 Johan N. Siebert1,2*
Johan N. Siebert1,2* Lutz Hamann3
Lutz Hamann3 Charlotte M. Verolet4Cécile Gameiro5Stéphane Grillet2
Charlotte M. Verolet4Cécile Gameiro5Stéphane Grillet2 Claire-Anne Siegrist2,4,6
Claire-Anne Siegrist2,4,6 Klara M. Posfay-Barbe4,6
Klara M. Posfay-Barbe4,6
- 1Department of Pediatric Emergency Medicine, Geneva Children’s Hospital, Geneva University Hospitals, Geneva, Switzerland
- 2Department of Pathology-Immunology and Pediatrics, Center for Vaccinology and Neonatal Immunology, Geneva Medical Center, Geneva, Switzerland
- 3Institute of Medical Microbiology and Hygiene, Charité-University Medical Center Berlin, Berlin, Germany
- 4Department of Pediatrics, Geneva Children’s Hospital, Geneva University Hospitals, Geneva, Switzerland
- 5Flow Cytometry Core Facility, Geneva University Medical Center, Geneva, Switzerland
- 6Faculty of Medicine, University of Geneva, Geneva, Switzerland
Lower respiratory tract infections (LRTI) are often caused by Streptococcus pneumoniae (Spn) and can be recurrent in 8% of children older than 2 years of age. Spn is recognized by pattern-recognition receptors (PRRs) of the innate immune system, in particular toll-like receptors (TLRs) 2 and 4. To assess whether a defect somewhere along this TLR signaling pathway increases susceptibility to recurrent pneumococcal LRTI, we conducted a prospective case–control study with 88 healthy individuals and 45 children with recurrent LRTI aged 2–5 years old. We examined cell surface expression of TLR2 and TLR4, as well as eight genetic variants of these receptors or associated co-receptors TLR1 and TLR6. Interleukin-6 production was measured after whole blood stimulation assays with specific agonists and heat-killed Spn. Our findings reveal that single-nucleotide polymorphisms within toll-interleukin 1 receptor domain-containing adaptor protein (TIRAP) alone or in combination with TLR1 N248S, TLR1 I602S, or TLR6 S249P polymorphisms contributes to various degree of susceptibility to recurrent pneumococcal LRTI in children by modulating the inflammatory response. In that respect, carriage of the TIRAP S180L heterozygous trait increases the likelihood to protect against pneumococcal LRTI, whereas children carrying the mutant homozygous TIRAP 180L polymorphism might be more likely susceptible to recurrent pneumococcal LRTI.
Introduction
Lower respiratory tract infections (LRTI), such as pneumonia and bronchitis are major causes of hospitalization in young children less than 5 years old, and are associated with significant morbidity and mortality (1, 2). Streptococcus pneumoniae (Spn) is the most common bacterial cause of community-acquired pneumonia (CAP) in childhood (3–6). Despite an early and high prevalence of nasopharyngeal pneumococcal colonization of the human upper respiratory tract, the immature immune system of young children responds poorly to the polysaccharidic antigens of Spn (7). This is reflected by an incidence of invasive pneumococcal disease (IPD) as high in young infants as in the elderly (8). After the second year of life, Spn infections become much rarer, which theoretically reflects immune maturation (8). However, approximately 8% of children older than 2 years of age hospitalized for CAP are recognized as having recurrent LRTI (9), of which a significant proportion is caused by Spn (10–14). While most children with recurrent infections probably have a normal immunity, it is important to recognize, and treat appropriately those with an underlying transient or permanent immune dysfunction.
The innate immune system is crucial for the control of colonization and for early recognition of invasive microorganisms to initiate an immediate protective response (15). Spn recognition starts at the cell surface of human monocytes through the pattern-recognition receptors (PRRs) family. Among them, the transmembrane glycoproteins toll-like receptors (TLRs) play a pivotal role (16). Upon this recognition, the TLRs regulates downstream intracellular MyD88-dependent and TRIF-dependent signaling pathways involving several adaptor proteins, including toll-interleukin 1 receptor domain-containing adaptor protein (TIRAP) (17). This activates transcription factors and results in the production of a diverse array of inflammatory mediators, such as interleukin-1 (IL-1), interleukin-6 (IL-6), and type I interferon. Previous studies have shown that TLR signaling defects can impair the innate immune responses and are associated with greatly enhanced susceptibility to invasive life-threatening infections (18–21). However, to the best of our knowledge, this has not been studied in young children with recurrent pneumococcal LRTI.
In this study, we investigate whether an abnormal or delayed maturation pattern of the innate immune response to pneumococcal pathogen-associated molecular patterns was present in young children with recurrent LRTI along the TLR signaling pathways. To achieve this, we explore the TLR2 and TLR4 monocytes’ cell surface expressions that are mainly involved in the host immune response against Spn (16). We also examine the influence of loss-of-function genetic variants in eight single and combined nucleotide polymorphisms (SNPs) in the corresponding TLRs, or their associated co-receptors, as well as in microRNA-146 reported to be a regulator of TLR4 expression and inflammatory response (22). The findings are then correlated with functional assays investigating the SNP’s influence on the individual’s ability to produce an inflammatory response through IL-6 production after whole blood stimulation by specific TLR2 and TLR4 ligands. We determine their role in susceptibility to recurrent pneumococcal pneumonia in children.
Materials and Methods
This prospective case–control study was conducted in a tertiary hospital with 30,000 outpatient consultations per year. The study was approved by the institutional ethics committee, and written consent was obtained from all parents or legal guardians. It was conducted in accordance with the principles of the Declaration of Helsinki, the standards of Good Clinical Practice, and Swiss regulatory requirements. Patients’ records were anonymized and de-identified before analysis. Health-related information, genetic data, and biological materials were stored in secured places at the Geneva University Medical Center and Geneva University Hospitals. They were at the disposition of the investigators for the sole purpose of the study. Only small samples of blood consistent with physiological minimal risk were harvested from children. Residual blood was destroyed.
Patients and Healthy Subjects
The patients were previously described (14). Briefly, cases were children between 2 and 5 years of age with recurrent LRTI, defined as having had two or more episodes of pneumonia in a single year or at least three episodes (12). The initial diagnosis had to be either confirmed clinically according to the WHO criteria (23) and/or by radiology, with complete clinical resolution between occurrences. Patients were referred through their pediatrician, the infectious diseases or the pulmonology outpatient clinic, the laboratory of vaccinology, or recruited at time of hospitalization for repeated pneumonia. Exclusion criteria were as follows: anatomic anomalies of the respiratory tract, primary ciliary dyskinesia, cystic fibrosis, foreign body in the respiratory tract, asthma or severe atopic disease, known congenital or acquired immunodeficiencies, lymphopenia, or intravenous immunoglobulin substitution therapy. Cases were followed prospectively and had blood samples on a yearly basis. Samples were taken outside of acute infectious episodes to avoid measurement of related cellular changes or inflammation. Healthy controls were age-matched children referred for elective surgery not related to the lungs, who provided a single blood sample at enrollment. They had no history of LRTI and had been immunized against Spn, in accordance with the Swiss recommendations.
Blood Sampling and Leukocytes Isolation
Fresh peripheral blood (PB) samples were placed into sterile heparinized-tubes (BD Vacutainer, Becton Dickinson, Franklin Lakes, NJ, USA) and processed on the day of collection. Plasma was isolated by centrifugation (1,500 rpm, 5 min, 4°C) and stored at −80°C. Twenty microliters of naïve mouse serum were added to 100 µl of whole blood to prevent Fc-mediated interactions, and incubated at room temperature (RT), in the dark. After 5 min, 20 µl of isotype control, anti-TLR2, or anti-TLR4 monoclonal antibodies was added to the blood samples. After five additional minutes, 20 µl of anti-CD14 monoclonal antibodies was added to each sample and incubated for another 20 min, at RT in the dark. Then, cells were washed once with PBS 0.1% sodium azide and centrifuged (1,500 rpm, 5 min, RT). Supernatant was decanted, and the contaminating erythrocytes were lysed by resuspending the cell pellet with 3 ml pH 7.4 BD Pharm Lyse™ lysing solution diluted in sterile pyrogen-free H2O (1:10). The samples were incubated 15 min, in the dark, at RT followed by washing twice with FACS Buffer (prepared by adding 50 µl NaN3 + 0.5 g BSA to 50 ml PBS 0.1% sodium azide) and centrifuged (1,500 rpm, 5 min, 4°C). Supernatant was decanted, and the cell pellet resuspended in 100 µl of FACS buffer and transferred into sterile Falcon 96-well Microplate U-bottom (BD Biosciences, VWR, Switzerland). Finally, the plate was centrifuged, supernatant was decanted, and the cell pellet resuspended by 120 µl of FACS buffer before going to the cytofluorometer. All experiments were conducted on the day of blood collection.
Flow Cytometry Analysis
PBMC were stained with appropriate combinations of phycoerythrin (PE) and allophycocyanin (APC) monoclonal antibodies. Optimal working dilutions of the antibodies were assessed in titration assays. PE anti-human TLR2 mAb (clone TL2.1, mouse IgG2a κ, RRID:AB_10667887), PE anti-human TLR4 mAb (clone HTA125, mouse IgG2a κ, RRID:AB_1603311), PE isotype control (clone eBM2a, mouse IgG2a κ, RRID:AB_470063), and APC anti-human CD14 mAb (clone 61D3, mouse IgG1, RRID:AB_10669167) were all purchased from eBioscience (THP Medical Products, Switzerland). Cells were all analyzed using a fluorescence-activated cell sorting FACSArray flow cytometer (BD Biosciences, San Diego, CA, USA) interfaced with BD FACSDIVA™ software (v.6.1.3, BD Biosciences). Side/forward scatter dot plot was used to exclude dead cells and to gate the live monocyte population. To refine the recognition of TLR expression on monocytes, double staining for CD14 and TLR2, CD14 and TLR4, or CD14 and IgG2a control isotype was used. The expression of these surface markers was calculated as median fluorescence intensity (MFI) in non-overlapping Yellow-A (PE fluorochrome) and Red-A channels (APC fluorochrome) and recorded for each patient.
We also used flow cytometry in a pilot experiment to determinate which pro-inflammatory cytokine would best reflect the TLR-induced inflammatory response following whole blood stimulation assay. Thanks to a multiplex cytometric bead array (CBA) kit (Human Inflammation Kit; BD Biosciences Pharmingen, San Diego, CA, USA), we simultaneously quantified IL-6, IL-8, IL-1β, IL-10, IL-12 p70, and tumor necrosis factor-α (TNF-α) production. This CBA kit consists of a mix of six microbead populations dyed to six different fluorescence intensities. Each microbead is coated with a monoclonal antibody against a given cytokine. Following incubation with the test sample, the bead-captured cytokines are simultaneously detected and quantified by direct immunoassay using six different antibodies labeled with phycoerythrin (PE). Our test sample was supernatant collected after centrifugation (1,000 rpm, 5 min, 4°C) of healthy human whole blood previously stimulated and incubated during 6 h with Ultrapure E. coli K12 LPS (LabForce, Switzerland). We found that IL-6 was the fastest and most discriminative cytokine in the supernatant. The experiment was performed according to the manufacturer’s instructions using the 4-colors FACSArray flow cytometer. Standard curves were generated for each cytokine using the standard provided in the kit. Each cytokine concentration within the supernatant was interpolated from these standard curves.
In Vitro Whole Blood Functional Assays
We performed measurements of cellular in vitro responses to four TLR agonists and to Spn in whole blood functional assays. It has been shown that TLR function, as assessed by the production of IL-6, is stable between birth and 60 years old (24) and that whole blood is the best way to study in vitro cytokine production (25). Because these are more strongly correlated with monocytes than cytokines from purified PBMC (26). In most studies, however, in vitro cytokine production has been studied with mononuclear cells isolated from PBMC. This method suppresses the complex physiological interactions normally expected between immune cells and mediators and does not faithfully reflect the natural milieu (24). All procedures were performed in sterile conditions using sterile instruments and fluids. Fresh PB samples were obtained and placed into heparinized syringes. On collection day (maximum 1 h delay), blood was diluted 1:2 in a reservoir (Corning Life Science Costar® reagent reservoirs, Vitaris AG, Switzerland) with heparinized culture medium RPMI 1640 with GlutaMAX™ I, 25 mM HEPES (Invitrogen, Switzerland) pre-treated with 50 µg/ml gentamicin (Sigma-Aldrich, Switzerland). Blood was plated at 60 µl/well in a 96-well U-bottom microplate (Nunc, Milian, Switzerland) and incubated at 37°C for 15 min with 5% CO2. The following TLR agonists were used at optimal concentrations that elicited cytokine responses as preliminarily determined by dose–response curves: (1) Ultrapure E. coli K12 LPS (200 ng/ml final), (2) Pam3CSK4 (10 µg/ml final), and (3) Muramyl Dipeptide (MDP, 200 µg/ml final) were all purchased from InvivoGen (LabForce, Switzerland). MDP (200 µg/ml final), which is recognized by the nucleotide-binding oligomerization domain (NOD2) protein immune receptor, was also used in combination with Pam3CSK4 (20 µg/ml final). (4) TLA4e/AF04 (400 ng/ml final) was kindly provided by Sanofi Pasteur. TLA4e is constituted with an oil-in-water emulsion as described in WO 07/006939 and the Eisai product ER 804057 (also known as E6020, described in US 7,683,200) which is a TLR4 agonist. In addition, 1 × 108 cfu/ml heat-killed Spn were used (see below). Each agonist was serially diluted 1:2 (eight dilutions available) with filtered (Steriflip Millipore 0.22 µm, Milian, Switzerland) RPMI 1640 supplemented with 0.25% human serum albumin 20% (CSL Behring AG, Switzerland), and 60 µl of each sample was added 1:2 to individual blood wells in duplicate. Non-stimulated blood (with RPMI alone) was used to measure the unspecific negative control. After 6 h of incubation at 37°C with 5% CO2, supernatants were collected after centrifugation (1,000 rpm, 5 min, 4°C), and IL-6 was measured by enzyme-linked immunosorbent assay (ELISA).
Preparation of Heat-Killed Spn
Streptococcus pneumoniae, serotype 22, was obtained from our hospital’s bacterial culture collection. Modified from a previous protocol (27), colonies were incubated overnight at 37°C on a Columbia AGAR (sheep blood 5%) plate with 5% CO2. Colonies were used to inoculate 10 ml of brain heart infusion (without AGAR). The inoculum was incubated for 16 h at 37°C and added to 1,000 ml of the same media. Bacteria were grown to logarithmic phase growth. To confirm that the bacterial growth has reached log phase, we used a spectrometer and read the culture broth at 600 nm to fall between OD600 = 0.4–0.6. Bacteria were stopped from growing further by incubating at 4°C. After centrifugation at 6,000 g for 10 min at 4°C, the entire bacteria pellet was washed two times with PBS and killed by incubation at 70°C for 1 h. Killing was confirmed by cultivation on sheep blood agar plates, and the presence of Spn was confirmed by microscopic examination and optochin inhibition test. Bacteria were stored at −80°C in 20% glycerol/80% PBS.
IL-6 Enzyme-Linked Immunosorbent Assay
Interleukin-6 was measured by performing an ELISA using 96-well flat-bottom plates (Nunc Maxisorp™, Milian, Switzerland), previously coated with 1.01 µg/ml purified mouse anti-human IL-6 (clone 5IL6, IgG1, κ) mAb (Thermo Fisher Scientific, Perbio Science, Switzerland). On the day of experiments, plates were washed four times with PBS/0.05% Tween 20 and then saturated with PBS/0.05% Tween 20/16% human serum albumin (Sigma-Aldrich, Switzerland) during 1 h at 37°C. Then, 0.5 µg/ml of mouse anti-human IL-6 biotinylated (clone 7IL6, IgG1) mAb (Thermo Fisher Scientific, Perbio Science, Switzerland) was added in each well (50 µl/well). The first International standard IL-6 reference (NIBSC no. 89/548) was added into the plate by using serial dilutions 1:2 (from 20 ng/ml to 156 pg/ml final, 50 µl/well). Supernatants obtained after whole blood stimulation assays were added to the remaining wells (50 µl/well), and the samples were incubated 2 h at 37°C in the dark. Two blank columns served as negative controls. Plates were then washed four times with PBS/0.05% Tween 20 and incubated 1 h at 37°C with 1:1,000 ExtrAvidin 50 µl/well (Sigma-Aldrich, Switzerland). Plates were again washed four times and were developed using ABTS (2,2′-azino-bis[3-ethylbenzothiazoline-6-sulfonic acid]-diammonium salt) as a substrate, plus 10 μl H2O2 after 30 min of incubation. The optical density was determined at a wavelength of 405 nm using a Spectramax plate reader interfaced with a SoftMax Pro software (Molecular Devices, Sunnyvale, CA, USA). All values were interpolated based on the standard IL-6 curve. IL-6 concentrations were expressed as picograms per milliliter and normalized for 104 monocytes (monocytes blood count was determined by flow cytometry); the values were logarithmically (log10) transformed.
DNA Preparation and Genotyping
Genomic DNA was prepared by standard procedures from whole blood. Genotyping for TLR1 (rs4833095, rs5743618), TLR2 (rs5743708), TLR4 (rs4986790, rs4986791), TLR6 (rs5743810), TIRAP (rs8177374), and miR-164a (rs2910164) was performed by PCR including fluorescence-labeled hybridization FRET probes followed by melting curve analysis employing the LightCycler 480 (Roche Diagnostics). Primer and probes used in this study are shown in Table 1. Details have been published previously (28–33).

Table 1. Primers and probes used in this study.
Statistical Analysis
Data were displayed as arithmetic mean and 95% CI or median and interquartile ranges (IQRs). Hardy–Weinberg equilibrium was examined using a goodness-of-fit χ2 test with 1 degree of freedom to compare the observed and expected genotype distributions among the study subjects. Association between SNPs and risk of recurrent LRTI were tested using four common genetic models of inheritance (codominant, dominant, recessive, and overdominant) analysis by multiple logistic regressions to obtain odds ratios (ORs), 95% confidence intervals (CIs), and P-values. Codominant1 [homozygous wild type (WT) versus heterozygous (HT)], codominant2 (WT versus homozygous mutant (HM)), codominant3 (HT versus HM), dominant (WT versus HM + HT), recessive (HM versus WT + HT), and overdominant (HT versus WT + HM) models were assessed (34). All variables with a P-value lower than 0.2 in the univariate models were used in multivariate logistic models to adjust our result for gender and origin. Akaike’s Information Criterion was applied to estimate the best-fit model for each SNP; the model with the least AIC value was the most probable. Pairwise linkage disequilibrium (LD), D′, and Pearson’s r correlation were calculated using a web-based software for SNP analysis (http://bioinfo.iconcologia.net/SNPstats/). Based on associated biological TLR pathways and on the results of the single gene associations, we also tested the joint effects of combinations of SNPs pairs. For group comparisons, Student’s t-test or Wilcoxon test was applied to compare continuous variables, depending on their distribution. The Anderson–Darling test for normality was used to assess the IL-6 distribution. Chi-square and Fisher’s exact tests were used for dichotomic variables. Linear regressions were used to analyze associations between variables. Data analyses were performed on STATA (version IC 14.0; StataCorp LP, College Station, TX, USA).
Results
Patient’s Characteristics
Forty-five children with recurrent pneumonia and 88 healthy controls aged 2–5 years were enrolled. The mean age (SD) was 42.7 (13.0) and 44.3 (11.9) months, respectively (P = NS). Age categories were arbitrarily distributed in two groups: group 1 (24–48 months) and group 2 (48–72 months). Among the 45 LRTI children, 24 were males and 21 were females. Among the 88 healthy children, there was a gender predominance of 54 males over 34 females. This predominance was the result of a selection bias (elective surgery for circumcision). Ethnicities were equally distributed among children, with 76% of LRTI children and 73% of healthy children being Caucasians. Among the remaining LRTI children, 6 (13%) were of mixed origins, 2 (4%) were Middle Eastern, 2 (4%) were South American and 1 (2%) was from Sri Lanka.
Hardy–Weinberg Equilibrium
Genotype distributions of TLR1 (rs4833095 and rs5743618), TLR2 (rs5743708), TLR4 (rs4986790 and rs4986791), TLR6 (rs5743810), and miR-146a (rs2910164) SNPs were consistent with Hardy–Weinberg equilibrium among both healthy and LRTI children (data not shown). TIRAP (rs8177374) SNP was not in Hardy–Weinberg equilibrium, both in LRTI and healthy children (P < 0.0001).
TLR Genotyping Reveals Homozygous TIRAP S180L Polymorphism Predominance Traits in LRTI Children
Wild-type (WT), heterozygous (HT), and homozygous (HM) traits were compared between healthy and LRTI children for eight SNPs. The results are summarized in Table 2. The differences in the number of patients screened for SNPs either in healthy or LRTI children were due to not usable DNA. Compared with WT, carriage of the TIRAP heterozygous trait in the codominant model was higher in the healthy population than in LRTI children (24.7 versus 7.3%, respectively, OR 0.27 [95% CI 0.07–1.01], P = 0.03). Compared with HM, carriage of the TIRAP HT trait was also predominantly observed in the healthy children (OR 0.18 [95% CI 0.045–0.74], P = 0.017). This association was also seen in the overdominant model, where the HT trait was more observed in the healthy population (P = 0.013). On the other hand, in the codominant model, the HM trait was more observed in the LRTI population when compared with HT (OR 5.44 [95% CI 1.35–22.01], P = 0.017). When compared with WT, carriage of the TIRAP HM trait was also higher in the LRTI children but without reaching significance (P = 0.36). No associations were observed between all the other single SNPs studied and recurrent pneumococcal LRTI. Neither gender nor origins have shown any influence on the results. The allelic frequencies compared between both healthy and LRTI children were not significant.
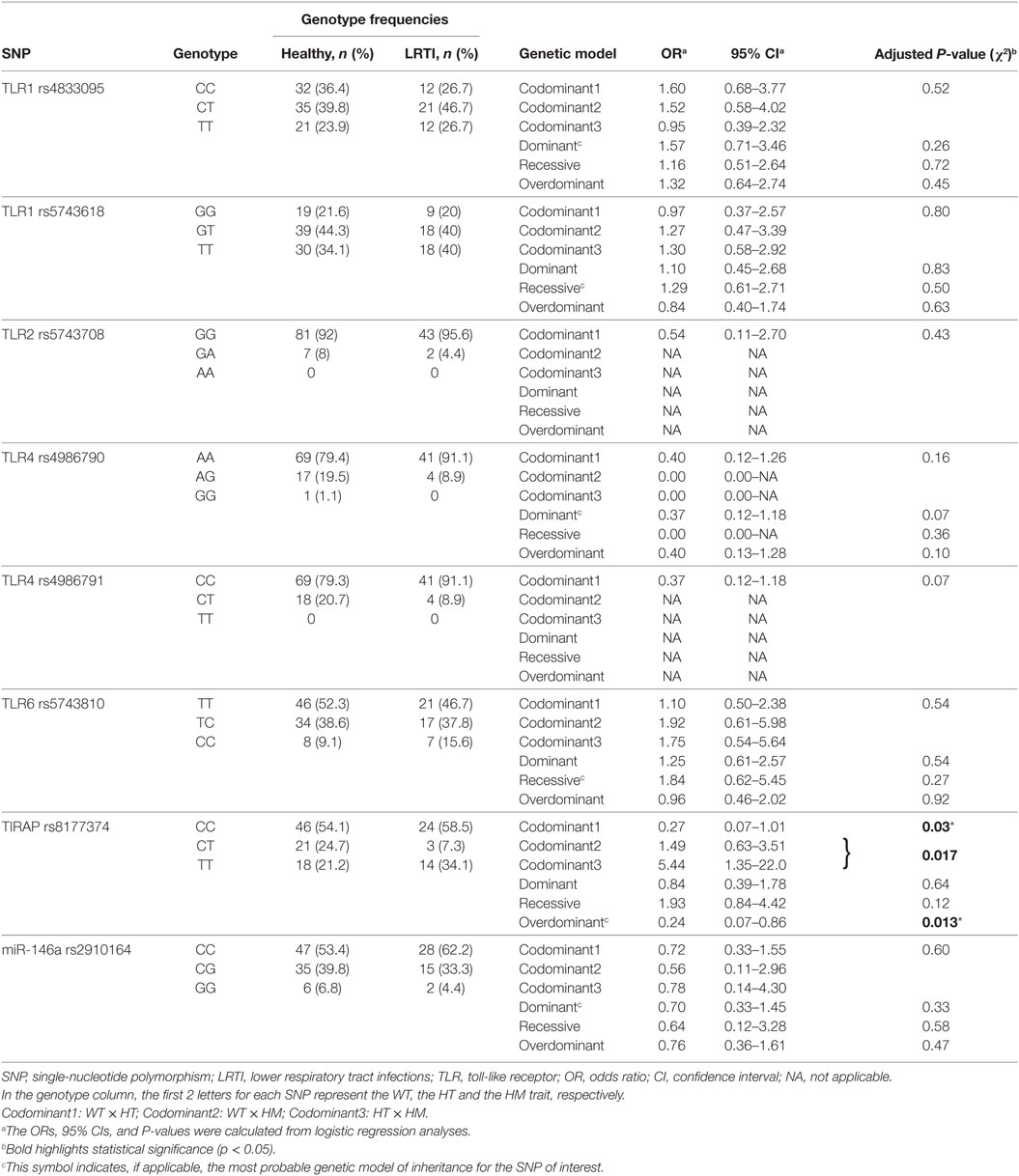
Table 2. Relationship between TLR single-nucleotide polymorphisms and recurrent LRTI in children.
TLR2 Overexpression in Children With Recurrent Pneumococcal LRTI, and TLR4 Downregulation in Those Carrying the Mutant Homozygous TIRAP 180L Polymorphism
TLR2 expression on monocytes was significantly higher in LRTI compared with healthy children, although this was a fairly moderate increase (Table 3). Stratified into two age groups (i.e., children <3 years old or children ≥3 years old) this higher TLR2 expression concerned LRTI children older than 3 years of age (P = 0.044) but did not reached significance for younger children (Table 3). Overall, there were no significant differences between the TLR4 MFI in healthy and LRTI children, even when stratified by age groups (Table 3). We next asked if the cell surface expression of TLR2 on monocytes was comparable to TLR4. TLR2 MFI were always higher than TLR4 in both healthy and LRTI children (P < 0.001) in all age groups. Based on a simple regression model, we computed correlation coefficients between both TLRs and found that TLR2 and TLR4 expressions were strongly correlated in LRTI children (Pearson r = 0.64 [95% CI 0.43–0.78], P < 0.0001) and moderately in healthy children (Spearman r = 0.52 [95% CI 0.17–0.75], P = 0.0045). The seven TLR1, TLR2, TLR4, TLR6, and miR-146a polymorphisms assessed above were not associated with changes of TLR2 and TLR4 expression levels at the cell surface of monocytes. Only HM LRTI children for the TIRAP variant showed a lower expression of TLR4 [median: 396.5 (IQR: 376.5–426.5)] compared with homozygous carriers of the wild-type TIRAP trait [438.0 (419.0–455.0), P = 0.028]. TLR2 and TLR4 expression did not vary with gender.
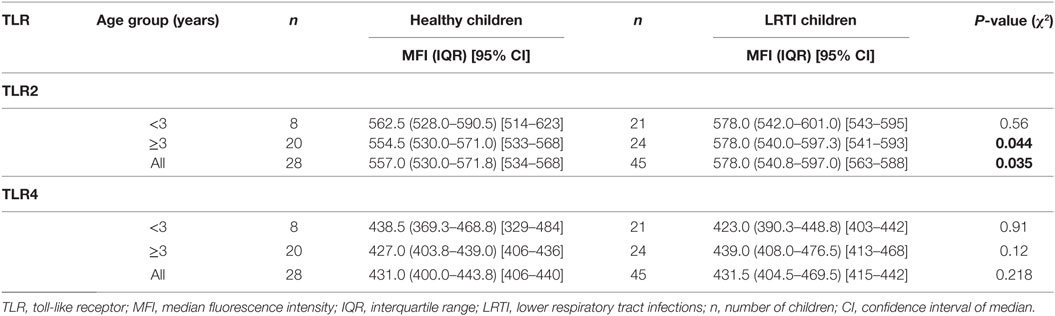
Table 3. TLR-2 and TLR-4 expression on monocytes in peripheral blood.
Increased IL-6 Release in LRTI Children After Whole-Blood Stimulation by Spn and TLR4 Ligands
To study functional response of TLR2 and TLR4, IL-6 secretion was measured as read-out for monocytes activation after stimulation by the corresponding TLR ligands: LPS, Pam3CSK4, Pam3CSK4 + MDP, TLA4e, and Spn (Figure 1). The amount of IL-6 secreted was significantly higher in LRTI than healthy children when stimulated with LPS (mean [95% CI]: 5,214.6 [3,852.7–6576.5] versus 3,591.4 [2,825.1–4357.7], P = 0.0087), TLA4e (5,982.8 [3,913.4–8052.3] versus 3,184.4 [1,112.8–5256.1], P = 0.019), and Spn (4,732.8 [3,425.0–6040.7] vs 1,870.0 [1,176.1–2563.8], P = 0.0004). No significant difference between LRTI and healthy children was observed in the overall amount of IL-6 secreted with Pam3CSK4 alone (1,937.4 [1,104.1–2770.7] versus 1,023.9 [761.0–1286.9]) or in combination with MDP (3,528.4 [2,126.2–4930.5] versus 2,562.9 [1,861.6–3264.2]). We found no correlation between IL-6 levels and TLR2 or TLR4 MFI. In particular, we found no association between IL-6 levels and TLR2 or TLR4 MFI after stimulation with seven serial dilutions of Spn ranging from 5 × 106 to 78 × 103 cfu/ml.
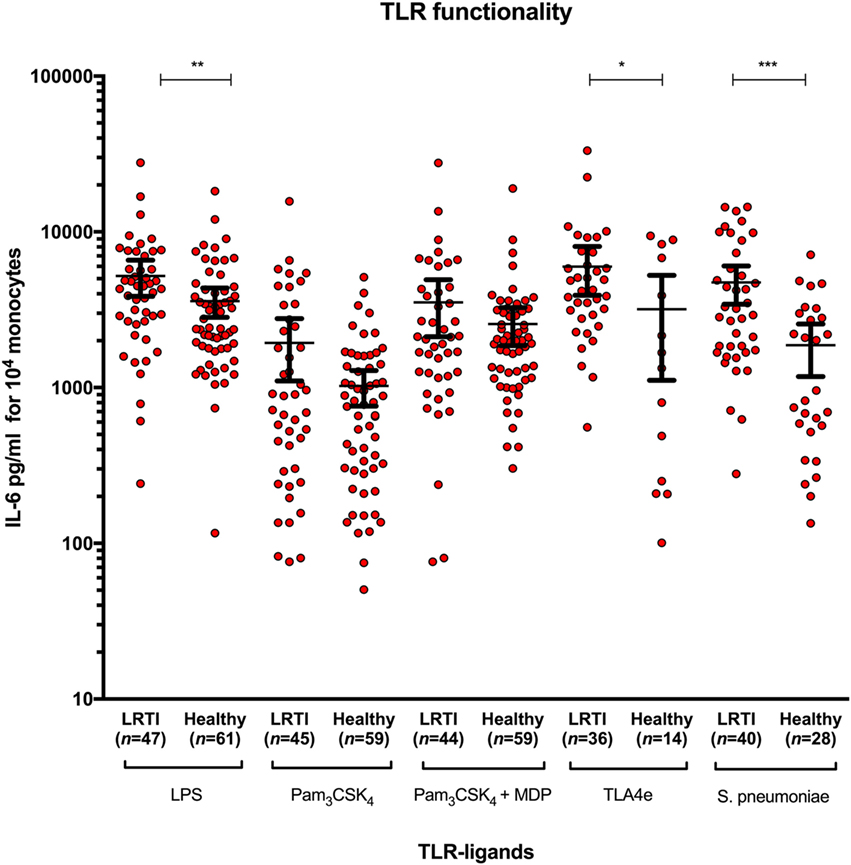
Figure 1. Increased IL-6 release in LRTI children after whole-blood stimulation by Streptococcus pneumoniae and TLR2 and TLR4 ligands. The diagram shows scatter dot plots of IL-6 secretion in LRTI and healthy children after whole blood stimulation by five TLR ligands. Data are presented as mean and 95% CI (black bars). IL-6 secretion is expressed in picograms per milliliter for 104 monocytes. *P < 0.05; **P < 0.01; and ***P < 0.001 by Mann–Whitney test. Abbreviations: TLR, toll-like receptor; LRTI, lower respiratory tract infections; n, number of children; IL-6, interleukin-6.
Increased Spn-Driven IL-6 Release in LRTI Children Expressing TIRAP Variants
We next investigated whether the susceptibility to LRTI was attributable to functional effects of different SNPs on IL-6 responses (Table S1 in Supplementary Material). In children with the TLR1 rs4833095 SNP (Figure S1A in Supplementary Material), the IL-6 production was significantly higher in both WT healthy and LRTI children than HM after either Pam3CSK4 or Pam3CSK4 + MDP stimulation. Similar results were obtained between HT and HM children. As expected, the synergistic effect of MDP combined with Pam3CSK4 showed increased levels of IL-6 production in all patients compared with Pam3CSK4 alone. Overall, there was no significant difference between LRTI and healthy children stimulated with Pam3CSK4 and Pam3CSK4 + MDP. Whole blood stimulation with heat-killed Spn yielded nearly significant increased IL-6 production in WT LRTI children compared with healthy children (P = 0.0502).
Regarding TLR1 rs5743618 SNP (Figure S1B in Supplementary Material), more IL-6 was produced in WT than in HM both in LRTI and healthy children after Pam3CSK4 stimulation, and between WT and HT healthy children. Pam3CSK4 + MDP stimulation induced increased IL-6 levels only in WT healthy children compared with HM. Similarly to TLR1 rs4833095 SNP, we noted for rs5743618 SNP a trend toward an increased IL-6 production after Spn stimulation in WT LRTI children compared with healthy children (P = 0.07). The difference was significant between HM LRTI and healthy children.
After stimulation with Spn, we observed increased IL-6 levels in WT LRTI compared with WT healthy children for TLR2 rs5743708, TLR4 rs4986790, and TLR4 rs4986791 (Figures S2A and 3A,B in Supplementary Material, respectively). When stimulated by Pam3CSK4, WT LRTI children with TLR6 rs5743810 SNP produced more IL-6 than HT and HM patients (Figure S2B in Supplementary Material). In addition, IL-6 tended to be higher in WT LRTI than in WT healthy children (P = 0.052). Pam3CSK4 + MDP showed only a difference between WT and HT LRTI children. Again, WT LRTI children produced more IL-6 than healthy children when challenged by Spn (Figure S2B in Supplementary Material).
Regarding TIRAP rs8177374 SNP (Figure 2), we found that LPS produced more IL-6 in WT LRTI than in healthy children (P = 0.011). Furthermore, TLA4e yielded more IL-6 in HM LRTI children (P = 0.0002). In addition, Spn produced more IL-6 in WT (P = 0.031) or HM LRTI children (P = 0.019) than healthy children.
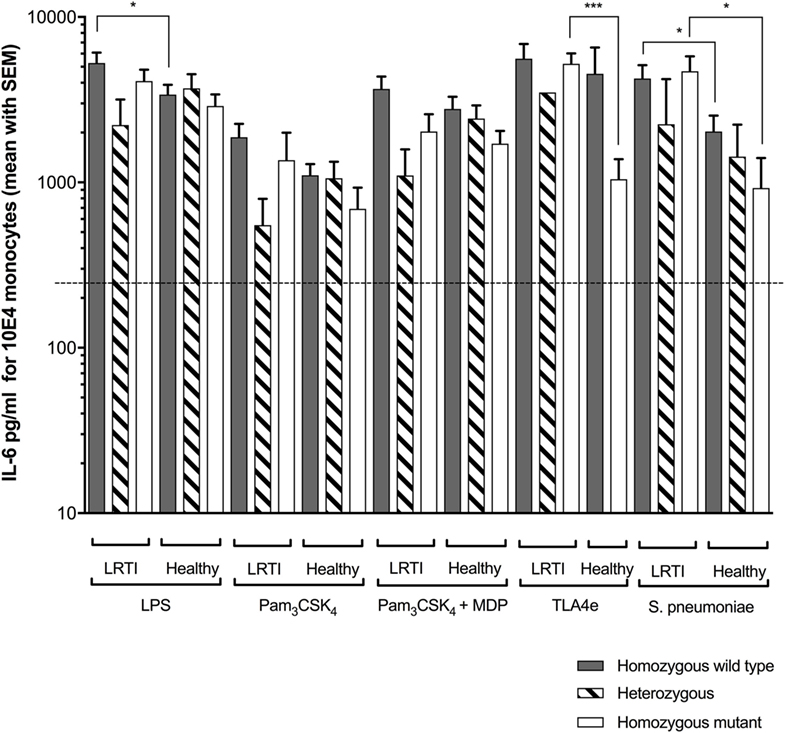
Figure 2. Increased Streptococcus pneumoniae (Spn)-driven IL-6 release in LRTI children expressing TIRAP variants. This figure shows the impact of TIRAP rs8177374 polymorphism on monocyte release of IL-6 following ex vivo whole blood stimulation in LRTI and healthy children by either E. coli K12 LPS (200 ng/ml final), Pam3CSK4 (10 µg/ml final), Pam3CSK4 (20 µg/ml final) + MDP (200 µg/ml final), TLA4e/AF04 (400 ng/ml final), or heat-killed Spn (1 × 108 cfu/ml). Gray histograms represent homozygous wild-type individuals, dashed histograms represent heterozygous, and white histograms represent homozygous mutant. Upper borders of histograms denote mean and solid horizontal lines denote SEM. The dashed lines represent the limit detection threshold (250 pg/ml). IL-6 secretion is expressed in picograms per milliliter for 104 monocytes. Number of children per histogram is provided in Table 4. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001 by Mann–Whitney test. Abbreviations: LRTI, lower respiratory tract infections; IL-6, interleukin-6.
Finally, with miR-146a rs2910164 SNP (Figure S4 in Supplementary Material), we found that IL-6 production was increased in HT LRTI children compared with WT LRTI children when stimulated by either Pam3CSK4 or Pam3CSK4 + MDP. We observed similar results in healthy children stimulated by Pam3CSK4 + MDP. Stimulation by TLA4e and Spn yielded more IL-6 production in HT LRTI than in HT healthy children. More IL-6 was produced in response to Spn in WT LRTI than in healthy children.
TLR4 Asp299Gly/Thr399Ile Haplotype Does Not Affect IL-6 Release in LRTI Children
Consistent with the 98% co-segregation level described in Caucasian populations (35), TLR4 Asp299Gly and Thr399Ile SNPs were, in our study, co-segregated in 44 of 45 LRTI children (97.8%) and in 86 of 87 healthy children (98.9%). IL-6 production was not influenced by the TLR4 haplotype with LPS or Spn stimulation. The IL-6 production was indeed similar between LRTI children with the double-homozygous wild-type haplotype (AA + CC) and the heterozygous haplotype (AG + CT) (Figure 3). Only healthy double WT children showed decreased levels of IL-6 compared with LRTI children when stimulated by Spn (mean [95% CI]: 4,444 [2,988–5,901] versus 1,917 [1,081–2,753], P = 0.0045).
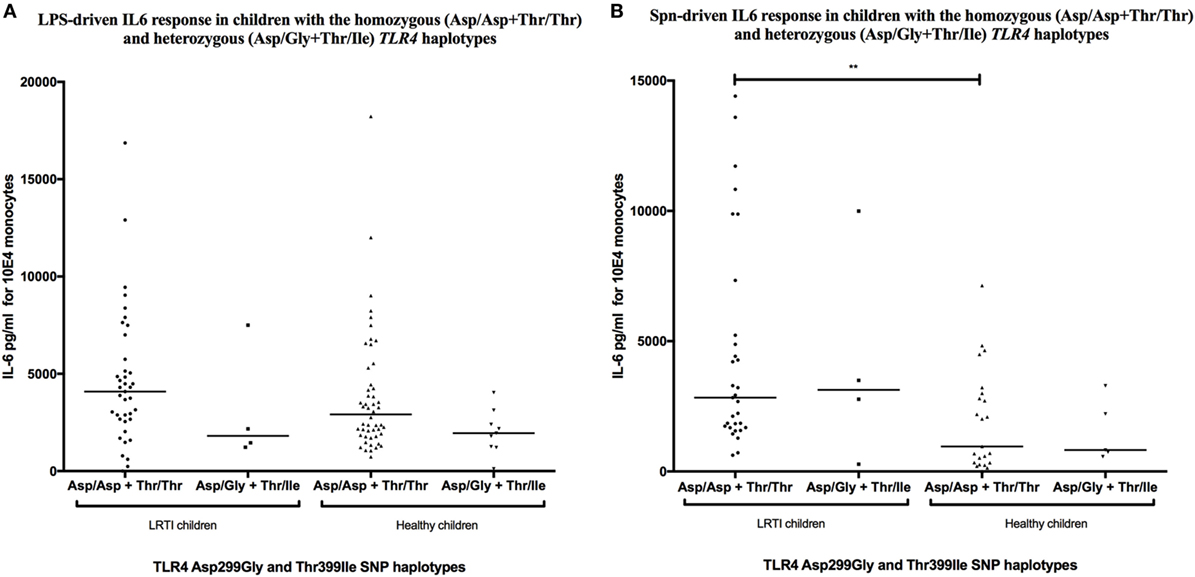
Figure 3. TLR4 Asp299Gly/Thr399Ile haplotype does not influence IL-6 release in LRTI children. This figure shows scatter dot plots of IL-6 secretion in TLR4 Asp299Gly and Thr399Ile haplotypes co-segregation after (A) LPS or (B) Streptococcus pneumoniae (Spn) stimulation. IL-6 secretion is expressed in picograms per milliliter for 104 monocytes. Solid line denotes mean. *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001 by Mann–Whitney test. Abbreviations: TLR, toll-like receptor; LRTI, lower respiratory tract infections; IL-6, interleukin-6.
Combined Nucleotide Polymorphisms in TLR1 or TLR6 and TIRAP Are Associated With a Decreased TLR2-Ligand-Driven IL-6 Release in LRTI Children
Combination of TLR4 and TIRAP SNPs has been described to be associated with low circulating levels of TNF-α and IL-6 in adult patients with ventilator-associated pneumonia following 1 ng/ml LPS-stimulation (36). We investigated in our study the frequency of this double mutation in LRTI children (genotypes distribution are summarized in Table 4) and whether it was similarly associated with low levels of IL-6. In our study, IL-6 circulating levels were not different in children with or without combined mutations in TLR4 and TIRAP genes after stimulation by 1.56 ng/ml LPS or 62,500 cfu of Spn (Figure 4A). After stimulation by Pam3CSK4, children with combined mutations in TLR1 rs4833095 or TLR1 rs5743618 and TIRAP exhibited a deficient IL-6 secretion compared with WT children (P < 0.001 and P < 0.01, respectively) or children carriers of only TLR1 or TIRAP polymorphisms (P < 0.05, Figures 4B,C). Similar results were found for combined mutations in TLR6 rs5743810 (P < 0.001) and TIRAP (P < 0.05, Figure 4D). Given that no LRTI children had TLR2 homozygous mutations, we were not able to similarly assess combination of TLR2 and TIRAP SNPs. We found no association with these combined mutations and IL-6 levels when monocytes from LRTI children were stimulated with Spn.
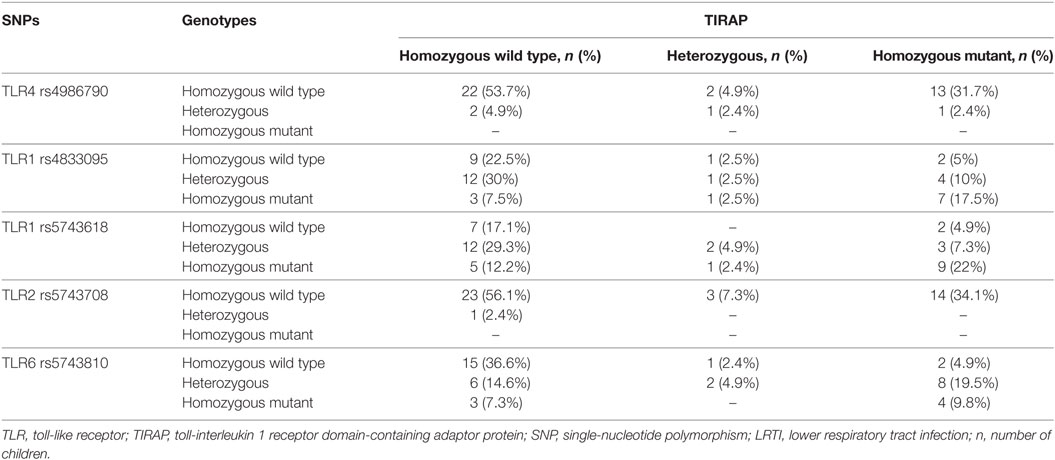
Table 4. Distribution of TLR1, 2, 4, or 6 and TIRAP genotypes in LRTI children.
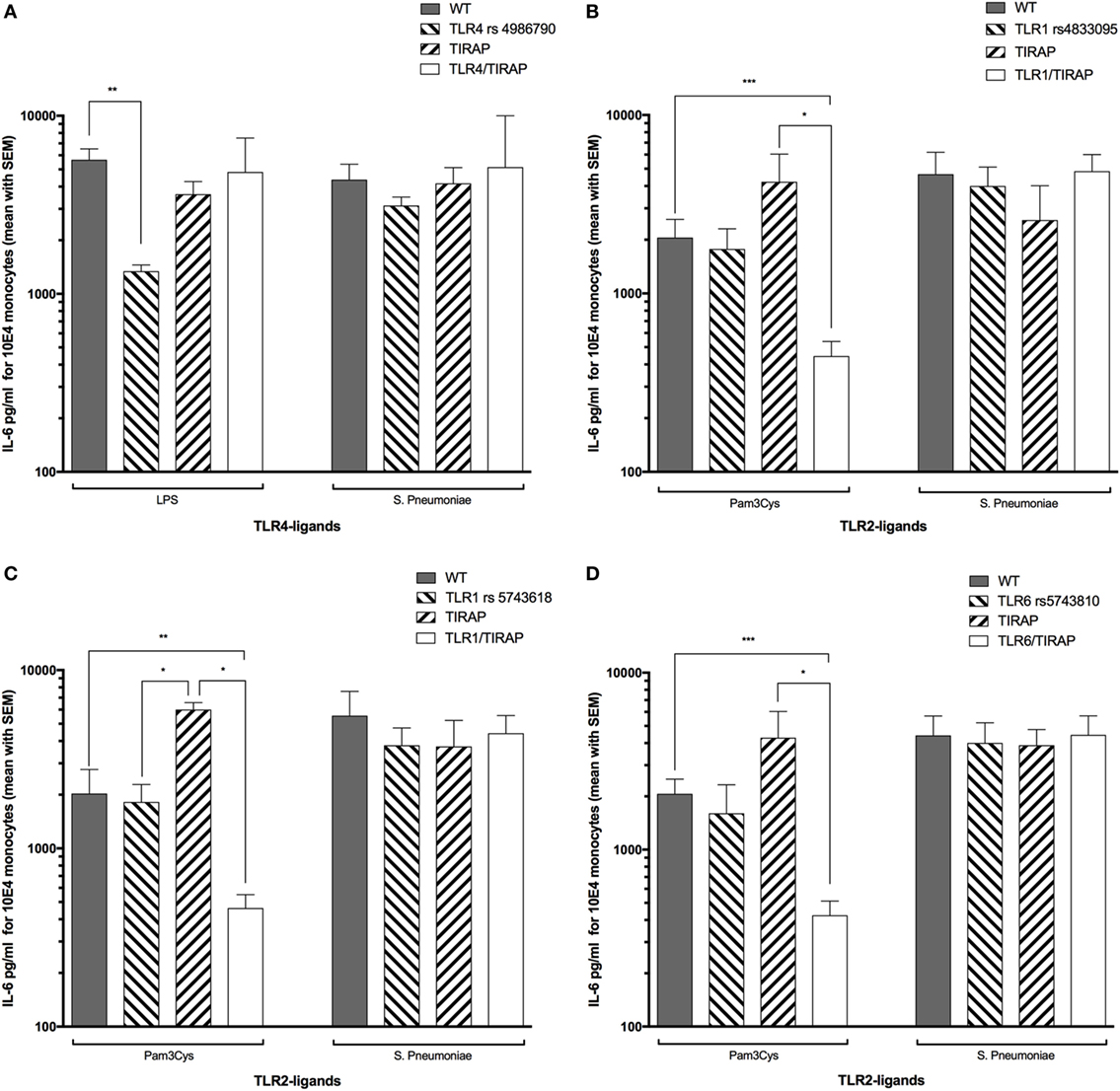
Figure 4. Combined nucleotide polymorphisms in TLR1 or TLR6 and TIRAP are associated with a decreased TLR2 ligand-driven IL-6 release in LRTI children. This figure shows the impact of TIRAP rs8177374, TLR1 rs4833095, TLR1 rs5743618, TLR4 rs4986790, or TLR6 rs5743810 polymorphisms or their combinations on monocyte release of IL-6 following ex vivo whole blood stimulation by LPS, Pam3Cys, or Streptococcus pneumoniae (Spn). Values are shown as mean with SEM. Number of children per histogram may differ from those in Table 4 (due to unavailable IL-6 values). (A) Number of patients in each group: wild type (WT) = 20, TIRAP [heterozygous (HT) and homozygous mutant (HM) reported together] = 15, and TLR4/TIRAP (HT and HM reported together) = 2, TLR4 = 2. (B) WT = 8, TIRAP (HT and HM reported together) = 3, TLR1 rs4833095/TIRAP (HT and HM reported together) = 12, and TLR1 rs4833095 (HT and HM reported together) = 14. (C) Controls = 6, TIRAP (HT and HM reported together) = 2, TLR1 rs5743618/TIRAP (HT and HM reported together) = 13, and TLR1 rs5743618 (HT and HM reported together) = 16. (D) Controls = 13, TIRAP (HT and HM reported together) = 3, TLR6/TIRAP (HT and HM reported together) = 12, and TLR6 (HT and HM reported together) = 9. Gray histograms represent homozygous wild-type individuals, dashed histograms represent heterozygous, and white histograms represent homozygous mutant. Upper borders of histograms denote mean, and solid horizontal lines denote SEM. IL-6 secretion is expressed in picograms per milliliter for 104 monocytes. *P < 0.05; **P < 0.01; and ***P < 0.001 by Mann–Whitney test. Abbreviations: TLR, toll-like receptor; WT, wild type; IL-6, interleukin-6.
Discussion
To the best of our knowledge, this is the first study to show an association between SNP in TIRAP and increased susceptibility to recurrent pneumococcal LRTI in children. Previous studies have reported that common genetic variations in TLRs affect host susceptibility to infectious diseases (37–39). In particular, some authors found a relationship between IPD and SNPs in TLRs (40) or Toll-IL1R (TIR) signaling pathway proteins (41), but none of them studied specifically TIRAP genes. In our study, the key finding was that the TIRAP S180L heterozygous trait contributes to protection to recurrent pneumococcal LRTI in children, whereas the 180L homozygous mutant trait, alone or in combination with TLR1 N248S, TLR1 I602S or TLR6 S249P polymorphisms, contributes to increased susceptibility by modulating the inflammatory response. Children carrying the mutant homozygous TIRAP 180L polymorphism were indeed more likely to have recurrent pneumococcal LRTI, while carrying the heterozygous S180L polymorphism decreased the likelihood to be in this category.
Interestingly, similar results have been described in adult studies where TIRAP polymorphism in its heterozygous S180L form conferred protection against primary immune deficiency, or septic shock (42). It decreased the risk of an excessive inflammatory response by attenuating TLR2 and TLR4 signal transduction through the impairment of cytokine responses and NF-κB activation following stimulation with TLR2 and TLR4 ligands (36, 43, 44). Our results extend these previous adult findings to pediatrics. Likewise, the same pattern of TIRAP associations was observed in other Gram-positive or Gram-negative bacterial infections, bacteremia, malaria, and tuberculosis in more than 6,100 individuals (44). Since TIRAP is essential and restricted to the TLR2- (with TLR1 and TLR6 as co-receptors) and TLR4-mediated MyD88-dependent signaling pathway (45), it is possible that a genetic variation at this central position explains the variable susceptibility of the host’s immune response to various infectious diseases. In accordance with previous human studies (19, 46), we observed no direct associations between susceptibility to pneumococcal LRTI and the other SNPs assessed in our study. Our results support the interpretation that the TIRAP S180L polymorphism might affect directly the clinical course of recurrent pneumococcal LRTI in children whereas SNPs in TRL2 or TLR4 are not directly associated with an increased risk of IPD. They are in line with those from other studies that have shown that individuals homozygous for the TIRAP 180L variant have an increased risk of infection, in particular sepsis (42) and invasive Haemophilus influenzae serotype b (Hib) infection in immunized children (47).
Controversial results have been reported regarding the impact of the TLR2 and TLR4 polymorphisms on the expression levels. First, we observed that TLR2 was significantly up-regulated in LRTI compared with healthy children, independently of any SNPs that we tested. In line with our results, TLR2 upregulation on monocytes during the clinical course of infections has already been observed in adults and seemed even to be a possible predictor of recurrence in patients with bacterial infectious diseases (48). The fact that this upregulation was particularly marked in LRTI children above 3 years old in our study might reflect their sustained recurrent episodes of pneumococcal infections over years. Second, conflicting results exist on whether TLR4 polymorphisms may impair trafficking and cell surface expression of TLR4 (11, 13, 49). In our study and others (50, 51), it was observed that TLR4 was only down-regulated in TIRAP homozygous mutant carriers. This reduced level of TLR4 expression might be one of the factors underlying the susceptibility to pneumococcal recurrent LRTI in this subpopulation. Future studies are required to further examine this question.
In addition to genetic association between TLRs polymorphisms and cell surface expression of TLR2 and TLR4, we also indirectly investigated the integrity of the children’s TLR2 and TLR4 signaling pathways by investigating various TLR ligands-driven IL-6 responses. With the use of a whole blood stimulation assay, we observed that the overall Spn-, LPS- and TLA4e-driven IL-6 levels were higher in children with recurrent pneumococcal LRTI than in healthy children. However, according to other studies (52, 53), we observed that the Asp299Gly/Thr399Ile haplotype had no different IL-6 production after stimulation with LPS and Spn. By stratifying the children according to the various SNPs studied, we found that TIRAP was the gene most commonly involved in these IL-6 levels differences. Although restricted by unavailable data in some SNPs subpopulations, especially HM genotypes, we found that Spn-driven IL-6 levels were specifically increased in LRTI children compared with healthy children for all SNPs studied. This was especially true in both WT and HM subpopulations with TIRAP polymorphisms.
In addition, we observed that other specific TLR ligands—but not Spn—yielded a significant reduction in IL-6 levels in HM children for the TLR1, TLR6 and TIRAP genes, but without a difference between the healthy and LRTI children. This reduction was even more obvious when individuals were assessed for combined double mutations with TIRAP. Unlike Kumpf et al. who showed that combination of TLR4 and TIRAP SNPs was associated with low circulating levels of IL-6 in adult patients with ventilator-associated pneumonia (36), we observed instead significantly reduced IL-6 levels in TLR1 or TLR6 and TIRAP double-mutant children following Pam3CSK4 stimulation. This difference might be related to the nature of the bacteria studied. Indeed, whereas the patients studied by Kumpf et al. developed pneumonia predominantly caused by Gram-negative bacteria, known to be specifically recognized by TLR4, the children in our study were supposedly infected by Gram-positive Spn recognized by both TLR2 (and TLR1 and TLR6 as co-receptors) and TLR4. The fact that Pam3CSK4, a specific TLR2 ligand, was unable to induce potent IL-6 production in double-mutated LRTI children (TIRAP 180Leu/TLR1 248S or TIRAP 180Leu/TLR1 602S or TIRAP 180Leu/TLR6 249P) might reflect a defect within the TLR2 or TLR1 and TLR6 co-receptors. On the other hand, given that Spn-induced IL-6 levels were preserved in double-mutant LRTI children might supposedly reflect the role played by the intact TLR4 pathway. This redundant TLR2–TLR4 recognition system might serve, at least in part when TIRAP is functional, as a rescue system to prevent the escape of some microorganisms from the innate immune system. However, these findings raised the possibility that the redundant signaling cascades using TLR2 and TLR4 paths may be overwhelmed in pneumococcal infections if TIRAP becomes less functional through homozygote mutations. This may explain why in our study, only children with TIRAP mutations showed abnormal IL-6 levels when compared with healthy children after a Spn challenge. Taken together, these results showed that the TLR signaling system needs to be tightly regulated to constantly balance between activation and inhibition to avoid inappropriate inflammatory responses (54). A plausible way by which TIRAP polymorphisms may explain varying susceptibilities to Spn in children might rely on an imbalance of downstream proinflammatory cytokines levels. Although the exact mechanism by which this imbalance leads in the end to pneumonia extends beyond the scope of this study and needs further investigations. However, whereas increased TLR-driven pro-inflammatory responses play a pivotal role in host defense against invading pathogens, altered TLR function might contribute to destructive inflammatory damages and increased susceptibility to infection. This hypothesis is supported by a study performed by Ferwarda et al. These authors observed that a moderately increased cytokine release during the primary stage of infection, as would be expected in heterozygous S180L individuals, is likely beneficial for the outcome of the infection and the early clearance of bacteria. However, if the cytokine release is reaching a certain threshold leading to systemic inflammatory effects, which was suggested to take place in homozygous 180L individuals, then this could ultimately prove deleterious to the host (42). In our study, LRTI children homozygous for the TIRAP 180L mutant trait appeared to be deprived of the attenuating effect provided by the S180L and more prone to excessive IL-6 levels production in response to Spn compared with healthy controls. According to Khor et al., this finding was consistent with increasing evidence that an excessive host inflammatory status might render individuals more susceptible to infectious diseases (44). However, other microbial, environmental, genetic and immune factors may also contribute to the susceptibility for infection, as children’s infection rates usually spontaneously improve after the age of 5.
Our study has some limitations. First, LRTI is defined clinically according to the WHO definitions. Because up to 50% of children with pneumonia have no bacterial proof despite thorough investigations (55), obtaining samples confirming the presence of Spn for each child would be difficult and probably unethical. However, local and international epidemiological studies recognize Spn as the most prevalent cause of LRTIs in young children (23). This limitation was already addressed earlier (14).
Second, we did not assess other adaptor molecules that have been described to be associated with an increased susceptibility to invasive bacterial infections (18, 40, 56–61). It would be interesting to compare the influence of these molecules to TIRAP polymorphism on recurrent pneumococcal LRTI in children in future studies. However, these defects can be a diagnostic challenge because the affected patients present narrow susceptibility ranges for invasive pyogenic bacterial infections (20), and their routine immunological screening is often normal.
Third, because blood sampling was performed only once in healthy children, it is possible that normal interindividual variations were missed. Since our results were obtained from a modest number of patients, further studies with larger population sizes and investigating defects along the TLR2 and 4 pathways in children with pneumococcal LRTI would be valuable. Moreover, although the children were shown to have no total Ig deficiency (14), we cannot totally exclude that some children had an unrecognized underlying rare immune deficiency.
Fourth, TLR4 requires the involvement of the MD-2 glycoprotein and CD14 as co-receptors on its extracellular domain to form a complex able to recognize and bind LPS, with the help of LPS binding protein (62). Because we did not assess polymorphism in these molecules, we cannot exclude that other SNPs may affect susceptibility to recurrent LRTI. To date, studies on MD-2 or CD14 polymorphisms and association to PID remain rare (40, 63).
Sixth, the quality of antibodies could have influenced the MFI results. However, fluorochrome-matched monoclonal anti-TLR2 and anti-TLR4 were purchased from the same company and prepared following manufacturer’s instructions at optimal dilution to ensure reliability of the results. These commercially available antibodies are expected to have undergone extensive quality control programs to meet the manufacturer’s strict release criteria. Median fluorescence intensity were chosen as a read-out instead of mean fluorescence intensity because median is generally considered a much better statistical approach in that median is less influenced by skew or outlier events.
Finally, we did not assess TLR1 and TLR6 expression at the cell surface of the monocytes. Given our results showing the impact of related SNPs on these TLRs, further analysis of SNPs in these TLRs would be interesting to fully understand these pathways and clarify their precise role in PID.
Restrictions Apply to the Datasets
The datasets for this manuscript are not publicly available to avoid patients’ identification and to preserve anonymization. Requests to access the anonymized datasets should be directed to Dr. Johan Siebert atam9oYW4uc2llYmVydEBoY3VnZS5jaA==.
Ethics Statement
The study was approved by the institutional ethics committee, and written consent was obtained from all parents or legal guardians. It was conducted in accordance with the principles of the Declaration of Helsinki, the standards of Good Clinical Practice, and Swiss regulatory requirements.
Author Contributions
Conceptualization: JS and KP-B; methodology: JS and KP-B; investigation: JS, KP-B, and SG; formal analysis: JS and CV; validation: CG and LH; writing—original draft: JS; visualization: JS; writing—review and editing: JS, LH, C-AS, and KP-B; funding acquisition: KP-B; resources: CG, LH, and C-AS; supervision: C-AS and KP-B. All authors discussed the results and commented on the manuscript. All authors have contributed to, seen and approved the final, submitted version of the manuscript, and had full access to all of the data (including statistical reports and tables) in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis. The corresponding author confirms that he had full access to participant’s data and endorsed the final responsibility for the submission, and affirms that the manuscript is an honest, accurate, and transparent account of the study being reported; that no important aspects of the study have been omitted; and that any discrepancies from the study as planned have been explained.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Technical help from the Plateforme de Recherche Clinique en Pédiatrie, part of the Geneva Children’s Hospital (Fabienne Maréchal, Maria Rodriguez, Carole Salomon, and Suzanne Duperret) and the Center of Vaccinology and Neonatal Immunology (Paolo Valenti, Suzanne Schlegel, Gianna Cadau, Elodie Belnoue, and Arun Kamath) is greatly acknowledged. Consent to publication was obtained from the participants.
Funding
KP-B has received grant support from the Swiss National Research Foundation (SCORE Grant no. 3232BO-111354 & 3200BO-111355).
Supplementary Material
The Supplementary Material for this article can be found online at https://www.frontiersin.org/articles/10.3389/fimmu.2018.01780/full#supplementary-material.
Abbreviations
SNP, single-nucleotide polymorphism; LRTI, lower respiratory tract infections; Spn, Streptococcus pneumoniae; TLR, toll-like receptor; TIRAP, toll-interleukin 1 receptor domain-containing adaptor protein; CAP, community-acquired pneumonia; IPD, invasive pneumococcal disease; MFI, median fluorescence intensity; WT, wild type; HT, heterozygous; HM, homozygous.
References
1. Nair H, Simoes EA, Rudan I, Gessner BD, Azziz-Baumgartner E, Zhang JS, et al. Global and regional burden of hospital admissions for severe acute lower respiratory infections in young children in 2010: a systematic analysis. Lancet (2013) 381(9875):1380–90. doi:10.1016/S0140-6736(12)61901-1
2. Liu L, Oza S, Hogan D, Chu Y, Perin J, Zhu J, et al. Global, regional, and national causes of under-5 mortality in 2000–15: an updated systematic analysis with implications for the Sustainable Development Goals. Lancet (2016) 388(10063):3027–35. doi:10.1016/S0140-6736(16)31593-8
3. Cevey-Macherel M, Galetto-Lacour A, Gervaix A, Siegrist CA, Bille J, Bescher-Ninet B, et al. Etiology of community-acquired pneumonia in hospitalized children based on WHO clinical guidelines. Eur J Pediatr (2009) 168(12):1429–36. doi:10.1007/s00431-009-0943-y
4. Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, et al. Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet (2010) 375(9730):1969–87. doi:10.1016/S0140-6736(10)60549-1
5. Harris M, Clark J, Coote N, Fletcher P, Harnden A, McKean M, et al. British Thoracic Society guidelines for the management of community acquired pneumonia in children: update 2011. Thorax (2011) 66(Suppl 2):ii1–23. doi:10.1136/thoraxjnl-2011-200598
6. Honkinen M, Lahti E, Osterback R, Ruuskanen O, Waris M. Viruses and bacteria in sputum samples of children with community-acquired pneumonia. Clin Microbiol Infect (2012) 18(3):300–7. doi:10.1111/j.1469-0691.2011.03603.x
7. Khan MN, Pichichero ME. The host immune dynamics of pneumococcal colonization: implications for novel vaccine development. Hum Vaccin Immunother (2014) 10(12):3688–99. doi:10.4161/21645515.2014.979631
8. Nelson JC, Jackson M, Yu O, Whitney CG, Bounds L, Bittner R, et al. Impact of the introduction of pneumococcal conjugate vaccine on rates of community acquired pneumonia in children and adults. Vaccine (2008) 26(38):4947–54. doi:10.1016/j.vaccine.2008.07.016
9. Montella S, Corcione A, Santamaria F. Recurrent pneumonia in children: a reasoned diagnostic approach and a single centre experience. Int J Mol Sci (2017) 18(2):E296. doi:10.3390/ijms18020296
10. O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet (2009) 374(9693):893–902. doi:10.1016/S0140-6736(09)61204-6
11. Everard ML. ‘Recurrent lower respiratory tract infections’ – going around in circles, respiratory medicine style. Paediatr Respir Rev (2012) 13(3):139–43. doi:10.1016/j.prrv.2012.03.003
12. Long SP, Prober C. Principles and Practice of Pediatric Infectious Disease. 4th ed. New York: Churchill Livingstone Elsevier (2012).
13. Patria MF, Esposito S. Recurrent lower respiratory tract infections in children: a practical approach to diagnosis. Paediatr Respir Rev (2013) 14(1):53–60. doi:10.1016/j.prrv.2011.11.001
14. Siebert JN, L’Huillier AG, Grillet S, Delhumeau C, Siegrist CA, Posfay-Barbe KM. Memory B cell compartment constitution and susceptibility to recurrent lower respiratory tract infections in young children. J Leukoc Biol (2013) 93(6):951–62. doi:10.1189/jlb.0312117
15. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature (2007) 449(7164):819–26. doi:10.1038/nature06246
16. Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol (2012) 14(4):460–6. doi:10.1111/j.1462-5822.2011.01746.x
17. O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in toll-like receptor signalling. Nat Rev Immunol (2007) 7(5):353–64. doi:10.1038/nri2079
18. Schroder NW, Schumann RR. Single nucleotide polymorphisms of toll-like receptors and susceptibility to infectious disease. Lancet Infect Dis (2005) 5(3):156–64. doi:10.1016/S1473-3099(05)01308-3
19. Brouwer MC, de Gans J, Heckenberg SG, Zwinderman AH, van der Poll T, van de Beek D. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis (2009) 9(1):31–44. doi:10.1016/S1473-3099(08)70261-5
20. Picard C, Casanova JL, Puel A. Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency. Clin Microbiol Rev (2011) 24(3):490–7. doi:10.1128/CMR.00001-11
21. Ellis MK, Elliott KS, Rautanen A, Crook DW, Hill AV, Chapman SJ. Rare variants in MYD88, IRAK4 and IKBKG and susceptibility to invasive pneumococcal disease: a population-based case-control study. PLoS One (2015) 10(4):e0123532. doi:10.1371/journal.pone.0123532
22. Lederhuber H, Baer K, Altiok I, Sadeghi K, Herkner KR, Kasper DC. MicroRNA-146: tiny player in neonatal innate immunity? Neonatology (2011) 99(1):51–6. doi:10.1159/000301938
23. Posfay-Barbe KM, Galetto-Lacour A, Grillet S, Ochs MM, Brookes RH, Kraehenbuhl JD, et al. Immunity to pneumococcal surface proteins in children with community-acquired pneumonia: a distinct pattern of responses to pneumococcal choline-binding protein A. Clin Microbiol Infect (2011) 17(8):1232–8. doi:10.1111/j.1469-0691.2010.03389.x
24. Hirschfeld AF, Bettinger JA, Victor RE, Davidson DJ, Currie AJ, Ansermino JM, et al. Prevalence of toll-like receptor signalling defects in apparently healthy children who developed invasive pneumococcal infection. Clin Immunol (2007) 122(3):271–8. doi:10.1016/j.clim.2006.10.012
25. De Groote D, Zangerle PF, Gevaert Y, Fassotte MF, Beguin Y, Noizat-Pirenne F, et al. Direct stimulation of cytokines (IL-1 beta, TNF-alpha, IL-6, IL-2, IFN-gamma and GM-CSF) in whole blood. I. Comparison with isolated PBMC stimulation. Cytokine (1992) 4(3):239–48. doi:10.1016/1043-4666(92)90062-V
26. Damsgaard CT, Lauritzen L, Calder PC, Kjaer TM, Frokiaer H. Whole-blood culture is a valid low-cost method to measure monocytic cytokines – a comparison of cytokine production in cultures of human whole-blood, mononuclear cells and monocytes. J Immunol Methods (2009) 340(2):95–101. doi:10.1016/j.jim.2008.10.005
27. Temple SE, Cheong KY, Almeida CM, Price P, Waterer GW. Polymorphisms in lymphotoxin alpha and CD14 genes influence TNFalpha production induced by Gram-positive and Gram-negative bacteria. Genes Immun (2003) 4(4):283–8. doi:10.1038/sj.gene.6363963
28. Hamann L, Hamprecht A, Gomma A, Schumann RR. Rapid and inexpensive real-time PCR for genotyping functional polymorphisms within the toll-like receptor-2, -4, and -9 genes. J Immunol Methods (2004) 285(2):281–91. doi:10.1016/j.jim.2003.12.005
29. Johnson CM, Lyle EA, Omueti KO, Stepensky VA, Yegin O, Alpsoy E, et al. Cutting edge: a common polymorphism impairs cell surface trafficking and functional responses of TLR1 but protects against leprosy. J Immunol (2007) 178(12):7520–4. doi:10.4049/jimmunol.178.12.7520
30. Hamann L, Kumpf O, Schuring RP, Alpsoy E, Bedu-Addo G, Bienzle U, et al. Low frequency of the TIRAP S180L polymorphism in Africa, and its potential role in malaria, sepsis, and leprosy. BMC Med Genet (2009) 10:65. doi:10.1186/1471-2350-10-65
31. Schuring RP, Hamann L, Faber WR, Pahan D, Richardus JH, Schumann RR, et al. Polymorphism N248S in the human toll-like receptor 1 gene is related to leprosy and leprosy reactions. J Infect Dis (2009) 199(12):1816–9. doi:10.1086/599121
32. Hamann L, Koch A, Sur S, Hoefer N, Glaeser C, Schulz S, et al. Association of a common TLR-6 polymorphism with coronary artery disease – implications for healthy ageing? Immun Ageing (2013) 10(1):43. doi:10.1186/1742-4933-10-43
33. Hamann L, Glaeser C, Schulz S, Gross M, Franke A, Nothlings U, et al. A micro RNA-146a polymorphism is associated with coronary restenosis. Int J Immunogenet (2014) 41(5):393–6. doi:10.1111/iji.12136
34. Clarke GM, Anderson CA, Pettersson FH, Cardon LR, Morris AP, Zondervan KT. Basic statistical analysis in genetic case-control studies. Nat Protoc (2011) 6(2):121–33. doi:10.1038/nprot.2010.182
35. Ferwerda B, McCall MB, Alonso S, Giamarellos-Bourboulis EJ, Mouktaroudi M, Izagirre N, et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc Natl Acad Sci U S A (2007) 104(42):16645–50. doi:10.1073/pnas.0704828104
36. Kumpf O, Giamarellos-Bourboulis EJ, Koch A, Hamann L, Mouktaroudi M, Oh DY, et al. Influence of genetic variations in TLR4 and TIRAP/Mal on the course of sepsis and pneumonia and cytokine release: an observational study in three cohorts. Crit Care (2010) 14(3):R103. doi:10.1186/cc9047
37. Netea MG, Wijmenga C, O’Neill LA. Genetic variation in toll-like receptors and disease susceptibility. Nat Immunol (2012) 13(6):535–42. doi:10.1038/ni.2284
38. Trejo-de la OA, Hernandez-Sancen P, Maldonado-Bernal C. Relevance of single-nucleotide polymorphisms in human TLR genes to infectious and inflammatory diseases and cancer. Genes Immun (2014) 15(4):199–209. doi:10.1038/gene.2014.10
39. Skevaki C, Pararas M, Kostelidou K, Tsakris A, Routsias JG. Single nucleotide polymorphisms of toll-like receptors and susceptibility to infectious diseases. Clin Exp Immunol (2015) 180(2):165–77. doi:10.1111/cei.12578
40. Yuan FF, Marks K, Wong M, Watson S, de Leon E, McIntyre PB, et al. Clinical relevance of TLR2, TLR4, CD14 and FcgammaRIIA gene polymorphisms in Streptococcus pneumoniae infection. Immunol Cell Biol (2008) 86(3):268–70. doi:10.1038/sj.icb.7100155
41. Carrasco-Colom J, Jordan I, Alsina L, Garcia-Garcia JJ, Cambra-Lasaosa FJ, Martin-Mateos MA, et al. Association of polymorphisms in IRAK1, IRAK4 and MyD88, and severe invasive pneumococcal disease. Pediatr Infect Dis J (2015) 34(9):1008–13. doi:10.1097/INF.0000000000000779
42. Ferwerda B, Alonso S, Banahan K, McCall MB, Giamarellos-Bourboulis EJ, Ramakers BP, et al. Functional and genetic evidence that the Mal/TIRAP allele variant 180L has been selected by providing protection against septic shock. Proc Natl Acad Sci U S A (2009) 106(25):10272–7. doi:10.1073/pnas.0811273106
43. Mansell A, Brint E, Gould JA, O’Neill LA, Hertzog PJ. Mal interacts with tumor necrosis factor receptor-associated factor (TRAF)-6 to mediate NF-kappaB activation by toll-like receptor (TLR)-2 and TLR4. J Biol Chem (2004) 279(36):37227–30. doi:10.1074/jbc.C400289200
44. Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, et al. A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet (2007) 39(4):523–8. doi:10.1038/ng1976
45. Yamamoto M, Sato S, Hemmi H, Sanjo H, Uematsu S, Kaisho T, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature (2002) 420(6913):324–9. doi:10.1038/nature01182
46. van Well GT, Sanders MS, Ouburg S, Kumar V, van Furth AM, Morre SA. Single nucleotide polymorphisms in pathogen recognition receptor genes are associated with susceptibility to meningococcal meningitis in a pediatric cohort. PLoS One (2013) 8(5):e64252. doi:10.1371/journal.pone.0064252
47. Ladhani SN, Davila S, Hibberd ML, Heath PT, Ramsay ME, Slack MP, et al. Association between single-nucleotide polymorphisms in Mal/TIRAP and interleukin-10 genes and susceptibility to invasive Haemophilus influenzae serotype b infection in immunized children. Clin Infect Dis (2010) 51(7):761–7. doi:10.1086/656236
48. Orihara K, Nagata K, Hamasaki S, Oba R, Hirai H, Ishida S, et al. Time-course of toll-like receptor 2 expression, as a predictor of recurrence in patients with bacterial infectious diseases. Clin Exp Immunol (2007) 148(2):260–70. doi:10.1111/j.1365-2249.2007.03352.x
49. Ziakas PD, Prodromou ML, El Khoury J, Zintzaras E, Mylonakis E. The role of TLR4 896 A>G and 1196 C>T in susceptibility to infections: a review and meta-analysis of genetic association studies. PLoS One (2013) 8(11):e81047. doi:10.1371/journal.pone.0081047
50. Dessing MC, Schouten M, Draing C, Levi M, von Aulock S, van der Poll T. Role played by toll-like receptors 2 and 4 in lipoteichoic acid-induced lung inflammation and coagulation. J Infect Dis (2008) 197(2):245–52. doi:10.1086/524873
51. Long H, O’Connor BP, Zemans RL, Zhou X, Yang IV, Schwartz DA. The toll-like receptor 4 polymorphism Asp299Gly but not Thr399Ile influences TLR4 signaling and function. PLoS One (2014) 9(4):e93550. doi:10.1371/journal.pone.0093550
52. Ferwerda B, McCall MB, Verheijen K, Kullberg BJ, van der Ven AJ, Van der Meer JW, et al. Functional consequences of toll-like receptor 4 polymorphisms. Mol Med (2008) 14(5–6):346–52. doi:10.2119/2007-00135.Ferwerda
53. Douville RN, Lissitsyn Y, Hirschfeld AF, Becker AB, Kozyrskyj AL, Liem J, et al. TLR4 Asp299Gly and Thr399Ile polymorphisms: no impact on human immune responsiveness to LPS or respiratory syncytial virus. PLoS One (2010) 5(8):e12087. doi:10.1371/journal.pone.0012087
54. Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol (2005) 5(6):446–58. doi:10.1038/nri1630
55. Durbin WJ, Stille C. Pneumonia. Pediatr Rev (2008) 29(5):147–158; quiz 159–160. doi:10.1542/pir.29-5-147
56. Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science (2003) 299(5615):2076–9. doi:10.1126/science.1081902
57. Currie AJ, Davidson DJ, Reid GS, Bharya S, MacDonald KL, Devon RS, et al. Primary immunodeficiency to pneumococcal infection due to a defect in toll-like receptor signaling. J Pediatr (2004) 144(4):512–8. doi:10.1016/j.jpeds.2003.10.034
58. Burgner D, Jamieson SE, Blackwell JM. Genetic susceptibility to infectious diseases: big is beautiful, but will bigger be even better? Lancet Infect Dis (2006) 6(10):653–63. doi:10.1016/S1473-3099(06)70601-6
59. Hill AV. Aspects of genetic susceptibility to human infectious diseases. Annu Rev Genet (2006) 40:469–86. doi:10.1146/annurev.genet.40.110405.090546
60. Ku CL, von Bernuth H, Picard C, Zhang SY, Chang HH, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med (2007) 204(10):2407–22. doi:10.1084/jem.20070628
61. von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, Ku CL, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science (2008) 321(5889):691–6. doi:10.1126/science.1158298
62. Re F, Strominger JL. Monomeric recombinant MD-2 binds toll-like receptor 4 tightly and confers lipopolysaccharide responsiveness. J Biol Chem (2002) 277(26):23427–32. doi:10.1074/jbc.M202554200
Keywords: toll-like receptors, Streptococcus pneumoniae, child, pneumonia, immunity, innate, polymorphism, single nucleotide
Citation: Siebert JN, Hamann L, Verolet CM, Gameiro C, Grillet S, Siegrist C-A and Posfay-Barbe KM (2018) Toll-Interleukin 1 Receptor Domain-Containing Adaptor Protein 180L Single-Nucleotide Polymorphism Is Associated With Susceptibility to Recurrent Pneumococcal Lower Respiratory Tract Infections in Children. Front. Immunol. 9:1780. doi: 10.3389/fimmu.2018.01780
Received: 12 January 2018; Accepted: 19 July 2018;
Published: 07 August 2018
Edited by:
Laurel L. Lenz, University of Colorado, United StatesReviewed by:
Yi Liu, Qilu Children’s Hospital of Shandong University, ChinaScott Alper, National Jewish Health, United States
Copyright: © 2018 Siebert, Hamann, Verolet, Gameiro, Grillet, Siegrist and Posfay-Barbe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan N. Siebert, am9oYW4uc2llYmVydEBoY3VnZS5jaA==