 Angela Rita Elia
Angela Rita Elia Sara Caputo
Sara Caputo Matteo Bellone
Matteo Bellone- Cellular Immunology Unit, Department of Immunology, Transplantation and Infectious Diseases, San Raffaele Scientific Institute, Milan, Italy
Prostate adenocarcinoma (PCa) and melanoma are paradigmatic examples of tumors that are either poorly or highly sensitive to therapies based on monoclonal antibodies directed against regulatory pathways in T lymphocytes [i.e., immune checkpoint blockade (ICB)]. Yet, approximately 40% of melanoma patients are resistant or acquire resistance to ICB. What characterize the microenvironment of PCa and ICB-resistant melanoma are a scanty cytotoxic T cell infiltrate and a strong immune suppression, respectively. Here, we compare the tumor microenvironment in these two subgroups of cancer patients, focusing on some among the most represented immune checkpoint molecules: cytotoxic T lymphocyte-associated antigen-4, programmed death-1, lymphocyte activation gene-3, and T cell immunoglobulin and mucin-domain containing-3. We also report on several examples of crosstalk between cancer and immune cells that are mediated by inhibitory immune checkpoints and identify promising strategies aimed at overcoming ICB resistance both in PCa and melanoma.
Introduction
Activated T lymphocytes require mechanisms that timely and properly shut them down to prevent excessive damage at the inflammation site. Inhibitory immune checkpoint molecules, such as cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed death-1 (PD-1), lymphocyte activation gene-3 (LAG-3), and T cell immunoglobulin and mucin-domain containing-3 (TIM-3), are progressively upregulated on activated T cells, and, by interacting with their ligands, switch inhibitory pathways on in T cells (1). Interactions between immune checkpoint molecules on T cells and their ligands on target cells may also signal in the latters, thus generating a crosstalk between T lymphocytes and other cells (2–4). These mechanisms are crucial for self-tolerance, but also represent the Achilles’ heel of cancer immunity, as ligands for inhibitory immune checkpoint molecules are expressed on neoplastic and other cells within the tumor microenvironment. In addition, a growing tumor may condition secondary lymphoid organs, thus limiting expansion of tumor-specific T cells (5).
Building on these evidences, monoclonal antibodies (mAbs) directed against regulatory pathways in T lymphocytes [i.e., immune checkpoint blockade (ICB) (6)] have been developed. Phase III clinical trials with anti-PD-1/programmed death-ligand 1 (PD-L1) or anti-CTLA-4 mAbs documented excellent efficacy, and ICB has been approved for the treatment of various solid and hematological malignancies (7). Because several inhibitory checkpoints act simultaneously, the combination of two or more mAbs can improve ICB therapeutic outcomes (8).
Although melanomas are generally sensitive to ICB, also because of their heavy cytotoxic T lymphocyte (CTL) infiltrate, approximately 40% of melanoma patients are resistant to ICB even when two mAbs are combined (9). ICB resistance was recently reviewed [e.g., Ref. (10, 11)]. Other tumors like prostate adenocarcinoma (PCa) are intrinsically resistant to ICB (12), and either anti-PD-1/PD-L1 or anti-CTLA-4 monotherapy did not impact PCa patients’ overall survival (13, 14). ICB resistance in PCa is attributed to tumor cell intrinsic mechanisms and a scanty immune infiltrate (15) dominated by macrophages. In addition, soon after ICB, immune cells upregulate other inhibitory molecules such as V-domain Ig suppressor of T cell activation [VISTA; (16)], a phenomenon not limited to PCa (17). Interestingly, orally available small molecules targeting both PD-L1 and VISTA are investigated in patients affected by advanced tumors (ClinicalTrials.gov Identifier: NCT02812875).
Therefore, melanoma and PCa epitomize two classes of ICB-resistant tumors, in which tumor cell-intrinsic mechanisms of ICB resistance associate with heavy but immunosuppressed or modest immune infiltrates, respectively. Thus, while in the former the combination of two or more ICB mAbs should succeed, in the latter strategies to improve tumor infiltration by CTLs will be needed to improve ICB sensitivity. We will analyze differences and similarities in ICB-resistant melanoma and PCa, focusing on immune checkpoint-mediated interactions between tumor and immune cells. We will also highlight strategies that might improve sensitivity to ICB.
T Cell Exhaustion
Prolonged antigen exposure progressively impairs T cell proliferation and effector functions (18) through epigenetic mechanisms (19). In the early dysfunctional state, which is plastic and reprogrammable, CD8+ T cells express PD-1 and LAG-3 and low TIM-3 levels. Later on, T cells enter fixed exhaustion characterized by TIM-3 upregulation, and the additional expression of high CD38 and CD101 and low CD5 levels. The latter cells are no longer reprogrammable by ICB (20). Partially exhausted CD8+ T cells, expressing high levels of PD-1 and CTLA-4 predicted response to anti-PD-1 in metastatic melanoma patients (21). Because also activated T cells express PD-1, this molecule cannot be used as marker of exhaustion, which should instead be functionally defined (22). Additional exhaustion markers (e.g., 2B4 and TIGIT) cannot be discussed here because of space constraint.
Also CD4+ T cells undergo exhaustion (23), loosing helper function and releasing IL-10 (24). CTLA-4 on CD4+ Tregs is an additional mechanism of immune suppression in cancer (25).
Cytotoxic T Lymphocyte-Associated Antigen-4
Cytotoxic T lymphocyte-associated antigen-4 has been the first immune checkpoint investigated in clinic. Because of higher affinity for CD80 and CD86 than CD28, CTLA-4 impairs T cell co-stimulation (26). Whereas CTLA-4 is expressed on activated effector T cells (27), it is constitutively expressed on Tregs and contributes to their immunosuppressive activity. Thus, anti-CTLA-4 mAbs mainly act in secondary lymphoid organs, also causing Treg depletion through antibody-dependent cellular toxicity (28).
IFN-γ signaling activates expression of CTLA-4 in melanoma cells, and after ipilimumab (anti-CTLA-4) treatment, human melanomas upregulated IFN-γ responsive genes, including CTLA-4, which associated with durable response (29). Thus, anti-CTLA-4 mAbs can directly affect melanoma cells (30). CTLA-4 on tumor cells might also act as local mechanism of immune escape. Of relevance, mutations in the IFN responsive genes associate with resistance to ICB (31). Restifo and collaborators found that also mutations in genes indirectly correlated to the IFN response (e.g., APLNR), profoundly affected in vivo sensitivity to both adoptive T cell therapy (ACT) and anti-CTLA-4 blockade (32). It is anticipated that investigations on such comprehensive lists of genes will identify new drugs overcoming ICB resistance.
An alternative strategy to increase sensitivity to ICB is to combine them with other therapeutic strategies, such as chemotherapy, hormonal therapy, vaccines, etc. (Tables 1 and 2). As an example, both in mice and humans, the combination of local chemotherapy and systemic ICB increased tumor infiltration by effector T cells, and clinical response rates (NCT01323517) (33). Others have shown that targeting myeloid-derived suppressor cells (MDSCs), which are relevant immunosuppressive populations in PCa (34, 35), with tyrosine kinase inhibitors increased sensitivity to ICB in castration-resistant PCa (36). Both in orthotopic melanoma and autochthonous PCa, even the combination of anti-CTLA-4 and anti-PD-1 exerted modest antitumor effects (37), and required the addition of fresh T cells (i.e., ACT) and minute amounts of TNF-α targeted to tumor-associated vessels to favor endothelial cell activation, tumor infiltration by fully effector T cells, and tumor debulking (38, 39). Interestingly, only this triple-combined treatment guaranteed a prolonged overall survival of the mice affected by autochthonous PCa, thus suggesting the treatment generated a potent tumor-specific memory response (37). Additional strategies can be implemented to favor access of both T cells and mAbs to the tumor (40).
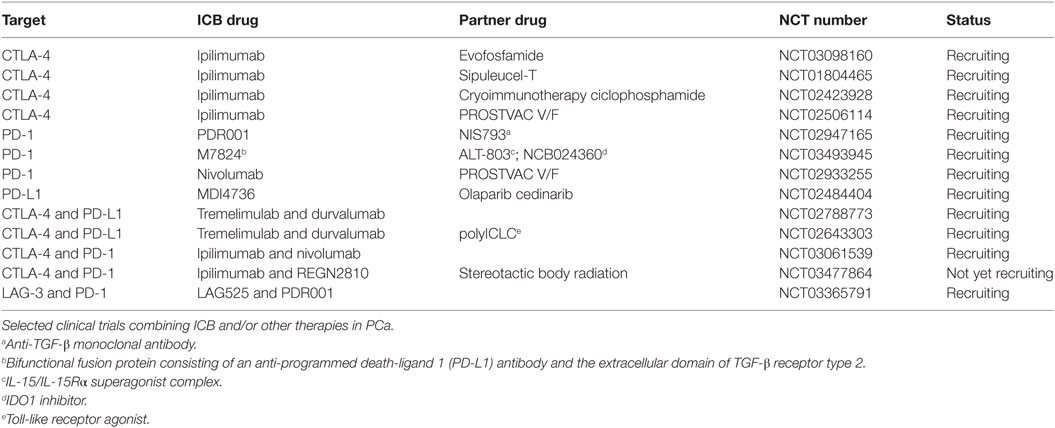
Table 1. Clinical trials of immune checkpoint blockade (ICB) combined with other strategies in prostate adenocarcinoma (PCa).
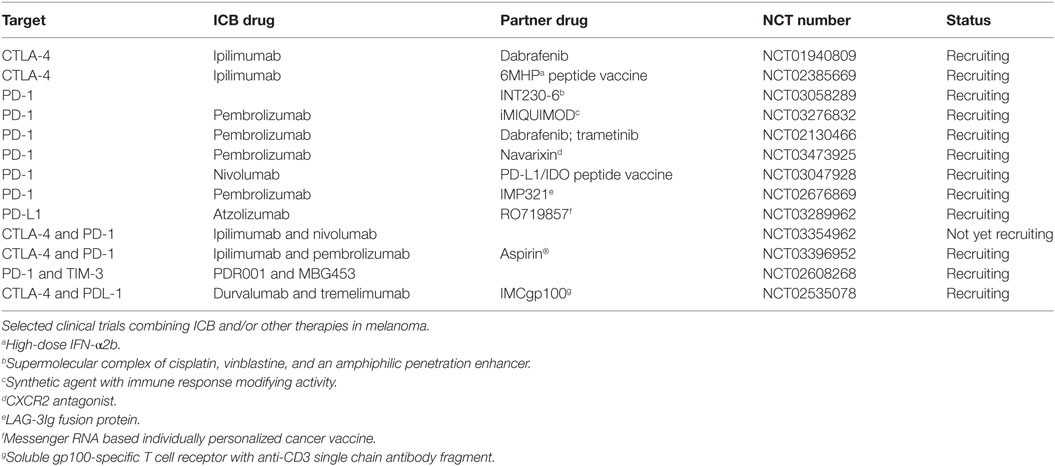
Table 2. Clinical trials of immune checkpoint blockade (ICB) combined with other strategies in melanoma.
Overall, these data support the concept that several therapeutic strategies need to be combined to overcome ICB resistance.
PD-1/PD-L1
Programmed death-1 is upregulated on T cells upon antigen recognition, and by interacting with either PD-L1 expressed on tumor, stromal and immune cells or PD-L2 expressed on myeloid cells, impairs T cell activation (41). An exhaustion-specific enhancer regulates PD-1 expression in T cells (42), and editing exhaustion-specific enhancers might improve the therapeutic efficacy of ACT. Similarly, blocking de novo DNA methylation in chronically stimulated CD8+ T cells allowed retention of their effector functions (43).
Programmed death-1 blockade with nivolumab, lambrolizumab, or pembrolizumab has led to relevant clinical benefits in cancer patients, mainly by rejuvenating cytotoxicity and cytokine secretion capability of T cells (44). However, as mentioned above, T cells undergoing fixed exhaustion are no longer reprogrammable by ICB. An interesting study compared the epigenetic regulation of tumor- or virus-specific T cells in melanoma-bearing mice. Only melanoma-infiltrating, tumor-specific T lymphocytes (TILs) upregulated PD-1, LAG-3, and TIM-3 and showed reduced TNF-α, IFN-γ, and IL-2 secretion ability when compared with virus-specific cells. Exhausted T cells displayed more accessible chromatin in proximity to PD-1 and LAG-3 gene promoters. Treatment with anti-PD-1 mAbs had a positive impact on effector functions of exhausted T cells and on tumor growth, but induced only limited changes in gene expression and chromatin accessibility (45). Similar findings have been reported in a transplantable model of PCa, in which tumor-specific CD8+ T cells showed de novo methylation in Tcf7, Ccr7, Myc, and IFN-γ genes, and impaired proliferation and effector functions that could not be restored by ICB. Only combination of decitabine, inhibiting the DNA methyltransferase DNMT3A, and anti-PD-1 mAbs re-established proliferation capability of exhausted T cells, thus resulting in delayed tumor growth (43).
Clinical trials evaluating the efficacy of pembrolizumab in combination with epigenetic drugs are ongoing in advanced melanoma patients (NCT03278665, NCT02816021, and NCT02437136). Also in PCa, PD-1 blockade is clinically investigated in combination with ipilimumab (NCT02601014), anti-PD-L1 (NCT03170960, NCT03061539), and other therapies including hormone, vaccine, and cryosurgery (NCT02787005, NCT02499835, and NCT02489357).
Programmed death-1 can be found expressed also on tumor cells, and PD-1 triggering on melanoma cells increases three-dimensional growth capability with concomitant activation of the mTOR pathway (3). Interestingly, treatment with BRAF and MEK inhibitors associated with increased frequency of PD-1+ tumor cells in melanoma patients, and PD-1 expression sensitized melanoma to PD-1 blockade in immunodeficient mice (46). The same authors also noticed a correlative expression of PD-1 and the stem cell marker Oct-4, thus linking PD-1 to cancer stem cells (46).
Also anti-PD-L1 mAbs may directly affect tumor cells by impacting tumor metabolism, reducing extracellular acidification, phosphorylation of mTOR, and glycolysis (4). mTORC1 expression has been associated with PD-L1 expression in melanoma cells, and PD-L1low cells showed decreased levels of mTORC1, and an altered autophagy pathway. Furthermore, treatment of immunodeficient mice with anti-PD-L1 mAbs delayed melanoma growth, reduced metastases, and prolonged animal survival (2). PD-L1 has also been found overexpressed in melanoma tumor-initiating cells, and the lack of PD-L1 significantly reduced the frequency of these cells in melanoma-bearing mice (47). Thus, interfering with the PD-1/PD-L1 axis may impact both tumor and immune cells.
Lymphocyte Activation Gene-3
Lymphocyte activation gene-3 is closely related to CD4, is expressed on dysfunctional T cells (48), and TILs in melanoma patients express LAG-3 (49). Because LAG-3 binding to MHC class II molecules activates myeloid cells (50), and MHC class II can be expressed by melanoma cells (51), engagement of LAG-3 with MHC class II might provide a survival signal to tumor cells. LAG-3 also binds LSECtin and Galectin-3 (Gal-3) (49, 52) and associates with the CD3/TCR complex, thus impairs TCR signaling (18, 52). Conversely, LAG-3 binding on Tregs increases their immunosuppressive activity (53).
Lymphocyte activation gene-3 may synergize with other immune checkpoints, and the combination of anti-LAG-3 and anti-PD-1 resulted in more potent inhibition of murine tumor growth than single treatments (54). Anti-LAG-3 mAbs or LAG-3 fusion proteins are being tested in melanoma patients resistant to anti-PD-1/PD-L1 ICB as single agent (NCT01968109), or in combination to anti-PD-1 (NCT02676869).
Drake and collaborators originally reported that in PCa, tumor-specific CD4+ and CD8+ T cells rapidly upregulate LAG-3 upon in vivo antigen encounter. Treatment with anti-LAG-3 mAbs enhanced the number and effector function of tumor-specific CD8+ T cells in TRAMP mice, and delayed tumor growth (55). Also Tregs in human PCa lesions upregulate both CTLA-4 and LAG-3 (56). The latter finding has been challenged by recent data showing low expression of LAG-3 in Tregs infiltrating PCa lesions (57). Further investigation is needed to better define the role of LAG-3 in T cell exhaustion and/or Treg function in PCa. One clinical trial is ongoing that investigates efficacy of anti-LAG-3 mAbs in combination with anti-PD-1 in castration-resistant PCa (NCT03365791).
T Cell Immunoglobulin and Mucin-Domain Containing-3
Programmed death-1 expression in TILs is often associated with TIM-3, and its transient or persistent expression relates to short or chronic antigen stimulations, respectively (58). Indeed, PD-1+TIM-3+ T cells are functionally more exhausted than PD-1+TIM-3−/low T cells (59), and TIM-3 can be considered a marker of terminally differentiated T cells.
T cell immunoglobulin and mucin-domain containing-3 is expressed on dysfunctional, tumor-specific CD8+ T cells in melanoma (60) and PCa patients (61), and in ipilimumab-treated melanoma patients, increased expression and frequency of TIM-3 and PD-1 on both peripheral NK and T cells associated with poor prognosis (62). Correlative data on TIM-3 in PCa patients are conflicting. Whereas one report showed that high TIM-3 expression on PCa cells predicted short recurrence-free and progression-free survival in chemotherapy and radiotherapy naïve PCa patients (63), others found that negative TIM-3 expression was an independent prognostic factor of poor prognosis in advanced metastatic PCa (64). Outcome differences might be brought back to the different subpopulations of PCa patients analyzed in the two studies. The latter also showed that silencing TIM-3 in PCa cell lines reduced tumor cell proliferation and invasion in vitro (63), thus, suggesting that TIM-3 has a functional role in PCa cells. Interestingly, the combined targeting of TIM-3 and PD-1 pathways is more effective in controlling tumor growth than targeting either pathway alone (59).
Mechanistically, the interaction between TIM-3 on T cells and one of its ligands [i.e., Galectin-9 (Gal-9)] triggers cell death in effector T cells (65). Ceacam-1, an additional TIM-3 ligand, is co-expressed on exhausted T cells, can bind TIM-3 both in cis and trans, and both interactions drive the inhibitory function of TIM-3 (66). TIM-3 also enhances FoxP3+ Tregs inhibitory functions (59), and is expressed and upregulated upon activation on human NK cells. In contrast to effector T cells, Gal-9-mediated TIM-3 triggering in NK cells induces IFN-γ production (67). Interestingly, it has been shown that MHC class I downregulation or deficiency in mouse tumors induces upregulation of PD-1 and TIM-3 on NK cells and their exhaustion. PD-1+TIM-3+ NK cells were also found in human melanoma samples, and correlated with low HLA expression (68). Because in vitro, TIM-3 blockade reversed NK cell exhaustion (69), it will be interesting to investigate the in vivo effects of mAbs against both PD-1 and TIM-3 on NK cells.
While TIM-3 is higher and more precociously upregulated on tumor-associated dendritic cells (DCs) than on CD8+ T cells, its role in innate immunity is controversial (70). By interacting with phosphatidylserine, TIM-3 favors DC uptake of apoptotic cells and cross-presentation (71). Conversely, interaction of the alarmin high mobility group protein B1 with Tim-3 on DCs limits their release of pro-inflammatory cytokines, thus blunting type-1 immunity (72). TIM-3 is also expressed on tumor-associated macrophages (72), and TIM-3 negatively modulates the production of pro-inflammatory cytokines in human CD14+ monocytes (73). Finally, TIM-3 can suppress the antitumor immunity by promoting induction of MDSCs (74).
Clinical trials are investigating safety and tolerability of anti-TIM-3 mAbs given either alone (NCT03489343) or in combination with anti-PD-1 (NCT02817633 and NCT02608268) or anti-PD-L1 (NCT03099109) in cancer patients.
Galectins
Apart from being ligands for LAG-3 and TIM-3, galectins also exert relevant pro-tumor functions (75). Increased expression of Gal-3 in melanoma lesions correlates with tumor progression (76), and Gal-3 activates NFAT1 (77), which also regulates IL-8 and MMP3 expression in melanoma cells, thus promoting a malignant phenotype (78). Gal-3 released by melanoma cells can also capture IFN-γ, thus reducing its antitumor activity (79). At odds, others reported that tumor cell expression of Gal-3 or myeloid cell expression of Gal-9 in melanoma lesions associated with a longer survival (80). The latter findings are counterintuitive and deserve further investigation.
Inhibiting Gal-3 together with anticancer vaccination restores the effector function of melanoma TILs (81). Therefore, Gal-3 not only contributes to melanoma tumor growth and metastasis but also dampens the antitumor immune response. Gal-3 inhibition is currently investigated in combination with ICB and vaccine in melanoma (NCT02575404, NCT02117362, and NCT01723813).
Galectin-3 is also expressed in PCa lesions, exerts direct pro-tumor and pro-metastatic functions, and correlates with biochemical recurrence (82). Indeed, administration of a Gal-3 inhibitor suppressed PCa lung metastasis (83).
Galectin-3 is a marker of cancer stem cells (84) and maintains stemness of carcinoma progenitor cells (85). In the TRAMP model, we found that PCa stem-like cells endowed with immunosuppressive activities express Gal-3 (86). We have also evidence that Gal-3 favors growth and metastasis of tumors generated by PCa stem-like cells (Caputo et al., manuscript in preparation). It will be interesting to investigate if Gal-3 also contributes to their immunosuppressive activity.
Concluding Remarks
Inhibitory immune checkpoint triggering in TILs cripples cancer immune surveillance. As consequence of local inflammation, immune checkpoints are also upregulated on cancer cells, supporting tumor growth and aggressiveness. Thus, the effect of ICB goes beyond rescuing of exhausted/dysfunctional TILs and may directly impact tumor cells.
To overcome TIL exhaustion, several promising combined approaches are envisioned among many others: coupling two or more mAbs against immune checkpoints; increase tumor immunogenicity by exploiting conventional chemotherapy and targeted anticancer agents (87); transiently modify the tumor vasculature to favor T cell infiltration (88–90); combine additional immunotherapeutic approaches such as vaccines and ACT (37); abolish additional mechanisms of local immune suppression (91). Several high throughput analyses (e.g., methylomics and metabolomics) and microbiota sequencing will likely define novel areas of therapeutic intervention in the field of ICB. Finally, it will be essential to focus on adverse events that increase along with therapeutic efficacy (92).
Author Contributions
AE, SC, and MB wrote and reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize with the many authors whose excellent work we could note cite due to space limitations.
Funding
The work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC; grant #IG16807 to MB). SC is supported by a fellowship from the Fondazione Italiana per la Ricerca sul Cancro/AIRC (grant #18314).
References
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
2. Clark CA, Gupta HB, Sareddy G, Pandeswara S, Lao S, Yuan B, et al. Tumor-intrinsic PD-L1 signals regulate cell growth, pathogenesis, and autophagy in ovarian cancer and melanoma. Cancer Res (2016) 76(23):6964–74. doi:10.1158/0008-5472.CAN-16-0258
3. Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell (2015) 162(6):1242–56. doi:10.1016/j.cell.2015.08.052
4. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162(6):1229–41. doi:10.1016/j.cell.2015.08.016
5. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39(1):1–10. doi:10.1016/j.immuni.2013.07.012
6. Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity (2016) 44(5):1069–78. doi:10.1016/j.immuni.2016.04.023
7. Melero I, Berman DM, Aznar MA, Korman AJ, Perez Gracia JL, Haanen J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer (2015) 15(8):457–72. doi:10.1038/nrc3973
8. Jardim DL, de Melo Gagliato D, Giles FJ, Kurzrock R. Analysis of drug development paradigms for immune checkpoint inhibitors. Clin Cancer Res (2018) 24(8):1785–94. doi:10.1158/1078-0432.CCR-17-1970
9. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi:10.1056/NEJMoa1504030
10. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer (2016) 16(2):121–6. doi:10.1038/nrc.2016.2
11. Bellone M, Elia AR. Constitutive and acquired mechanisms of resistance to immune checkpoint blockade in human cancer. Cytokine Growth Factor Rev (2017) 36:17–24. doi:10.1016/j.cytogfr.2017.06.002
12. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell (2017) 168(4):707–23. doi:10.1016/j.cell.2017.01.017
13. Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol (2017) 35(1):40–7. doi:10.1200/JCO.2016.69.1584
14. Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15(7):700–12. doi:10.1016/S1470-2045(14)70189-5
15. Petitprez F, Fossati N, Vano Y, Freschi M, Becht E, Luciano R, et al. PD-L1 expression and CD8+ T cell infiltrate are associated with clinical progression in patients with node positive prostate cancer. Eur Urol Focus (2017). doi:10.1016/j.euf.2017.05.013
16. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med (2017) 23(5):551–5. doi:10.1038/nm.4308
17. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi:10.1038/ncomms10501
18. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest (2011) 121(6):2350–60. doi:10.1172/JCI46102
19. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity (2016) 45(2):389–401. doi:10.1016/j.immuni.2016.07.011
20. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature (2017) 545(7655):452–6. doi:10.1038/nature22367
21. Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest (2016) 126(9):3447–52. doi:10.1172/JCI87324
22. Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, et al. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating T cells. Cell (2016) 166(6):1500–11.e9. doi:10.1016/j.cell.2016.08.052
23. Severson JJ, Serracino HS, Mateescu V, Raeburn CD, McIntyre RC Jr, Sams SB, et al. PD-1+Tim-3+ CD8+ T lymphocytes display varied degrees of functional exhaustion in patients with regionally metastatic differentiated thyroid cancer. Cancer Immunol Res (2015) 3(6):620–30. doi:10.1158/2326-6066.CIR-14-0201
24. Parish IA, Marshall HD, Staron MM, Lang PA, Brustle A, Chen JH, et al. Chronic viral infection promotes sustained Th1-derived immunoregulatory IL-10 via BLIMP-1. J Clin Invest (2014) 124(8):3455–68. doi:10.1172/JCI66108
25. Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol (2005) 23:515–48. doi:10.1146/annurev.immunol.23.021704.115611
26. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7-CD28 ligand-receptor family. Immunity (2016) 44(5):955–72. doi:10.1016/j.immuni.2016.05.002
27. Qureshi OS, Kaur S, Hou TZ, Jeffery LE, Poulter NS, Briggs Z, et al. Constitutive clathrin-mediated endocytosis of CTLA-4 persists during T cell activation. J Biol Chem (2012) 287(12):9429–40. doi:10.1074/jbc.M111.304329
28. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell (2018) 33(4):649–63.e4. doi:10.1016/j.ccell.2018.02.010
29. Mo X, Zhang H, Preston S, Martin K, Zhou B, Vadalia N, et al. Interferon-gamma signaling in melanocytes and melanoma cells regulates expression of CTLA-4. Cancer Res (2018) 78(2):436–50. doi:10.1158/0008-5472.CAN-17-1615
30. Laurent S, Queirolo P, Boero S, Salvi S, Piccioli P, Boccardo S, et al. The engagement of CTLA-4 on primary melanoma cell lines induces antibody-dependent cellular cytotoxicity and TNF-alpha production. J Transl Med (2013) 11:108. doi:10.1186/1479-5876-11-108
31. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167(2):397–404.e9. doi:10.1016/j.cell.2016.08.069
32. Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature (2017) 548(7669):537–42. doi:10.1038/nature23477
33. Ariyan CE, Brady MS, Siegelbaum RH, Hu J, Bello DM, Rand J, et al. Robust antitumor responses result from local chemotherapy and CTLA-4 blockade. Cancer Immunol Res (2018) 6(2):189–200. doi:10.1158/2326-6066.CIR-17-0356
34. Rigamonti N, Bellone M. Prostate cancer, tumor immunity and a renewed sense of optimism in immunotherapy. Cancer Immunol Immunother (2012) 61(4):453–68. doi:10.1007/s00262-012-1216-6
35. Lopez-Bujanda Z, Drake CG. Myeloid-derived cells in prostate cancer progression: phenotype and prospective therapies. J Leukoc Biol (2017) 102(2):393–406. doi:10.1189/jlb.5VMR1116-491RR
36. Lu X, Horner JW, Paul E, Shang X, Troncoso P, Deng P, et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature (2017) 543(7647):728–32. doi:10.1038/nature21676
37. Elia AR, Grioni M, Basso V, Curnis F, Freschi M, Corti A, et al. Targeting tumor vasculature with TNF leads effector T cells to the tumor and enhances therapeutic efficacy of immune checkpoint blockers in combination with adoptive cell therapy. Clin Cancer Res (2018) 24(9):2171–81. doi:10.1158/1078-0432.CCR-17-2210
38. Calcinotto A, Grioni M, Jachetti E, Curnis F, Mondino A, Parmiani G, et al. Targeting TNF-alpha to neoangiogenic vessels enhances lymphocyte infiltration in tumors and increases the therapeutic potential of immunotherapy. J Immunol (2012) 188(6):2687–94. doi:10.4049/jimmunol.1101877
39. Manzo T, Sturmheit T, Basso V, Petrozziello E, Hess Michelini R, Riba M, et al. T cells redirected to a minor histocompatibility antigen instruct intratumoral TNFalpha expression and empower adoptive cell therapy for solid tumors. Cancer Res (2017) 77(3):658–71. doi:10.1158/0008-5472.CAN-16-0725
40. Marcucci F, Bellone M, Rumio C, Corti A. Approaches to improve tumor accumulation and interactions between monoclonal antibodies and immune cells. MAbs (2013) 5(1):34–46. doi:10.4161/mabs.22775
41. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi:10.1038/nri.2017.108
42. Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (2016) 354(6316):1165–9. doi:10.1126/science.aae0491
43. Ghoneim HE, Fan Y, Moustaki A, Abdelsamed HA, Dash P, Dogra P, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell (2017) 170(1):142–57.e19. doi:10.1016/j.cell.2017.06.007
44. Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature (2017) 545(7652):60–5. doi:10.1038/nature22079
45. Mognol GP, Spreafico R, Wong V, Scott-Browne JP, Togher S, Hoffmann A, et al. Exhaustion-associated regulatory regions in CD8(+) tumor-infiltrating T cells. Proc Natl Acad Sci U S A (2017) 114(13):E2776–85. doi:10.1073/pnas.1620498114
46. Sanlorenzo M, Vujic I, Floris A, Novelli M, Gammaitoni L, Giraudo L, et al. BRAF and MEK inhibitors increase PD1-positive melanoma cells leading to a potential lymphocyte-independent synergism with anti-PD1 antibody. Clin Cancer Res (2018) 24(14):3377–85. doi:10.1158/1078-0432.CCR-17-1914
47. Gupta HB, Clark CA, Yuan B, Sareddy G, Pandeswara S, Padron AS, et al. Tumor cell-intrinsic PD-L1 promotes tumor-initiating cell generation and functions in melanoma and ovarian cancer. Signal Transduct Target Ther (2016) 1:16030. doi:10.1038/sigtrans.2016.30
48. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi:10.1084/jem.171.5.1393
49. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res (2014) 74(13):3418–28. doi:10.1158/0008-5472.CAN-13-2690
50. Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood (2003) 102(6):2130–7. doi:10.1182/blood-2003-01-0273
51. Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol (2011) 186(9):5173–83. doi:10.4049/jimmunol.1002050
52. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res (2015) 3(4):412–23. doi:10.1158/2326-6066.CIR-14-0150
53. Hannier S, Tournier M, Bismuth G, Triebel F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol (1998) 161(8):4058–65.
54. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2012) 72(4):917–27. doi:10.1158/0008-5472.CAN-11-1620
55. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117(11):3383–92. doi:10.1172/JCI31184
56. Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, et al. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res (2008) 14(11):3254–61. doi:10.1158/1078-0432.CCR-07-5164
57. Davidsson S, Andren O, Ohlson AL, Carlsson J, Andersson SO, Giunchi F, et al. FOXP3(+) regulatory T cells in normal prostate tissue, postatrophic hyperplasia, prostatic intraepithelial neoplasia, and tumor histological lesions in men with and without prostate cancer. Prostate (2018) 78(1):40–7. doi:10.1002/pros.23442
58. Sanchez-Fueyo A, Tian J, Picarella D, Domenig C, Zheng XX, Sabatos CA, et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat Immunol (2003) 4(11):1093–101. doi:10.1038/ni987
59. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi:10.1084/jem.20100643
60. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med (2010) 207(10):2175–86. doi:10.1084/jem.20100637
61. Japp AS, Kursunel MA, Meier S, Malzer JN, Li X, Rahman NA, et al. Dysfunction of PSA-specific CD8+ T cells in prostate cancer patients correlates with CD38 and Tim-3 expression. Cancer Immunol Immunother (2015) 64(11):1487–94. doi:10.1007/s00262-015-1752-y
62. Tallerico R, Cristiani CM, Staaf E, Garofalo C, Sottile R, Capone M, et al. IL-15, TIM-3 and NK cells subsets predict responsiveness to anti-CTLA-4 treatment in melanoma patients. Oncoimmunology (2017) 6(2):e1261242. doi:10.1080/2162402X.2016.1261242
63. Piao YR, Piao LZ, Zhu LH, Jin ZH, Dong XZ. Prognostic value of T cell immunoglobulin mucin-3 in prostate cancer. Asian Pac J Cancer Prev (2013) 14(6):3897–901. doi:10.7314/APJCP.2013.14.6.3897
64. Wu J, Lin G, Zhu Y, Zhang H, Shi G, Shen Y, et al. Low TIM3 expression indicates poor prognosis of metastatic prostate cancer and acts as an independent predictor of castration resistant status. Sci Rep (2017) 7(1):8869. doi:10.1038/s41598-017-09484-8
65. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep (2015) 5:15659. doi:10.1038/srep15659
66. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517(7534):386–90. doi:10.1038/nature13848
67. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood (2012) 119(13):3064–72. doi:10.1182/blood-2011-06-360321
68. Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun (2017) 8:15776. doi:10.1038/ncomms15776
69. da Silva IP, Gallois A, Jimenez-Baranda S, Khan S, Anderson AC, Kuchroo VK, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res (2014) 2(5):410–22. doi:10.1158/2326-6066.CIR-13-0171
70. Han G, Chen G, Shen B, Li Y. Tim-3: an activation marker and activation limiter of innate immune cells. Front Immunol (2013) 4:449. doi:10.3389/fimmu.2013.00449
71. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood (2009) 113(16):3821–30. doi:10.1182/blood-2008-10-185884
72. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol (2012) 13(9):832–42. doi:10.1038/ni.2376
73. Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY, Moorman JP, et al. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J Leukoc Biol (2012) 91(2):189–96. doi:10.1189/jlb.1010591
74. Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol (2010) 185(3):1383–92. doi:10.4049/jimmunol.0903275
75. Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer (2005) 5(1):29–41. doi:10.1038/nrc1527
76. Prieto VG, Mourad-Zeidan AA, Melnikova V, Johnson MM, Lopez A, Diwan AH, et al. Galectin-3 expression is associated with tumor progression and pattern of sun exposure in melanoma. Clin Cancer Res (2006) 12(22):6709–15. doi:10.1158/1078-0432.CCR-06-0758
77. Braeuer RR, Zigler M, Kamiya T, Dobroff AS, Huang L, Choi W, et al. Galectin-3 contributes to melanoma growth and metastasis via regulation of NFAT1 and autotaxin. Cancer Res (2012) 72(22):5757–66. doi:10.1158/0008-5472.CAN-12-2424
78. Shoshan E, Braeuer RR, Kamiya T, Mobley AK, Huang L, Vasquez ME, et al. NFAT1 directly regulates IL8 and MMP3 to promote melanoma tumor growth and metastasis. Cancer Res (2016) 76(11):3145–55. doi:10.1158/0008-5472.CAN-15-2511
79. Gordon-Alonso M, Hirsch T, Wildmann C, van der Bruggen P. Galectin-3 captures interferon-gamma in the tumor matrix reducing chemokine gradient production and T-cell tumor infiltration. Nat Commun (2017) 8(1):793. doi:10.1038/s41467-017-00925-6
80. Melief SM, Visconti VV, Visser M, van Diepen M, Kapiteijn EH, van den Berg JH, et al. Long-term survival and clinical benefit from adoptive T-cell transfer in stage IV melanoma patients is determined by a four-parameter tumor immune signature. Cancer Immunol Res (2017) 5(2):170–9. doi:10.1158/2326-6066.CIR-16-0288
81. Demotte N, Wieers G, Van Der Smissen P, Moser M, Schmidt C, Thielemans K, et al. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res (2010) 70(19):7476–88. doi:10.1158/0008-5472.CAN-10-0761
82. Knapp JS, Lokeshwar SD, Vogel U, Hennenlotter J, Schwentner C, Kramer MW, et al. Galectin-3 expression in prostate cancer and benign prostate tissues: correlation with biochemical recurrence. World J Urol (2013) 31(2):351–8. doi:10.1007/s00345-012-0925-y
83. Pienta KJ, Naik H, Akhtar A, Yamazaki K, Replogle TS, Lehr J, et al. Inhibition of spontaneous metastasis in a rat prostate cancer model by oral administration of modified citrus pectin. J Natl Cancer Inst (1995) 87(5):348–53. doi:10.1093/jnci/87.5.348
84. Ilmer M, Mazurek N, Byrd JC, Ramirez K, Hafley M, Alt E, et al. Cell surface galectin-3 defines a subset of chemoresistant gastrointestinal tumor-initiating cancer cells with heightened stem cell characteristics. Cell Death Dis (2016) 7(8):e2337. doi:10.1038/cddis.2016.239
85. Tummala KS, Brandt M, Teijeiro A, Grana O, Schwabe RF, Perna C, et al. Hepatocellular carcinomas originate predominantly from hepatocytes and benign lesions from hepatic progenitor cells. Cell Rep (2017) 19(3):584–600. doi:10.1016/j.celrep.2017.03.059
86. Jachetti E, Caputo S, Mazzoleni S, Brambillasca CS, Parigi SM, Grioni M, et al. Tenascin-C protects cancer stem-like cells from immune surveillance by arresting T-cell activation. Cancer Res (2015) 75(10):2095–108. doi:10.1158/0008-5472.CAN-14-2346
87. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28(6):690–714. doi:10.1016/j.ccell.2015.10.012
88. Bellone M, Calcinotto A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front Oncol (2013) 3:231. doi:10.3389/fonc.2013.00231
89. Bellone M, Calcinotto A, Corti A. Won’t you come on in? How to favor lymphocyte infiltration in tumors. Oncoimmunology (2012) 1(6):986–8. doi:10.4161/onci.20213
90. Bellone M, Mondino A, Corti A. Vascular targeting, chemotherapy and active immunotherapy: teaming up to attack cancer. Trends Immunol (2008) 29(5):235–41. doi:10.1016/j.it.2008.02.003
91. Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nat Commun (2018) 9(1):741. doi:10.1038/s41467-017-02696-6
Keywords: prostate cancer, melanoma, immunity, immune checkpoint, immunotherapy, cytotoxic T lymphocytes
Citation: Elia AR, Caputo S and Bellone M (2018) Immune Checkpoint-Mediated Interactions Between Cancer and Immune Cells in Prostate Adenocarcinoma and Melanoma. Front. Immunol. 9:1786. doi: 10.3389/fimmu.2018.01786
Received: 21 May 2018; Accepted: 19 July 2018;
Published: 31 July 2018
Edited by:
Amedeo Amedei, Università degli Studi di Firenze, ItalyReviewed by:
Kawaljit Kaur, University of California, Los Angeles, United StatesAlessandro Poggi, Ospedale Policlinico San Martino, Italy
Carlos Alfaro, NavarraBiomed, Spain
Dennis O. Adeegbe, Moffitt Cancer Center, United States
Giulio Cesare Spagnoli, Universität Basel, Switzerland
Derre Laurent, Centre Hospitalier Universitaire Vaudois (CHUV), Switzerland
Copyright: © 2018 Elia, Caputo and Bellone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matteo Bellone, YmVsbG9uZS5tYXR0ZW9AaHNyLml0