβ T cell functions. (2) At the initiation of tumor formation, circulating γδ T cells can recognize and kill cancer cells, but they are reprogrammed with cancer development. (3) At stages of tumor progression, IL-17 and granulocyte macrophage colony-stimulating factor (GM-CSF), produced by γδ T cells in response to IL-23 and transforming growth factor beta 1 (TGF-β1) from inflammatory dendritic cells (inf-DCs) and cancer cells, are essential for the recruitment of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs), consequently inducing a cascade of suppressive immune responses. CTLA4, cytotoxic T lymphocyte-associated protein-4; PD-1, programmed cell death protein 1; TNF-α, tumor necrosis factor α; IL-17, interleukin-17.
Taken together, γδ T cells seem to maintain tissue architecture during chronic inflammation, probably via IL-17 production and MDSC accumulation. However, it is noteworthy that these mediators have the ability to support tumor development by enhancing angiogenesis and promoting immunological tolerance to tumor cells. While these mechanisms have not been demonstrated in patients with IBD, the complex interactions between γδ T cells and MDSCs hint at the possible regulatory pathways in humans.
The Facilitative Role of γδ T Cells in CRC
In tumor initiation, circulating Vγ9Vδ2 T cells, activated by the over-produced IPP in cancer cells, express the inflammatory homing chemokine receptors (e.g., CXCR3 and CCR5), guiding them into tumor sites (69, 70). By identifying the various upregulated ligands in cancer cells (e.g., MHC class I-related chains A/B and UL16-binding proteins), Vγ9Vδ2 T cells can either kill tumor cells indirectly by releasing abundant cytokines (e.g., IFN-γ and TNF-α), thereby displaying a Th1 cell-like property, or do so directly via death receptor signal (e.g., Fas-FasL and TNF-TNFR), secreting cytotoxic molecules such as granzymes (71, 72). By contrast, with tumor progression, tumor-associated macrophages and MDSCs are enriched in TME, acting as endless suppliers of IL-1β, IL-23, and transforming growth factor beta (TGF-β). Therefore, the recruited Vγ9Vδ2 T cells are reprogrammed by the local TME and may be polarized to γδT17 cells or regulatory γδ T cells (γδTregs). The polarized γδT17 cells or γδTregs produce abundant IL-17 or TGF-β, triggering a cascade of suppressive immune responses to promote CRC progression (Figure 2) (32, 73, 74).
The local TME has a considerable impact on tissue-infiltrating Vδ1+ T cells as well. Interestingly, γδ T cells were found to be the major source of IL-17 and majority of tumor-infiltrating γδT17 cells were Vδ1+ (21). Epithelial barrier damage was found to cause liberation of microbial products, resulting in activation of inflammatory DCs (inf-DCs), which polarized γδT17 cells in an IL-23-dependent manner (21). Tumor-infiltrating γδT17 cells could secrete IL-17, IL-8, TNF-α, and granulocyte macrophage colony-stimulating factor to promote the proliferation and survival of polymorphonuclear MDSCs, thus transforming the CRC-triggered inflammation into an immunosuppressive condition (21). In addition to indirect immunosuppressive role of γδT17 cells, a novel population of CD39+γδTregs was identified, which mediated a direct and robust immunosuppressive effect on human CRC (75). CD39+γδTregs took up nearly 50% of the total γδ T cells and expressed high levels of suppressive molecules (such as Forkhead box p3, cytotoxic T lymphocyte-associated protein-4, and programmed cell death protein 1), as well as cytokines (including IL-10 and IL-17), contributing to the formation of a potent immunosuppressive microenvironment. Stimulated by TGF-β1, CD39+γδTregs could secrete adenosine to foster CRC progression (Figure 2) (75). More importantly, the frequency of γδT17 and CD39+γδTregs was related to clinicopathological factors in patients with CRC (e.g., TNM stages, tumor size, and invasion), suggesting them to be potential prognostic factors (21, 75). Similarly, Rong et al. described a disproportionate distribution of Vδ1+ and Vδ2+ T cells in rectal cancer tissues (76). A higher percentage of Vδ1+ T cells and a lower percentage of Vδ2+ T cells were found in the tumor tissues compared to that in the adjacent normal tissues. Tumor-infiltrating Vδ1+ T cells had strong inhibitory functions and positively associated with the T stage of patients (76).
Conclusion and Future Direction
γδ T cell is a key mediator of barrier surveillance, chronic inflammation regulation, and immunosuppressive TME formation. In this review, we focused on the dynamic changes of γδ T cell infiltration during the prolonged period of intestinal architectural disruption, suggesting that milieu alteration is a decisive factor for cell polarization. Based on recent reports, a potential mechanistic framework has been elucidated (Figures 1 and 2). In fact, we have just begun to uncover the implication of γδ T cells in the CRC sequence. For example, although utilization of γδ T cells (mainly Vγ9Vδ2) (adoptive transfer of in vitro expanded cells or in vivo activation) has been employed in a series of clinical trials, the clinical responses of CRC treatment are barely satisfactory (77–80). Several additional factors should be considered for future γδ T cell immunotherapy: primarily, the impact of local TME should be seriously considered. Despite transmutation of Vγ9Vδ2 T cells from anti-tumoral to pro-tumoral activity can occur in TME, the CRC-derived Vδ1+ tumor-infiltrating lymphocytes have been shown to kill colonic cancer cells in vitro (81–83). Therefore, it confirms that environmental changes can lead to remodeling of cell function. Moreover, the difference of microbial community structure between individuals, as well as the interaction between gut microbiota and γδ T cells is also worth considering. Questions such as: whether alteration of a certain species of microorganisms is the cause or consequence in the alteration of γδ T cell subsets, which microbiota-derived metabolites can be certainly related to the polarization of γδ T cells, what are the exact mechanisms resulting from microbiota dysbiosis or chronic inflammation that cause skewed responses of γδ T lymphocytes, whether pro-tumoral activities of γδ T cells benefit from commensal penetration, and in turn facilitate uncontrolled inflammation and tumor formation, or whether γδ T cells can be reprogrammed by precise regulation of the microbiota composition to avoid chronic inflammatory microenvironment transformation, will need to be addressed, for the manipulation of γδ T cell-based immunotherapy to improve the efficacy of CRC treatment.
Author Contributions
Conception and design: FQ and JH. Write, review, and revision of the manuscript: YY, CX, DW, and ZW. Supervision: PW and LL.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LW declared a shared affiliation, though no other collaboration, with the authors to the handling Editor.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81472640, 81672802, FQ; 81520108024, JH; 81602692, DW; 81572800, PW) and the Zhejiang Provincial Natural Science Foundation of China (No. LZ17H160004, FQ).
References
1. Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin (2017) 67:177–93. doi:10.3322/caac.21395
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin (2017) 67:7–30. doi:10.3322/caac.21387
3. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin (2015) 65:87–108. doi:10.3322/caac.21262
4. Belkaid Y, Harrison OJ. Homeostatic immunity and the microbiota. Immunity (2017) 46:562–76. doi:10.1016/j.immuni.2017.04.008
5. Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal cancer. Annu Rev Microbiol (2016) 70:395–411. doi:10.1146/annurev-micro-102215-095513
7. Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer (2013) 13:800–12. doi:10.1038/nrc3610
8. Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe (2014) 15:317–28. doi:10.1016/j.chom.2014.02.007
9. Flemer B, Lynch DB, Brown JM, Jeffery IB, Ryan FJ, Claesson MJ, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut (2017) 66:633–43. doi:10.1136/gutjnl-2015-309595
10. Goedert JJ, Gong Y, Hua X, Zhong H, He Y, Peng P, et al. Fecal microbiota characteristics of patients with colorectal adenoma detected by screening: a population-based study. EBioMedicine (2015) 2:597–603. doi:10.1016/j.ebiom.2015.04.010
11. Zackular JP, Rogers MA, Ruffin MTT, Schloss PD. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res (Phila) (2014) 7:1112–21. doi:10.1158/1940-6207.CAPR-14-0129
12. Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, et al. Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology (2017) 153:1621–33.e1626. doi:10.1053/j.gastro.2017.08.022
13. Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, et al. The gut microbiome modulates colon tumorigenesis. MBio (2013) 4:e00692–13. doi:10.1128/mBio.00692-13
14. Baxter NT, Zackular JP, Chen GY, Schloss PD. Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome (2014) 2:20. doi:10.1186/2049-2618-2-20
15. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe (2013) 14:207–15. doi:10.1016/j.chom.2013.07.007
16. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140:883–99. doi:10.1016/j.cell.2010.01.025
17. Hold GL, Garrett WS. Gut microbiota. Microbiota organization – a key to understanding CRC development. Nat Rev Gastroenterol Hepatol (2015) 12:128–9. doi:10.1038/nrgastro.2015.25
18. Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol (2014) 12:661–72. doi:10.1038/nrmicro3344
19. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi:10.1038/nature12726
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
21. Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity (2014) 40:785–800. doi:10.1016/j.immuni.2014.03.013
22. Born WK, Reardon CL, O’Brien RL. The function of gammadelta T cells in innate immunity. Curr Opin Immunol (2006) 18:31–8. doi:10.1016/j.coi.2005.11.007
23. Carding SR, Egan PJ. Gammadelta T cells: functional plasticity and heterogeneity. Nat Rev Immunol (2002) 2:336–45. doi:10.1038/nri797
24. Chien YH, Meyer C, Bonneville M. Gammadelta T cells: first line of defense and beyond. Annu Rev Immunol (2014) 32:121–55. doi:10.1146/annurev-immunol-032713-120216
25. Constantinides MG. Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol (2018) 103:409–19. doi:10.1002/JLB.3RI0917-378R
26. Hayday AC. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol (2000) 18:975–1026. doi:10.1146/annurev.immunol.18.1.975
27. Fleming C, Morrissey S, Cai Y, Yan J. Gammadelta T cells: unexpected regulators of cancer development and progression. Trends Cancer (2017) 3:561–70. doi:10.1016/j.trecan.2017.06.003
28. Papotto PH, Ribot JC, Silva-Santos B. IL-17+ gammadelta T cells as kick-starters of inflammation. Nat Immunol (2017) 18:604–11. doi:10.1038/ni.3726
29. Sun X, Cai Y, Fleming C, Tong Z, Wang Z, Ding C, et al. Innate gammadeltaT17 cells play a protective role in DSS-induced colitis via recruitment of Gr-1+CD11b+ myeloid suppressor cells. Oncoimmunology (2017) 6:e1313369. doi:10.1080/2162402X.2017.1313369
30. Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol (2013) 13:88–100. doi:10.1038/nri3384
31. Zheng J, Liu Y, Lau YL, Tu W. Gammadelta-T cells: an unpolished sword in human anti-infection immunity. Cell Mol Immunol (2013) 10:50–7. doi:10.1038/cmi.2012.43
32. Xiang Z, Tu W. Dual face of Vgamma9Vdelta2-T cells in tumor immunology: anti-versus pro-tumoral activities. Front Immunol (2017) 8:1041. doi:10.3389/fimmu.2017.01041
33. Silva-Santos B, Serre K, Norell H. Gammadelta T cells in cancer. Nat Rev Immunol (2015) 15:683–91. doi:10.1038/nri3904
34. Nielsen MM, Witherden DA, Havran WL. Gammadelta T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol (2017) 17(12):733–45. doi:10.1038/nri.2017.101
35. Chen Y, Chou K, Fuchs E, Havran WL, Boismenu R. Protection of the intestinal mucosa by intraepithelial gamma delta T cells. Proc Natl Acad Sci U S A (2002) 99:14338–43. doi:10.1073/pnas.212290499
36. Ismail AS, Severson KM, Vaishnava S, Behrendt CL, Yu X, Benjamin JL, et al. Gammadelta intraepithelial lymphocytes are essential mediators of host-microbial homeostasis at the intestinal mucosal surface. Proc Natl Acad Sci U S A (2011) 108:8743–8. doi:10.1073/pnas.1019574108
37. Ismail AS, Behrendt CL, Hooper LV. Reciprocal interactions between commensal bacteria and gamma delta intraepithelial lymphocytes during mucosal injury. J Immunol (2009) 182:3047–54. doi:10.4049/jimmunol.0802705
38. Hoytema van Konijnenburg DP, Reis BS, Pedicord VA, Farache J, Victora GD, Mucida D. Intestinal epithelial and intraepithelial T cell crosstalk mediates a dynamic response to infection. Cell (2017) 171:783–94.e713. doi:10.1016/j.cell.2017.08.046
39. Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of intestinal epithelial cell turnover by commensal bacteria: role of short-chain fatty acids. PLoS One (2016) 11:e0156334. doi:10.1371/journal.pone.0156334
40. Boismenu R, Havran WL. Modulation of epithelial cell growth by intraepithelial gamma delta T cells. Science (1994) 266:1253–5. doi:10.1126/science.7973709
41. Chien YH, Zeng X, Prinz I. The natural and the inducible: interleukin (IL)-17-producing gammadelta T cells. Trends Immunol (2013) 34:151–4. doi:10.1016/j.it.2012.11.004
42. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity (2009) 31:331–41. doi:10.1016/j.immuni.2009.08.001
43. Martin B, Hirota K, Cua DJ, Stockinger B, Veldhoen M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity (2009) 31:321–30. doi:10.1016/j.immuni.2009.06.020
44. Dar AA, Patil RS, Chiplunkar SV. Insights into the relationship between toll like receptors and gamma delta T cell responses. Front Immunol (2014) 5:366. doi:10.3389/fimmu.2014.00366
45. Duan J, Chung H, Troy E, Kasper DL. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells. Cell Host Microbe (2010) 7:140–50. doi:10.1016/j.chom.2010.01.005
46. Fleming C, Cai Y, Sun X, Jala VR, Xue F, Morrissey S, et al. Microbiota-activated CD103(+) DCs stemming from microbiota adaptation specifically drive gammadeltaT17 proliferation and activation. Microbiome (2017) 5:46. doi:10.1186/s40168-017-0263-9
47. Eberl M, Roberts GW, Meuter S, Williams JD, Topley N, Moser B. A rapid crosstalk of human gammadelta T cells and monocytes drives the acute inflammation in bacterial infections. PLoS Pathog (2009) 5:e1000308. doi:10.1371/journal.ppat.1000308
48. Devilder MC, Allain S, Dousset C, Bonneville M, Scotet E. Early triggering of exclusive IFN-gamma responses of human Vgamma9Vdelta2 T cells by TLR-activated myeloid and plasmacytoid dendritic cells. J Immunol (2009) 183:3625–33. doi:10.4049/jimmunol.0901571
49. McCarthy NE, Bashir Z, Vossenkamper A, Hedin CR, Giles EM, Bhattacharjee S, et al. Proinflammatory Vdelta2+ T cells populate the human intestinal mucosa and enhance IFN-gamma production by colonic alphabeta T cells. J Immunol (2013) 191:2752–63. doi:10.4049/jimmunol.1202959
50. McCarthy NE, Hedin CR, Sanders TJ, Amon P, Hoti I, Ayada I, et al. Azathioprine therapy selectively ablates human Vdelta2(+) T cells in Crohn’s disease. J Clin Invest (2015) 125:3215–25. doi:10.1172/JCI80840
51. Tyler CJ, McCarthy NE, Lindsay JO, Stagg AJ, Moser B, Eberl M. Antigen-presenting human gammadelta T cells promote intestinal CD4+ T cell expression of IL-22 and mucosal release of calprotectin. J Immunol (2017) 198:3417–25. doi:10.4049/jimmunol.1700003
52. Choi CR, Bakir IA, Hart AL, Graham TA. Clonal evolution of colorectal cancer in IBD. Nat Rev Gastroenterol Hepatol (2017) 14:218–29. doi:10.1038/nrgastro.2017.1
53. Rogler G. Chronic ulcerative colitis and colorectal cancer. Cancer Lett (2014) 345:235–41. doi:10.1016/j.canlet.2013.07.032
54. Feagins LA, Souza RF, Spechler SJ. Carcinogenesis in IBD: potential targets for the prevention of colorectal cancer. Nat Rev Gastroenterol Hepatol (2009) 6:297–305. doi:10.1038/nrgastro.2009.44
55. Catalan-Serra I, Sandvik AK, Bruland T, Andreu-Ballester JC. Gammadelta T cells in Crohn’s disease: a new player in the disease pathogenesis? J Crohns Colitis (2017) 11:1135–45. doi:10.1093/ecco-jcc/jjx039
56. Fay NS, Larson EC, Jameson JM. Chronic inflammation and gammadelta T cells. Front Immunol (2016) 7:210. doi:10.3389/fimmu.2016.00210
57. Fukushima K, Masuda T, Ohtani H, Sasaki I, Funayama Y, Matsuno S, et al. Immunohistochemical characterization, distribution, and ultrastructure of lymphocytes bearing T-cell receptor gamma/delta in inflammatory bowel disease. Gastroenterology (1991) 101:670–8. doi:10.1016/0016-5085(91)90524-O
58. McVay LD, Li B, Biancaniello R, Creighton MA, Bachwich D, Lichtenstein G, et al. Changes in human mucosal gamma delta T cell repertoire and function associated with the disease process in inflammatory bowel disease. Mol Med (1997) 3:183–203.
59. Cuvelier CA, De Wever N, Mielants H, De Vos M, Veys EM, Roels H. Expression of T cell receptors alpha beta and gamma delta in the ileal mucosa of patients with Crohn’s disease and with spondylarthropathy. Clin Exp Immunol (1992) 90:275–9. doi:10.1111/j.1365-2249.1992.tb07942.x
60. Bucht A, Soderstrom K, Esin S, Grunewald J, Hagelberg S, Magnusson I, et al. Analysis of gamma delta V region usage in normal and diseased human intestinal biopsies and peripheral blood by polymerase chain reaction (PCR) and flow cytometry. Clin Exp Immunol (1995) 99:57–64. doi:10.1111/j.1365-2249.1995.tb03472.x
61. Andreu-Ballester JC, Amigo-Garcia V, Catalan-Serra I, Gil-Borras R, Ballester F, Almela-Quilis A, et al. Deficit of gammadelta T lymphocytes in the peripheral blood of patients with Crohn’s disease. Dig Dis Sci (2011) 56:2613–22. doi:10.1007/s10620-011-1636-8
62. Benet-Campos C, Cuellar C, Garcia-Ballesteros C, Zamora V, Gil-Borras R, Catalan-Serra I, et al. Determination of anti-anisakis simplex antibodies and relationship with alphabeta and gammadelta lymphocyte subpopulations in patients with Crohn’s disease. Dig Dis Sci (2017) 62:934–43. doi:10.1007/s10620-017-4473-6
63. Kadivar M, Petersson J, Svensson L, Marsal J. CD8alphabeta+ gammadelta T cells: a novel T cell subset with a potential role in inflammatory bowel disease. J Immunol (2016) 197:4584–92. doi:10.4049/jimmunol.1601146
64. Hoffmann JC, Peters K, Henschke S, Herrmann B, Pfister K, Westermann J, et al. Role of T lymphocytes in rat 2,4,6-trinitrobenzene sulphonic acid (TNBS) induced colitis: increased mortality after gammadelta T cell depletion and no effect of alphabeta T cell depletion. Gut (2001) 48:489–95. doi:10.1136/gut.48.4.489
65. Inagaki-Ohara K, Chinen T, Matsuzaki G, Sasaki A, Sakamoto Y, Hiromatsu K, et al. Mucosal T cells bearing TCRgammadelta play a protective role in intestinal inflammation. J Immunol (2004) 173:1390–8. doi:10.4049/jimmunol.173.2.1390
66. Kuhl AA, Loddenkemper C, Westermann J, Hoffmann JC. Role of gamma delta T cells in inflammatory bowel disease. Pathobiology (2002) 70:150–5. doi:10.1159/000068147
67. Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol (2009) 183:4169–75. doi:10.4049/jimmunol.0901017
68. Lee JS, Tato CM, Joyce-Shaikh B, Gulen MF, Cayatte C, Chen Y, et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity (2015) 43:727–38. doi:10.1016/j.immuni.2015.09.003
69. Glatzel A, Wesch D, Schiemann F, Brandt E, Janssen O, Kabelitz D. Patterns of chemokine receptor expression on peripheral blood gamma delta T lymphocytes: strong expression of CCR5 is a selective feature of V delta 2/V gamma 9 gamma delta T cells. J Immunol (2002) 168:4920–9. doi:10.4049/jimmunol.168.10.4920
70. Brandes M, Willimann K, Lang AB, Nam KH, Jin C, Brenner MB, et al. Flexible migration program regulates gamma delta T-cell involvement in humoral immunity. Blood (2003) 102:3693–701. doi:10.1182/blood-2003-04-1016
71. Hannani D, Ma Y, Yamazaki T, Dechanet-Merville J, Kroemer G, Zitvogel L. Harnessing gammadelta T cells in anticancer immunotherapy. Trends Immunol (2012) 33:199–206. doi:10.1016/j.it.2012.01.006
72. Kong Y, Cao W, Xi X, Ma C, Cui L, He W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood (2009) 114:310–7. doi:10.1182/blood-2008-12-196287
73. Mao Y, Yin S, Zhang J, Hu Y, Huang B, Cui L, et al. A new effect of IL-4 on human gammadelta T cells: promoting regulatory Vdelta1 T cells via IL-10 production and inhibiting function of Vdelta2 T cells. Cell Mol Immunol (2016) 13:217–28. doi:10.1038/cmi.2015.07
74. Peters C, Oberg HH, Kabelitz D, Wesch D. Phenotype and regulation of immunosuppressive Vdelta2-expressing gammadelta T cells. Cell Mol Life Sci (2014) 71:1943–60. doi:10.1007/s00018-013-1467-1
75. Hu G, Wu P, Cheng P, Zhang Z, Wang Z, Yu X, et al. Tumor-infiltrating CD39+gammadeltaTregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology (2017) 6:e1277305. doi:10.1080/2162402X.2016.1277305
76. Rong L, Li K, Li R, Liu HM, Sun R, Liu XY. Analysis of tumor-infiltrating gamma delta T cells in rectal cancer. World J Gastroenterol (2016) 22:3573–80. doi:10.3748/wjg.v22.i13.3573
77. Bennouna J, Levy V, Sicard H, Senellart H, Audrain M, Hiret S, et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vgamma9Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol Immunother (2010) 59:1521–30. doi:10.1007/s00262-010-0879-0
78. Noguchi A, Kaneko T, Kamigaki T, Fujimoto K, Ozawa M, Saito M, et al. Zoledronate-activated Vgamma9gammadelta T cell-based immunotherapy is feasible and restores the impairment of gammadelta T cells in patients with solid tumors. Cytotherapy (2011) 13:92–7. doi:10.3109/14653249.2010.515581
79. Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, et al. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. Br J Cancer (2011) 105:778–86. doi:10.1038/bjc.2011.293
80. Izumi T, Kondo M, Takahashi T, Fujieda N, Kondo A, Tamura N, et al. Ex vivo characterization of gammadelta T-cell repertoire in patients after adoptive transfer of Vgamma9Vdelta2 T cells expressing the interleukin-2 receptor beta-chain and the common gamma-chain. Cytotherapy (2013) 15:481–91. doi:10.1016/j.jcyt.2012.12.004
81. Maeurer MJ, Martin D, Walter W, Liu K, Zitvogel L, Halusczcak K, et al. Human intestinal Vdelta1+ lymphocytes recognize tumor cells of epithelial origin. J Exp Med (1996) 183:1681–96. doi:10.1084/jem.183.4.1681
82. Groh V, Steinle A, Bauer S, Spies T. Recognition of stress-induced MHC molecules by intestinal epithelial gammadelta T cells. Science (1998) 279:1737–40. doi:10.1126/science.279.5357.1737
Keywords: γδ T cells, microbiota, chronic inflammation, colorectal cancer, tumor microenvironment
Citation: Yang Y, Xu C, Wu D, Wang Z, Wu P, Li L, Huang J and Qiu F (2018) γδ T Cells: Crosstalk Between Microbiota, Chronic Inflammation, and Colorectal Cancer. Front. Immunol. 9:1483. doi: 10.3389/fimmu.2018.01483
Received: 27 February 2018; Accepted: 14 June 2018;
Published: 26 June 2018
Edited by:
Vladimir Brusic, The University of Nottingham Ningbo China, ChinaReviewed by:
Lie Wang, Zhejiang University School of Medicine, ChinaHelal Fouad Hetta, University of Cincinnati College of Medicine, United States
Copyright: © 2018 Yang, Xu, Wu, Wang, Wu, Li, Huang and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fuming Qiu, cWl1ZnVtaW5nQHpqdS5lZHUuY24=;
Jian Huang, ZHJodWFuZ2ppYW5Aemp1LmVkdS5jbg==
†These authors have contributed equally to this work.