 Jelka Pohar
Jelka Pohar Quentin Simon
Quentin Simon Simon Fillatreau
Simon Fillatreau- 1Institut Necker-Enfants Malades, INSERM U1151-CNRS UMR 8253, Paris, France
- 2Faculté de Médecine, Université Paris Descartes, Sorbonne Paris Cité, Paris, France
- 3AP-HP, Hôpital Necker Enfants Malades, Paris, France
CD4+Foxp3+ T regulatory cells (Treg) are essential for the life of the organism, in particular because they protect the host against its own autoaggressive CD4+Foxp3− T lymphocytes (Tconv). Treg distinctively suppress autoaggressive immunity while permitting efficient defense against infectious diseases. This split effect indicates that Treg activity is controlled in an antigen-specific manner. This specificity is achieved first by the formation of the Treg repertoire during their development, and second by their activation in the periphery. This review presents novel information on the antigen-specificity of Treg development in the thymus, and Treg function in the periphery. These aspects have so far remained imprecisely understood due to the lack of knowledge of the actual antigens recognized by Treg during the different steps of their life, so that most previous studies have been performed using artificial antigens. However, recent studies identified some antigens mediating the positive selection of autoreactive Treg in the thymus, and the function of Treg in the periphery in autoimmune and allergic disorders. These investigations emphasized the remarkable specificity of Treg development and function. Indeed, the development of autoreactive Treg in the thymus was found to be mediated by single autoantigens, so that the absence of one antigen led to a dramatic loss of Treg reacting toward that antigen. The specificity of Treg development is important because the constitution of the Treg repertoire, and especially the presence of holes in this repertoire, was found to crucially influence human immunopathology. Indeed, it was found that the development of human immunopathology was permitted by the lack of Treg against the antigens driving the autoimmune or allergic T cell responses rather than by the impairment of Treg activation or function. The specificity of Treg suppression in the periphery is therefore intimately associated with the mechanisms shaping the formation of the Treg repertoire during their development. This novel information refines significantly our understanding of the antigen-specificity of Treg protective function, which is required to envision how these cells distinctively regulate unwanted immune responses as well as for the development of appropriate approaches to optimally harness them therapeutically in autoimmune, malignant, and infectious diseases.
Introduction
CD4+Foxp3+ T regulatory cells (Treg) are required throughout life to maintain a healthy immune system because conventional CD4+ T cells (Tconv) pose a permanent threat of deadly autoimmune attack if inappropriately controlled (1, 2). In human, this is illustrated by the early development of the severe immune dysregulation polyendocrinopathy, enteropathy X-linked (IPEX) syndrome in patients lacking Treg due to a FOXP3 gene deficiency (3). IPEX can be fatal within the first year of life, and is often associated with insulin-dependent diabetes mellitus, enteropathy, eczema, thrombocytopenia, anemia, as well as cachexia (3). The generation of Treg occurs both in the thymus (thymus-derived tTreg account for around 95% of peripheral Treg), and in the periphery including at mucosal surfaces where some Tconv convert into peripherally generated Treg (pTreg) (4, 5). These separate ontogenic pathways might account for the existence of Treg reacting toward distinct categories of antigens including self-antigens (6–8), innocuous environmental antigens, or pathogens (9–12). Both tTreg and pTreg are required for the maintenance of immune homeostasis (13, 14).
There is clear indication that the generation of Treg is decisively influenced by the reactivity of their T cell receptor (TCR) for antigen (4). In mice with monoclonal T cells, the differentiation of tTreg is only observed with some TCR (15), and the TCR repertoires of Tconv, tTreg, and pTreg are largely different (16–19). This raises the question of the mode of antigen recognition by the TCR that is associated with the commitment of T cell precursors to the tTreg lineage. An initial study showed that the ectopic expression of a model antigen in the thymus resulted in the commitment of T cell precursors carrying a transgenic TCR of high affinity for this antigen (but not of those bearing a TCR of lower affinity) into tTreg (20). Since multiple tissue-restricted antigens are promiscuously expressed in the thymus (21, 22), this led to the concept that the recognition of promiscuously expressed self-antigens via TCR–major histocompatibility complex (MHC-II) interactions of high affinity drove autoreactive tTreg development. However, other data obtained using distinct systems for the expression of transgenic antigens did not agree with this model (23). Furthermore, studies on the negative selection of T cell progenitors have shown that preventing the negative selection of highly autoreactive T cells in the thymus was not always associated with their diversion into the tTreg lineage (24). These conflicting observations led to some uncertainty in the field. TCR signals are also important for the function of Treg in the periphery (25, 26), and the modulation of TCR signaling has been used to increase or decrease their regulatory activity (24, 27). However, the relevance of antigen-specificity for the function of Treg in human disease is poorly understood, largely due to the lack of knowledge of the antigen pertinent for Treg function in human disease.
It was therefore important to use novel approaches in order to directly address how “real” endogenous (or exogenous) antigens actually select tTreg, and how antigen-specificity contributes to Treg function in the periphery. Such investigations have for a long time been hampered by the lack of the knowledge of the endogenous antigens recognized by Treg, and the difficulty of identifying them. Recent studies overcame this obstacle and identified physiological agonists recognized by Treg in the thymus, and in the periphery. These recent advances help understanding the relationship between the specificity of tTreg selection in the thymus and the specificity of tTreg-mediated suppression in the periphery, as well as how the antigen-specificity of Treg relates to the prevention or the failure to prevent autoimmune disorders and allergies. This review discusses the role of antigen-specificity in Treg development and function in the light of these recent studies.
Endogenous Self-Antigens and the Selection of Autoreactive Treg in the Thymus
The role of the thymus in dominant immune tolerance was brought to light from the observation that thymectomy at day 3, but not at day 7, after birth led to the development of fatal autoimmune attacks affecting various tissues (28). This phenomenon is due to the delayed emergence of tTreg relatively to Tconv during development (29). Indeed, the thymus produces almost exclusively Tconv before day 3, so that thymectomy at this time results in a situation of Treg deficiency. As already mentioned, the importance of cognate antigen recognition for the development of tTreg was initially indicated by experiments involving double transgenic mice ectopically expressing the hemagglutinin (HA) from influenza virus in the thymus, and a TCR specific for this antigen in T cells (30). In the thymus, T cell progenitors expressing a transgenic TCR of high affinity for HA gave rise to tTreg while those expressing a specific TCR of lower affinity for this antigen did not, suggesting that the development of tTreg was instructed by TCR interactions of high affinity for their cognate antigen (30). Observations made using an Nur77-eGFP reporter mice, in which the relative level of TCR signaling could be inferred from the level of eGFP expression, also indicated in the context of a wild-type TCR repertoire that Treg underwent stronger TCR signaling than Tconv during their thymic development (31). However, the concept that the autoimmune reactivity of a TCR rules its capacity to instruct Treg development was not supported by data obtained using other transgenic systems, as discussed in other reviews (20, 23). Moreover, some studies of the signaling processes regulating T cell fate decision in the thymus indicated that higher affinity was not always associated with increased tTreg selection (24). It therefore appeared important to identify endogenous antigens mediating tTreg selection, and to characterize the TCR–MHC-II–antigen peptide interaction through which they generated tTreg, in order to refine this model.
Several endogenous antigens implicated in tTreg development have recently been identified. A first study analyzed the development and function of Treg specific for myelin oligodendrocyte glycoprotein (MOG), a minor component of the myelin sheath that is targeted by T cells and autoantibodies in autoimmune diseases of the central nervous system (CNS) such as multiple sclerosis and its animal model experimental autoimmune encephalomyelitis (EAE) (32). After immunization with rodent MOG, C57BL/6 mice develop an EAE driven by MOG-reactive CD4+ T cells. This T cell response is dominated by a public TCRβ rearrangement found in both pathogenic Tconv and protective Treg in the CNS of sick mice (33, 34). Using mice carrying this TCRβ as a transgene, it was possible to compare the repertoire and antigen-recognition properties of the TCR carried by disease-relevant MOG-reactive Treg and Tconv, which were taken from the CNS at the peak of EAE, following the characterization of their TCRα chain (34). The analysis of 17 TCR from Treg and 11 TCR from Tconv obtained in such a way showed that Treg expressed TCR of markedly higher functional affinity for MOG than Tconv (34). The TCR from Treg and Tconv also interacted with MOG–MHC-II complexes in a qualitatively different manner (34). This was shown using MOG peptide variants differing from the endogenous MOG peptide sequence by alanine substitutions at various positions. The substitution of a particular histidine by an alanine in this peptide increased the stimulation of all the TCR from MOG-reactive Tconv by about 10-fold while having little effect on the response triggered by the TCR from MOG-reactive Treg. These data suggest that these TCR from Treg and Tconv differently engage the MOG peptide–MHC-II complex, as previously observed for in vitro generated Tr1 cells (35). If this is the case and is a general property, it might be possible to identify small molecules that preferentially interfere with antigen recognition by Tconv but not Treg, and vice versa. Another difference between these two types of TCR was that the wild-type MOG peptide was among the best agonists for Treg-derived TCR while this was not the case for Tconv-derived TCR, which displayed stronger responses when stimulated with some altered peptides than with the wild-type one (34). Thus, the best agonists for autoreactive tTreg might be self-peptides, while the best agonists for Tconv might be non-self-peptides, as expected from the divergent functions of these cells. The profound loss of responsiveness observed with MOG peptide variants for Treg-derived TCR suggests that Treg might be able to respond efficiently only to their cognate self-peptide, whereas Tconv might be able to react against a broader diversity of non-self sequences.
These TCR from MOG-reactive Treg and Tconv were then exploited to investigate the role of endogenous MOG in the selection of MOG-reactive tTreg (34). Retrogenic mice were prepared by reconstituting irradiated wild-type mice and Mog-deficient mice, which did not show any neurological abnormality (36), with hematopoietic stem cells (HSC) genetically engineered to express a single MOG-reactive TCR (37). In retrogenic mice made using wild-type mice as recipients, donor-derived tTreg only emerged when HSC expressed TCR from MOG-reactive Treg, whereas TCR from MOG-reactive Tconv only generated Tconv, confirming the instructive role of the TCR for tTreg development (34). Remarkably, the production of MOG-reactive tTreg was severely reduced when HSC expressing MOG-reactive TCR from Treg were transferred into Mog-deficient recipient mice, while the generation of MOG-reactive Tconv was not affected by the absence of MOG (34). Thus, autoreactive tTreg are positively selected on endogenous self-antigen, and the absence of a single endogenous antigen can lead to a severe loss of tTreg positive selection. By contrast, the selection of MOG-reactive Tconv was not affected by the absence of endogenous MOG, implying that MOG-reactive Tconv were positively selected on distinct antigen in the thymus. The high specificity of the Treg positive selection process is surprising given that the TCR recognition of antigen is viewed as degenerate (38, 39). It was therefore unexpected that none of the other antigen–MHC-II complexes available in the thymus could compensate for the absence of MOG. Similar findings were obtained for autoreactive tTreg reactive against the TRPM8 channel-associated factor 3 (TCAF3) antigen (7). This antigen is exclusively expressed in the prostate, and additionally in the thymus in an Aire-dependent manner (7). tTreg directed toward this antigen accumulate in the prostate when this organ develops tumor or autoimmune lesions (7). The analysis of genetically modified mice engineered to lack only the recognized TCAF3 epitope revealed that the absence of this single antigen was sufficient to abrogate the positive selection of TCAF3-reactive tTreg. Thus, tTreg development was also mediated by a single endogenous antigen in this case (7). As a third example, tTreg reacting toward the interphotoreceptor retinoid-binding protein (IRBP) might also be selected in a highly specific manner on IRBP because IRBP-specific Treg-mediated suppression was functionally detectable in wild-type mice but not in Irbp-deficient mice, suggesting that IRBP-reactive Treg were absent in these mice (40, 41). Altogether, these independent studies demonstrate that autoreactive tTreg are positively selected in an exquisitely specific manner on natural self-antigen through TCR interaction of high affinity. This modality of selection is relevant for the protective function of Treg in the periphery, and the design of antigen-specific Treg immunotherapy. Indeed, in adoptive cell therapy, polyclonal Treg engineered by TCR gene transfer to express TCR of high affinity for MOG (initially cloned from MOG-reactive Treg) almost completely protected recipient mice from EAE, while Treg manufactured with TCR of low affinity for MOG barely had any protective effect (34). The protective efficacy of engineered autoreactive Treg was also proportional to the affinity of their TCR for the target self-antigen in a model of type 1 diabetes (42). This implies that the antigen-reactive TCR has to be carefully chosen to optimize the beneficial effect of Treg engineered by TCR gene transfer in adoptive cell therapy. Treg expressing antigen-reactive TCR with suboptimal affinity for the target antigen might have no effect on the course of the disease.
In the thymus, TCR–antigen interactions of different strengths can lead to the development of distinct tTreg subsets. A study performed using transgenic mice expressing ovalbumin in the thymus, and six TCR reacting toward ovalbumin with different strength, showed that a broad range of TCR affinity varying by 1,000-fold drove tTreg development with the TCR of highest affinity for ovalbumin giving rise to the largest tTreg production (6). Several tTreg subsets were produced including GITRhiPD-1hiCD25hi and GITRloPD-1loCD25lo tTreg, which also differed by their expression of Helios, ICOS, CD44, and CD62L (43). Both are selected by self-antigens, being present in similar amount in germ-free mice, antigen-free mice [i.e., germ-free mice fed with a minimal antigen-free diet consisting of glucose and amino acids (44)], and mice kept in standard specific-pathogen-free conditions (43). They differ by their affinity for self-antigens: GITRhiPD-1hiCD25hi Treg express TCR of higher affinity for self-antigens than GITRloPD-1loCD25lo Treg, consistently with their higher expression of CD5 and Nur77. Remarkably, these two Treg subsets display different functions in vivo. GITRhiPD-1hiCD25hi Treg control T and B cell activation in secondary lymphoid organs, while GITRloPD-1loCD25lo Treg protect recipient mice from colitis induced by T cell transfer (43). Thus, the TCR-mediated recognition of antigen in the thymus not only directs the partitioning of T cell precursors along the tTreg fate but also fixes the functional status of these cells.
The role of endogenous antigens as the selecting moieties for autoreactive tTreg relates to the promiscuous expression of multiple self-antigens, which are otherwise restricted to defined tissues, in medullary thymic epithelial cells (mTEC). This promiscuous antigen expression in mTEC occurs partly in an Aire-dependent manner (22, 45, 46). AIRE controls the expression of more than 3,900 genes in mTEC providing a source of endogenous antigens for the positive selection of tTreg and the negative selection of Tconv in the thymus (47). The ectopic expression of tissue-restricted antigens in mTEC is strongly regulated. There are about 100,000–200,000 mTEC in a mouse thymus (48), and each tissue-specific antigen is expressed by only 1–3% of them at a given time (49–51). Although Aire-dependent antigens are implicated in the partition of some TCR into the Treg compartment, Malchow and colleagues estimated that only 5% of peripheral Treg develop in an Aire-dependent manner (22). Such a modest role for AIRE in Treg selection is in keeping with the finding that Treg from Aire-deficient mice can control autoreactive Tconv from wild-type mice (22). It is also consistent with the fact that Aire-deficient mice on a C57BL/6 background develop only a mild autoimmune phenotype (52) and survive for more than a year, whereas Foxp3-deficient mice become moribund within 4 weeks of age (53). Similarly, humans with loss-of-function mutation in AIRE develop the disease autoimmune polyendocrinopathy-candidiasis ectodermal dysplasia (APECED) yet these presents with a different clinical profile than IPEX (54). Nonetheless, APECED patients show a modest decrease in Treg (55–57). These experimental and clinical observations might be explained by the fact that AIRE controls only a fraction of the genes promiscuously expressed in mTEC (47, 58, 59). Another fraction is controlled by the transcription factor FEZF2, which in the thymus is exclusively expressed by mTEC (59). AIRE and FEZF2 together control about 60% of the antigens promiscuously expressed in mTEC (59). Mice with a deletion of Fezf2 in mTEC display a reduced frequency of Treg in the thymus as well as in secondary lymphoid organs, and develop a more severe autoimmune phenotype than Aire-deficient mice (59). In that study, 30% of mice with a deletion of Fezf2 specifically in mTEC succumbed within 12 months from a lethal inflammatory syndrome, while all Aire-deficient mice survived for more than 16 months after birth (59). The lack of Fezf2 expression in mTEC results in a different autoimmune profile than the one observed with Aire deficiency, leading to an infiltration of lung, liver, kidney, stomach, small intestine, salivary gland, brain, and testis but not of the pancreas and retina, which are the prominent targets in Aire-deficient individuals (59). Thus, FEZF2 controls the establishment of immune tolerance toward different antigens than AIRE (59, 60). The function of FEZF2 might, however, be more complex because, beyond regulating the promiscuous expression of tissue-restricted antigens in mTEC, it also influences the number and distribution of these cells within the thymic medulla (59, 60). Noteworthy, about 37% of the genes promiscuously expressed in mTEC are regulated neither by AIRE nor FEZF2, implying the existence of additional mechanisms.
Medullary thymic epithelial cells can present promiscuously expressed antigens to T cell progenitors directly or transfer these antigens to neighboring dendritic cells (DC) that can then act as antigen-presenting cells (61). These two routes of antigen presentation are required to generate the complete Treg repertoire (61). There are three major DC subsets in the thymus, resident CD8α+ DC and two subsets of migratory DC, namely plasmacytoid DC and Sirp1α+ DC that are maintained in the thymus through the migration of cells from the periphery (62–64). These three subsets rely on different chemokine receptors to reach their thymic niche. The resident Batf3-dependent CD8α+ DC express XCR1 that is the chemokine receptor for XCL1, a chemokine expressed in mTEC in an Aire-dependent manner (65). This chemotactic axis might promote the positioning of CD8α+ DC next to mTEC, and thus facilitate the capture of promiscuously expressed antigens to generate tTreg (61, 65). Sirp1α+ DC and pDC accumulate in the thymus in CCR2- and CCR9-dependent manners, respectively (66, 67). These two migratory DC subsets contribute to the ontogeny of tTreg since TCAF3-specific Treg develop in a DC-dependent manner and are still found in mice lacking resident CD8α+ DC (68). Sirp1α+ DC can process antigen obtained from mTEC in vivo, and promote Treg development in vitro (61, 69–72). Some pDC co-localize with Treg in the thymic medulla and can induce the development of T cell progenitors into Treg in vitro (71, 73). In addition to handling antigens obtained from mTEC, these migratory DC subsets might bring tissue-restricted or systemic antigens to the thymus and promote locally the formation of tTreg. Such pathway might reduce the distinction between tTreg and pTreg repertoires, as well as offer environmental antigens the possibility of contributing to tTreg selection. Importantly, the capacity of these DC to migrate to thymus, and hence to induce tTreg formation, is reduced following their stimulation via toll-like receptors, limiting the possibility for microbes to harness this immune suppressive axis to their advantage (63).
In sum, the available studies concur to show that autoreactive tTreg are selected on endogenous self-antigens in a highly specific manner through TCR interaction of high affinity in the thymus. Defects in the presentation of self-antigen, either due to impaired expression of this antigen or MHC polymorphism can thus create holes in the Treg repertoire. The process of tTreg selection is not monolithic: distinct molecular mechanisms control the expression and presentation of different (auto)antigens in the thymus, involving different sets of transcription factors and antigen-presenting cells (Figure 1). This raises the question of the redundancy between these pathways, and of the additional signals each of them provides to T cell progenitors besides the antigen–MHC-II complex, with the ensuing consequences this may have for T cell development and function. It is conceivable that this diversity explains the observations highlighting cases in which TCR affinity is not directly proportional to tTreg selection. Further studies of the cellular and biochemical events associated with the selection of T cell progenitors specific for distinct endogenous antigens such as MOG, TCAF3, and IRBP into tTreg might help to clarify these issues. Further investigation of these distinct pathways might also help to better understand the biochemical mechanisms that orient T cell progenitors toward either tTreg positive selection or clonal deletion when they encounter MHC-II peptide ligands of high affinity.
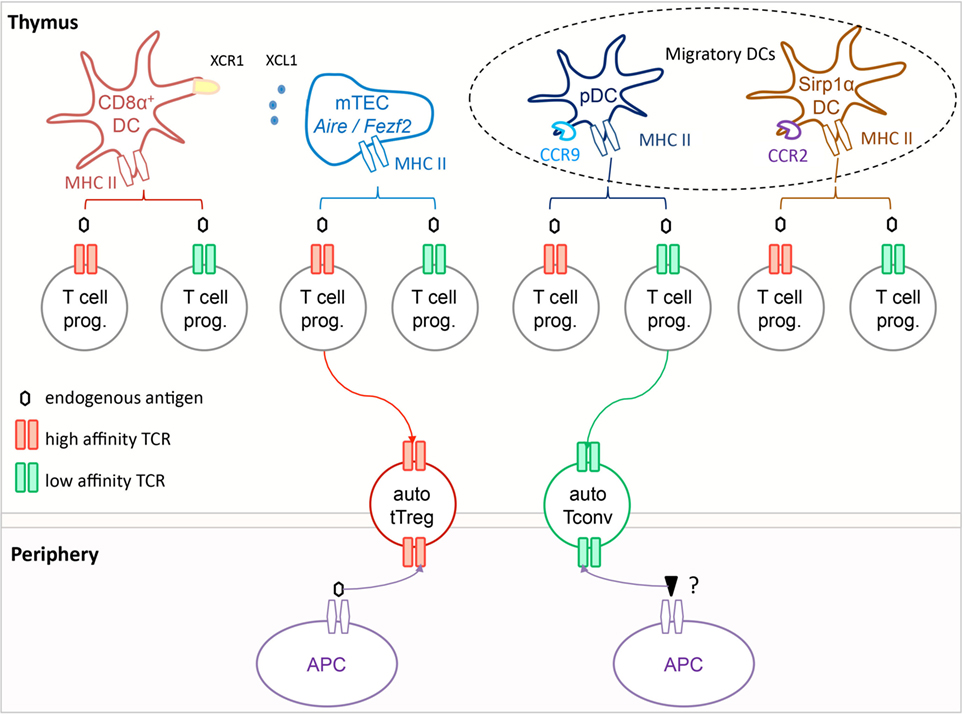
Figure 1. Autoreactive tTreg are selected in the thymus on self-antigens via T cell receptor (TCR) interactions of high functional affinity. In the thymus, T cell progenitors expressing TCR of high functional affinity (red) for self-antigens are selected into the tTreg compartment, while those expressing TCR of low functional affinity (green) are selected to become Tconv. The antigen-presenting cell can be a medullary thymic epithelial cells (mTEC), a resident CD8α+ dendritic cells (DC), or a migratory pDC or SIRP1α+ DC, or possibly a combination of these if the selection process involves multiple T cell–APC contacts. In the periphery, tTreg appear to react primarily toward the self-antigen that mediated their development, while Tconv respond most efficiently toward distinct non-self-antigens. It is noteworthy that all Tconv are positively selected, and hence have a certain degree of autoreactivity.
How does Antigen-Specificity Contribute to Treg Suppressive Function in Human Diseases?
The suppressive activity of mature Treg involves the engagement of their TCR by antigen (25, 26). Treg require TCR–antigen–MHC-II interactions of high functional affinity to achieve significant suppressive effect in the periphery (34). Thus, autoreactive tTreg-mediated suppression may operate in a highly specific manner, due to the low degree of degeneracy in the antigen-reactivity of these cells, so that the repertoire of Treg available plays an important role for the control of immunity. A high degree of specificity would help explaining how Treg suppress unwanted immune responses, while permitting efficient defense against infectious diseases. A highly specific system, however, raises the risk of holes in the Treg repertoire, and a lack of control of some deleterious immune responses. In opposition to a high degree of specificity, it has been shown that once activated in an antigen-specific manner via their TCR, Treg can then suppress Tconv specific for other antigens in a bystander manner. The breadth of this bystander suppression and the mechanisms controlling its amplitude have remained imprecisely defined. There is a need to elucidate the mechanisms controlling the breadth versus specificity of Treg-mediated suppression in human diseases, which requires the possibility of following disease-relevant Treg and Tconv concomitantly in an antigen-specific manner. Recent studies have successfully provided some first insight on this key aspect of adaptive immunity.
A quantitative analysis of relevant antigen-specific Treg and Tconv was performed in Goodpasture’s disease, a rare autoimmune disorder affecting kidneys and lungs with an incidence of about one case per million individuals per year (74). A hallmark of this disease is the presence of autoantibodies against the noncollagenous domain 1 (NC1) of the α3 chain of type 4 collagen [α3(IV)NC1] (75, 76). These autoantibodies can transfer disease, as shown in monkeys infused with antibodies from patients with Goodpasture’s disease (77). Autoantibodies might cause immunopathology directly and indirectly by promoting autoaggressive T cell responses. In support of the latter, the injection of such antibodies induced disease in wild-type mice, but not in T cell-deficient mice (78). CD4+ T cells have direct pathogenic roles (79) and can even induce disease independently of autoantibody in rodents (80). T cells directed against α3(IV)NC1 were found in increased numbers in patients with Goodpasture’s disease during acute flares compared to control individuals (81, 82). Supporting the notion that CD4+ T cells are key players in this human disease, its incidence shows a strong association with HLA alleles, particularly HLA-DRB*1501 and HLA-DRB*1502 (83). Thus, about 80% of patients with Goodpasture’s disease are positive for HLA-DRB*1501 or HLA-DRB*1502, while the frequency of these alleles is only around 20% in the control population (83). By contrast, the presence of HLA-DR1 or HLA-DR7 markedly reduces the risk of disease in HLA-DRB*1501+ individuals (83). An important feature of this disease is that α3(IV)NC1 is the major autoantigen, so that it is possible to study the most important disease-relevant T cells by focusing only on those reacting toward this antigen. The impact of these deleterious and beneficial HLA alleles on the selection of α3(IV)NC1-reactive CD4+ T cells was analyzed using HLA class 2 tetramers loaded with the α3135–145 peptide (8). CD4+ T cells reacting against HLA-DR15-α3135–145 were present at a 100-fold higher frequency in HLA-DR15+ patients with Goodpasture’s disease compared to healthy HLA-DR15+ individuals (8). The vast majority of these cells were FOXP3− and produced pro-inflammatory cytokines upon antigenic stimulation, suggesting that they represented a pathogenic population. By contrast, HLA-DR1-α3135–145-reactive CD4+ T cells were mostly FOXP3+CD25hiCD127−, and thus presented with a typical Treg phenotype (8). These Treg were possibly tTreg selected on α3(IV)NC1 peptides, since this protein was found to be expressed in the thymus (84). Remarkably, in HLA-DR15+HLA-DR1+ individuals, HLA-DR15-α3135–145-reactive CD4+ T cells were mostly FOXP3−, while HLA-DR1-α3135–145-reactive CD4+ T cells were FOXP3+ (8). Thus, the HLA allele associated with a higher risk of developing disease selects autoreactive Tconv, while the protective allele elicits autoreactive Treg targeting the same antigen. The relevance of this dichotomy for the development of pathology was evaluated in a model of Goodpasture’s disease using mice carrying transgenic HLA-DR15 and/or HLA-DR1 molecules on an FcgRIIb-deficient background. In these mice, HLA-DR1-α3135–145-reactive CD4+ T cells were, as observed in human, mostly Treg, and HLA-DR15-α3135–145-reactive CD4+ T cells were in majority Tconv. After immunization with the α3135–145 peptide, HLA-DR15+ mice developed a disease that was not affected by the prior depletion of Treg, implying a lack of protective Treg on this background (8). Other Treg were incapable of preventing disease via bystander suppression. By contrast, HLA-DR15+HLA-DR1+ mice remained healthy after immunization, unless Treg were depleted, demonstrating that protective Treg were restricted to HLA-DR1 (8). Thus, the HLA-DR1 allele afforded dominant protection by favoring the selection of autoreactive Treg, which counteracted the autoreactive Tconv directed against HLA-DR15-α3135–145. The protective function of Treg in this disease is therefore operating in a specific manner. This is striking since HLA-DR15 and HLA-DR1 differ only by few amino acids (8). These data further underline the specificity of Treg development, and of their function in the periphery. It also demonstrates that a hole in the Treg repertoire, related to the HLA profile of the individual, can lead to an unbalanced pool of autoreactive Tconv, which are selected following distinct modalities, and thus increase the susceptibility of the individual to a specific autoimmune disease. This underlines the role of HLA in the development of the Treg repertoire.
T regulatory cells are also important for limiting pathological immunity toward environmental antigens including allergens (85). IPEX patients display severe allergic manifestations including food allergy, and eczema, which are associated with increased TH2 responses, IgE levels, as well as eosinophilia (86, 87). Some early reports provided evidence that in young children the resolution of allergy toward cow milk products was related to the accumulation of allergen-reactive Treg (88, 89). A limitation of these studies, however, was that they only assessed Treg indirectly by comparing the proliferation of peripheral blood mononuclear cells depleted or not from CD25+ cells in response to an antigen stimulation, rather than by directly enumerating and characterizing antigen-reactive Treg. The direct analysis of disease-relevant Treg and Tconv can be performed using MHC-II-peptide tetramers when the antigen-specificity of these cells is known at the epitope level, and if such tetramers are available. However, the knowledge of the antigen-reactivity of disease-relevant Treg is limited in most cases. To identify disease-relevant Tconv and Treg in diseases for which the epitope landscape is not known, several laboratories developed assays that distinguish antigen-reactive Treg and Tconv according to their distinct upregulation of cell surface receptors after a short-term antigen stimulation ex vivo (90). A considerable asset of such methodology is that it requires neither the knowledge of the epitope(s) within the relevant antigen, nor of its MHC restriction, to make the identification of antigen-reactive cells feasible. Such an approach has recently been used to examine the antigen-reactivity of Treg and Tconv against aeroantigens, in particular birch pollen, in healthy and allergic individuals (85). This study used the fact that antigen-reactive Treg and Tconv distinctively upregulate CD137 and CD154, respectively, after a short antigen-specific stimulation of several hours (91). Although the low abundance of antigen-reactive cells makes their direct characterization based on their upregulation of these markers by flow cytometry difficult, their enrichment immediately after the stimulation by magnetic purification of CD154+ and CD137+ cells makes such cytometric analysis subsequently possible. This protocol has been used to identify, enumerate, and characterize T cells reactive toward food antigens, commensal bacteria, infectious antigens, and aeroantigens in the naïve repertoire (85, 92, 93). These analyses revealed the existence of birch-reactive Treg in the blood of healthy adults, which showed an increased proliferation during the allergen season (85). These cells were not found in cord blood, and possibly represented peripherally induced Treg. By contrast, birch-reactive Tconv did not proliferate during this season in healthy individuals, suggesting that they were efficiently regulated by Treg. Of note, the Treg expressed TCR of higher functional avidity for birch antigens than memory CD4+ Tconv, which possibly contributed to their dominant effect (85). Remarkably, birch-reactive Treg were present in similar numbers, and showed comparable functional properties in allergic individuals compared to the control group (85). These findings argue against the notion that allergy develops as a result of an acquired defect in the Treg compartment. However, as expected, allergic donors displayed a distinct birch-reactive Tconv profile: these cells proliferated during the allergen season and showed elevated TH2 cytokine expression, indicating that they escaped immune regulation (85). Treg and TH2 Tconv recognized different proteins within the allergen, and TH2 responses against a protein of the allergen were observed in an individual only when this protein was not efficiently recognized by Treg (85). This example illustrates the specificity of Treg-mediated suppression, and provides a case where it may be possible to analyze some of the factors controlling Treg-mediated bystander suppression in human. Why did Treg directed against some other proteins of the allergen not inhibit immunity toward all the proteins of this allergen? Interestingly, TH2 Tconv preferentially recognized water-soluble antigens rapidly released from the allergen particles when these were incubated in solution, while Treg reacted predominantly against proteins that remained associated with the non-soluble fraction of the particles (85). It is therefore possible that these distinct antigens showed a distinct bio-distribution in vivo, so that those generating pro-allergenic TH2 cells could not be subjected to Treg-mediated bystander suppression. The fact that Treg were directed against a distinguishable fraction of allergen antigens supports further the hypothesis that these were pTreg rather than tTreg. As elegantly discussed in that study, it might be possible to induce antigen-specific human pTreg by associating selected antigens with non-soluble particles alike those of allergens, which could then be delivered via the airways (85). Could such therapeutically induced pTreg control antigen-specific TH2 cells and disease in allergic patients?
Discussion
The studies described above have underlined the importance of the composition of the Treg repertoire for the suppression of unwanted immunity in human, and hence of the mechanisms presiding at the development of this repertoire.
The positive selection of autoreactive tTreg seems to be highly specific because the removal of single endogenous antigens led to the profound loss of tTreg toward these antigens. This suggests that there is little compensation between selecting self-antigens, meaning that tTreg selected on one autoantigen usually do not recognize efficiently another autoantigen. This low degree of degeneracy could be explained by the process of negative selection, i.e., the removal of T cell progenitors that receive excessive signals during their development in the thymus. Indeed, Treg are selected by TCR–antigen interaction that are already of high affinity, close to the threshold leading to negative selection, so that the efficient recognition of additional antigens in the thymus might lead to the elimination of such cells. In support of this model, several mechanisms implicated in the selection of Treg have been shown to be important for negative selection. Thus, Aire contributes to the establishment of immune tolerance through the negative selection of autoreactive T cells. For instance, the Aire-dependent expression of IRBP in the thymus is necessary for the deletion of pathogenic IRBP-reactive Tconv cells that provoke autoimmune uveitis (94). The autoreactive tTreg repertoire might therefore be composed of independent units of tTreg (each unit being selected by a given autoantigen) with little inter-unit connectivity. According to this view, the size of the autoreactive tTreg repertoire is limited, at the functional level, by the number of selecting agonist peptides presented by MHC-II molecules. This number has been estimated to lie between 103 and 105 (39). The composition of this repertoire is also influenced by the HLA alleles expressed by the individual. It may thus be possible that the repertoire of tTreg is in some cases limited, and that some polymorphisms in HLA molecules might have some strong impact on the functional property of the tTreg repertoire by determining the absence or presence of tTreg directed toward specific antigens. This appears to be the case in Goodpasture’s disease, in which the presence of α3135–145-reactive Treg was dependent on the HLA-DR1 allele. Holes in the Treg repertoire create windows of opportunity for the development of deleterious immune responses. The studies described in this review validate this concept for Goodpasture’s disease and airway allergy. It is important in this context to emphasize that a hole in the Treg repertoire does not immediately lead to a pathological immune reaction. This may account for the fact that only a minority of HLA-DR15+HLA-DR1− individuals develop Goodpasture’s disease, indicating that other mechanisms have a decisive influence on the onset of autoimmune disease, which may include other immune regulating mechanisms.
Such a high specificity in antigen-guided development and function might be unique among T cells to the Treg subset. Indeed, the development and function of Tconv appears to be more degenerate. In fact, Tconv are first selected on self-antigens in the thymus to then react against distinct foreign antigens in the periphery, implying that degenerated antigen recognition is a principle of their biology. Several studies previously attempted to estimate how many TCR can be selected in the Tconv compartment by a single positively selecting peptide using mice with restricted peptide-presenting capacity (95, 96), yet such an estimation appeared difficult to realize using such an experimental strategy (97). Other studies investigated the diversity of peptides that could positively select Tconv expressing a single TCR using fetal thymic organ culture. Testing 95 peptides selected from a set of peptides separated from I-Ek presented on a B cell line, Ebert and colleagues found that six could positively select CD4+ T cells carrying the 5C.C7 TCR, which is known to respond to a fragment of moth cytochrome c (MCC) in the context of I-Ek (98). In a separate study performed using the same strategy, Lo and colleagues found that one out of the same 95 peptides mediated the positive selection of T cell progenitors carrying the AND TCR, which also recognizes and can be activated by MCC in the periphery (99). Considering that the set of 95 peptides used in these two studies was estimated to represent 5% of the different peptide species presented by I-Ek molecules these findings suggest that a limited number of peptides yet more than one are able to positively select T cell progenitors expressing a given TCR into the Tconv compartment. Similar findings were obtained for CD8+ T cells, with the identification of two naturally occurring H-2Kb-bound self-peptides that were able to promote the positive selection of OT-I ovalbumin-reactive CD8+ T cells (100). The selection of Tconv might thus be more degenerate than for Treg. There is evidence that the recognition of antigen by Tconv shows some degeneracy in the periphery, in the sense that one Tconv can react toward multiple epitopes. Some autoreactive TCR can recognize several antigens expressed in the same tissue, a situation of cumulative autoimmunity that might facilitate the activation and acquisition of effector activity by autoreactive T cells (101). The degeneracy of T cell recognition is also important during heterologous immunity, that is the contribution of memory or effector cells specific for a pathogen to the control of a distinct pathogen (102). Heterologous immunity has been observed for virus-specific CD8+ T cells reacting toward influenza and Epstein–Barr virus (103), and has been suggested to play a role in the development of severe acute infectious mononucleosis (104). Given this apparently higher degree of degeneracy and the higher number of Tconv present in the periphery compared to Treg (which represent only a small fraction of the T cell compartment) it seems more likely to find holes in the Treg repertoire than in the Tconv repertoire. One may speculate that this relates to the primary function of the adaptive immune system, which is to defend the host against infection.
In conclusion, the novel experimental models and technologies available to study the antigen-specificity of Treg development and function will undoubtedly lead to a great improvement of our understanding of the specificity of this vital suppressive mechanism, and illuminate how to best manipulate it therapeutically. These exciting developments are eagerly awaited.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
SF lab is supported by ERC PREG-LAB 647696, AXA Chair Translational Immunology, Agence Nationale de la recherche (ANR-16-CE18-0007-01), and Chair of Excellence (Université Sorbonne Paris Cité).
References
1. Chinen T, Volchkov PY, Chervonsky AV, Rudensky AY. A critical role for regulatory T cell-mediated control of inflammation in the absence of commensal microbiota. J Exp Med (2010) 207:2323–30. doi:10.1084/jem.20101235
2. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet (2001) 27:68–73. doi:10.1038/83784
3. Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet (2001) 27:18–20. doi:10.1038/83707
4. Lathrop SK, Santacruz NA, Pham D, Luo J, Hsieh CS. Antigen-specific peripheral shaping of the natural regulatory T cell population. J Exp Med (2008) 205:3105–17. doi:10.1084/jem.20081359
5. Haribhai D, Ziegelbauer J, Jia S, Upchurch K, Yan K, Schmitt EG, et al. Alternatively activated macrophages boost induced regulatory T and Th17 cell responses during immunotherapy for colitis. J Immunol (2016) 196:3305–17. doi:10.4049/jimmunol.1501956
6. Lee HM, Bautista JL, Scott-Browne J, Mohan JF, Hsieh CS. A broad range of self-reactivity drives thymic regulatory T cell selection to limit responses to self. Immunity (2012) 37:475–86. doi:10.1016/j.immuni.2012.07.009
7. Leonard JD, Gilmore DC, Dileepan T, Nawrocka WI, Chao JL, Schoenbach MH, et al. Identification of natural regulatory T cell epitopes reveals convergence on a dominant autoantigen. Immunity (2017) 47:107–17.e8. doi:10.1016/j.immuni.2017.06.015
8. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature (2017) 545:243–7. doi:10.1038/nature22329
9. Suffia IJ, Reckling SK, Piccirillo CA, Goldszmid RS, Belkaid Y. Infected site-restricted Foxp3+ natural regulatory T cells are specific for microbial antigens. J Exp Med (2006) 203:777–88. doi:10.1084/jem.20052056
10. Moon JJ, Dash P, Oguin TH, Mcclaren JL, Chu HH, Thomas PG, et al. Quantitative impact of thymic selection on Foxp3+ and Foxp3- subsets of self-peptide/MHC class II-specific CD4+ T cells. Proc Natl Acad Sci U S A (2011) 108:14602–7. doi:10.1073/pnas.1109806108
11. Zhao J, Fett C, Trandem K, Fleming E, Perlman S. IFN-γ- and IL-10-expressing virus epitope-specific Foxp3(+) Treg cells in the central nervous system during encephalomyelitis. J Exp Med (2011) 208:1571–7. doi:10.1084/jem.20110236
12. Shafiani S, Dinh C, Ertelt JM, Moguche AO, Siddiqui I, Smigiel KS, et al. Pathogen-specific Treg cells expand early during Mycobacterium tuberculosis infection but are later eliminated in response to interleukin-12. Immunity (2013) 38:1261–70. doi:10.1016/j.immuni.2013.06.003
13. Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity (2011) 35:109–22. doi:10.1016/j.immuni.2011.03.029
14. Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature (2012) 482:395–9. doi:10.1038/nature10772
15. Itoh M, Takahashi T, Sakaguchi N, Kuniyasu Y, Shimizu J, Otsuka F, et al. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol (1999) 162:5317–26.
16. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity (2004) 21:267–77. doi:10.1016/j.immuni.2004.07.009
17. Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity (2006) 25:249–59. doi:10.1016/j.immuni.2006.05.016
18. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature (2011) 478:250–4. doi:10.1038/nature10434
19. Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs – role in immune homeostasis and autoimmunity. Front Immunol (2013) 4:232. doi:10.3389/fimmu.2013.00232
20. Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol (2012) 12:157–67. doi:10.1038/nri3155
21. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science (2013) 339:1219–24. doi:10.1126/science.1233913
22. Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA. Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity (2016) 44:1102–13. doi:10.1016/j.immuni.2016.02.009
23. Kraj P, Ignatowicz L. The mechanisms shaping the repertoire of CD4+ Foxp3+regulatory T cells. Immunology (2018) 153:290–6. doi:10.1111/imm.12859
24. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol (2016) 16:220–33. doi:10.1038/nri.2016.26
25. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15:1070–8. doi:10.1038/ni.3004
26. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity (2014) 41:722–36. doi:10.1016/j.immuni.2014.10.012
27. Schmidt AM, Lu W, Sindhava VJ, Huang Y, Burkhardt JK, Yang E, et al. Regulatory T cells require TCR signaling for their suppressive function. J Immunol (2015) 194:4362–70. doi:10.4049/jimmunol.1402384
28. Nishizuka Y, Sakakura T. Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science (1969) 166:753–5. doi:10.1126/science.166.3906.753
29. Asano M, Toda M, Sakaguchi N, Sakaguchi S. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med (1996) 184:387–96. doi:10.1084/jem.184.2.387
30. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol (2001) 2:301–6. doi:10.1038/86302
31. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med (2011) 208:1279–89. doi:10.1084/jem.20110308
32. Fillatreau S, Anderton SM. B-cell function in CNS inflammatory demyelinating disease: a complexity of roles and a wealth of possibilities. Expert Rev Clin Immunol (2007) 3:565–78. doi:10.1586/1744666X.3.4.565
33. Fazilleau N, Delarasse C, Sweenie CH, Anderton SM, Fillatreau S, Lemonnier FA, et al. Persistence of autoreactive myelin oligodendrocyte glycoprotein (MOG)-specific T cell repertoires in MOG-expressing mice. Eur J Immunol (2006) 36:533–43. doi:10.1002/eji.200535021
34. Kieback E, Hilgenberg E, Stervbo U, Lampropoulou V, Shen P, Bunse M, et al. Thymus-derived regulatory T cells are positively selected on natural self-antigen through cognate interactions of high functional avidity. Immunity (2016) 44:1114–26. doi:10.1016/j.immuni.2016.04.018
35. Beringer DX, Kleijwegt FS, Wiede F, Van Der Slik AR, Loh KL, Petersen J, et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat Immunol (2015) 16:1153–61. doi:10.1038/ni.3271
36. Delarasse C, Daubas P, Mars LT, Vizler C, Litzenburger T, Iglesias A, et al. Myelin/oligodendrocyte glycoprotein-deficient (MOG-deficient) mice reveal lack of immune tolerance to MOG in wild-type mice. J Clin Invest (2003) 112:544–53. doi:10.1172/JCI15861
37. Kieback E, Hilgenberg E, Fillatreau S. A method for the generation of TCR retrogenic mice. Methods Mol Biol (2014) 1193:117–26. doi:10.1007/978-1-4939-1212-4_12
38. Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today (1998) 19:395–404. doi:10.1016/S0167-5699(98)01299-7
39. Vrisekoop N, Monteiro JP, Mandl JN, Germain RN. Revisiting thymic positive selection and the mature T cell repertoire for antigen. Immunity (2014) 41:181–90. doi:10.1016/j.immuni.2014.07.007
40. Avichezer D, Grajewski RS, Chan CC, Mattapallil MJ, Silver PB, Raber JA, et al. An immunologically privileged retinal antigen elicits tolerance: major role for central selection mechanisms. J Exp Med (2003) 198:1665–76. doi:10.1084/jem.20030413
41. Grajewski RS, Silver PB, Agarwal RK, Su SB, Chan CC, Liou GI, et al. Endogenous IRBP can be dispensable for generation of natural CD4+CD25+ regulatory T cells that protect from IRBP-induced retinal autoimmunity. J Exp Med (2006) 203:851–6. doi:10.1084/jem.20050429
42. Yeh WI, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and bystander suppressive capacity of human regulatory T cells expressing. Front Immunol (2017) 8:1313. doi:10.3389/fimmu.2017.01313
43. Wyss L, Stadinski BD, King CG, Schallenberg S, Mccarthy NI, Lee JY, et al. Affinity for self antigen selects Treg cells with distinct functional properties. Nat Immunol (2016) 17:1093–101. doi:10.1038/ni.3522
44. Kim KS, Hong SW, Han D, Yi J, Jung J, Yang BG, et al. Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science (2016) 351:858–63. doi:10.1126/science.aac5560
45. Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science (2002) 298:1395–401. doi:10.1126/science.1075958
46. Anderson MS, Su MA. AIRE expands: new roles in immune tolerance and beyond. Nat Rev Immunol (2016) 16:247–58. doi:10.1038/nri.2016.9
47. Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res (2014) 24:1918–31. doi:10.1101/gr.171645.113
48. Gray DH, Seach N, Ueno T, Milton MK, Liston A, Lew AM, et al. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood (2006) 108:3777–85. doi:10.1182/blood-2006-02-004531
49. Derbinski J, Pinto S, Rösch S, Hexel K, Kyewski B. Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc Natl Acad Sci U S A (2008) 105:657–62. doi:10.1073/pnas.0707486105
50. Brennecke P, Reyes A, Pinto S, Rattay K, Nguyen M, Küchler R, et al. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat Immunol (2015) 16:933–41. doi:10.1038/ni.3246
51. Meredith M, Zemmour D, Mathis D, Benoist C. Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol (2015) 16:942–9. doi:10.1038/ni.3247
52. Hubert FX, Kinkel SA, Crewther PE, Cannon PZ, Webster KE, Link M, et al. Aire-deficient C57BL/6 mice mimicking the common human 13-base pair deletion mutation present with only a mild autoimmune phenotype. J Immunol (2009) 182:3902–18. doi:10.4049/jimmunol.0802124
53. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi:10.1038/ni904
54. Fischer A. Human primary immunodeficiency diseases: a perspective. Nat Immunol (2004) 5:23–30. doi:10.1038/ni1023
55. Ryan KR, Lawson CA, Lorenzi AR, Arkwright PD, Isaacs JD, Lilic D. CD4+CD25+ T-regulatory cells are decreased in patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J Allergy Clin Immunol (2005) 116:1158–9. doi:10.1016/j.jaci.2005.08.036
56. Kekäläinen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pöntynen N, et al. A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol (2007) 178:1208–15. doi:10.4049/jimmunol.178.2.1208
57. Wolff AS, Oftedal BE, Kisand K, Ersvaer E, Lima K, Husebye ES. Flow cytometry study of blood cell subtypes reflects autoimmune and inflammatory processes in autoimmune polyendocrine syndrome type I. Scand J Immunol (2010) 71:459–67. doi:10.1111/j.1365-3083.2010.02397.x
58. Derbinski J, Gäbler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, et al. Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J Exp Med (2005) 202:33–45. doi:10.1084/jem.20050471
59. Takaba H, Morishita Y, Tomofuji Y, Danks L, Nitta T, Komatsu N, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell (2015) 163:975–87. doi:10.1016/j.cell.2015.10.013
60. Klein L. Aire gets company for immune tolerance. Cell (2015) 163:794–5. doi:10.1016/j.cell.2015.10.057
61. Perry JSA, Lio CJ, Kau AL, Nutsch K, Yang Z, Gordon JI, et al. Distinct contributions of Aire and antigen-presenting-cell subsets to the generation of self-tolerance in the thymus. Immunity (2014) 41:414–26. doi:10.1016/j.immuni.2014.08.007
62. Li J, Park J, Foss D, Goldschneider I. Thymus-homing peripheral dendritic cells constitute two of the three major subsets of dendritic cells in the steady-state thymus. J Exp Med (2009) 206:607–22. doi:10.1084/jem.20082232
63. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14:377–91. doi:10.1038/nri3667
64. Mildner A, Jung S. Development and function of dendritic cell subsets. Immunity (2014) 40:642–56. doi:10.1016/j.immuni.2014.04.016
65. Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J Exp Med (2011) 208:383–94. doi:10.1084/jem.20102327
66. Baba T, Nakamoto Y, Mukaida N. Crucial contribution of thymic Sirp alpha+ conventional dendritic cells to central tolerance against blood-borne antigens in a CCR2-dependent manner. J Immunol (2009) 183:3053–63. doi:10.4049/jimmunol.0900438
67. Hadeiba H, Lahl K, Edalati A, Oderup C, Habtezion A, Pachynski R, et al. Plasmacytoid dendritic cells transport peripheral antigens to the thymus to promote central tolerance. Immunity (2012) 36:438–50. doi:10.1016/j.immuni.2012.01.017
68. Leventhal DS, Gilmore DC, Berger JM, Nishi S, Lee V, Malchow S, et al. Dendritic cells coordinate the development and homeostasis of organ-specific regulatory T cells. Immunity (2016) 44:847–59. doi:10.1016/j.immuni.2016.01.025
69. Proietto AI, Van Dommelen S, Zhou P, Rizzitelli A, D’amico A, Steptoe RJ, et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc Natl Acad Sci U S A (2008) 105:19869–74. doi:10.1073/pnas.0810268105
70. Koble C, Kyewski B. The thymic medulla: a unique microenvironment for intercellular self-antigen transfer. J Exp Med (2009) 206:1505–13. doi:10.1084/jem.20082449
71. Wirnsberger G, Mair F, Klein L. Regulatory T cell differentiation of thymocytes does not require a dedicated antigen-presenting cell but is under T cell-intrinsic developmental control. Proc Natl Acad Sci U S A (2009) 106:10278–83. doi:10.1073/pnas.0901877106
72. Guerri L, Peguillet I, Geraldo Y, Nabti S, Premel V, Lantz O. Analysis of APC types involved in CD4 tolerance and regulatory T cell generation using reaggregated thymic organ cultures. J Immunol (2013) 190:2102–10. doi:10.4049/jimmunol.1202883
73. Martín-Gayo E, Sierra-Filardi E, Corbí AL, Toribio ML. Plasmacytoid dendritic cells resident in human thymus drive natural Treg cell development. Blood (2010) 115:5366–75. doi:10.1182/blood-2009-10-248260
74. Tang W, Mcdonald SP, Hawley CM, Badve SV, Boudville NC, Brown FG, et al. Anti-glomerular basement membrane antibody disease is an uncommon cause of end-stage renal disease. Kidney Int (2013) 83:503–10. doi:10.1038/ki.2012.375
75. Kalluri R, Wilson CB, Weber M, Gunwar S, Chonko AM, Neilson EG, et al. Identification of the alpha 3 chain of type IV collagen as the common autoantigen in antibasement membrane disease and Goodpasture syndrome. J Am Soc Nephrol (1995) 6:1178–85.
76. Pedchenko V, Bondar O, Fogo AB, Vanacore R, Voziyan P, Kitching AR, et al. Molecular architecture of the Goodpasture autoantigen in anti-GBM nephritis. N Engl J Med (2010) 363:343–54. doi:10.1056/NEJMoa0910500
77. Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med (1967) 126:989–1004. doi:10.1084/jem.126.6.989
78. Kalluri R, Danoff TM, Okada H, Neilson EG. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice. J Clin Invest (1997) 100:2263–75. doi:10.1172/JCI119764
79. Wu J, Hicks J, Ou C, Singleton D, Borillo J, Lou YH. Glomerulonephritis induced by recombinant collagen IV alpha 3 chain noncollagen domain 1 is not associated with glomerular basement membrane antibody: a potential T cell-mediated mechanism. J Immunol (2001) 167:2388–95. doi:10.4049/jimmunol.167.4.2388
80. Wu J, Hicks J, Borillo J, Glass WF, Lou YH. CD4(+) T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J Clin Invest (2002) 109:517–24. doi:10.1172/JCI13876
81. Salama AD, Chaudhry AN, Ryan JJ, Eren E, Levy JB, Pusey CD, et al. In Goodpasture’s disease, CD4(+) T cells escape thymic deletion and are reactive with the autoantigen alpha3(IV)NC1. J Am Soc Nephrol (2001) 12:1908–15.
82. Zou J, Hannier S, Cairns LS, Barker RN, Rees AJ, Turner AN, et al. Healthy individuals have Goodpasture autoantigen-reactive T cells. J Am Soc Nephrol (2008) 19:396–404. doi:10.1681/ASN.2007050546
83. Phelps RG, Rees AJ. The HLA complex in Goodpasture’s disease: a model for analyzing susceptibility to autoimmunity. Kidney Int (1999) 56:1638–53. doi:10.1046/j.1523-1755.1999.00720.x
84. Wong D, Phelps RG, Turner AN. The Goodpasture antigen is expressed in the human thymus. Kidney Int (2001) 60:1777–83. doi:10.1046/j.1523-1755.2001.00014.x
85. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell (2016) 167:1067–78.e16. doi:10.1016/j.cell.2016.09.050
86. Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr (1982) 100:731–7. doi:10.1016/S0022-3476(82)80573-8
87. Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, et al. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J Clin Invest (2000) 106:R75–81. doi:10.1172/JCI11679
88. Karlsson MR, Rugtveit J, Brandtzaeg P. Allergen-responsive CD4+CD25+ regulatory T cells in children who have outgrown cow’s milk allergy. J Exp Med (2004) 199:1679–88. doi:10.1084/jem.20032121
89. Shreffler WG, Wanich N, Moloney M, Nowak-Wegrzyn A, Sampson HA. Association of allergen-specific regulatory T cells with the onset of clinical tolerance to milk protein. J Allergy Clin Immunol (2009) 123:43–52.e7. doi:10.1016/j.jaci.2008.09.051
90. Bacher P, Scheffold A. New technologies for monitoring human antigen-specific T cells and regulatory T cells by flow-cytometry. Curr Opin Pharmacol (2015) 23:17–24. doi:10.1016/j.coph.2015.04.005
91. Schoenbrunn A, Frentsch M, Kohler S, Keye J, Dooms H, Moewes B, et al. A converse 4-1BB and CD40 ligand expression pattern delineates activated regulatory T cells (Treg) and conventional T cells enabling direct isolation of alloantigen-reactive natural Foxp3+ Treg. J Immunol (2012) 189:5985–94. doi:10.4049/jimmunol.1201090
92. Bacher P, Schink C, Teutschbein J, Kniemeyer O, Assenmacher M, Brakhage AA, et al. Antigen-reactive T cell enrichment for direct, high-resolution analysis of the human naive and memory Th cell repertoire. J Immunol (2013) 190:3967–76. doi:10.4049/jimmunol.1202221
93. Bacher P, Kniemeyer O, Schönbrunn A, Sawitzki B, Assenmacher M, Rietschel E, et al. Antigen-specific expansion of human regulatory T cells as a major tolerance mechanism against mucosal fungi. Mucosal Immunol (2014) 7:916–28. doi:10.1038/mi.2013.107
94. DeVoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med (2006) 203:2727–35. doi:10.1084/jem.20061864
95. Ignatowicz L, Kappler J, Marrack P. The repertoire of T cells shaped by a single MHC/peptide ligand. Cell (1996) 84:521–9. doi:10.1016/S0092-8674(00)81028-4
96. Surh CD, Lee DS, Fung-Leung WP, Karlsson L, Sprent J. Thymic selection by a single MHC/peptide ligand produces a semidiverse repertoire of CD4+ T cells. Immunity (1997) 7:209–19. doi:10.1016/S1074-7613(00)80524-5
97. Barton GM, Rudensky AY. Requirement for diverse, low-abundance peptides in positive selection of T cells. Science (1999) 283:67–70. doi:10.1126/science.283.5398.67
98. Ebert PJ, Jiang S, Xie J, Li QJ, Davis MM. An endogenous positively selecting peptide enhances mature T cell responses and becomes an autoantigen in the absence of microRNA miR-181a. Nat Immunol (2009) 10:1162–9. doi:10.1038/ni.1797
99. Lo WL, Felix NJ, Walters JJ, Rohrs H, Gross ML, Allen PM. An endogenous peptide positively selects and augments the activation and survival of peripheral CD4+ T cells. Nat Immunol (2009) 10:1155–61. doi:10.1038/ni.1796
100. Santori FR, Kieper WC, Brown SM, Lu Y, Neubert TA, Johnson KL, et al. Rare, structurally homologous self-peptides promote thymocyte positive selection. Immunity (2002) 17:131–42. doi:10.1016/S1074-7613(02)00361-8
101. Krishnamoorthy G, Saxena A, Mars LT, Domingues HS, Mentele R, Ben-Nun A, et al. Myelin-specific T cells also recognize neuronal autoantigen in a transgenic mouse model of multiple sclerosis. Nat Med (2009) 15:626–32. doi:10.1038/nm.1975
102. Welsh RM, Selin LK. No one is naive: the significance of heterologous T-cell immunity. Nat Rev Immunol (2002) 2:417–26. doi:10.1038/nri820
103. Clute SC, Watkin LB, Cornberg M, Naumov YN, Sullivan JL, Luzuriaga K, et al. Cross-reactive influenza virus-specific CD8+ T cells contribute to lymphoproliferation in Epstein-Barr virus-associated infectious mononucleosis. J Clin Invest (2005) 115:3602–12. doi:10.1172/JCI25078
Keywords: T regulatory cells, T cell, Tconv, Foxp3, autoimmunity, allergy, Goodpasture’s disease, experimental autoimmune encephalomyelitis
Citation: Pohar J, Simon Q and Fillatreau S (2018) Antigen-Specificity in the Thymic Development and Peripheral Activity of CD4+FOXP3+ T Regulatory Cells. Front. Immunol. 9:1701. doi: 10.3389/fimmu.2018.01701
Received: 16 April 2018; Accepted: 10 July 2018;
Published: 23 July 2018
Edited by:
Mireia Sospedra, UniversitätsSpital Zürich, SwitzerlandReviewed by:
Ashutosh Chaudhry, Memorial Sloan Kettering Cancer Center, United StatesAri Waisman, Johannes Gutenberg-Universität Mainz, Germany
Copyright: © 2018 Pohar, Simon and Fillatreau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Simon Fillatreau, c2ltb25maWxsYXRyZWF1QGdvb2dsZW1haWwuY29t