 E. Jane Homan
E. Jane Homan Robert D. Bremel
Robert D. Bremel- EigenBio LLC, Madison, WI, United States
Helminth infections, by nematodes, trematodes, or cestodes, can lead to the modulation of host immune responses. This allows long-duration parasite infections and also impacts responses to co-infections. Surface, secreted, excreted, and shed proteins are thought to play a major role in modulation. A commonly reported feature of such immune modulation is the role of T regulatory (Treg) cells and IL-10. Efforts to identify helminth proteins, which cause immunomodulation, have identified candidates but not provided clarity as to a uniform mechanism driving modulation. In this study, we applied a bioinformatics systems approach, allowing us to analyze predicted T-cell epitopes of 17 helminth species and the responses to their surface proteins. In addition to major histocompatibility complex (MHC) binding, we analyzed amino acid motifs that would be recognized by T-cell receptors [T-cell-exposed motifs (TCEMs)]. All the helminth species examined have, within their surface proteins, peptides, which combine very common TCEMs with predicted high affinity binding to many human MHC alleles. This combination of features would result in large cognate T cell and a high probability of eliciting Treg responses. The TCEMs, which determine recognition by responding T-cell clones, are shared to a high degree between helminth species and with Plasmodium falciparum and Mycobacterium tuberculosis, both common co-infecting organisms. The implication of our observations is not only that Treg cells play a significant role in helminth-induced immune modulation but also that the epitope specificities of Treg responses are shared across species and genera of helminth. Hence, the immune response to a given helminth cannot be considered in isolation but rather forms part of an epitope ecosystem, or microenvironment, in which potentially immunosuppressive peptides in the helminth network via their common T-cell receptor recognition signals with T-cell epitopes in self proteins, microbiome, other helminths, and taxonomically unrelated pathogens. Such a systems approach provides a high-level view of the antigen-immune system signaling dynamics that may bias a host’s immune response to helminth infections toward immune modulation. It may indicate how helminths have evolved to select for peptides that favor long-term parasite host coexistence.
Introduction
Parasitic helminth infections affect over 2 billion people, mostly in tropical and subtropical countries, causing a vast burden of clinical and subclinical disease (1–3). To allow them to infect their hosts for extended periods, sometimes over decades, helminths have evolved to modulate their host’s immune responses (4–6). Down-regulation of the immune response results in many infected individuals being asymptomatic carriers and thus ensures perpetuation of the parasite life cycle.
Immune modulation by helminths extends to other immune stimulants; in infected individuals the immune response to secondary parasite infections, to other pathogens, and to vaccines is also down-regulated (7–11). While epidemiologic and socioeconomic factors also contribute to the prevalence of co-infections, helminth-induced immune modulation may predispose to infection by Mycobacterium tuberculosis or malaria (7, 12–14). A number of helminth infections have been associated with increased risks of cancer. The oriental liver flukes, Clonorchis sinensis and Opisthorchis viverrini, are risk factors for development of cholangiocarcinoma, typically many decades after initial infection (15). Infection with Schistosoma haematobium is associated with an increased risk of bladder carcinoma (16). Intestinal helminths have been thought to reduce risk of Helicobacter pylori associated adenocarcinoma (17), but Opisthorchis flukes may serve as risk factor by vectoring H. pylori and thus increasing associated cancers (18).
Helminth immune modulation has some beneficial effects as allergies, and inflammatory and autoimmune diseases are less common in populations infected with helminths (11, 19–21). Treatment with anthelminthics removes this effect and is reported to increase the incidence of inflammatory, autoimmune and allergic diseases (22, 23). The observation that helminth infection modulates inflammation and allergic responses has raised interest in the use of helminth infections, helminth extracts, or recombinant helminth-derived proteins as therapeutic interventions (24–27).
The mechanisms of immunomodulation arising from helminth infections have been extensively researched and are the subject of a large body of literature and many authoritative reviews (4, 11, 27–29). Among the many reports of possible modes of action (30, 31), two common themes emerge. First, certain groups of proteins appear to play a key role in bringing about changes in the host immune response. This includes proteins, which are secreted or excreted (32–34), proteins that are present in outer surface tegument or gastrointestinal surfaces, or proteins that are continually shed into the environs of the worm, either in isolation or as components of extracellular vesicles (35, 36). Extracts of secreted and surface proteins have been shown experimentally to elicit some of the immunomodulatory effects (32, 34). This has been extended to testing a number of individual proteins and identifying several proteins that can affect immune function (24, 28, 31, 37). The second unifying theme is that T regulatory (Treg) cells, and induction of IL-10, play a central role in helminth-induced host immune modulation (38–44), whether by classical Foxp3+ CD4 cells or other IL-10 secreting populations of CD4+ or CD8+ cells (45–50).
Helminth infections are not uniformly immunosuppressive. Protective immunity does emerge over time and provides the impetus for research toward vaccines (1, 51, 52), although reinfection may follow anthelmintic treatment (53, 54). Allergic responses to some helminths also occur and may also predispose to asthma (55, 56).
Studies of helminth immune modulation have largely focused on how cytokine-mediated effector mechanisms may impact the immune response. However, they have not addressed the question of whether the initial signaling in immune recognition of the parasite causes components of the immune response to be biased toward a suppressive or regulatory pathway.
In this study, we use a computational systems approach to evaluate the initial signal recognition patterns between helminth and host T cells. We evaluate the complete proteomes of 17 representative helminths and three reported co-infections based on the pattern of amino acids that would be exposed to T cells, which determine the interaction of parasite antigen and T cells. Essentially, we are attempting to see the helminth antigens as a T-cell receptor would see them, based on the minimal differentiating signal patterns from major histocompatibility complex (MHC)-bound helminth peptides that would be exposed from the MHC histotope. We have previously examined such interactions between T-cell receptor and amino acid motifs in a variety of bacterial pathogens and commensals and found distinct patterns that appear to relate to the pathogenesis of the organisms (57).
A single T-cell does not recognize, or respond to, a whole helminth; it instead engages with the few amino acids in a bound peptide MHC (pMHC) protruding from a MHC histotope, which is determined by the host human leucocyte allele (HLA) genotype. Protein antigens are processed by antigen-presenting cells (APCs) and presented to T cells as peptides bound in the groove of MHC molecules. The bound peptides (8–11 amino acids for MHC I and 13–22 amino acids for MHC II) comprise inward facing amino acids which determine peptide binding in the groove pockets [the groove-exposed motif (GEM) or pocket positions]. The intervening amino acids protrude outwards from the MHC surface or histotope and are thus exposed to the T-cell receptor, determining binding to that cognate receptor (58, 59). We refer to these amino acid motifs as the T-cell-exposed motif (TCEM). In MHC I-bound peptides, the TCEM comprises a continuous pentamer comprising positions 4, 5, 6, 7, and 8 of a nonamer, whereas in MHC II-bound peptides, the TCEM is a discontinuous pentamer in two possible recognition patterns, denoted here as TCEM IIA and IIB and comprised amino acid positions ~2,3~5~7,8~ or −1~~3~5~7,8~, respectively (where ~ is the intervening positions), relative to the central nonamer core of a longer bound peptide (60, 61). Hence, the signal that determines the outcome of the interaction with the cognate T cell is determined by the pentameric TCEM motif. This enables T cells to be polyspecific, responding to the same TCEM recognition signal, even though the MHC-bound peptide may be derived from antigens of different sources (62). The duration of TCEM signaling is determined by the stability of the pMHC complex and thus the dwell time of the peptide in the MHC groove, a function of the amino acids making up the MHC GEM. The frequency of signaling is determined by the size of the cognate T-cell population encountering the signal, which in turn is influenced by the frequency of occurrence of that TCEM in self proteins, commensal microbiome, and other pathogens.
The proteins of the human microbiome, especially the gastrointestinal microbiome, the human proteome, and the immunoglobulin repertoire are also continually processed by APCs and presented to T cells (63, 64). In examining the immunoglobulinome, it emerged that there is a frequency hierarchy of TCEM. This includes, at one extreme, common motifs found in most immunoglobulin variable regions. These are not limited to motifs encoded by the germline but also include motifs produced by somatic mutation. At the other extreme, very rare motifs are encountered only once in several million B-cell clones (65, 66). This frequency distribution is representative of that in the rest of the human proteome and gastrointestinal microbiome (57). The frequency of appearance of a TCEM determines the frequency of stimulation of a clone or clones of cognate T cells. Hence, more common motifs would be expected to beget larger cognate T-cell populations than rare motifs. A highly bound peptide that recruits a large T-cell population and a high frequency of signal is more likely to generate a suppressive or Treg response (67–72). The human immunoglobulinome comprises the greatest diversity of motifs relative to the rest of the human proteome and microbiome and undergoes considerably more turnover. We use the immunoglobulinome variable regions as a reference scale of TCEM motif frequency (65). Here, we show that a feature of helminths is their content of common TCEM that, when in the context of a binding GEM, is potentially immunosuppressive and to which the infected host is chronically exposed.
Helminth parasites have large genomes and complex life cycles (73, 74). Considerable progress has been made in helminth genome sequencing, but many proteins remain unannotated, and transcriptomic studies that relate proteins to life stage, while more advanced for a few helminths (75–79) are mostly incomplete. With these limitations, we cannot define an exact combination of T-cell epitope modulation signals functioning within the human host, but the results we present are indicative of the complexity of potential T-cell epitope interactions within and among helminth species and with potential co-infections. Such a systems approach provides a high-level view of the antigen-immune system signaling dynamics that may bias a host immune response to helminth infections toward immune modulation and may suggest approaches to develop a further understanding of the host:parasite interrelationship.
Materials and Methods
Helminth Proteomes
Protein FASTA files for 17 complete helminth proteomes were downloaded from Wormbase (www.wormbase.org) (80) as shown in Table 1. All infect humans; most have human as their primary definitive host; some (e.g., Echinococcus granulosus, Trichinella spiralis, Fasciola hepatica, and Loa loa infect humans as an accidental zoonotic host). Proteomes identified and curated by different originating laboratories varied in the size criteria applied for inclusion as did criteria for inclusion of multiple splice variants. Proteins under 50 amino acids were excluded from our analysis as end effects of the sliding analysis windows preclude making meaningful comparisons to larger proteins.
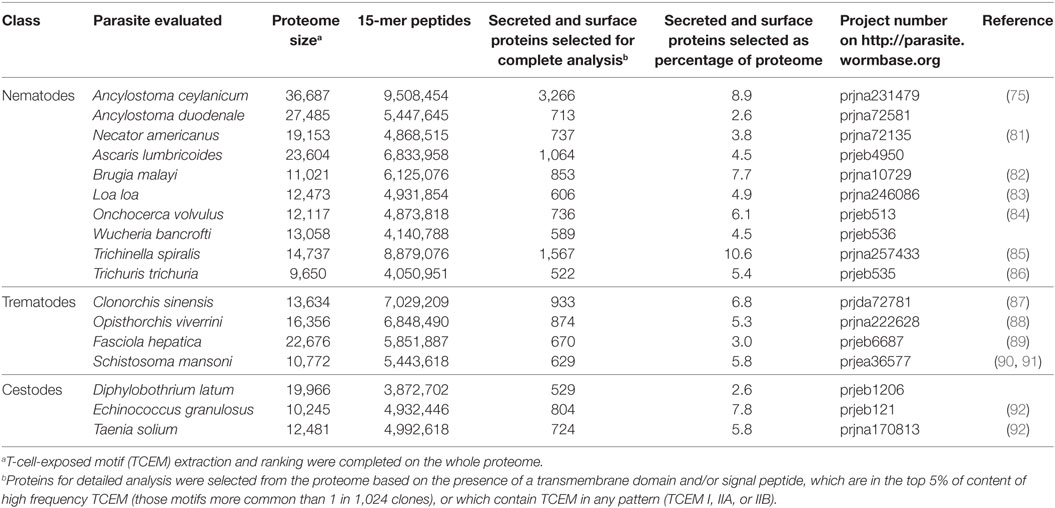
Table 1. Helminth proteomes analyzed.
Comparative Proteomes
The proteins of the human immunoglobulinome, comprising the variable region sequences of 40 million distinct immunoglobulins, were assembled as previously described, supplemented with sequences from clinical samples and other published immunoglobulin sequences, and the TCEM frequency distributions were precomputed as previously described (61, 66, 93). Examination of the frequency of occurrence of TCEM in the large immunoglobulin dataset allows us to assign a frequency category (FC) to each TCEM motif as it occurs in the immunoglobulinome clonotypes. Each motif in each recognition pattern (TCEM I for MHC I and TCEM IIA and IIB for MHC II) is assigned a log base 2 FC based on whether it occurs in half (FC1), 1 in 4 (FC2), and 1 in 8 (FC3) immunoglobulin variable regions, up to FC23, which represents 1 in 8.39 million (223) clonotypes, or approximately one T-cell clone in the entire repertoire that a single human carries. A further category of FC24 designates those motifs not yet encountered in immunoglobulin variable regions analyzed to date. The mean frequency in the human immunoglobulinome is FC10, a motif which is found in 1 in 1,024 (210) variable regions. Motifs of FC1-3 are referred to as very high frequency and those of FC10 or less as high frequency.
The human proteome, including all known isoforms of each protein (totaling 88,000 proteins), but excluding immunoglobulin sequences, was assembled from Uniprot (www.uniprot.org) and its TCEMs precomputed. The proteomes of constituents of the GI microbiome were assembled and precomputed as previously described (57). This database comprised 67 bacterial species.
Proteomes of reference sequences of Plasmodium falciparum (3D7), M. tuberculosis (H37Rv), and H. pylori (26695) were downloaded from EupathDb (http://eupathdb.org) and Patric (https://www.patricbrc.org/) (94, 95). These proteomes were screened in their entirety for high frequency TCEM and not filtered by SP and transmembrane helix (TMH) proteins.
Determination of TCEMs and Predicted MHC Binding
T-cell-exposed motif patterns were extracted from the complete proteomes and ranked as previously described for each of three recognition patterns (61, 65). Frequencies of motif occurrence were determined with respect to the immunoglobulinome, the human proteome, and the gastrointestinal microbiome (57). MHC binding was predicted using neural network ensembles trained to the LN ic50 (96) for approximately 230,000 binding reactions using the neural platform of JMP® (SAS Institute). This is an updated version of a system described previously (61). Predicted MHC binding was computed for each sequential 9-mer and 15-mer peptides in selected protein sets, wherein the predicted binding affinity to 37 MHC I and 28 MHC II alleles was determined. To estimate population behavior comprising multiple alleles with varying affinities for any peptide, the LN ic50 binding data estimates were transformed and standardized to a zero mean unit variance within each protein using a Johnson Sb distribution (97). This transformation enables the combination of binding results from different alleles, which is akin to using the principle of the additivity of variance commonly used in quantitative genetics (98, 99). The probability of cleavage of each protein by human cathepsin B, L, or S was determined. Both binding affinity and cleavage predictions were accomplished using previously described methods by neural network predictors based on principal component analysis of amino acid physical properties (100).
Suppressive Indices
To derive an indicator of the probability of a particular peptide generating a Treg response, we combined the prediction of standardized MHC binding and the frequency of occurrence of the TCEM. The combination of these two parameters provides a metric that is directly related to the probability that a collision between a cognate T cell and a pMHC on the surface of an APC will occur. For each peptide, where the standardized predicted binding affinity is greater than the mean for the protein and allele combination, the predicted standardized affinity of each allele (A) is weighted by its cognate encounter number (nA × FC16-FC) and the sum that product provides the suppressive index. Thus, the final suppressive index is a composite metric of affinity and how often cognate T-cell encounters will occur in the human population (Figure S1 in Supplementary Material).
The suppressive index is therefore an indicator of the predicted response of the human population of diverse immunogenetics; the response of any particular HLA allele will vary around this mean index. Hence the suppressive index, as computed here, does not distinguish the differences in modulation, which may arise between different ethnic or immunogenetic population subsets or between individual immunogenetically distinct hosts.
Bacteria and viruses, with small proteomes, typically comprise peptides with suppressive indices of up to a few hundred and usually under about 8,000, rare motifs have indices of 20,000–50,000 (data not shown). Only exceptional proteins in bacteria and viruses have motifs with indices >200,000. A peptide scoring a suppressive index over 1 million is thus extremely rare and would only be possible with an extremely common motif (FC1-3) that binds to almost all human alleles (in either frames IIA and IIB or MHC I A and B).
Selection of Proteins Comprising Potential Suppressor Motifs
Given the large size of each helminth proteome, we focused attention particularly on proteins of the secreted and surface proteins and on those most likely to elicit a suppressive response. To this end, all proteins with signal peptides and/or transmembrane helices in an organism proteome were selected. To identify transmembrane regions and signal peptides in the primary amino acid sequence of proteins, the online resource Phobius http://phobius.sbc.su.se/was used. This combines the two types of domain identification tools generally used in genome annotation in one resource (101). Signal peptides were predicted for eukaryotic organisms.
From these, in order to focus on the likely immunomodulatory protein candidates, two subsets of proteins were then compiled from each species based on the following criteria: the proteins having the highest content of common TCEM (less than or equal to FC 10) per 100 amino acid length were selected; and those proteins having any TCEM (TCEM I, IIA, or IIB) of FC1-3 (motifs occurring at least as frequently as in one in eight immunoglobulin variable regions). These subsets were combined, duplicates were eliminated, and a full computation of predicted MHC binding and cathepsin excision sites was completed. This comprised from 2.5 to 10% of each proteome. All peptides for each protein in each species set (15-mer for MHC II and 9-mer for MHC I) were ranked by suppressive index. Any peptide having a suppressive index over 300,000 and its source protein were identified.
Proteins Reported to Be Associated With Immunomodulation
A set of sequences for proteins previously reported to be associated with immunomodulation (24, 28, 37) was assembled, using Reference sequences from GenBank. These proteins are listed in Table S1 in Supplementary Material.
Statistical Analysis
All data processing, pattern analysis, and statistical analysis were done with JMP® v13 from SAS Institute (Cary, NC, USA).
Results
Analyses were conducted for all three TCEM recognition patterns in the complete proteomes. To conserve space, in some cases, we show data for TCEM IIA in the body of the paper and provide corresponding TCEM I and TCEM IIB data in Supplementary Tables. Very similar results are seen for each of the TCEM patterns.
TCEM Characteristics of the Entire Organism Proteome
Sharing of TCEMs Is Common
For each TCEM pentameric motif pattern, the maximum possible configurations of the 20 amino acid is 3.2 million (205). All possible motifs are found in the immunoglobulinome. The human proteome and GI microbiome comprise approximately 75 and 91%, respectively, of the possible motifs in each recognition pattern.
Table 2 shows the content the TCEM IIA motifs in the helminth species studied and the degree of sharing of motifs (Table S2 in Supplementary Material shows additional motif patterns TCEM I and IIB). The repertoire of each helminth comprises from approximately 53 to 71% of the total possible motifs in each TCEM pattern, indicating the overlap of repertoire with the immunoglobulinome. Among all 17 species of helminth, >96% of all possible TCEMs are collectively represented.
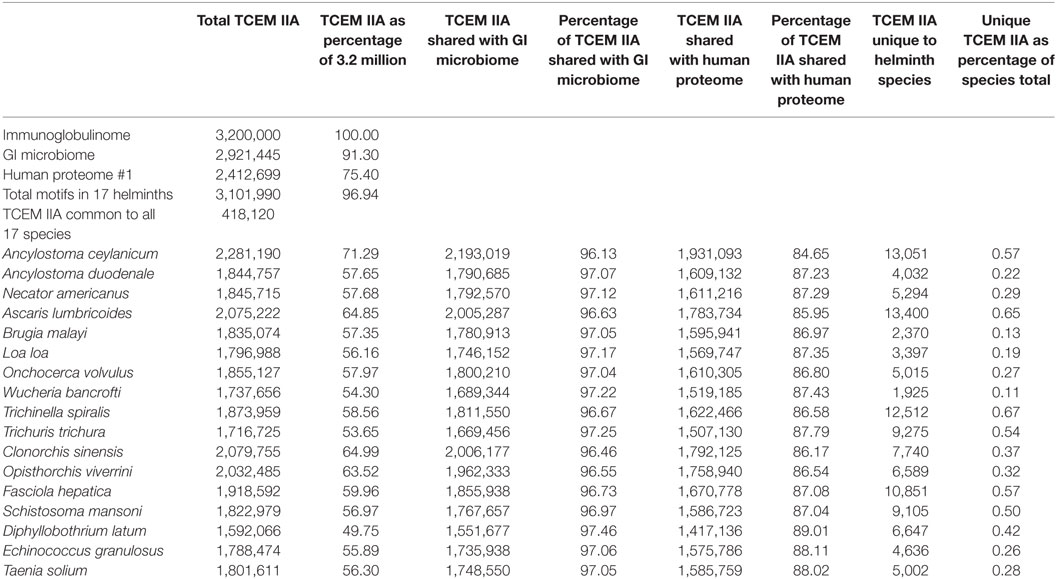
Table 2. Sharing of total T-cell-exposed motif (TCEM) IIA between helminths, gastrointestinal microbiome, and human proteome.
Sharing Between Helminth Species
Given the breadth of the TCEM repertoires of each species of helminth there is inevitably overlap in repertoires between the species. Overall ~415,000 TCEM motifs of each pattern are present in all of the 17 species analyzed. The remaining ~2.6 million motifs are shared in varying combinations among the species. Figure 1 shows the motif sharing patterns. Each tile represents a unique combination of sharing of the 3.2 million TCEM IIA motifs among the 17 species, in all comprising 112,225 different motif sharing combinations. This indicates the complexity of the potential T-cell epitope sharing and cross-reactivities between species.
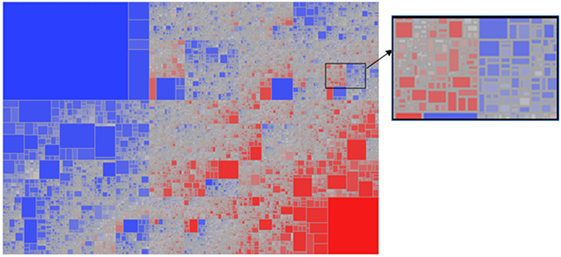
Figure 1. Treemap showing the complexity of sharing patterns of T-cell-exposed motifs (TCEMs) (shown for TCEM IIA) among the 17 helminth species analyzed. A total of 112,225 unique sharing patterns were identified. The sharing pattern refers to the combination of the 17 species of helminths that carries that motif. Some sharing patterns comprised a single TCEM motif represented by a very small tile; other sharing patterns comprise large groups of shared motifs and are shown by large tiles. The tiles are colored from highly shared motif groups (blue) to minimally shared motif groups (red). The large blue box at top left represents the TCEM IIA common to all 17 species; the red box at bottom right represents the TCEM IIA absent from all species. The intervening tiles each have a unique combination of motifs and species sharing pattern. The expanded high resolution cutout provides an appreciation of the number and complexity of different patterns embedded in the lower resolution picture.
Each helminth species has a small set of motifs, less than 0.6%, that are unique to that species relative to the other 16 species (last column Table 2). The exact number of these motifs is unlikely to be significant given the different approaches to sequencing and curation adopted for each species, but it is notable that each species does have a unique signature. However, it must be recognized that such unique motifs are overlapped with amino acids comprising non-unique motifs, so their utility in directing a unique immune response is limited.
Sharing of TCEM Between Helminths and Co-Infecting Pathogens
Three unrelated pathogens cited as common co-infections with heminthiases were selected for comparison. The extent of TCEM IIA sharing with P. falciparum, M. tuberculosis, and H. pylori is shown in Table S3 in Supplementary Material. Across all species of helminth, approximately 50% of TCEM have matches in P. falciparum, 28% have matches in M. tuberculosis, and 16% have matches in H. pylori.
Distribution of High-Frequency TCEMs Within the Helminth Proteome
The frequency of occurrence of TCEMs in each helminth proteome was compared to their frequency in the immunoglobulinome. There is no significant difference in the distribution of motifs found in helminths among the frequency categories observed in the immunoglobulinome (correlation coefficients all >0.9); those motifs that are common in immunoglobulins are commonly found in helminths and rare immunoglobulin motifs are also rare in helminths.
We then examined the distribution of high frequency TCEM in the proteins of each helminth proteome to evaluate whether particular proteins might be contributing more to an immunosuppressive response. Figure 2 shows examples of the distribution of proteins based on their content of TCEM of FC10 or lower, i.e., those motifs more common than the mean frequency in the immunoglobulin reference database. Each protein is plotted according to its size and the number of the frequent motifs. Comparable plots for the other helminths, and the three comparative co-infections, are provided in Figure S2 in Supplementary Material. While the distribution is not dissimilar from that seen in many other pathogens (57), the scatter of outlier proteins with a high content of frequent or rare TCEM is distinctive. As shown in Figure 2, the outliers are not proteins with signal peptides or transmembrane domains. Having a high content of high frequency TCEM motifs would be an indicator that a protein may have more immunosuppressive peptides, if such peptides are appropriately processed in APC and bind to MHC. On closer examination, the outliers were found to be proteins which have a high content of repetitive residues, either homorepeats of amino acids or repeats of simple amino acid motifs. Homorepeats of L, S, G, T, and Y emerge as frequent FC categories while those homorepeats of M, C, H, Q, and W are rare motifs in immunoglobulins. Figure S3 in Supplementary Material shows the sequences of those Onchocerca volvulus examples labeled in Figure 2, which are examples of proteins comprising amino acid repeats. L. loa is particularly striking with respect to the number of proteins with extended homorepeats that show as having a high content of common TCEM (Figure S1 in Supplementary Material).
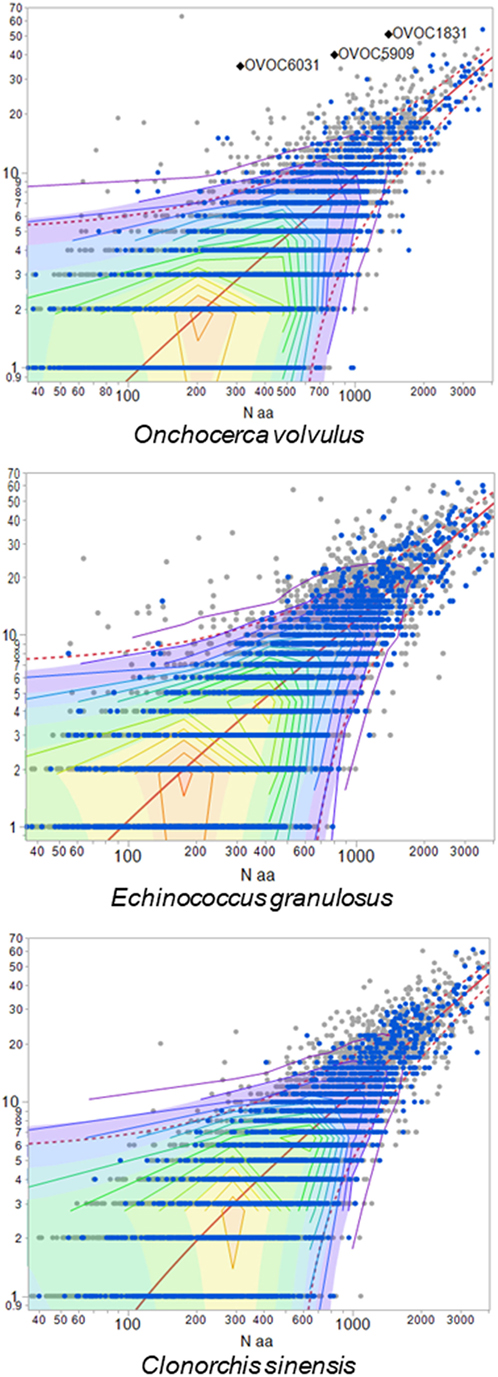
Figure 2. Proteins with high content of common T-cell-exposed motif (TCEM). Each dot represents one protein of the species indicated. X-axis shows size of the protein (log10 amino acid number). Y-axis shows number of TCEM IIA motifs of higher than FC10, i.e., occurring more often than the mean in an immunoglobulinome reference database. Proteins shown in blue are secreted and surface proteins. Three example Onchocerca volvulus proteins are labeled, and sequences for these provided in Figure S3 in Supplementary Material.
TCEM Characteristics of Surface and TMH Proteins
Sharing of TCEM Associated With High Suppressive Index Between Helminth Species
Given the focus in the literature on the secreted, surface, and shed proteins as potential contributors to immunomodulation (32–35), we elected to focus further analysis on the subset of proteins which have a signal peptide, one or more transmembrane helices or both, along with the highest frequency TCEM. For each helminth species, the top 2.5–10% of protein with respect to their likelihood of comprising suppressive motifs were analyzed, comprising 15,816 proteins overall (Table 1). The number of peptides with extremely high suppressive indices over 300,000 was determined and included 903 peptides with high MHC I suppressive indices, of which 627 were unique, and 1,377 peptides with high MHC II suppressive indices, of which 977 were unique. Such highly suppressive index peptides were found in all the helminth species examined. Table 3 shows the counts by species and suppressive index groupings. Among these, 70 proteins were identified containing more than one high suppressive index peptide at separate positions. Given the differing criteria in helminth proteome assembly, and the criteria imposed for the selection of analysis of a secreted or surface subset of proteins, the absolute numbers shown are not of particular significance except to indicate that such high indices are quite common findings in helminths.
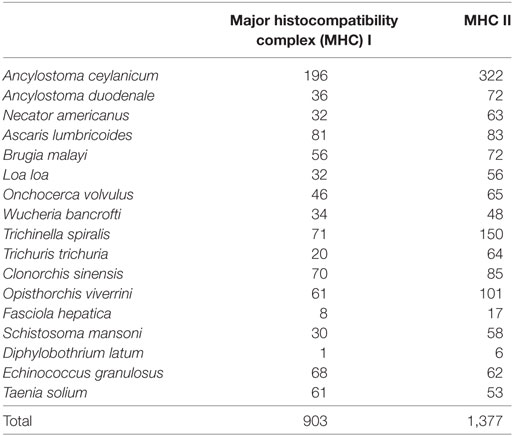
Table 3. Content of extremely high suppressive index peptides in secreted and surface proteins, showing peptides with suppressive index over 300,000.
Several helminths have peptides of high suppressive index, which comprise shared TCEM motifs, but in association with the same or different flanking peptides. Not surprisingly, the same flanking sequences are found in closely related helminths, such as Clonorchis and Opisthorchis as well as in proteins with multiple different transcripts, as shown for Ancylostoma ceylanicum. It should also be noted however that the same TCEM motifs may occur in the same parasites in the context of flanking peptides which are not high affinity binders. Depending on their binding affinity, these would have a shorter or even a transitory dwell time within the MHC groove. However, to the degree they may attract cognate Treg cells whose phenotype has already been determined, they may reinforce Treg signaling. Figure 3 shows an example of peptides from O. viverrini and A. lumbricoides each comprising the same FC3 TCEM IIA motif SL~L~LV but within an array of peptides, which show different predicted MHC II binding affinities. It also indicates how binding may vary between alleles to generate different outcomes in different hosts.
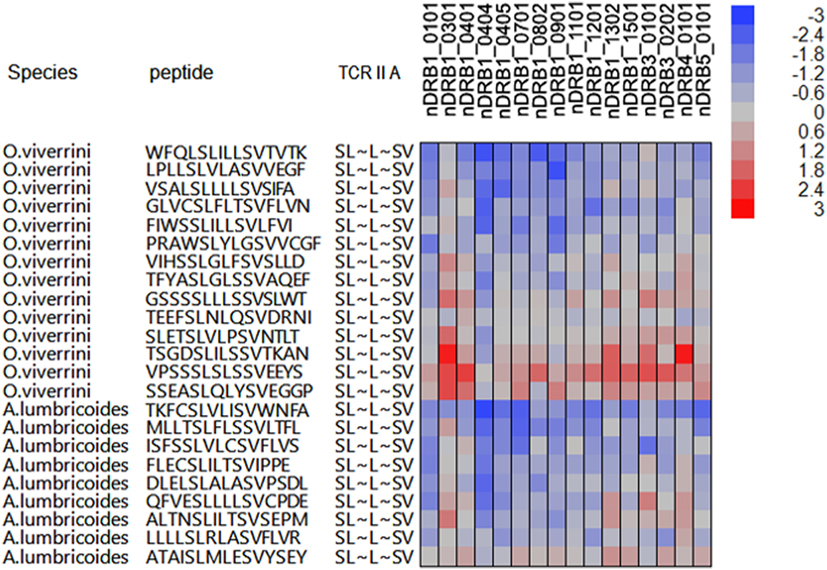
Figure 3. Example of peptides of Opisthorchis viverrini and Ascaris lumbricoides, which have shared T-cell-exposed motifs (TCEMs) but in the context of differing groove-exposed motifs. X-axis of cell plot indicates multiple different major histocompatibility complex (MHC) II DRB alleles. Coloration of the cell plot indicates predicted binding affinity in standard deviation units below the mean for that protein. Blue indicates high binding affinity; red indicates low affinity. The peptides represent a range of suppressive indices: O. viverrini ranges from 621,829 to 20,788 and A. lumbricoides from 371,273 to 36,698 based on binding to all MHC II alleles evaluated. As the TCEM motif SL~L~LV is a common motif (FC3), these are all relatively high indices.
Sharing of TCEM Associated With High Suppressive Index Between Helminths and Co-Infections
High-frequency TCEM associated with high suppressive indices was also shared between helminths and the three co-infecting pathogens examined. Table 4 shows the count of shared motifs and the TCEM associated with highest suppressive indices in helminths and their matches in the three co-infecting agents. Shared highly suppressive motifs were found particularly for M. tuberculosis and P. falciparum. We have focused on those with suppressive indices over 300,000. Figure 4 shows a conceptual model for how such T-cell networking among multiple helminths and co-infections may occur, based on a small illustrative group of five TCEM IIA motifs, which would each engage a finite number of T-cell clonotypes.
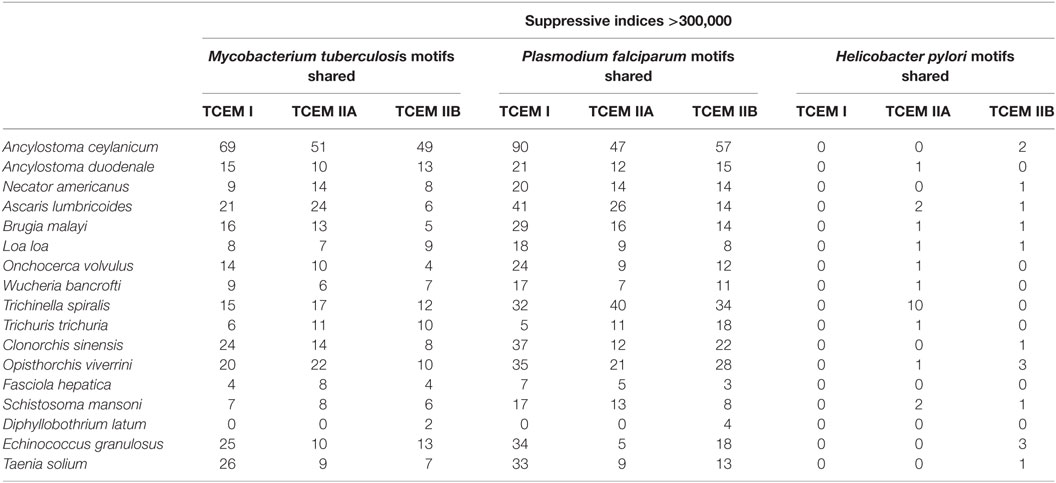
Table 4. Sharing of helminth T-cell-exposed motifs (TCEMs) associated with extremely high suppressive index motifs with selected co-infections.
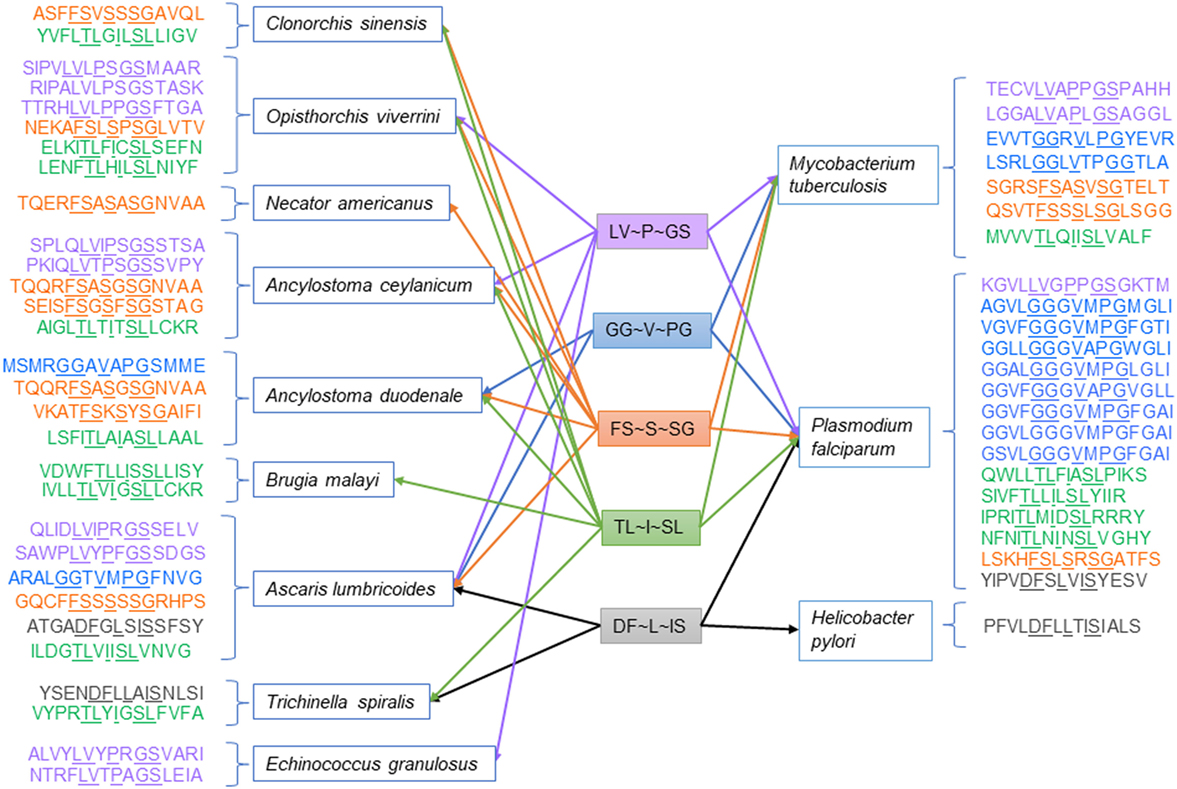
Figure 4. Illustration of networking among high suppressive index T-cell-exposed motifs in helminths and selected co-infections.
Proteins Previously Reported to Be Associated With Immunomodulation
Analysis of suppressive indices in 21 proteins reported to be associated with immunomodulation (Table S1 in Supplementary Material) identified only one, T. spiralis p53 with a peptide having a suppressive index over 300,000; the 15-mer at index position 456 has a suppressive index of 305,422, driven by the motif AV~Y~AR, which is found in 25% of all immunoglobulin variable regions. Suppressive indices of the other proteins are noted in Table S1 in Supplementary Material. Several proteins have suppressive indices in the range of 20,000–80,000.
Discussion
Reports of immunomodulation by helminths have led to a search for individual proteins, which may cause this effect. A comprehensive explanation of helminth associated immunomodulation has been elusive, but a recurring theme is that Treg cells play a central role (38, 40). In the present analysis, we apply a systems approach examining the potential magnitude of networking of T-cell responses between parasites, host, and co-infecting microrganisms and offering a conceptual model, which may shed new light on the immunomodulation phenomenon. The goal of this study was not to identify a definitive set of peptides that are solely responsible for immunomodulation, but rather to highlight potential interplay of T-cell epitopes in an ecosystem in which helminths are present, and also to know how such networking may contribute to immunomodulation of the host.
Our study has several important limitations. Both annotation of helminth proteomes and transcriptomic studies of helminths infecting their hosts are incomplete and challenging (102, 103). Indeed, we do not know in which life stage of the parasite, many proteins are expressed or in which host microenvironment they may have an effect. Thus, we cannot anticipate the quantitative or temporal impact of each protein. In a highly expressed protein, a suppressive motif may be multiplied many times or a moderately suppressive peptide motif may carry more weight if that protein is highly upregulated. Each parasite proteome is large, and to facilitate analysis, we selected certain subsets (surface and secreted proteins) for more detailed evaluation. For most of the helminths studied, only a single proteome is available. We only studied one isolate or reference proteome for each organism, both helminth and the three selected co-infecting pathogens. Polymorphisms of proteins between isolates based on single amino acid changes are sufficient to change MHC binding of peptides and the TCEM presented (104). Furthermore, single amino acid changes impact T-cell interactions across up to 46 potential binding peptides, by changing any of cathepsin processing, MHC binding, or the TCEMs presented (65). Hence, different isolates will have somewhat different networking patterns, but the overall conceptual model will apply.
The starting point for such a networking model is the premise that a T-cell receptor is agnostic as to the source of the amino acid motif of a bound peptide, which the TCR engages in the context of the MHC histotope. A cognate TCR engages a particular pentameric motif whether that occurs in a host protein, a microbiome protein, a helminth, or a co-infecting pathogen. While the flanking and intercalated amino acids in the groove or pocket positions determine MHC binding affinity (and thus the peptide dwell time), they are hidden from the TCR. Only three variables determine the signaling directed to the TCR: whether the TCR engages the pentameric motif or not; how frequently that motif is encountered and hence the population of cognate T cells stimulated to engage it; and the dwell time of the peptide bound in the MHC groove. A precondition is that the peptide is efficiently processed by peptidases in the APC to allow excision of a peptide of appropriate length to bind in the MHC.
The second, and rather obvious, key point is that the repertoire of pentameric amino acid motifs in each recognition pattern is limited to 205 or 3.2 million permutations. These may be distributed in at least three different patterns: two recognition patterns based on binding in MHC II and one pattern based on binding in MHC I (58, 60). This limits the maximal repertoire of T-cell differentiation to 9.6 million. This is entirely consistent with estimates of a human’s total T-cell count and the essential role of polyspecificity (62, 105). Given this relatively small number, it is not surprising that the large helminth proteomes we examined embodied a large percentage of the total pentameric possibilities, including the common motifs, and showed a high degree of overlap with the other proteomes and organisms examined. This mirrors findings of motif sharing among bacteria, both pathogens and commensal microbiome (57).
A further layer of variables, not addressed in this analysis, lies in the fact that multiple different clones of T cells may have TCR that bind the same TCEM peptide motif but with different affinities, and hence different dwell times, and that may trigger different cytokine outcomes (106). Higher binding affinity, consistent with higher suppressive indices, is reportedly more prone to generate IL-10 upregulation (106, 107). Binding based on less than five amino acid residues, or so-called “near neighbor” binding, further expands the possibilities for lesser binding affinities (108). The multiplicity of T-cell clones, which respond to any specific TCEM with differing binding affinities, do not change the importance of the TCEM cross-reactivity patterns but do expand the degrees of modulation possible.
The examples in Figures 3 and 4 show, for a small number of TCEM IIA motifs, that shared motifs may be embedded within many different peptides. The differing MHC GEMs, those amino acids that are directed into the MHC pockets (i.e. the non-TCEM amino acids), each determine the affinity of binding to a different set of HLA. We have focused here only on those peptides, which generate the most extreme high suppressive indices. To achieve such a score, a peptide has to be able to bind to almost all HLA alleles with high affinity. Peptides that bind with high affinity to only two or three alleles would not achieve such a high score but would have a potentially suppressive effect in individuals carrying those alleles. This reinforces two points. First, the data shown are illustrations based on the most extreme cases of epitope sharing leading to potential shared immunosuppression, when in fact a much greater span of complexity of epitope networking patterns exists across lesser degrees of binding affinity. Second, regional/ethnic population adaptation to parasites – and by the parasites to their local hosts – may be a selection function of those alleles carried by the native subpopulation binding potentially suppressive peptides with greater affinity, while those of foreign hosts do not. In addition, peptide binding to mouse, or other animal, alleles may differ from human alleles, in some cases limiting the relevance of animal models.
The analyses, even with the limitations noted, suggest that a systems approach in which the immune response is seen as a network of shared T-cell engagement may be needed to fully understand the immunomodulatory functions of helminthiasis. What is extraordinary among the helminths, which initially drew our attention, is the presence of many extremely high suppressive indices, indicative of common TCEM combined with high binding by most or many HLA alleles. In this regard, the helminths, on an individual peptide basis, far outstrip viruses and bacteria we have previously examined, where any suppressive index over 100,000 would be considered very high and where we have only in extremely rare instances observed an index of >1,000,000. Overall, this is likely due to the large helminth proteomes that increase the probability of common TCEM motifs and, when these also bind to MHC, and potentially beneficial modulatory peptides will be selected over evolutionary time.
Where shared TCEM motifs occur between helminth species, it implies that the cognate T-cell population responsive to the first helminth TCEM would also be responsive, or cross-reactive, to the same TCEM where it occurs in a second helminth. If the MHC binding of the first motif is such that it has a long dwell time and is likely to elicit a Treg response (67–70); when that same motif occurs in a second helminth and is bound by the same T-cell clones, the effect of the Treg phenotype would carry over to the response to the second helminth. Induction of relatively few Treg cells may impact the effector cells within the local microenvironment through bystander suppression (109, 110). The lengthy association of the parasite with the host, and the motif sharing among multiple parasites, would ensure a chronic presence of motifs, which have high suppressive indices, thereby fulfilling another requirement for Treg induction, repeated stimulation (111). Whereas protozoal parasites employ antigenic variation as a defensive mechanism to maintain infection (73), helminths employ the continuity of antigenic signaling to modulate the host T cell as a defense, while the surface proteins retain their invasive and other functions. It appears that relatively few Treg responders are needed to overcome a stimulatory response (67).
Mycobacterium tuberculosis, P. falciparum, and H. pylori were selected as examples of co-infections for analysis based on literature reports of interaction with helminths (7, 13, 14, 18). The widespread sharing of TCEMs among helminths, host, and the three co-infections indicates that there is potential for T-cell repertoires conditioned by exposure to a co-infecting pathogen proteome to be cross-reactive with helminths and vice versa. TCEMs provide the minimal prerequisite for T-cell polyspecificity or cross-reactivity. The shared motif counts in Table 2 are the maximum possible count of cross-reactions. This count must be further conditioned by MHC binding and peptidase processing and contemporaneous transcription. The degree of sharing is in part a function of the size of the proteome of each organism; the larger the proteome the more chance that rare motifs will be present and also that there will be more shared common motifs. However, such shared motifs can lead to mutual immunomodulatory consequences. Among the motifs that helminths share with the selected co-infections are potentially highly suppressive peptides, indicating, for example, that a Treg response elicited by M. tuberculosis may be shared with helminths, or vice versa. Notably, in M. tuberculosis, TCEM IIA matches resulting in high suppressive indices were found in 27 proteins of the PE and PE PGRS families, which have been reported to be associated with Treg induction (112, 113). In P. falciparum, shared high suppressive index motifs were most frequently found in rifins and in proteins annotated as conserved plasmodium proteins. Rifins are a family of proteins expressed on P. falciparum-infected erythrocytes, which have been implicated in immune evasion through binding to immune inhibitory receptors and the development of severe malaria (114, 115). Less sharing was seen in H. pylori, which has a smaller proteome and where the interaction with helminths may not be dependent on immune mechanisms (18).
A networking model in which a helminth infection creates a Treg cell-rich microenvironment and simultaneously causes chronic tissue damage, and thus a high rate of host cell division prone to mutations, may also create an environment tolerant to the progression of neoplasia. Treg cells have been demonstrated to be increased in Opisthorchis infections (40) and may lead to impaired response to neoepitopes. While multiple mechanisms may trigger carcinogenesis, such a model could explain the emergence of cholangiocarcinoma following prolonged infection with Clonorchis or Opisthorchis (15, 18). Conversely, the increase in autoimmune, inflammatory, and allergic disease observed in the absence of helminths, or following anthelminthic application, would be consistent with removal of the peptides driving an immunosuppressive Treg-dominated network.
Helminth proteins contain a large number of homopolymers of amino acids along with repetitious simple amino acid motifs. Only a few homopolymers (glycines, leucines, serines, threonines, and tyrosines) contribute to the counts of higher frequency TCEMs; of these, only leucines and tyrosines result in fairly high binding affinity for multiple alleles. While such repeats are a more common feature of eukaryotes than prokaryotes (116) and may offer some selective advantages (117), they do not emerge as a driver of very high suppressive indices in the helminths examined. However, their presence does raise the question of whether multiple occurrences, especially of leucine or tyrosine observed in L. loa, might have an additive suppressive effect.
We omitted from consideration proteins under 50 amino acids in length. Some helminth proteomes included a large number of small open reading frames (sORFs). Whether sORFs which generate peptides large enough to bind MHCs may also contribute to the overall epitope landscape is unknown (118, 119). Given species differences in the curation of proteins for inclusion, a comparative analysis is not possible.
In addressing networking as a factor in immunomodulation, we have focused here on the most common TCEM and those that are shared by many helminths. We also noted that a very small percentage of TCEM motifs are unique to any single helminth among those evaluated; these may or may not be motifs rarely found in the immunoglobulinome but are unlikely to be among the most common motifs. Figure 1 reflects the sharing patterns of motifs among helminth species. There is a range of less common motifs, which also have sharing patterns among the species (as reflected by the grey and red tiles in Figure 1). These are motifs that may elicit immune stimulation and potentially have utility in a vaccine. The challenge in designing a vaccine comprising T-cell epitopes is to identify proteins comprising conserved stimulatory epitopes, appropriately processed by cathepsins for MHC presentation, which are not negated by an immunosuppressive motif (120).
It is likely that many mechanisms are contributing simultaneously to immunomodulation. The proteins previously reported (24, 27, 28, 37) may have a primary or secondary role in immune regulation. The cystatins, which have been reported to play an immunomodulatory role in several parasites (121–123), would likely inhibit cathepsin function and, hence, peptide excision for presentation on the MHC. Others may act through the induction of Treg cells or as a downstream consequence of such regulation. Among the previously reported immunomodulators that we analyzed, only T. spiralis p53 was found to have a suppressive index over 300,000. Several others had suppressive indices below this level, indicating that they might have a suppressive effect in individuals of certain alleles, or when present in large amounts; the major egg antigen of S. mansoni is one example. However, it is important to keep in mind that several of the proteins previously examined for their immunomodulatory role are from helminths that are not human pathogens and they have been examined in animal models, so our analysis of suppressive index based on human alleles examines those out of the host context in which they were tested.
There has been increasing interest in exploiting the immunomodulatory effects of helminths (23, 26), by administering low level live parasite infections, extracts, or recombinant proteins (24, 25, 31). The TCEM networking model put forth here offers an approach to defining predicted immunomodulatory peptides, which have benefited from selection through an evolutionary filter and which may then be tested in a therapeutic model at less risk than a live parasite.
Given the lack of annotation of many individual proteins in the helminth proteomes, and the recognition that we have only worked with one isolate of each species, we have not focused here on highlighting the particular potential highly suppressive peptides for each parasite. Rather we have addressed this as a conceptual model. For every motif example for which we show an unambiguous predicted outcome for most HLA, there are many more subtle effects based on lower suppressive indices or particular host HLAs. The systems approach we have taken suggests that the immune response to a given parasite cannot be considered in isolation but should be seen as a part of an epitope ecosystem, or microenvironment, in which the potentially highly suppressive peptides in the helminths network through their common T-cell receptor recognition signals with T-cell epitopes in self proteins, microbiome, in other helminths and in taxonomically unrelated pathogens. It indicates the complexity of T-cell interactions, which may have allowed helminths to evolve to select for the peptides that drive an immunosuppressive repertoire of T cells. Such a repertoire would favor long-term coexistence with the host, or an immunogenetic subpopulation thereof, and facilitate polyparasitism and co-infections. This approach suggests that a different paradigm, in which epitope networking is considered, can contribute to understanding helminth associated immunomodulation. It is a paradigm, which is both very simple in conception and profoundly complex to test through a reductionist approach.
Author Contributions
RB designed the algorithms, EH planned the study and performed the analyses, and EH and RB wrote the article.
Conflict of Interest Statement
Both authors are employees and equity holders in ioGenetics LLC, the parent company of EigenBio LLC.
The reviewer PT and handling Editor declared their shared affiliation.
Acknowledgments
Systems analysis of this type is not possible without the assembly of genomes and proteomes into accessible databases such as EuPathDB, Patric, Wormbase, Uniprot, and the Human Microbiome Project. We are indebted to the consortia, which maintain such databases. The authors thank Erich Marquard Schwarz for insightful discussion and Alleen Hager for computational and editorial assistance.
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01763/full#supplementary-material.
Abbreviations
APC, antigen-presenting cell; FC, frequency category; GEM, groove-exposed motif; HLA, human leucocyte allele; MHC, major histocompatibility complex; pMHC, peptide MHC; sORF, small open reading frame; TCEM, T-cell-exposed motif; TMH, transmembrane helix.
References
1. Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, Jacobson J. Helminth infections: the great neglected tropical diseases. J Clin Invest (2008) 118(4):1311–21. doi:10.1172/JCI34261
2. Brooker S, Bethony J, Hotez PJ. Human hookworm infection in the 21st century. Adv Parasitol (2004) 58:197–288. doi:10.1016/S0065-308X(04)58004-1
3. Herricks JR, Hotez PJ, Wanga V, Coffeng LE, Haagsma JA, Basanez MG, et al. The global burden of disease study 2013: what does it mean for the NTDs? PLoS Negl Trop Dis (2017) 11(8):e0005424. doi:10.1371/journal.pntd.0005424
4. McSorley HJ, Maizels RM. Helminth infections and host immune regulation. Clin Microbiol Rev (2012) 25(4):585–608. doi:10.1128/CMR.05040-11
5. Harris AR, Russell RJ, Charters AD. A review of schistosomiasis in immigrants in Western Australia, demonstrating the unusual longevity of Schistosoma mansoni. Trans R Soc Trop Med Hyg (1984) 78(3):385–8. doi:10.1016/0035-9203(84)90129-9
6. Wright EP, Jain S. Human infestation by Taenia saginata lasting over 20 years. Postgrad Med J (1984) 60(705):495–6. doi:10.1136/pgmj.60.705.495
7. Wammes LJ, Hamid F, Wiria AE, de Gier B, Sartono E, Maizels RM, et al. Regulatory T cells in human geohelminth infection suppress immune responses to BCG and Plasmodium falciparum. Eur J Immunol (2010) 40(2):437–42. doi:10.1002/eji.200939699
8. Cooper PJ, Chico M, Sandoval C, Espinel I, Guevara A, Levine MM, et al. Human infection with Ascaris lumbricoides is associated with suppression of the interleukin-2 response to recombinant cholera toxin B subunit following vaccination with the live oral cholera vaccine CVD 103-HgR. Infect Immun (2001) 69(3):1574–80. doi:10.1128/IAI.69.3.1574-1580.2001
9. Stelekati E, Wherry EJ. Chronic bystander infections and immunity to unrelated antigens. Cell Host Microbe (2012) 12(4):458–69. doi:10.1016/j.chom.2012.10.001
10. Supali T, Verweij JJ, Wiria AE, Djuardi Y, Hamid F, Kaisar MM, et al. Polyparasitism and its impact on the immune system. Int J Parasitol (2010) 40(10):1171–6. doi:10.1016/j.ijpara.2010.05.003
11. Maizels RM, Yazdanbakhsh M. T-cell regulation in helminth parasite infections: implications for inflammatory diseases. Chem Immunol Allergy (2008) 94:112–23. doi:10.1159/000154944
12. Babu S, Bhat SQ, Kumar NP, Jayantasri S, Rukmani S, Kumaran P, et al. Human type 1 and 17 responses in latent tuberculosis are modulated by coincident filarial infection through cytotoxic T lymphocyte antigen-4 and programmed death-1. J Infect Dis (2009) 200(2):288–98. doi:10.1086/599797
13. George PJ, Anuradha R, Kumaran PP, Chandrasekaran V, Nutman TB, Babu S. Modulation of mycobacterial-specific Th1 and Th17 cells in latent tuberculosis by coincident hookworm infection. J Immunol (2013) 190(10):5161–8. doi:10.4049/jimmunol.1203311
14. Chatterjee S, Nutman TB. Helminth-induced immune regulation: implications for immune responses to tuberculosis. PLoS Pathog (2015) 11(1):e1004582. doi:10.1371/journal.ppat.1004582
15. Sripa B, Kaewkes S, Sithithaworn P, Mairiang E, Laha T, Smout M, et al. Liver fluke induces cholangiocarcinoma. PLoS Med (2007) 4(7):e201. doi:10.1371/journal.pmed.0040201
16. Mostafa MH, Sheweita SA, O’Connor PJ. Relationship between schistosomiasis and bladder cancer. Clin Microbiol Rev (1999) 12(1):97–111.
17. Whary MT, Sundina N, Bravo LE, Correa P, Quinones F, Caro F, et al. Intestinal helminthiasis in Colombian children promotes a Th2 response to Helicobacter pylori: possible implications for gastric carcinogenesis. Cancer Epidemiol Biomarkers Prev (2005) 14(6):1464–9. doi:10.1158/1055-9965.EPI-05-0095
18. Brindley PJ, da Costa JM, Sripa B. Why does infection with some helminths cause cancer? Trends Cancer (2015) 1(3):174–82. doi:10.1016/j.trecan.2015.08.011
19. Cooper PJ. Intestinal worms and human allergy. Parasite Immunol (2004) 26(11–12):455–67. doi:10.1111/j.0141-9838.2004.00728.x
20. Correale J, Farez M. Association between parasite infection and immune responses in multiple sclerosis. Ann Neurol (2007) 61(2):97–108. doi:10.1002/ana.21067
21. Libbey JE, Cusick MF, Fujinami RS. Role of pathogens in multiple sclerosis. Int Rev Immunol (2014) 33(4):266–83. doi:10.3109/08830185.2013.823422
22. Sanya RE, Nkurunungi G, Andia Biraro I, Mpairwe H, Elliott AM. A life without worms. Trans R Soc Trop Med Hyg (2017) 111(1):3–11. doi:10.1093/trstmh/trx010
23. Wammes LJ, Mpairwe H, Elliott AM, Yazdanbakhsh M. Helminth therapy or elimination: epidemiological, immunological, and clinical considerations. Lancet Infect Dis (2014) 14(11):1150–62. doi:10.1016/S1473-3099(14)70771-6
24. Nascimento Santos L, Carvalho Pacheco LG, Silva Pinheiro C, Alcantara-Neves NM. Recombinant proteins of helminths with immunoregulatory properties and their possible therapeutic use. Acta Trop (2017) 166:202–11. doi:10.1016/j.actatropica.2016.11.016
25. Smallwood TB, Giacomin PR, Loukas A, Mulvenna JP, Clark RJ, Miles JJ. Helminth immunomodulation in autoimmune disease. Front Immunol (2017) 8:453. doi:10.3389/fimmu.2017.00453
26. Harnett MM, Harnett W. Can parasitic worms cure the modern world’s ills? Trends Parasitol (2017) 33(9):694–705. doi:10.1016/j.pt.2017.05.007
27. Finlay CM, Walsh KP, Mills KH. Induction of regulatory cells by helminth parasites: exploitation for the treatment of inflammatory diseases. Immunol Rev (2014) 259(1):206–30. doi:10.1111/imr.12164
28. Maizels RM, McSorley HJ. Regulation of the host immune system by helminth parasites. J Allergy Clin Immunol (2016) 138(3):666–75. doi:10.1016/j.jaci.2016.07.007
29. McSorley HJ, Loukas A. The immunology of human hookworm infections. Parasite Immunol (2010) 32(8):549–59. doi:10.1111/j.1365-3024.2010.01224.x
30. McSorley HJ, Hewitson JP, Maizels RM. Immunomodulation by helminth parasites: defining mechanisms and mediators. Int J Parasitol (2013) 43(3–4):301–10. doi:10.1016/j.ijpara.2012.11.011
31. Navarro S, Pickering DA, Ferreira IB, Jones L, Ryan S, Troy S, et al. Hookworm recombinant protein promotes regulatory T cell responses that suppress experimental asthma. Sci Transl Med (2016) 8(362):362ra143. doi:10.1126/scitranslmed.aaf8807
32. Geiger SM, Fujiwara RT, Freitas PA, Massara CL, Dos Santos Carvalho O, Correa-Oliveira R, et al. Excretory-secretory products from hookworm l(3) and adult worms suppress proinflammatory cytokines in infected individuals. J Parasitol Res (2011) 2011:512154. doi:10.1155/2011/512154
33. Maizels RM, Philipp M, Ogilvie BM. Molecules on the surface of parasitic nematodes as probes of the immune response in infection. Immunol Rev (1982) 61:109–36. doi:10.1111/j.1600-065X.1982.tb00375.x
34. Ferreira I, Smyth D, Gaze S, Aziz A, Giacomin P, Ruyssers N, et al. Hookworm excretory/secretory products induce interleukin-4 (IL-4)+ IL-10+ CD4+ T cell responses and suppress pathology in a mouse model of colitis. Infect Immun (2013) 81(6):2104–11. doi:10.1128/IAI.00563-12
35. Lightowlers MW, Rickard MD. Excretory-secretory products of helminth parasites: effects on host immune responses. Parasitology (1988) 96(Suppl):S123–66. doi:10.1017/S0031182000086017
36. Coakley G, Maizels RM, Buck AH. Exosomes and other extracellular vesicles: the new communicators in parasite infections. Trends Parasitol (2015) 31(10):477–89. doi:10.1016/j.pt.2015.06.009
37. Morais SB, Figueiredo BC, Assis NRG, Alvarenga DM, de Magalhaes MTQ, Ferreira RS, et al. Schistosoma mansoni SmKI-1 serine protease inhibitor binds to elastase and impairs neutrophil function and inflammation. PLoS Pathog (2018) 14(2):e1006870. doi:10.1371/journal.ppat.1006870
38. Maizels RM, Smith KA. Regulatory T cells in infection. Adv Immunol (2011) 112:73–136. doi:10.1016/B978-0-12-387827-4.00003-6
39. Ricci ND, Fiuza JA, Bueno LL, Cancado GG, Gazzinelli-Guimaraes PH, Martins VG, et al. Induction of CD4(+)CD25(+)FOXP3(+) regulatory T cells during human hookworm infection modulates antigen-mediated lymphocyte proliferation. PLoS Negl Trop Dis (2011) 5(11):e1383. doi:10.1371/journal.pntd.0001383
40. Kaewraemruaen C, Sermswan RW, Wongratanacheewin S. Induction of regulatory T cells by Opisthorchis viverrini. Parasite Immunol (2016) 38(11):688–97. doi:10.1111/pim.12358
41. Taylor MD, Harris A, Babayan SA, Bain O, Culshaw A, Allen JE, et al. CTLA-4 and CD4+ CD25+ regulatory T cells inhibit protective immunity to filarial parasites in vivo. J Immunol (2007) 179(7):4626–34. doi:10.4049/jimmunol.179.7.4626
42. Sawant DV, Gravano DM, Vogel P, Giacomin P, Artis D, Vignali DA. Regulatory T cells limit induction of protective immunity and promote immune pathology following intestinal helminth infection. J Immunol (2014) 192(6):2904–12. doi:10.4049/jimmunol.1202502
43. Doetze A, Satoguina J, Burchard G, Rau T, Loliger C, Fleischer B, et al. Antigen-specific cellular hyporesponsiveness in a chronic human helminth infection is mediated by T(h)3/T(r)1-type cytokines IL-10 and transforming growth factor-beta but not by a T(h)1 to T(h)2 shift. Int Immunol (2000) 12(5):623–30. doi:10.1093/intimm/12.5.623
44. Metenou S, Dembele B, Konate S, Dolo H, Coulibaly SY, Coulibaly YI, et al. At homeostasis filarial infections have expanded adaptive T regulatory but not classical Th2 cells. J Immunol (2010) 184(9):5375–82. doi:10.4049/jimmunol.0904067
45. Khan IA, Hakak R, Eberle K, Sayles P, Weiss LM, Urban JF Jr. Coinfection with Heligmosomoides polygyrus fails to establish CD8+ T-cell immunity against Toxoplasma gondii. Infect Immun (2008) 76(3):1305–13. doi:10.1128/IAI.01236-07
46. Marple A, Wu W, Shah S, Zhao Y, Du P, Gause WC, et al. Cutting Edge: helminth coinfection blocks effector differentiation of CD8 T cells through alternate host Th2- and IL-10-mediated responses. J Immunol (2017) 198(2):634–9. doi:10.4049/jimmunol.1601741
47. Walsh CM, Smith P, Fallon PG. Role for CTLA-4 but not CD25+ T cells during Schistosoma mansoni infection of mice. Parasite Immunol (2007) 29(6):293–308. doi:10.1111/j.1365-3024.2007.00947.x
48. Sachdev D, Gough KC, Flynn RJ. The chronic stages of bovine Fasciola hepatica are dominated by CD4 T-cell exhaustion. Front Immunol (2017) 8:1002. doi:10.3389/fimmu.2017.01002
49. Huang Y, Mao K, Chen X, Sun MA, Kawabe T, Li W, et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science (2018) 359(6371):114–9. doi:10.1126/science.aam5809
50. Logan J, Navarro S, Loukas A, Giacomin P. Helminth-induced regulatory T cells and suppression of allergic responses. Curr Opin Immunol (2018) 54:1–6. doi:10.1016/j.coi.2018.05.007
51. Loukas A, Bethony J, Brooker S, Hotez P. Hookworm vaccines: past, present, and future. Lancet Infect Dis (2006) 6(11):733–41. doi:10.1016/S1473-3099(06)70630-2
52. Noon JB, Aroian RV. Recombinant subunit vaccines for soil-transmitted helminths. Parasitology (2017) 144(14):1845–70. doi:10.1017/S003118201700138X
53. Jia TW, Melville S, Utzinger J, King CH, Zhou XN. Soil-transmitted helminth reinfection after drug treatment: a systematic review and meta-analysis. PLoS Negl Trop Dis (2012) 6(5):e1621. doi:10.1371/journal.pntd.0001621
54. Albonico M, Smith PG, Ercole E, Hall A, Chwaya HM, Alawi KS, et al. Rate of reinfection with intestinal nematodes after treatment of children with mebendazole or albendazole in a highly endemic area. Trans R Soc Trop Med Hyg (1995) 89(5):538–41. doi:10.1016/0035-9203(95)90101-9
55. Ahumada V, Garcia E, Dennis R, Rojas MX, Rondon MA, Perez A, et al. IgE responses to Ascaris and mite tropomyosins are risk factors for asthma. Clin Exp Allergy (2015) 45(7):1189–200. doi:10.1111/cea.12513
56. Loukas A, Prociv P. Immune responses in hookworm infections. Clin Microbiol Rev (2001) 14(4):689–703. doi:10.1128/CMR.14.4.689-703.2001
57. Bremel RD, Homan J. Extensive T-cell epitope repertoire sharing among human proteome, gastrointestinal microbiome, and pathogenic bacteria: implications for the definition of self. Front Immunol (2015) 6:538. doi:10.3389/fimmu.2015.00538
58. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC Specificity of T cell recognition. Cell (2014) 157(5):1073–87. doi:10.1016/j.cell.2014.03.047
59. Wucherpfennig KW. The structural interactions between T cell receptors and MHC-peptide complexes place physical limits on self-nonself discrimination. Curr Top Microbiol Immunol (2005) 296:19–37.
60. Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol (2006) 24:419–66. doi:10.1146/annurev.immunol.23.021704.115658
61. Bremel RD, Homan EJ. An integrated approach to epitope analysis II: a system for proteomic-scale prediction of immunological characteristics. Immunome Res (2010) 6(1):8. doi:10.1186/1745-7580-6-8
62. Wucherpfennig KW, Allen PM, Celada F, Cohen IR, De Boer R, Garcia KC, et al. Polyspecificity of T cell and B cell receptor recognition. Semin Immunol (2007) 19(4):216–24. doi:10.1016/j.smim.2007.02.012
63. Bogen B, Weiss S. Processing and presentation of idiotypes to MHC-restricted T cells. Int Rev Immunol (1993) 10:337–55. doi:10.3109/08830189309061709
64. Weiss S, Bogen B. B-lymphoma cells process and present their endogenous immunoglobulin to major histocompatibility complex-restricted T cells. Proc Natl Acad Sci U S A (1989) 86(1):282–6. doi:10.1073/pnas.86.1.282
65. Bremel RD, Homan EJ. Frequency patterns of T-cell exposed amino acid motifs in immunoglobulin heavy chain peptides presented by MHCs. Front Immunol (2014) 5:541. doi:10.3389/fimmu.2014.00541
66. Hoglund RA, Lossius A, Johansen JN, Homan J, Benth JS, Robins H, et al. In silico prediction analysis of idiotope-driven T-B cell collaboration in multiple sclerosis. Front Immunol (2017) 8:1255. doi:10.3389/fimmu.2017.01255
67. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol (2016) 16(4):220–33. doi:10.1038/nri.2016.26
68. Valitutti S. The serial engagement model 17 years after: from TCR triggering to immunotherapy. Front Immunol (2012) 3:272. doi:10.3389/fimmu.2012.00272
69. Faroudi M, Zaru R, Paulet P, Muller S, Valitutti S. Cutting edge: T lymphocyte activation by repeated immunological synapse formation and intermittent signaling. J Immunol (2003) 171(3):1128–32. doi:10.4049/jimmunol.171.3.1128
70. Gabrysova L, Wraith DC. Antigenic strength controls the generation of antigen-specific IL-10-secreting T regulatory cells. Eur J Immunol (2010) 40(5):1386–95. doi:10.1002/eji.200940151
71. Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol (2003) 4(4):355–60. doi:10.1038/ni908
72. Feng Y, Brazin KN, Kobayashi E, Mallis RJ, Reinherz EL, Lang MJ. Mechanosensing drives acuity of alphabeta T-cell recognition. Proc Natl Acad Sci U S A (2017) 114(39):E8204–13. doi:10.1073/pnas.1703559114
73. Zarowiecki M, Berriman M. What helminth genomes have taught us about parasite evolution. Parasitology (2015) 142(Suppl 1):S85–97. doi:10.1017/S0031182014001449
74. Blaxter M, Koutsovoulos G. The evolution of parasitism in Nematoda. Parasitology (2015) 142(Suppl 1):S26–39. doi:10.1017/S0031182014000791
75. Schwarz EM, Hu Y, Antoshechkin I, Miller MM, Sternberg PW, Aroian RV. The genome and transcriptome of the zoonotic hookworm Ancylostoma ceylanicum identify infection-specific gene families. Nat Genet (2015) 47(4):416–22. doi:10.1038/ng.3237
76. Mulvenna J, Sripa B, Brindley PJ, Gorman J, Jones MK, Colgrave ML, et al. The secreted and surface proteomes of the adult stage of the carcinogenic human liver fluke Opisthorchis viverrini. Proteomics (2010) 10(5):1063–78. doi:10.1002/pmic.200900393
77. Young ND, Campbell BE, Hall RS, Jex AR, Cantacessi C, Laha T, et al. Unlocking the transcriptomes of two carcinogenic parasites, Clonorchis sinensis and Opisthorchis viverrini. PLoS Negl Trop Dis (2010) 4(6):e719. doi:10.1371/journal.pntd.0000719
78. Almeida CR, Stoco PH, Wagner G, Sincero TC, Rotava G, Bayer-Santos E, et al. Transcriptome analysis of Taenia solium cysticerci using open reading frame ESTs (ORESTES). Parasit Vectors (2009) 2(1):35. doi:10.1186/1756-3305-2-35
79. Bennuru S, Cotton JA, Ribeiro JM, Grote A, Harsha B, Holroyd N, et al. Stage-specific transcriptome and proteome analyses of the filarial parasite Onchocerca volvulus and Its Wolbachia endosymbiont. mBio (2016) 7(6):e2028–2016. doi:10.1128/mBio.02028-16
80. Harris TW, Lee R, Schwarz E, Bradnam K, Lawson D, Chen W, et al. WormBase: a cross-species database for comparative genomics. Nucleic Acids Res (2003) 31(1):133–7. doi:10.1093/nar/gkg053
81. Tang YT, Gao X, Rosa BA, Abubucker S, Hallsworth-Pepin K, Martin J, et al. Genome of the human hookworm Necator americanus. Nat Genet (2014) 46(3):261–9. doi:10.1038/ng.2875
82. Ghedin E, Wang S, Spiro D, Caler E, Zhao Q, Crabtree J, et al. Draft genome of the filarial nematode parasite Brugia malayi. Science (2007) 317(5845):1756–60. doi:10.1126/science.1145406
83. Tallon LJ, Liu X, Bennuru S, Chibucos MC, Godinez A, Ott S, et al. Single molecule sequencing and genome assembly of a clinical specimen of Loa loa, the causative agent of loiasis. BMC Genomics (2014) 15:788. doi:10.1186/1471-2164-15-788
84. Cotton JA, Bennuru S, Grote A, Harsha B, Tracey A, Beech R, et al. The genome of Onchocerca volvulus, agent of river blindness. Nat Microbiol (2016) 2:16216. doi:10.1038/nmicrobiol.2016.216
85. Korhonen PK, Pozio E, La Rosa G, Chang BC, Koehler AV, Hoberg EP, et al. Phylogenomic and biogeographic reconstruction of the Trichinella complex. Nat Commun (2016) 7:10513. doi:10.1038/ncomms10513
86. Foth BJ, Tsai IJ, Reid AJ, Bancroft AJ, Nichol S, Tracey A, et al. Whipworm genome and dual-species transcriptome analyses provide molecular insights into an intimate host-parasite interaction. Nat Genet (2014) 46(7):693–700. doi:10.1038/ng.3010
87. Huang Y, Chen W, Wang X, Liu H, Chen Y, Guo L, et al. The carcinogenic liver fluke, Clonorchis sinensis: new assembly, reannotation and analysis of the genome and characterization of tissue transcriptomes. PLoS One (2013) 8(1):e54732. doi:10.1371/journal.pone.0054732
88. Young ND, Nagarajan N, Lin SJ, Korhonen PK, Jex AR, Hall RS, et al. The Opisthorchis viverrini genome provides insights into life in the bile duct. Nat Commun (2014) 5:4378. doi:10.1038/ncomms5378
89. Cwiklinski K, Dalton JP, Dufresne PJ, La Course J, Williams DJ, Hodgkinson J, et al. The Fasciola hepatica genome: gene duplication and polymorphism reveals adaptation to the host environment and the capacity for rapid evolution. Genome Biol (2015) 16:71. doi:10.1186/s13059-015-0632-2
90. Berriman M, Haas BJ, LoVerde PT, Wilson RA, Dillon GP, Cerqueira GC, et al. The genome of the blood fluke Schistosoma mansoni. Nature (2009) 460(7253):352–8. doi:10.1038/nature08160
91. Protasio AV, Tsai IJ, Babbage A, Nichol S, Hunt M, Aslett MA, et al. A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni. PLoS Negl Trop Dis (2012) 6(1):e1455. doi:10.1371/journal.pntd.0001455
92. Tsai IJ, Zarowiecki M, Holroyd N, Garciarrubio A, Sanchez-Flores A, Brooks KL, et al. The genomes of four tapeworm species reveal adaptations to parasitism. Nature (2013) 496(7443):57–63. doi:10.1038/nature12031
93. Bremel RD, Homan EJ. An integrated approach to epitope analysis I: dimensional reduction, visualization and prediction of MHC binding using amino acid principal components and regression approaches. Immunome Res (2010) 6:7. doi:10.1186/1745-7580-6-7
94. Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res (2014) 42(Database issue):D581–91. doi:10.1093/nar/gkt1099
95. Aurrecoechea C, Barreto A, Basenko EY, Brestelli J, Brunk BP, Cade S, et al. EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Res (2017) 45:D581–91. doi:10.1093/nar/gkw1105
96. Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res (2015) 43(Database issue):D405–12. doi:10.1093/nar/gku938
97. Johnson NL. Systems of frequency curves generated by methods of translation. Biometrika (1949) 36(Pt 1–2):149–76. doi:10.2307/2332539
98. Crow JF. Perspective: here’s to fisher, additive genetic variance, and the fundamental theorem of natural selection. Evolution (2002) 56(7):1313–6. doi:10.1111/j.0014-3820.2002.tb01445.x
99. Lessard S. Fisher’s fundamental theorem of natural selection revisited. Theor Popul Biol (1997) 52(2):119–36. doi:10.1006/tpbi.1997.1324
100. Bremel RD, Homan EJ. Recognition of higher order patterns in proteins: immunologic kernels. PLoS One (2013) 8(7):e70115. doi:10.1371/journal.pone.0070115
101. Kall L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol (2004) 338(5):1027–36. doi:10.1016/j.jmb.2004.03.016
102. Gilabert A, Curran DM, Harvey SC, Wasmuth JD. Expanding the view on the evolution of the nematode dauer signalling pathways: refinement through gene gain and pathway co-option. BMC Genomics (2016) 17:476. doi:10.1186/s12864-016-2770-7
103. Viney M. The genomic basis of nematode parasitism. Brief Funct Genomics (2018) 17(1):8–14. doi:10.1093/bfgp/elx010
104. Flanagan KL, Wilson KL, Plebanski M. Polymorphism in liver-stage malaria vaccine candidate proteins: immune evasion and implications for vaccine design. Expert Rev Vaccines (2016) 15(3):389–99. doi:10.1586/14760584.2016.1125785
105. Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today (1998) 19(9):395–404.
106. Sprouse ML, Shevchenko I, Scavuzzo MA, Joseph F, Lee T, Blum S, et al. Cutting edge: low-affinity TCRs support regulatory T cell function in autoimmunity. J Immunol (2018) 200(3):909–14. doi:10.4049/jimmunol.1700156
107. Sprouse ML, Scavuzzo MA, Blum S, Shevchenko I, Lee T, Makedonas G, et al. High self-reactivity drives T-bet and potentiates Treg function in tissue-specific autoimmunity. JCI Insight (2018) 3(2):97322. doi:10.1172/jci.insight.97322
108. Petrova GV, Gorski J. Cross-reactive responses to modified M1(5)(8)-(6)(6) peptides by CD8(+) T cells that use noncanonical BV genes can describe unknown repertoires. Eur J Immunol (2012) 42(11):3001–8. doi:10.1002/eji.201242596
109. Belkaid Y, Tarbell K. Regulatory T cells in the control of host-microorganism interactions. Annu Rev Immunol (2009) 27:551–89. doi:10.1146/annurev.immunol.021908.132723
110. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol (2008) 9(3):239–44. doi:10.1038/ni1572
111. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol (2014) 15(11):1070–8. doi:10.1038/ni.3004
112. Delogu G, Brennan MJ. Comparative immune response to PE and PE_PGRS antigens of Mycobacterium tuberculosis. Infect Immun (2001) 69(9):5606–11. doi:10.1128/IAI.69.9.5606-5611.2001
113. Chatrath S, Gupta VK, Dixit A, Garg LC. PE_PGRS30 of Mycobacterium tuberculosis mediates suppression of proinflammatory immune response in macrophages through its PGRS and PE domains. Microbes Infect (2016) 18(9):536–42. doi:10.1016/j.micinf.2016.04.004
114. Saito F, Hirayasu K, Satoh T, Wang CW, Lusingu J, Arimori T, et al. Immune evasion of Plasmodium falciparum by RIFIN via inhibitory receptors. Nature (2017) 552(7683):101–5. doi:10.1038/nature24994
115. Goel S, Palmkvist M, Moll K, Joannin N, Lara P, Akhouri RR, et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat Med (2015) 21(4):314–7. doi:10.1038/nm.3812
116. Sim KL, Creamer TP. Abundance and distributions of eukaryote protein simple sequences. Mol Cell Proteomics (2002) 1(12):983–95. doi:10.1074/mcp.M200032-MCP200
117. Haerty W, Golding GB. Low-complexity sequences and single amino acid repeats: not just “junk” peptide sequences. Genome (2010) 53(10):753–62. doi:10.1139/G10-063
118. Plaza S, Menschaert G, Payre F. In search of lost small peptides. Annu Rev Cell Dev Biol (2017) 33:391–416. doi:10.1146/annurev-cellbio-100616-060516
119. Couso JP, Patraquim P. Classification and function of small open reading frames. Nat Rev Mol Cell Biol (2017) 18(9):575–89. doi:10.1038/nrm.2017.58
120. Morais SB, Figueiredo BC, Assis NRG, Homan J, Mambelli FS, Bicalho RM, et al. Schistosoma mansoni SmKI-1 orIts C-terminal fragment induces partial protection against S. mansoni infection in mice. Front Immunol (2018) 9:1762. doi:10.3389/fimmu.2018.01762
121. Jang SW, Cho MK, Park MK, Kang SA, Na BK, Ahn SC, et al. Parasitic helminth cystatin inhibits DSS-induced intestinal inflammation via IL-10(+)F4/80(+) macrophage recruitment. Korean J Parasitol (2011) 49(3):245–54. doi:10.3347/kjp.2011.49.3.245
122. Sun Y, Liu G, Li Z, Chen Y, Liu Y, Liu B, et al. Modulation of dendritic cell function and immune response by cysteine protease inhibitor from murine nematode parasite Heligmosomoides polygyrus. Immunology (2013) 138(4):370–81. doi:10.1111/imm.12049
Keywords: helminth, immunomodulation, bioinformatics, T regulatory, networking, T-cell-exposed motifs
Citation: Homan EJ and Bremel RD (2018) A Role for Epitope Networking in Immunomodulation by Helminths. Front. Immunol. 9:1763. doi: 10.3389/fimmu.2018.01763
Received: 15 April 2018; Accepted: 17 July 2018;
Published: 31 July 2018
Edited by:
Wenbin Tuo, Agricultural Research Service (USDA), United StatesReviewed by:
Susanne Hartmann, Freie Universität Berlin, GermanyPeter C. Thompson, Agricultural Research Service (USDA), United States
Copyright: © 2018 Homan and Bremel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: E. Jane Homan, amFuZV9ob21hbkBlaWdlbmJpby5jb20=