 Shu Horiuchi
Shu Horiuchi Hideki Ueno
Hideki Ueno- 1Department of Microbiology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Global Health and Emerging Pathogens Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
Convincing lines of evidence in both mice and humans show that exaggerated T follicular helper (Tfh) responses is pathogenic in autoimmune diseases. However, the cause of exaggerated Tfh response in humans is still much less clear than in mouse models where genetic factors can be manipulated for in vivo testing. Nonetheless, recent advances in our understanding on the mechanisms of human Tfh differentiation and identification of multiple risk loci in genome-wide association studies have revealed several pathways potentially associated with exaggerated Tfh response in human autoimmune diseases. In this review, we will first briefly summarize the differentiation mechanisms of Tfh cells in humans. We describe the features of “Tfh-like” cells recently identified in inflamed tissues of human autoimmune diseases. Then we will discuss how risk loci identified in GWAS are potentially involved in exaggerated Tfh response in human autoimmune diseases.
Introduction: T Follicular Helper (Tfh) Cells
T follicular helper cells represent a CD4+ T cell subset that plays fundamental roles for antibody responses against protein antigens [see also previous reviews for Ref. (1–4)]. Tfh cells are critical for the generation of antibody responses as well as for the establishment of long-lived and high-affinity B cell clones. The most mature Tfh cells reside in germinal centers (GC) in secondary lymphoid organs and provide help specifically to high-affinity B cells that have undergone somatic hypermutations within the GCs. The selected B cells undergo clonal expansion and eventually differentiate into long-lived plasma cells and memory B cells. Less mature Tfh cells also interact with antigen-presenting B cells outside GCs and induce B cell proliferation and differentiation into antibody producing cells. Among them, extrafollicular helper cells, another Tfh-lineage CD4+ T cell subset, induce the differentiation of extrafollicular plasma cells. This extrafollicular mechanism mainly contributes to the early generation of specific antibodies after antigen challenge. Extrafollicular helper cells share the phenotype, gene profiles, and the functions with GC Tfh cells.
T follicular helper cells and their precursors are endowed with features to perform these tasks (1–4). After interacting with antigen-presenting dendritic cells (DCs) in T cell-rich area of secondary lymphoid organs, Tfh precursors increase the expression of the chemokine receptor CXCR5 and the G protein-coupled receptor S1PR2, while decreasing the expression of CCR7 and the selectin ligand PSGL1. These changes in the cell surface molecular profile are required for their migration toward and retention within B cell follicles enriched with the chemokine CXCL13. Tfh cells produce large amounts of IL-21, which potently promotes the growth, differentiation, and class-switching of B cells (5, 6). Tfh cells in GCs express high levels of inducible co-stimulator (ICOS), a co-stimulatory molecule crucial for their interactions with B cells. CD40 ligand (CD40L) expressed by Tfh cells provides signals to B cells through CD40 for their differentiation and class-switching. The surface markers commonly used to define human GC Tfh cells and their precursors in lymphoid organs are CXCR5, ICOS, and PD-1. GC Tfh cells express these molecules at high levels, whereas their precursors express at low levels (Table 1). CD127 and CCR7 are expressed by Tfh precursors, but not by GC Tfh cells (7). The combination of CXCR5 and PD-1 is often used to define GC Tfh cells and their precursors also in mice, but ICOS can be broadly expressed by activated CD4+ T cells in lymphoid organs (8) and is rarely used to define mouse GC Tfh cells.

Table 1. Cell surface makers commonly used to define T follicular helper (Tfh) subsets in lymphoid organs.
Prolonged Tfh response results in enhanced generation of mature Tfh cells and/or extrafollicular helper cells, and eventually leads to autoimmunity (3, 4). This is convincingly demonstrated in many autoimmune-prone mouse models including Roquinsan/san mice, in which CD4+ T cells overexpress ICOS and excessive signals through ICOS cause exaggerated Tfh and GC responses (9). Molecular mechanisms for Tfh cell differentiation and the causal link of genetic mutations to exaggerated Tfh responses are relatively well defined in mice (3, 4). However, less is known for human autoimmune diseases. In this review, we will first summarize the differentiation mechanisms of Tfh cells in humans briefly. We will also summarize the features of pathogenic “Tfh-like” cells recently characterized in inflamed tissues of human autoimmune diseases. Last, we will discuss how risk loci identified in GWAS overlap with Tfh-differentiation pathway and how they are potentially associated with exaggerated Tfh response in human autoimmune diseases.
Differentiation of Tfh Cells in Humans
When interacting with antigen-presenting DCs, naïve CD4+ T cells receive signals via three major receptor families, which ultimately determine the fate of T cell differentiation: the T cell receptor (TCR, signal 1), receptors for co-stimulatory molecules (signal 2), and receptors for cytokines (signal 3). Recent studies show that each signal provides parameters that negatively and positively affect Tfh differentiation in humans and mice (Figure 1).
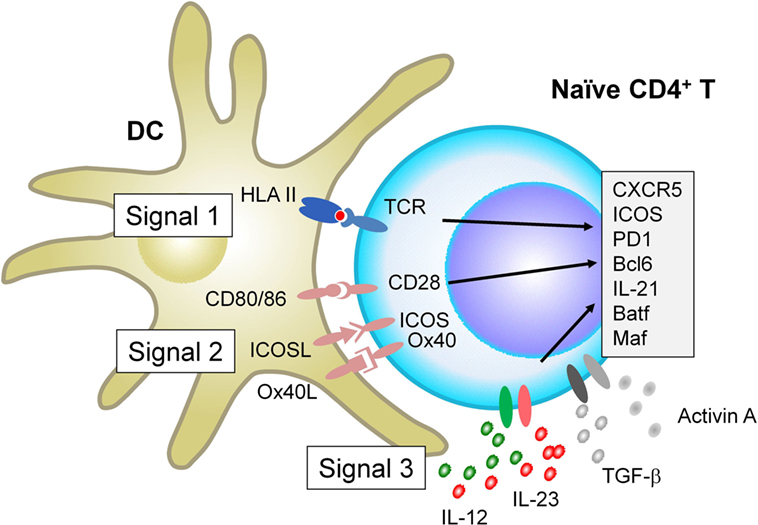
Figure 1. Factors that positively regulate human T follicular helper (Tfh) cell differentiation. When interacting with antigen-presenting dendritic cells (DCs), naïve CD4+ T cells receive signals via three major receptor families: the T cell receptor (TCR, signal 1), receptors for co-stimulatory molecules (signal 2), and receptors for cytokines (signal 3). For signal 1, evidence in both mice and humans shows that strong TCR signals promote Tfh cell differentiation. For signal 2, in addition to CD28 signals which is essential for optimal T cell activation, signals via inducible co-stimulator and Ox40 promote human naïve CD4+ T cells to express multiple Tfh molecules. For Signal 3, among inflammatory cytokines that activated DCs produce, IL-12 and IL-23 play dominant roles for human naïve CD4+ T cells to express Tfh molecules. The effect of IL-12 and IL-23 is further enhanced by the co-presence of TGF-β family molecules, TGF-β and Activin A. Given that TGF-β and Activin A are often highly expressed in human inflamed tissues, the source of these cytokines might be both from interacting DCs and from microenvironment.
Signal 1: TCR
Studies in mice demonstrated that strong TCR signals are required for the differentiation of fully mature Tfh cells (8, 10). Consistent with this, stimulation with stronger TCR signals in vitro induces human naïve CD4+ T cells to express higher levels of multiple Tfh molecules, including CXCR5, Bcl6, IL-21, and Ox40 (11). As demonstrated in experimental mouse models (8, 10), it is possible that human Tfh cell clones display relatively higher TCR affinity than non-Tfh cell clones, yet this remains to be tested.
Signal 2: Co-Stimulatory Molecules
Inducible co-stimulator is critically involved in Tfh cell biology at multiple levels, including the differentiation program at early stages (12, 13), their migration into B cell follicles (14), and the functions when interacting with B cells (15, 16). Patients with ICOS deficiency display severely impaired Tfh response accompanied by severely impaired memory B cell formation, indicating the essential role of ICOS in humans (17). Ox40 is another important co-stimulatory molecule promoting human Tfh cell differentiation. Ox40 signals together with TCR and CD28 signals promote human naïve and memory CD4+ T cells to express multiple Tfh molecules, including CXCR5, ICOS, PD-1, and Bcl6 (11). The direct contribution of Ox40 signals to Tfh cell differentiation was also recently demonstrated in mice with vaccinia viral infection (18). Unlike ICOS deficiency, however, loss-of-function (LOF) mutations in human Ox40 do not affect Tfh and antibody responses (19), suggesting that deficiency of Ox40 signals can be compensated by other mechanisms for Tfh differentiation. Instead, excessive Ox40 signals drive human CD4+ T cell differentiation toward Tfh cells and contribute to autoimmunity by increasing Tfh responses (11).
Signal 3: Cytokines
In humans, IL-12 produced by DCs is the major DC-derived cytokine driving human naïve CD4+ T cells to become Tfh-like cells (20–24). This is in contrast to mouse CD4+ T cells which acquire multiple features of Tfh cells in response to IL-6 and IL-21 (25, 26). Children who lack the expression of functional IL-12 receptor β1 chain, a common receptor for IL-12 and IL-23, display less Tfh cells and memory B cells in blood circulation and impaired GC formation in lymph nodes, providing in vivo evidence of the significance of this pathway for intact Tfh response in humans (21). Another important set of cytokines for human Tfh cell differentiation is TGF-β family cytokines TGF-β and Activin A, which activate the Smad signaling pathways including Smad2 and Smad3. Although only marginally effective by themselves, TGF-β and Activin A co-operate with IL-12 and IL-23 to promote human naïve CD4+ T cell differentiation toward the Tfh lineage (23, 27). TGF-β signals render STAT4 and STAT3 (activated by IL-12 and IL-23) to promote human naïve CD4+ T cells to acquire Tfh gene signature, while suppressing Th2 and regulatory T cell gene signatures (23). Furthermore, both TGF-β and Activin A also induce human CD4+ T cells to produce CXCL13 (27, 28), the major chemokine that human mature Tfh cells produce (7). TGF-β and Activin A are often strongly expressed in inflammatory sites, such as synovial fluid in rheumatoid arthritis (RA) (29, 30). Of note, neither TGF-β nor Activin A, even in the presence of Tfh-promoting cytokines, such as IL-6 and IL-21, induces Tfh molecules in mouse CD4+ T cells, and, therefore, this pathway is not shared in mice (23, 27).
Several cytokines are known to inhibit human Tfh cell differentiation. Type I (IFN-α, β, and ω) and type III (IFN-λ1 and λ2) interferons are potent inhibitors of Tfh cell differentiation in humans, and strongly diminish the expression of Tfh markers and gene signature by human naïve CD4+ T cells (23). This suggests that exaggerated Tfh cell responses in human autoimmune diseases with dominant IFN signature, such as systemic lupus erythematosus (SLE), is not mediated by the direct effect of type I IFNs on T cells, but by an indirect effect on APCs. Type I IFNs promote human DCs to produce Tfh-promoting cytokines, such as IL-12, IL-23, and IL-6 (31). Similarly, mouse studies demonstrated that type I IFN signals act as an adjuvant for antibody responses by promoting DCs to produce IL-6 (32, 33).
In mice, the IL-2–STAT5–Blimp1 axis inhibits Tfh cell differentiation at multiple differentiation stages (34, 35), and deprivation of IL-2 signals in the microenvironment is important for Tfh cell differentiation (36). There is evidence that IL-2 suppresses also human Tfh cell differentiation. IL-2 inhibition by a neutralizing Ab increased CXCR5 expression by human naïve CD4+ T cells cultured in Tfh-promoting culture conditions in vitro (27). Furthermore, treatment with low dose IL-2 decreased CXCR5+PD-1+CCR7lo cTfh cells in SLE accompanied with marked decrease of disease activity (37). It is of note, however, that it is unclear whether decrease of disease activity was dependent on decreased Tfh activity and whether decreased Tfh activity was the primary effect of IL-2 on Tfh cells. Current evidence highly suggests that the major target of low dose IL-2 treatment was Tregs. Low dose IL-2 treatment selectively induced STAT5 activation in Tregs but not conventional CD4+ T cells in vitro (38). In chronic GVHD patients, low dose IL-2 expanded circulating Treg frequency and their suppressive functions (38, 39). Therefore, it is possible that decrease of Tfh activity was secondary to increased activity of Tregs.
“Tfh-Like” Cells in Inflamed Tissues of Human Autoimmune Diseases
Recent studies have identified “Tfh-like” cells involved in autoantibody production in inflamed tissues of human autoimmune diseases. These T cells share properties with Tfh cells only partly. Characterization of CD4+ T cells in inflamed synovial tissues in RA patients revealed a massive expansion of CXCR5−PD-1+ CD4+ T cells. While these cells expressed many human Tfh markers, including IL-21, CXCL13, ICOS, CD84, TIGIT, and c-Maf, and were able to induce memory B cells to produce IgG in vitro, they lacked the expression of CXCR5 and Bcl6. CXCR5−PD-1+ cells instead expressed Blimp-1 and the chemokine receptors CCR2, CCR5, and CX3CR1 (40). Analysis of inflamed salivary glands in Sjogren’s syndrome showed an increase of CXCR5− ICOS+ CCR9+ CD4+ T cells. CCR9+ CD4+ T cells isolated from blood samples of patients produced IL-21 upon activation and were capable of inducing memory B cells to produce IgG (41). These studies suggest that the precise nature of B helper T cells associated with autoantibody production in inflamed tissues might differ among diseases, likely reflecting the differences in microenvironment. The chemokine receptors and integrins expressed by these “Tfh-like” cells might be critically involved for their migration to and retention in inflammatory tissues. It will be important to clarify how the developmental mechanisms and the functional regulations of “Tfh-like” cells are different from Tfh cells in lymphoid organs.
Transcription Factor Network in Tfh Cell Differentiation
CD4+ T cell differentiation is controlled by transcription factors which govern the expression of a set of target genes. Recent studies in mice identified a number of transcription factors and pathways that affect Tfh cell differentiation. Transcription factors positively regulate Tfh cell differentiation include Bcl6, Ascl2, Batf, cMaf, IRF4, STAT3, STAT1, STAT4, NOTCH1, NOTCH2, Tcf1, and Lef1 (42–48). Among these, Bcl6 is specifically required for the differentiation of Tfh cells, but not other CD4+ T cell subsets (7, 44, 49–51). The transcription factor Blimp-1, an antagonist of Bcl6, inhibits Tfh cell differentiation (51). Genome-wide analysis of Bcl6-binding sites by using ChIP-Seq demonstrated that Bcl6 binds to genes associated with the differentiation of other CD4+ T cell lineages, including Th1, Th2, Th17, and Tregs, and genes that negatively affect CD4+ T cell localization in GCs (52). Thus, Bcl6 primarily acts as a repressor to inhibit the differentiation toward other CD4+ T cell lineages and to diminish the molecules that exclude CD4+ T cells from GCs (49–52). Interestingly, many genes repressed by Bcl6 contained AP1 and STAT motifs but lacked Bcl6 motif, suggesting that AP1 and STAT recruit Bcl6 to the target sites and repress the target genes (52).
Risk Loci Identified in GWAS of Autoimmune Diseases and Their Potential Association with Exaggerated Tfh Response
The genome-wide association studies (53) are an experimental design used to detect associations between genetic variants and traits including diseases in samples from populations (54). GWAS in autoimmune diseases have successfully identified loci associated with an increase or a decrease of risk (53–55). A fraction of the identified risk loci is located within or in the proximity of genes encoding transcription factors and cell surface molecules expressed by T cells and/or antigen-presenting cells. How risk haplotypes are associated with their dysregulated function and linked to the immunological consequences largely remains to be established. Nonetheless, several key risk loci are located in the proximity of genes that regulate Tfh differentiation in humans, and thus might contribute to the exaggerated Tfh response in human autoimmune diseases. Here we focus on three genes, STAT4, IRF5, and TNFSF4, which are identified and validated as risk loci in GWAS in multiple autoantibody mediated autoimmune diseases with a strong disease association at the genome-wide p value of at least 5 × 10−8 (Figure 2).
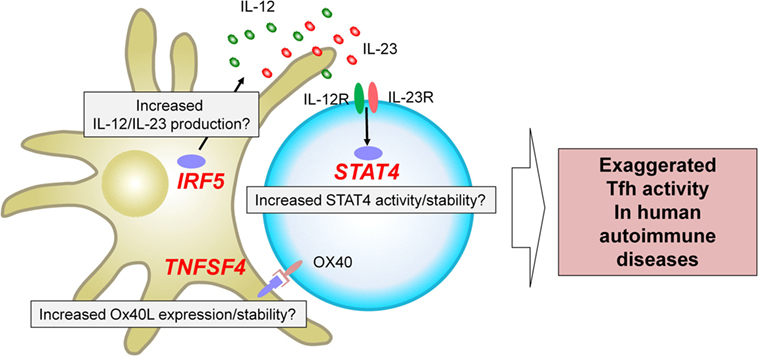
Figure 2. Three risk loci potentially associated with exaggerated T follicular helper (Tfh) response in human autoantibody-mediated autoimmune diseases. GWAS in autoimmune diseases have identified multiple loci associated with disease risk. STAT4, IRF5, and TNFSF4 have been identified and validated in multiple studies on autoantibody mediated autoimmune diseases as risk loci with a strong disease association at the genome-wide p value of at least 5 × 10−8. These risk loci might contribute to the exaggerated Tfh response in human autoimmune diseases. For example, IRF5 risk loci might be associated with an increased production of Tfh-promoting cytokines, including IL-12, IL-23, and IL-6. TNFSF4 risk loci might contribute to increased Ox40 signals that promote the expression of Tfh molecules by human CD4+ T cells. STAT4 risk loci might affect the activity and/or stability of STAT4 and might enhance the signals of IL-12 and IL-23.
STAT4
STAT4 is among the top risk loci (other than HLA genes) identified in GWAS on SLE, RA, and Sjogren’s syndrome (56). STAT4 is dominantly activated by IL-12 and to a lesser extent by IL-23 and type I IFNs (57). As discussed above, the IL-12–STAT4 pathway strongly promotes Tfh cell differentiation in humans. Human naïve CD4+ T cells primed in the presence of IL-12 highly upregulate expression of multiple Tfh molecules (21, 58), which are inhibited by decreasing STAT4 expression by specific siRNA transfection (22, 23). STAT4 in CD4+ T cells in the proximity of GCs in inflamed human tonsils is strongly phosphorylated, indicating that the IL-12–STAT4 pathway is active in situ (23). Previous studies showed that DCs and macrophages in inflamed lymphoid organs and tissues of autoimmune diseases express IL-12 and IL-23 (31). It is plausible that STAT4 allele variants [such as rs11889341-T in SLE (59)] might be directly involved in the exaggeration of Tfh response, for example, by increasing their signaling efficacy and/or stability of STAT4 in CD4+ T cells stimulated by IL-12 and/or IL-23. Of note, the IL-12–STAT4 pathway contributes to the Tfh differentiation also in mice, and STAT4 directly binds to multiple Tfh genes in IL-12-stimulated mouse CD4+ T cells, including Il21, Cxcr5, Pdcd1 (encoding PD-1), and Icos (60, 61).
A recent report showed that ustekinumab, a human recombinant antibody against IL-12p40 (a component of IL-12 and IL-23), was effective for active SLE in a phase II trial (presented in ACR 2017 meeting). Although this encouraging outcome remains to be confirmed, it is of great interest to determine whether the treatment with ustekinumab reduced Tfh responses, and more importantly whether the reduction of Tfh response correlated with the improvement of clinical parameters. Alternatively, treatment with ustekinumab might target other immune cells including Th1 and Th17 cells, which are also proposed to play pathogenic roles in SLE (62, 63). Determining the mode of action of ustekinumab will provide critical insights into the type of pathogenic CD4+ T cells and the pathways in SLE.
Other STAT molecules STAT3 and STAT1 also positively regulate Tfh cell differentiation in mice (4). STAT3 is also critical for human Tfh cell differentiation, as patients with STAT3 LOF mutations as well as patients with IL-21 receptor deficiency display significantly less cTfh cells (64). Furthermore, CD4+ T cells from patients with STAT3 LOF mutations and IL-21 receptor deficiency produce substantially less IL-21 upon IL-12 stimulation (24), suggesting that the IL-21–STAT3 axis is critical for enhancement of IL-21 expression. Interestingly, despite less IL-21 production, STAT3 LOF human CD4+ T cells stimulated with IL-12 express normal levels of CXCR5, ICOS, and Bcl-6 (26). Thus, STAT3 and STAT4 might differently contribute to the expression of each Tfh molecule in human CD4+ T cells, and IL-21 expression might be more dependent on the IL-21–STAT3 axis than the expression of CXCR5, ICOS, and Bcl-6. In this line, it is likely that decreased cTfh cells in STAT3 LOF are also due to severely impaired B cell response. STAT3 LOF display significantly less memory B cells (65), and the differentiation of STAT3 LOF B cells into plasmablast is severely impaired due to failure to respond to IL-6, IL-10, or IL-21 (65). Notably, unlike STAT4, STAT3 has been identified as risk loci in GWAS in autoinflammatory diseases, such as multiple sclerosis, Crohn’s disease, and ulcerative colitis, which are less dependent on autoantibodies (53). STAT1 signals seems to play less significant roles in Tfh cell differentiation in humans than in mice, as patients with both STAT1 LOF and gain-of-function (GOF) mutations display normal frequency of cTfh cells (24, 58, 64). No GWAS on autoimmune diseases have identified STAT1 as a risk allele. These observations suggest that genetic variants of STAT1 and STAT3 do not contribute much to pathogenic Tfh and GC responses in autoimmune diseases. However, this notion does not preclude the involvement of STAT3 mutations in human autoimmune diseases. Patients with STAT3 GOF mutations show massive multiorgan autoimmunity, autoimmune cytopenia, lymphoproliferation, and immunodeficiency, likely due to impaired Treg differentiation and functions (66–68). Whether STAT3 GOF increases Tfh response or not is currently unclear. However, many patients display hypogammaglobulinemia (68), and thus that autoimmunity in STAT3 GOF seems to be caused by dysregulation of antibody response rather than a general increase of Tfh response.
IRF5
IRF5 is another transcription factor whose haplotype is highly associated with an increased risk of SLE, RA, and Sjogren’s syndrome (53). IRF5 is expressed by a broad range of APCs, including monocytes, macrophages, DCs, plasmacytoid DCs, and B cells, and is critically involved in the production of inflammatory cytokines, including IL-12, IL-23, IL-6, and type I IFN (69–71). These cytokines are enriched in inflamed tissues in autoimmune diseases, and such inflammatory cytokine milieu causes a positive-feedback loop for the expression of IL-12 and IL-23 by promoting the generating M1 macrophages that highly express IRF5 (69). Thus, it is possible that IRF5 risk haplotype is linked to the enhanced production of Tfh-promoting cytokines in subjects with autoimmune diseases traits. IRF5 is required in normal human B cell proliferation and differentiation into plasmablasts (72), and therefore, IRF5 risk allele may also intrinsically affect B cells. The importance of IRF5 for lupus pathogenesis was also demonstrated by the attenuation of the disease in mouse models deficient of IRF5, which was accompanied with decreased activated CD4+ T cells and autoantibodies (73, 74).
TNFSF4
TNFSF4 encodes Ox40L, a TNF ligand family molecule that delivers signals promoting human Tfh cell differentiation (11) as described earlier. TNFSF4 has been identified as another prominent risk allele in SLE and RA (53). In active SLE, blood CD14+ monocytes and tissue CD11c+ myeloid cells upregulate the expression of Ox40L (11). TNFSF4 risk allele might enhance signals via Ox40 and contribute to enhanced Tfh responses. The importance of the Ox40–Ox40L axis for antibody response (75–77) and autoimmune disease was also demonstrated in studies with mouse models (18, 78, 79). TNFSF4 is also found as a risk allele in GWAS on allergies, such as asthma, hay fever, and eczema; an observation consistent with the importance of Ox40–Ox40L pathway for Th2 response (80).
In contrast, it is somewhat surprising that ICOS, another key co-stimulatory molecule for Tfh cell differentiation in humans and mice, and its ligand ICOS-ligand (encoded by ICOSLG) are not among the risk loci identified in GWAS on autoantibody mediated autoimmune diseases. ICOSLG was instead found to be highly associated with Crohn’s disease and ulcerative colitis (53). Thus, while ICOS and ICOS-ligand are involved in pathogenic Tfh response, as demonstrated in Roquinsan/san mice, the haplotype ICOS and ICOSLG themselves per se do not seem to contribute to pathogenic Tfh responses.
Concluding Remarks
Although the mode of actions remains to be established, a positive outcome of ustekinumab in active SLE patients provides a new proof of principle that T cells can be a therapeutic target for autoantibody-mediated autoimmune diseases. Another evidence came from a clinical trial performed some 15 years ago with anti-CD40L (BG9588) on lupus nephritis. Although the trial was prematurely terminated due to the development of thrombotic events and associated fatality in some patients, the early results were encouraging and the treatment reduced serum anti-dsDNA titers, increased serum complement levels, and reduced nephritis score (81). Thus, targeting B helper T cells can be beneficial for treatment of autoimmune diseases. It will be important to define the differentiation mechanisms and the functions of Tfh and Tfh-like cells that directly contribute to the production of autoantibodies in lymphoid organs as well as inflamed tissues. Determining how risk loci in autoimmune diseases are associated with the molecular mechanisms for exaggerated Tfh responses is also of great importance. These studies will enhance our understanding in human autoimmune diseases and also might provide novel therapeutic targets.
Author Contributions
HU and SH, have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by the National Institutes of Health grant U19-AI082715 and grants from Lupus Research Alliance.
References
1. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med (2012) 209(7):1241–53. doi:10.1084/jem.20120994
2. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi:10.1016/j.immuni.2014.10.004
3. Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol (2015) 16(2):142–52. doi:10.1038/ni.3054
4. Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol (2016) 34:335–68. doi:10.1146/annurev-immunol-041015-055605
5. Avery DT, Bryant VL, Ma CS, de Waal Malefyt R, Tangye SG. IL-21-induced isotype switching to IgG and IgA by human naive B cells is differentially regulated by IL-4. J Immunol (2008) 181(3):1767–79. doi:10.4049/jimmunol.181.3.1767
6. Zotos D, Coquet JM, Zhang Y, Light A, D’Costa K, Kallies A, et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med (2010) 207(2):365–78. doi:10.1084/jem.20091777
7. Bentebibel SE, Schmitt N, Banchereau J, Ueno H. Human tonsil B-cell lymphoma 6 (BCL6)-expressing CD4+ T-cell subset specialized for B-cell help outside germinal centers. Proc Natl Acad Sci U S A (2011) 108(33):E488–97. doi:10.1073/pnas.1100898108
8. Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol (2009) 10(4):375–84. doi:10.1038/ni.1704
9. Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature (2005) 435(7041):452–8. doi:10.1038/nature03555
10. Tubo NJ, Pagan AJ, Taylor JJ, Nelson RW, Linehan JL, Ertelt JM, et al. Single naive CD4+ T cells from a diverse repertoire produce different effector cell types during infection. Cell (2013) 153(4):785–96. doi:10.1016/j.cell.2013.04.007
11. Jacquemin C, Schmitt N, Contin-Bordes C, Liu Y, Narayanan P, Seneschal J, et al. OX40 ligand contributes to human lupus pathogenesis by promoting T follicular helper response. Immunity (2015) 42(6):1159–70. doi:10.1016/j.immuni.2015.05.012
12. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175(4):2340–8. doi:10.4049/jimmunol.175.4.2340
13. Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, et al. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity (2011) 34(6):932–46. doi:10.1016/j.immuni.2011.03.023
14. Xu H, Li X, Liu D, Li J, Zhang X, Chen X, et al. Follicular T-helper cell recruitment governed by bystander B cells and ICOS-driven motility. Nature (2013) 496(7446):523–7. doi:10.1038/nature12058
15. Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, et al. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature (1999) 397(6716):263–6. doi:10.1038/16717
16. Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C. A fundamental role for interleukin-21 in the generation of T follicular helper cells. Immunity (2008) 29(1):127–37. doi:10.1016/j.immuni.2008.06.001
17. Bossaller L, Burger J, Draeger R, Grimbacher B, Knoth R, Plebani A, et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J Immunol (2006) 177(7):4927–32. doi:10.4049/jimmunol.177.7.4927
18. Tahiliani V, Hutchinson TE, Abboud G, Croft M, Salek-Ardakani S. OX40 cooperates with ICOS to amplify follicular Th cell development and germinal center reactions during infection. J Immunol (2017) 198(1):218–28. doi:10.4049/jimmunol.1601356
19. Byun M, Ma CS, Akcay A, Pedergnana V, Palendira U, Myoung J, et al. Inherited human OX40 deficiency underlying classic Kaposi sarcoma of childhood. J Exp Med (2013) 210(9):1743–59. doi:10.1084/jem.20130592
20. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol (2009) 87(8):590–600. doi:10.1038/icb.2009.64
21. Schmitt N, Bustamante J, Bourdery L, Bentebibel SE, Boisson-Dupuis S, Hamlin F, et al. IL-12 receptor beta1 deficiency alters in vivo T follicular helper cell response in humans. Blood (2013) 121(17):3375–85. doi:10.1182/blood-2012-08-448902
22. Schmitt N, Morita R, Bourdery L, Bentebibel SE, Zurawski SM, Banchereau J, et al. Human dendritic cells induce the differentiation of interleukin-21-producing T follicular helper-like cells through interleukin-12. Immunity (2009) 31(1):158–69. doi:10.1016/j.immuni.2009.04.016
23. Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol (2014) 15(9):856–65. doi:10.1038/ni.2947
24. Ma CS, Wong N, Rao G, Nguyen A, Avery DT, Payne K, et al. Unique and shared signaling pathways cooperate to regulate the differentiation of human CD4+ T cells into distinct effector subsets. J Exp Med (2016) 213(8):1589–608. doi:10.1084/jem.20151467
25. Suto A, Kashiwakuma D, Kagami S, Hirose K, Watanabe N, Yokote K, et al. Development and characterization of IL-21-producing CD4+ T cells. J Exp Med (2008) 205(6):1369–79. doi:10.1084/jem.20072057
26. Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood (2012) 119(17):3997–4008. doi:10.1182/blood-2011-11-392985
27. Locci M, Wu JE, Arumemi F, Mikulski Z, Dahlberg C, Miller AT, et al. Activin A programs the differentiation of human TFH cells. Nat Immunol (2016) 17(8):976–84. doi:10.1038/ni1016-1235d
28. Kobayashi S, Watanabe T, Suzuki R, Furu M, Ito H, Ito J, et al. TGF-beta induces the differentiation of human CXCL13-producing CD4(+) T cells. Eur J Immunol (2016) 46(2):360–71. doi:10.1002/eji.201546043
29. Fava R, Olsen N, Keski-Oja J, Moses H, Pincus T. Active and latent forms of transforming growth factor beta activity in synovial effusions. J Exp Med (1989) 169(1):291–6. doi:10.1084/jem.169.1.291
30. Soler Palacios B, Estrada-Capetillo L, Izquierdo E, Criado G, Nieto C, Municio C, et al. Macrophages from the synovium of active rheumatoid arthritis exhibit an activin A-dependent pro-inflammatory profile. J Pathol (2015) 235(3):515–26. doi:10.1002/path.4466
31. Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol Rev (2008) 226:112–31. doi:10.1111/j.1600-065X.2008.00700.x
32. Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, Tough DF. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity (2001) 14(4):461–70. doi:10.1016/S1074-7613(01)00126-1
33. Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity (2009) 31(3):491–501. doi:10.1016/j.immuni.2009.07.005
34. Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, et al. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity (2012) 36(5):847–56. doi:10.1016/j.immuni.2012.02.012
35. Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med (2012) 209(2):243–50. doi:10.1084/jem.20111174
36. Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature (2016) 533(7601):110–4. doi:10.1038/nature17947
37. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med (2016) 22(9):991–3. doi:10.1038/nm.4148
38. Hirakawa M, Matos TR, Liu H, Koreth J, Kim HT, Paul NE, et al. Low-dose IL-2 selectively activates subsets of CD4(+) Tregs and NK cells. JCI Insight (2016) 1(18):e89278. doi:10.1172/jci.insight.89278
39. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365(22):2055–66. doi:10.1056/NEJMoa1108188
40. Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature (2017) 542(7639):110–4. doi:10.1038/nature20810
41. Blokland SLM, Hillen MR, Kruize AA, Meller S, Homey B, Smithson GM, et al. Increased CCL25 and T helper cells expressing CCR9 in the salivary glands of patients with primary Sjogren’s syndrome: potential new axis in lymphoid neogenesis. Arthritis Rheumatol (2017) 69(10):2038–51. doi:10.1002/art.40182
42. Liu X, Chen X, Zhong B, Wang A, Wang X, Chu F, et al. Transcription factor achaete-scute homologue 2 initiates follicular T-helper-cell development. Nature (2014) 507(7493):513–8. doi:10.1038/nature12910
43. Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nat Immunol (2011) 12(6):536–43. doi:10.1038/ni.2037
44. Kroenke MA, Eto D, Locci M, Cho M, Davidson T, Haddad EK, et al. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J Immunol (2012) 188(8):3734–44. doi:10.4049/jimmunol.1103246
45. Bollig N, Brustle A, Kellner K, Ackermann W, Abass E, Raifer H, et al. Transcription factor IRF4 determines germinal center formation through follicular T-helper cell differentiation. Proc Natl Acad Sci U S A (2012) 109(22):8664–9. doi:10.1073/pnas.1205834109
46. Xu L, Cao Y, Xie Z, Huang Q, Bai Q, Yang X, et al. The transcription factor TCF-1 initiates the differentiation of T(FH) cells during acute viral infection. Nat Immunol (2015) 16(9):991–9. doi:10.1038/ni.3229
47. Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, et al. TCF1 Is required for the T follicular helper cell response to viral infection. Cell Rep (2015) 12(12):2099–110. doi:10.1016/j.celrep.2015.08.049
48. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol (2015) 16(9):980–90. doi:10.1038/ni.3226
49. Yu D, Rao S, Tsai LM, Lee SK, He Y, Sutcliffe EL, et al. The transcriptional repressor Bcl-6 directs T follicular helper cell lineage commitment. Immunity (2009) 31(3):457–68. doi:10.1016/j.immuni.2009.07.002
50. Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, et al. Bcl6 mediates the development of T follicular helper cells. Science (2009) 325(5943):1001–5. doi:10.1126/science.1176676
51. Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science (2009) 325(5943):1006–10. doi:10.1126/science.1175870
52. Hatzi K, Nance JP, Kroenke MA, Bothwell M, Haddad EK, Melnick A, et al. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J Exp Med (2015) 212(4):539–53. doi:10.1084/jem.20141380
53. GWAS Catalog The NHGRI-EBI Catalog of Published Genome-wide Association Studies. Available from: https://www.ebi.ac.uk/gwas/home (Accessed: February 7, 2018).
54. Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet (2017) 101(1):5–22. doi:10.1016/j.ajhg.2017.06.005
55. Stranger BE, De Jager PL. Coordinating GWAS results with gene expression in a systems immunologic paradigm in autoimmunity. Curr Opin Immunol (2012) 24(5):544–51. doi:10.1016/j.coi.2012.09.002
56. Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases — connecting risk alleles with molecular traits of the immune system. Nat Rev Genet (2016) 17(3):160–74. doi:10.1038/nrg.2015.33
57. O’Shea John J, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity (2012) 36(4):542–50. doi:10.1016/j.immuni.2012.03.014
58. Schmitt N, Liu Y, Bentebibel SE, Ueno H. Molecular mechanisms regulating T helper 1 versus T follicular helper cell differentiation in humans. Cell Rep (2016) 16(4):1082–95. doi:10.1016/j.celrep.2016.06.063
59. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet (2015) 47(12):1457–64. doi:10.1038/ng.3434
60. Wei L, Vahedi G, Sun H-W, Watford WT, Takatori H, Ramos HL, et al. Discrete Roles of STAT4 and STAT6 transcription factors in tuning epigenetic modifications and transcription during T helper cell differentiation. Immunity (2010) 32(6):840–51. doi:10.1016/j.immuni.2010.06.003
61. Nakayamada S, Kanno Y, Takahashi H, Jankovic D, Lu KT, Johnson TA, et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity (2011) 35(6):919–31. doi:10.1016/j.immuni.2011.11.012
62. Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol (2008) 127(3):385–93. doi:10.1016/j.clim.2008.01.019
63. Li D, Guo B, Wu H, Tan L, Chang C, Lu Q. Interleukin-17 in systemic lupus erythematosus: a comprehensive review. Autoimmunity (2015) 48(6):353–61. doi:10.3109/08916934.2015.1037441
64. Ma CS, Wong N, Rao G, Avery DT, Torpy J, Hambridge T, et al. Monogenic mutations differentially affect the quantity and quality of T follicular helper cells in patients with human primary immunodeficiencies. J Allergy Clin Immunol (2015) 136(4):993–1006.e1. doi:10.1016/j.jaci.2015.05.036
65. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med (2010) 207(1):S1–5. doi:10.1084/jem.20091706
66. Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, Franco ED, et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet (2014) 46(8):812–4. doi:10.1038/ng.3040
67. Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood (2015) 125(4):591–9. doi:10.1182/blood-2014-09-602763
68. Haapaniemi EM, Kaustio M, Rajala HLM, Adrichem AJv, Kainulainen L, Glumoff V, et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood (2015) 125(4):639–48. doi:10.1182/blood-2014-04-570101
69. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol (2011) 12(3):231–8. doi:10.1038/ni.1990
70. Takaoka A, Yanai H, Kondo S, Duncan G, Negishi H, Mizutani T, et al. Integral role of IRF-5 in the gene induction programme activated by toll-like receptors. Nature (2005) 434(7030):243–9. doi:10.1038/nature03308
71. Bergstrom B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, et al. TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-beta production via a TAK1-IKKbeta-IRF5 signaling pathway. J Immunol (2015) 195(3):1100–11. doi:10.4049/jimmunol.1403176
72. De S, Zhang B, Shih T, Singh S, Winkler A, Donnelly R, et al. B cell-intrinsic role for IRF5 in TLR9/BCR-induced human B cell activation, proliferation, and plasmablast differentiation. Front Immunol (2018) 8:1938. doi:10.3389/fimmu.2017.01938
73. Tada Y, Kondo S, Aoki S, Koarada S, Inoue H, Suematsu R, et al. Interferon regulatory factor 5 is critical for the development of lupus in MRL/lpr mice. Arthritis Rheum (2011) 63(3):738–48. doi:10.1002/art.30183
74. Xu Y, Lee PY, Li Y, Liu C, Zhuang H, Han S, et al. Pleiotropic IFN-dependent and -independent effects of IRF5 on the pathogenesis of experimental lupus. J Immunol (2012) 188(8):4113–21. doi:10.4049/jimmunol.1103113
75. Brocker T, Gulbranson-Judge A, Flynn S, Riedinger M, Raykundalia C, Lane P. CD4 T cell traffic control: in vivo evidence that ligation of OX40 on CD4 T cells by OX40-ligand expressed on dendritic cells leads to the accumulation of CD4 T cells in B follicles. Eur J Immunol (1999) 29(5):1610–6. doi:10.1002/(SICI)1521-4141(199905)29:05<1610::AID-IMMU1610>3.0.CO;2-8
76. Walker LS, Gulbranson-Judge A, Flynn S, Brocker T, Raykundalia C, Goodall M, et al. Compromised OX40 function in CD28-deficient mice is linked with failure to develop CXC chemokine receptor 5-positive CD4 cells and germinal centers. J Exp Med (1999) 190(8):1115–22. doi:10.1084/jem.190.8.1115
77. Kim MY, Gaspal FM, Wiggett HE, McConnell FM, Gulbranson-Judge A, Raykundalia C, et al. CD4(+)CD3(-) accessory cells costimulate primed CD4 T cells through OX40 and CD30 at sites where T cells collaborate with B cells. Immunity (2003) 18(5):643–54. doi:10.1016/S1074-7613(03)00110-9
78. Sitrin J, Suto E, Wuster A, Eastham-Anderson J, Kim JM, Austin CD, et al. The Ox40/Ox40 ligand pathway promotes pathogenic Th cell responses, plasmablast accumulation, and lupus nephritis in NZB/W F1 mice. J Immunol (2017) 199(4):1238–49. doi:10.4049/jimmunol.1700608
79. Flynn S, Toellner KM, Raykundalia C, Goodall M, Lane P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. J Exp Med (1998) 188(2):297–304. doi:10.1084/jem.188.2.297
80. Liu Y-J. Thymic stromal lymphopoietin and OX40 ligand pathway in the initiation of dendritic cell–mediated allergic inflammation. J Allergy Clin Immunol (2007) 120(2):238–44. doi:10.1016/j.jaci.2007.06.004
Keywords: T follicular helper, autoimmune diseases, IRF5, TNFSF4, STAT4, ustekinumab, IL-23, IL-12
Citation: Horiuchi S and Ueno H (2018) Potential Pathways Associated With Exaggerated T Follicular Helper Response in Human Autoimmune Diseases. Front. Immunol. 9:1630. doi: 10.3389/fimmu.2018.01630
Received: 22 February 2018; Accepted: 02 July 2018;
Published: 16 July 2018
Edited by:
Raffi Gugasyan, Burnet Institute, AustraliaReviewed by:
Tam Quach, Feinstein Institute for Medical Research, United StatesStuart G. Tangye, Garvan Institute of Medical Research, Australia
Copyright: © 2018 Horiuchi and Ueno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideki Ueno, aGlkZWtpLnVlbm9AbXNzbS5lZHU=