 Viktor Reshetnikov1
Viktor Reshetnikov1 Jonas Hahn2
Jonas Hahn2 Christian Maueröder3,4
Christian Maueröder3,4 Christine Czegley2
Christine Czegley2 Luis Enrique Munoz2
Luis Enrique Munoz2 Martin Herrmann2
Martin Herrmann2 Markus H. Hoffmann2*
Markus H. Hoffmann2* Andriy Mokhir1*
Andriy Mokhir1*
- 1Department of Chemistry and Pharmacy, Organic Chemistry II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 2Department of Internal Medicine 3 – Rheumatology and Immunology, University Hospital Erlangen, Friedrich-Alexander-Universität Erlangen-Nürnberg, Erlangen, Germany
- 3Cell Clearance in Health and Disease Lab, VIB Center for Inflammation Research, Ghent, Belgium
- 4Department of Biomedical Molecular Biology, Ghent university, Ghent, Belgium
A number of chemical compounds are known, which amplify the availability of reactive oxygen species (ROS) in neutrophils both in vitro and in vivo. They can be roughly classified into NADPH oxidase 2 (NOX2)-dependent and NOX2-independent reagents. NOX2 activation is triggered by protein kinase C agonists (e.g., phorbol esters, transition metal ions), redox mediators (e.g., paraquat) or formyl peptide receptor (FPR) agonists (e.g., aromatic hydrazine derivatives). NOX2-independent mechanisms are realized by reagents affecting glutathione homeostasis (e.g., l-buthionine sulfoximine), modulators of the mitochondrial respiratory chain (e.g., ionophores, inositol mimics, and agonists of peroxisome proliferator-activated receptor γ) and chemical ROS amplifiers [e.g., aminoferrocene-based prodrugs (ABPs)]. Since a number of inflammatory and autoimmune diseases, as well as cancer and bacterial infections, are triggered or enhanced by aberrant ROS production in neutrophils, it is tempting to use ROS amplifiers as drugs for the treatment of these diseases. However, since the known reagents are not cell specific, their application for treatment likely causes systemic enhancement of oxidative stress, leading to severe side effects. Cell-targeted ROS enhancement can be achieved either by using conjugates of ROS amplifiers with ligands binding to receptors expressed on neutrophils (e.g., the GPI-anchored myeloid differentiation marker Ly6G or FPR) or by designing reagents activated by neutrophil function [e.g., phagocytic activity or enzymatic activity of neutrophil elastase (NE)]. Since binding of an artificial ligand to a receptor may trigger or inhibit priming of neutrophils the latter approach has a smaller potential for severe side effects and is probably better suitable for therapy. Here, we review current approaches for the use of ROS amplifiers and discuss their applicability for treatment. As an example, we suggest a possible design of neutrophil-specific ROS amplifiers, which are based on NE-activated ABPs.
Introduction
Neutrophils are formed from stem cells in the bone marrow and constitute roughly 40–75% of the leukocyte population in humans, which makes them the most abundant white blood cells. They play a key role in the response of the innate immune system toward both infectious and sterile agents. These cells possess high mobility due to their characteristic segmented nuclei that coined them the name polymorphonuclear (PMN) cells. Therefore, neutrophils can quickly migrate from blood to the site of inflammation, where they respond with either phagocytosis of the inflammatory trigger or with degranulation, finally resulting in disintegration of pathogens. Alternatively, neutrophils release DNA-rich neutrophil extracellular traps (NETs), which entrap and neutralize infectious pathogens and sterile factors (1, 2). Additionally, neutrophils recruit macrophages, activate dendritic cells, trigger production of antibodies, and stimulate CD4+ and CD8+ T cells, thereby affecting the adaptive immune system (3–5). In many of these crucial processes the generation of reactive oxygen species (ROS) plays a key role, either because ROS act directly as cytotoxic agents against pathogens or as important regulators of inflammatory responses including, e.g., NET formation, secretion of various proteases, redox enzymes, and antimicrobial factors. In fully functional primed neutrophils, ROS are mainly produced by NADPH oxidase 2 (NOX2) resulting in the so-called oxidative burst. In contrast to its pro-inflammatory role during the early phase of the fight against infections, ROS can also inhibit inflammatory responses (6), e.g., by deactivation of T cells (7–10) or by degradation of inflammatory mediators in NETs (11–13) (Figure 1). In agreement with these functions, insufficient ROS production by neutrophils, e.g., due to NOX2 deficiency results in persistent infections but also in autoimmunity and non-resolving inflammation, as can be observed in chronic granulomatous disease (CGD) (14–17).
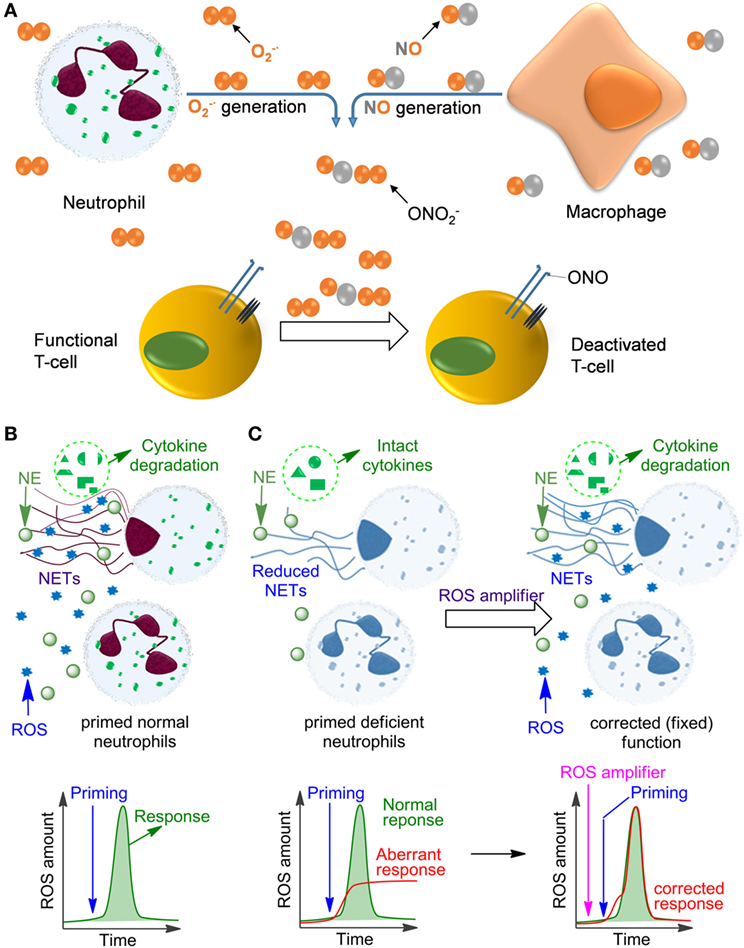
Figure 1. Influence of reactive oxygen species (ROS) on immune cell function. (A) The mechanism of deactivation of T-cells by ROS and reactive nitrogen species produced by neutrophils and monocytes/macrophages. This process is facilitated by the proximity of macrophages and T-cells at the sites of infection due to binding of the T-cells to antigens presented on the macrophage surface. (B) Oxidative burst in normal primed neutrophils leading to ROS production and neutrophil extracellular trap (NET) formation. (C) Aberrant response of NADPH oxidase 2-deficient neutrophils (e.g., in chronic granulomatous disease) leading to low ROS and insufficient NET formation. This deficiency can be fixed by applying ROS amplifiers.
Despite modern clinical management, long-term outcomes in patients with CGD are still bleak, especially for those individuals with fewer than 10% neutrophils with normal oxidase activity (18, 19). Hematopoietic stem cell transplantations, as the first-line definitive therapy in CGD, are often hampered by limitations in available matched bone marrow donors and the risk of graft-versus-host disease (20). In the last years, technical advantages have also paved the way for specific gene editing to restore proteins encoded by genes carrying loss-of-function mutations [reviewed in Ref. (21)]. Particularly, gene therapy using lentiviral vectors enabling specific expression in myeloid cells (22) and approaches employing the clustered regularly interspaced short palindromic repeats/Cas9 system (23) have obtained promising results.
An alternative approach would be the use of drugs that are capable to trigger ROS generation in NOX2-deficient neutrophils. Such compounds would have the potential to both stimulate the immune system and prevent chronic inflammation (24). In this review, we summarize chemical substances that reportedly can be used for stimulation of ROS production in neutrophils. We also discuss mechanisms of action as well as problems in application of these compounds as drugs and suggest possible solutions.
Formation of ROS Upon Priming of Functional Neutrophils and Their Mutual Transformations in Live Organisms
The multienzymatic NOX2 is activated upon neutrophil priming. In the active state it is able to catalyze one-electron reduction of molecular oxygen (3O2) with the formation of a superoxide anion radical (Figure 2). This reactive and, therefore, short-lived anion is one of the key precursors of other ROS in cells and the extracellular space, including hydrogen peroxide (H2O2), hydroxyl radicals (HO•), hypochlorous acid (HOCl) and its anion hypochlorite (ClO−), singlet oxygen (1O2), as well as a series of reactive nitrogen species, e.g., peroxynitrite (ONOO−) and carbon-centered (R•), alkoxy- (RO•) and alkylperoxy-radicals (ROO•). In aqueous solution is dismutated spontaneously with formation of H2O2 and 3O2. This reaction is accelerated over 104-fold in the presence of superoxide dismutase. In contrast to , H2O2 is a stable molecule. For example, concentrated aqueous solutions of H2O2 are commercially available, can be safely delivered over long distances and stored over extended time. Apart from NOX2, the protein folding machinery in the endoplasmic reticulum (ER) also causes generation of ROS (25).
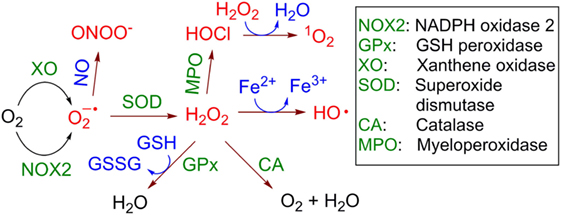
Figure 2. Generation and transformation of reactive oxygen species in live cells. Reactive species are red colored.
H2O2 is able to cross the cellular membrane, e.g., via the aquaporin-mediated pathway (26, 27). Therefore, it is distributed over both the intracellular and extracellular space independently of its site of generation. Though H2O2 is itself not a toxic molecule, in the presence of electron donors, e.g., Cu+ or Fe2+, it is reduced with cleavage of the O-O bond leading to formation of the hydroxide anion HO− and the HO• radical (Fenton reaction, Figure 2). HO• radicals are extremely reactive and, therefore, short-lived. They are capable of subtracting a hydrogen atom (“H”) even from very stable (bio)molecules, e.g., lipids, nucleic acids, proteins that leads to formation of a variety of organic radicals (e.g., R•, RO•, and ROO•), deactivation of the biomolecules, and ultimately induction of cell death via different pathways, e.g., apoptosis, necrosis, or the formation of NETs. Other reactions leading to H2O2 elimination in cells include catalase (CA)-induced conversion of H2O2 to water and molecular oxygen (3O2) and glutathione peroxidase-catalyzed reduction of H2O2 in the presence of glutathione (GSH) with the formation of glutathione disulfide (GSSG) and water. Interestingly, H2O2 is accumulated in some organelles, e.g., lysosomes (LY) and the ER. This can be explained by the low catalase activity in LY (28) and the low concentration of reduced GSH in ER relatively to its concentration in cytoplasm (29).
In neutrophils, H2O2 is also used as a substrate of myeloperoxidase (MPO), which transforms Cl− anions to highly reactive HOCl. At pH 7 the latter acid (pKa~7.5) is partially dissociated, forming ClO− anions. In the MPO-catalyzed reaction both H2O2 and ClO− co-exist for some time in solution. At these conditions electronically excited form of molecular oxygen 1O2 is formed with high yield (30). In contrast to highly reactive HO• and , which act locally at the site of their generation, 1O2 exhibits extended lifetime in aqueous solution of ~3 μs and can migrate over 100 nm after its generation (31).
Furthermore, nitric oxide (NO•), which is a rather reactive inorganic radical, is generated from l-arginine in the presence of inducible NO-synthase in activated macrophages (32) and to the lesser degree in primed neutrophils themselves (33). Since macrophages are located at the infection site in close proximity to neutrophils, NO• produced by these cells can combine with generated by neutrophils in an extremely quick reaction with the formation of highly reactive ONOO−. All these species (ROS, NO•, and ONOO−) chemically modify and in this way deactivate extracellular receptors of neighboring T cells or even cause their apoptosis and necrosis (8–10).
Stimulation of ROS Production in Neutrophils by Chemical Compounds
Chemical and natural compounds amplifying the ROS amount in neutrophils can be classified as NOX2-dependent modulators, which are more common, and NOX2-independent ones.
NOX2-Dependent ROS Modulators
Protein Kinase C (PKC) Agonists
After its activation within cells, PKC catalyzes phosphorylation of the p47phox subunit of the protein associate p47phox/SH3/p7phox (Figure 3A). The resulting product migrates to the cellular membrane, where it is assembled due to the interaction between SH3 and p22phox subunits with formation of the functional NOX2 system (34). Therefore, compounds enhancing the PKC activity (PKC agonists) are expected to stimulate NOX2 formation and correspondingly increase ROS generation. Examples of such agonists include a variety of hydrophobic phorbol esters, e.g., phorbol myristate acetate (PMA) and phorbol dibutyrate (PDB). These compounds act in this way due to their similarity to the natural activator of PKC, diacylglycerol (DAC) (Figure 3A). They are broadly used in immunological research. However, these esters exhibit a number of undesired side effects in vivo, which prevents their therapeutic applications. For example, PMA is oncogenic and can cause fever (35). The effects of phorbol esters are attenuated by diphenyleneiodonium (DPI), a commonly used unspecific covalent inhibitor of NOX2, which binds to reduced flavin adenine dinucleotide in the gp91phox subunit (36). These data confirm that the increased ROS production in neutrophils in response PMA is derived from the NOX2 activity.
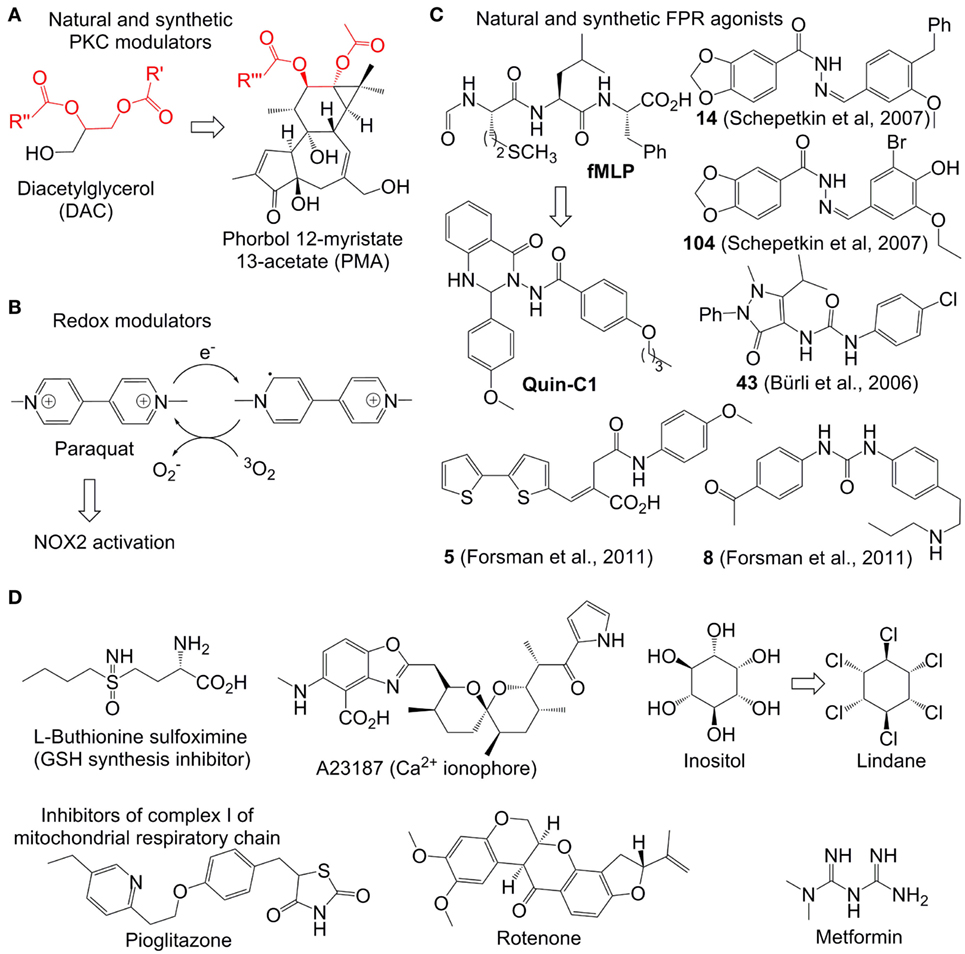
Figure 3. Representative reactive oxygen species (ROS) modulators in neutrophils. (A–C): NADPH oxidase 2(NOX2)-dependent modulators. Quin-C1 is an formyl peptide receptor agonist, but does not induce ROS in cells. (D) NOX2-independent modulators. Structurally related fragments in DAC and phorbol myristate acetate are indicated with red color.
The Ca2+-ionophore A23187 (Figure 3D) was found to further enhance the ROS-production in PDB-treated neutrophils (37). This synergy is logical, since the increased amount of intracellular Ca2+, caused by the treatment with Ca2+-ionophores such as A23187 and ionomycin, is expected to further stimulate PKC. Additionally to that, it has been found that Ca2+ strengthens binding of phorbol esters to their receptors (37). A number of other natural and synthetic agonists of PKC, which are either analogs of phorbol esters or unrelated structures, have been reported. However, clinical application of all known agonists is limited by substantial side effects (38).
Fully inorganic PKC activators, which are known to increase ROS in neutrophils in vitro, include ionic salts of the soft transition metal ions Zn2+, Cd2+, and Ni2+ ions. They are usually only active at the high concentration of ≥1 mM (39, 40). The metal ion effect is strongly dependent on the presence of chelating agents in the medium. Otherwise, the mechanism of activation is not known.
Redox Mediators
Paraquat is an intensely colored, dicationic 4,4′-bipyridinium salt (other name: methyl viologen) (Figure 3B). This compound is used as a broad spectrum herbicide. Its mode of action relies on its powerful electron acceptor properties. For example, in plants it accepts an electron from photosystem I with formation of a resonance-stabilized organic radical (Figure 3B). The latter species transfer the electron further to molecular oxygen with formation of followed by generation of other ROS (as described above). The toxic effects of ROS generation in plants explain the herbicidal properties of paraquat. In humans this drug is accumulated in lungs, where it catalyzes ROS production by mediation of the electron transfer to oxygen, analogously to its effect in plants, that leads to acute lung injury (41). In mammalian cells, NAPDH can potentially act as donor of electrons for paraquat (42). It has been reported that in neutrophils paraquat-induced ROS generation activates p38 MAPK and NF-kB signaling pathways thereby delaying neutrophil apoptosis. This effect is fully blocked by NOX2 inhibitors and partially blocked by PKC inhibitors confirming the involvement of the latter two enzymes in the paraquat-induced activation of neutrophils (43, 44).
Formyl Peptide Receptor (FPR) Agonists
Formyl peptide receptor is a G-protein coupled receptor expressed on the neutrophil membrane (45). Its stimulation by the bacteria-specific peptide formylmethionyl-leucyl-phenylalanine (fMLF) activates NOX2 thereby inducing ROS generation (Figure 3C). A number of organic molecules have been discovered, which act as fMLF mimics and, therefore, are able to stimulate ROS production in functional neutrophils. They include compounds 14, 104 (46), 43 (A) (47, 48), 5, and 10 (49). In contrast to the parent stimulant fMLF, all synthetic ligands are heterocyclic compounds. It is worth noting that many potent synthetic FPR ligands reported in the literature contain one N-N bond (Figure 3C). The role of this structural element still remains to be clarified.
Not all FPR agonists induce ROS production in neutrophils. One such example is the quinazolinone C derivative Quin-C1 (50, 51). Though it does not generate ROS, it induces mobilization of Ca2+, chemotaxis, and secretion of beta-glucuronidase.
Other known NOX2-activators include alkanes CnH2n+2 (n = 10–13) and phytol, which are active in the millimolar concentration range (24, 52) as well as a series of patented quinolinone derivatives (53).
NOX2-Independent ROS Modulators
Electron-Deficient Compounds Reactive With Sulfur- and Selen-Containing Biomolecules
A number of electron-deficient (electrophilic) chemical compounds increasing intracellular ROS amount in transformed and proliferating cells have been reported (Figure 3D). However, they are not necessarily applicable for modulation of ROS in neutrophils, which are terminally differentiated cells. For example, arsenic trioxide (As2O3, Trisenox) is a clinically approved inorganic drug for the treatment of acute promyelocytic leukemia. It is a relatively soft electrophile, which can coordinate soft nucleophilic sulfhydryl and selen-containing biomolecules in cells (54). For example, glutathione (GSH)-depleted cancer cells are especially sensitive to As2O3 indicating the important role of this tripeptide in As2O3-detoxification (55). The sulfhydryl- and selen-containing biomolecules including, e.g., GSH, glutathione reductase, and thioredoxin reductase participate in neutralization of ROS in the cells. Their deactivation by As2O3 usually leads to ROS increase in mammalian cells, which is believed to be one of the reasons of the anticancer activity of this drug. However, though the treatment of neutrophils with As2O3 causes their apoptosis, the latter is not associated with increase of the intracellular ROS amount (56). One possible explanation of this fact is the higher antioxidant capacity in neutrophils than that of As2O3– sensitive cancer cells. In particular, it has been found that even during the oxidative burst the amount of bulk antioxidants in neutrophils including GSH/GSSG, ascorbate, and vitamin E is not significantly altered (57).
Some highly potent drugs affecting homeostasis of intracellular thiols were found to induce oxidative stress also in myeloid cells. For example, l-buthionine sulfoximine (BSO, Figure 3D), an inhibitor of gamma-glutamyl-cysteine-synthase (γGCS) and suppressor of GSH synthesis, was found to enhance the oxidative stress in neutrophils and in cultured myeloid progenitors (58). This effect was also reproduced in vivo both for wild-type and X-linked CGD mice (59). Since NOX2 is not functional in the CGD mice, the BSO-induced ROS increase in the latter case should be NOX2 independent. In general, the effect of BSO and other GSH modulators on ROS production in neutrophils is substantially weaker than that in cancer cells. One possible explanation of that is the low level of GSH-dependent antioxidant enzymes in neutrophils, whose antioxidative protection seems to rely more strongly on the catalase activity (60).
Modulators of the Intracellular Concentration of Ca2+ ions
Ca2+ ions are present in high concentrations in extracellular space and in the intracellular organelle ER. Release of this metal ion into the cytoplasm triggers a number of biochemical processes including, e.g., NET-formation and mitochondrial permeability transition (61–63). Ca2+ transfer across the membrane can be induced by Ca2+ ionophores, e.g., A23187 or ionomycin. The ROS-enhancing effects of these ionophores were demonstrated for neutrophils as well as other cells (61–63). However, ROS produced by NOX2 are not essential for the effects of both ionophores, e.g., on formation of NETs (61).
Inositol Mimics
γ-Hexaclorocyclohexane (common abbreviation HCCH, other name Lindane) is a potent insecticide. It is also used for the treatment of scabies and lice infestation. However, these pharmaceutical applications have been restricted in many western countries due to pronounced side effects of Lindane, including neuro-, nephro-, and hepatotoxicity. Moreover, this compound was classified as carcinogenic for humans (group 1). Since Lindane is structurally related to inositol, it affects the phosphatidylinositol (PI) cycle in many cells (64), including those of myeloid origin. In particular, this drug triggers ROS amplification in pulmonary alveolar macrophages that is accompanied by increase of the PI turnover and intracellular Ca2+ concentration (65). The modulation of ROS and intracellular Ca2+ by Lindane was also observed in neutrophils. Additionally, this reagent was found to stimulate degranulation, but did not act as a chemotactic agent, which distinguishes it from stimulants like PMA (66). Apart from being an inositol mimic, HCCH is a hydrophobic molecule. It seems to be aggregated in aqueous solution as evidenced by its concentration-dependent octanol-water partition coefficients ranging from logP = 3.7 at 10 mg/L to logP = 3.9 at <0.1 μg/L (67). Due to these hydrophobic properties, Lindane can interact efficiently with cellular membranes leading to reorganization of phospholipids. In such altered membranes the phospholipids are more exposed to the external factors and, therefore, prone to degradation (68). One of the products formed during this degradation process is arachidonic acid (AA). AA can stimulate ROS production at least via two alternative pathways. In particular, AA as well as its metabolites can potentially activate NOX2 (69). Moreover, AA inhibits complex I and III of the mitochondrial respiratory chain that causes electron leakage and generation of superoxide anion radicals as well as other ROS (70). Since NOX2 inhibitors do not attenuate the HCCH-induced ROS-modulation in neutrophils, one can conclude that the mitochondrial AA-mediated pathway predominates in the Lindane-induced oxidative stress in the cells of this type.
Effectors of Mitochondrial ROS
Thioglitazones, e.g., pioglitazone and related derivatives, are drugs exhibiting insulin-sensitizing properties (71). They are approved for the treatment of type 2 diabetes. Moreover, these compounds are known to have moderate antiproliferative and anti-inflammatory activity (72). The mechanism of action of thioglitazones relies on activation (agonist properties) of peroxisome proliferator-activated receptor γ, which is a transcription factor of the nuclear receptor family. Moreover, they are known to induce production of mitochondrial ROS both in neutrophils and other cell types (73, 74) due to inactivation of complex I of the mitochondrial respiratory chain (75). Other common inhibitors of the complex I including rotenone (a natural compound, used as insecticide and pesticide) and metformin (a synthetic compound, first-line drug for the treatment of diabetes type 2) also induce ROS generation in neutrophils (76). As expected, this effect is not NOX2-dependent. Therefore, these drugs can be used for ROS modulation in NOX2-deficient states, as it has been demonstrated for pioglitazone in gp91phox−/− mice (a CGD model) and ex vivo for primary cells from X-linked CGD patients (74).
Neutrophil-Specific ROS Modulation
It has been convincingly confirmed in vitro and in vivo in several animal models of human inflammatory and autoimmune diseases that controlled increase of ROS can be beneficial for the pathological conditions caused by insufficient NOX2 activity in neutrophils, e.g., CGD (74), RA (7, 24), and systemic lupus erythematosus (77). Therefore, it would be appealing to use ROS amplifiers for the treatment of these and related diseases. However, known drugs of this type exhibit a number of undesired side effects due to their influence on other cells than neutrophils. Therefore, for the further clinical development of ROS-enhancing therapies, drugs specific toward neutrophils and able to trigger ROS production at the desired time points, including, e.g., pathogen challenge in CGD, are desirable.
The targeting of the therapeutic agents can in principle be conducted by using approaches developed for non-invasive tracking of neutrophils in vivo. For example, neutrophils were successfully labeled and monitored using quantum dots conjugated to a monoclonal antibody, raised against GPI-anchored myeloid differentiation marker (Ly6G or Gr1).
Though the labeled neutrophils remained fully functional in this case (78), higher loading of the drug would be necessary for a functional therapy approach. At these conditions antibodies against Ly6G (per example clone 1A8) are known to cause neutropenia in vivo (79). Another known approach for neutrophil labeling in mice makes use of targeting FPR. In particular, it has been demonstrated that conjugates of non-natural peptide ligand cinnamoyl-F-(D)L-F-(D)-L-FK (cFLFLF) with either the radioactive 64Cu complex (80) or the near-infrared fluorescence imaging probe Cy7 (81) can be used for neutrophil labeling and monitoring in mice: binding affinity of the Cu-conjugate to FPR was found to be ~18 nM. The weak agonistic effect of cFLFLF on FPR is not crucial in this case, since low loading of the detectable moiety is sufficient for the labeling. However, higher loading, required for functional therapy, is expected to cause unspecific neutrophil activation triggering an unwanted immune response.
Albertine and Gee (82) have reported on an alternative, receptor-independent approach for neutrophil labeling in vivo. In particular, they applied the hydrophobic cationic dye PKH26, which is commercially available from Merck (previously Sigma-Aldrich). Under optimized conditions this dye forms micro-aggregates, which are preferentially taken up by neutrophils in vivo. However, it has been found later on that these aggregates are also taken by other phagocytic cells, e.g., macrophages. Though at the concentrations required for labeling PKH26 is not toxic, in the presence of day light the toxicity is dramatically increased (83). Furthermore, PKH26 and related structures containing a positively charged polar head and two hydrophobic tail groups at higher concentrations, which would be required for the therapy, can potentially induce lysis of red blood cells. Thus, we conclude that all known neutrophil-targeting approaches are not ideal for therapeutic applications.
This warrants further research efforts in this field. A possible solution of the problems addressed above is the use of aminoferrocene-based prodrugs (ABP), which were originally developed for targeting cancer cells (84–91). A structure of the parent ABP is shown in Figure 4. Here the ferrocene fragment is a functional unit, which is covalently attached to an electron-acceptor protecting group (arylmethyloxycarbonylamino). Therefore, the molecules of this type are relatively electron-deficient and, correspondingly, do not act as electron donors both in the extracellular space and in cells (84). Furthermore, the ABP contains a triggering moiety (TM). Its removal leads to formation of unstable phenol or aniline derivatives, which spontaneously undergo 1,6-elimination with formation of substituted aminoferrocenes (AmFc). The AmFc derivatives are substantially more electron-rich than the parent ABPs (the redox potential ΔE1/2 is shifted by ca. 0.3V). Therefore, in contrast to the ABPs, they act as electron donors for endogenous H2O2 and 3O2 leading to formation of highly reactive ROS (HO• and ) as well as ferrocenium cations [AmFc]+. The latter product can be reduced back to the AmFc by endogenous bulk reducing agents (GSH, ascorbate, and NADPH), thereby closing the catalytic cycle. The ABPs were found to be powerful ROS amplifiers both in vitro (experiments with cell cultures and primary cells) and in vivo (experiments with mice and rats). By the variation of the TM moiety one can potentially design any cell-specific prodrug providing the cell-specific enzymatic activity is known. For example, already validated TM’s include aryl boronic acid esters (cancer cell-specific and activated under conditions of enhanced oxidative stress), arylazides (hypoxia-specific activation), and carboxylic acid ester (activation in the presence of esterases). Therefore, it is of high interest to explore the possibility of using these versatile prodrugs for design of neutrophil-specific ROS amplifiers.
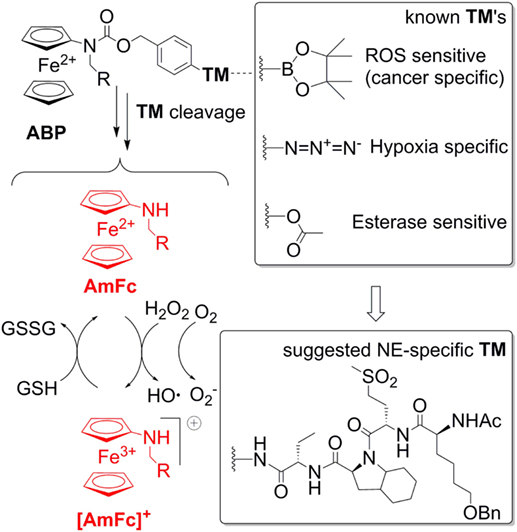
Figure 4. A general structure of aminoferrocene-based prodrugs (ABPs) and the mechanisms of their activation after cleavage of the triggering moiety (TM). The NE-sepcific TM is based on the peptide reported in Ref. (92).
Current Ideas and Future Perspectives
In pilot studies, we confirmed that a representative ABP compound, MIS43 (81), is able to enhance ROS in normal neutrophils. This effect is not inhibited by DPI, which indicates that it is not NOX2-dependent. We observed that at the selected experimental conditions MIS43 is a more potent ROS amplifier than known ones including Lindane and BSO, whereas the effect of PMA was found to be stronger (unpublished results).
To create a neutrophil-specific ROS amplifier, we suggest incorporating neutrophil elastase (NE)-specific moieties as TMs in the ABP structure (Figure 4). We selected NE as a neutrophil target, since it is released in the extracellular space and in phagosome and activated only upon neutrophil stimulation, whereas its activity is absent in inactive neutrophils. Moreover, it is known that NOX2-deficient neutrophils release large amounts of NE (93). Therefore, potentially such NE-specific ABPs can be used to enhance ROS specifically in NOX2-deficient neutrophils, which are relevant for CGD, RA, and lupus. As a NE-specific TM one can select a reactive fragment from the number of reported NE-specific fluorogenic substrates. For example, the non-natural tetrapeptide Ac-Nle(OBzl)-Met(O)2-Oic-Abu- could be suitable. In particular, this tetrapeptide is NE-specific. Moreover, since it is an artificial structure, it is not expected to be unspecifically degraded in vivo (92).
Author Contributions
VR helped in drafting the manuscript and the figures. JH, CM, CC, LM, MaHe, and MaHo provided scientific input to the manuscript. AM provided the first draft and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
MaHe and MaHo were supported by the German Research Foundation (DFG) (grant number SFB 1181-C03). MaHo further received support by the EU-project RISE-Redoxit (H2020-MSCA-RISE-2014, Project 644035). AM thanks German Research Council (DFG, grant MO 1418/7-1), Emerging Field Initiative of Friedrich-Alexander-University of Erlangen-Nuremberg (grant 3_Nat_01, “Chemistry in live cells”), and Interdisciplinary Center of Molecular Materials (ICMM, a Ph.D.-fellowship for VR) for funding this project. AM also acknowledges the partial support from the EU-project RISE-CLATHROPROBES (H2O2-MSCA-RISE-2017, Project 778245).
References
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi:10.1126/science.1092385
2. Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol (2016) 12:402–13. doi:10.1038/nrneph.2016.71
3. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6:173–82. doi:10.1038/nri1785
4. Kalyan S, Kabelitz D. When neutrophils meet T cells: beginnings of a tumultuous relationship with underappreciated potential. Eur J Immunol (2014) 44:627–33. doi:10.1002/eji.201344195
5. Holmdahl R, Sareila O, Olsson LM, Bäckdahl L, Wing K. Ncf1 polymorphism reveals oxidative regulation of autoimmune chronic inflammation. Immunol Rev (2016) 269(1):228–47. doi:10.1111/imr.12378
6. Hoffmann MH, Griffiths HR. The dual role of ROS in autoimmune and inflammatory diseases: evidence from preclinical models. Free Radic Biol Med (2018). doi:10.1016/j.freeradbiomed.2018.03.016
7. Hultqvist M, Olsson LM, Gelderman KA, Holmdahl R. The protective role of ROS in autoimmune disease. Trends Immunol (2009) 30(5):201–8. doi:10.1016/j.it.2009.03.004
8. Gabrilovich DI, Nagaraj S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9(3):162–74. doi:10.1038/nri2506
9. Franchina DG, Dostert C, Brenner D. Reactive oxygen species: involvement in T cell signalling and metabolism. Trends Immunol (2018) 39(6):489–502. doi:10.1016/j.it.2018.01.005
10. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol (2018) 19:108–19. doi:10.1038/s41590-017-0022-x
11. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med (2014) 20(5):511–7. doi:10.1038/nm.3547
12. Reinwald C, Schauer C, Csepregi JZ, Kienhöfer D, Weidner D, Malissen M, et al. Reply to "neutrophils are not required for resolution of acute gouty arthritis in mice". Nat Med (2016) 22(12):1384–6. doi:10.1038/nm.4217
13. Maueröder C, Kienhöfer D, Hahn J, Schauer C, Manger B, Schett G, et al. How neutrophil extracellular traps orchestrate the local immune response in gout. J Mol Med (2015) 93(7):727–34. doi:10.1007/s00109-015-1295-x
14. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (2000) 79(3):170–200. doi:10.1097/00005792-200005000-00004
15. De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquessen M, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol (2008) 122(6):1097–103. doi:10.1016/j.jaci.2008.07.050
16. Magnani A, Brosselin P, Beauté J, de Vergnes N, Mouy R, Debré M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol (2014) 134(3):655–62. doi:10.1016/j.jaci.2014.04.014
17. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Holland SM, Ochs H, Quie P, Buckley RH, Foster CB, Chanock SJ, Dickler H. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (2000) 79(3):155–69. doi:10.1097/00005792-200005000-00003
18. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med (2010) 363(27):2600–10. doi:10.1056/NEJMoa1007097
19. Marciano BE, Zerbe CS, Falcone EL, Ding L, DeRavin SS, Daub J, et al. X-linked carriers of chronic granulomatous disease: illness, lyonization, and stability. J Allergy Clin Immunol (2018) 141(1):365–71. doi:10.1016/j.jaci.2017.04.035
20. Kang EM, Marciano BE, DeRavin S, Zarember KA, Holland SM, Malech HL. Chronic granulomatous disease: overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol (2011) 127(6):1319–26. doi:10.1016/j.jaci.2011.03.028
21. Keller MD, Notarangelo LD, Malech HL. Future of care for patients with chronic granulomatous disease: gene therapy and targeted molecular medicine. J Pediatric Infect Dis Soc (2018) 7(Suppl_1):S40–4. doi:10.1093/jpids/piy011
22. Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP, et al. Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells. Mol Ther (2011) 19(1):122–32. doi:10.1038/mt.2010.226
23. De Ravin SS, Li L, Wu X, Choi U, Allen C, Koontz S, et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med (2017) 9(372):eaah3480. doi:10.1126/scitranslmed.aah3480
24. Hultqvist M, Olofsson P, Geldermann KA, Holmberg J, Holmdahl R. A new arthritis therapy with oxidative burst inducers. PLoS Med (2006) 3(9):e348. doi:10.1371/journal.pmed.0030348
25. Chaudhari N, Talwar P, Parimisetty A, Lefebvre d’Hellencourt C, Ravanan P. A molecular web: endoplasmic reticulum stress, inflammation, and oxidative stress. Front Cell Neurosci (2014) 8:213. doi:10.3389/fncel.2014.00213
26. Bienert GP, Moller ALB, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, et al. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J Biol Chem (2007) 282:1183–92. doi:10.1074/jbc.M603761200
27. Dynowski M, Schaaf G, Loque D, Moran O, Ludewig U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem J (2008) 414:53–61. doi:10.1042/BJ20080287
28. Terman A, Kurz T, Gustafsson B, Brunk UT. Lysosomal labilization. IUBMB Life (2006) 58(9):531–9. doi:10.1080/15216540600904885
29. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science (1992) 257:1496–502. doi:10.1126/science.1523409
30. Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol (2005) 77:598–625. doi:10.1189/jlb.1204697
31. Hatz S, Lambert JD, Ogilby PR. Measuring the lifetime of singlet oxygen in a single cell: addressing the issue of cell viability. Photochem Photobiol Sci (2007) 6(10):1106–16. doi:10.1039/b707313e
32. Fang FC, Vazquez-Torres A. Nitro oxide production by human macrophages: there is NO doubt about it. Am J Physiol Lung Cell Mol Physiol (2002) 282:941–3. doi:10.1152/ajplung.00017.2002
33. Wright CD, Mülsch A, Busse R, Osswald H. Generation of nitric oxide by human neutrophils. Biochem Biophys Res Commun (1989) 160(2):813–9. doi:10.1016/0006-291X(89)92506-0
34. Rastogi R, Geng X, Li F, Ding Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front Cell Neurosci (2017) 10:301. doi:10.3389/fncel.2016.00301
35. McKernan LN, Momjian D, Kulkosky J. Protein kinase C: one pathway towards the eradication of latent HIV-1 reservoirs. Adv Virol (2012) 2012:805347. doi:10.1155/2012/805347
36. Ellis JA, Mayer SJ, Jones OT. The effect of the NADPH oxidase inhibitor diphenyleneiodonium on aerobic and anaerobic microbicidal activities of human neutrophils. Biochem J (1988) 251(3):887–91. doi:10.1042/bj2510887
37. French JK, Hurst NP, Zalewski PD, Valente L, Forbes IJ. Calcium ionophore A23187 enhances human neutrophil superoxide release, stimulated by phorbol dibutyrate, by converting phorbol ester receptors from a low- to high-affinity state. FEBS Lett (1987) 212(2):242–6. doi:10.1016/0014-5793(87)81353-4
38. Jiang G, Dandekar S. Targeting NF-kB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. Aids Res Hum Retrovir (2015) 31:4–12. doi:10.1089/aid.2014.0199
39. Freitas M, Gomes A, Porto G, Fernandes E. Nickel induces oxidative burst, NF-kB activation and interleukin-8 production in human neutrophils. J Biol Inorg Chem (2010) 15:1275–83. doi:10.1007/s00775-010-0685-3
40. Freitas M, Porto G, Lima JLFC, Fernandes E. Zinc activates neutrophils’ oxidative burst. Biometals (2010) 23:31–41. doi:10.1007/s10534-009-9264-x
41. Dinis-Oliveira RJ, Sarmento A, Reis P, Amaro A, Remião F, Bastos ML, et al. Acute paraquat poisoning: report of a survival case following intake of a potential lethal dose. Pediatr Emerg Care (2006) 22(7):537–40. doi:10.1097/01.pec.0000223179.07633.8a
42. Bus JS, Aust SD, Gibson JE. Superoxide- and singlet oxygen-catalyzed lipid peroxidation as a possible mechanism for paraquat (methyl viologen) toxicity. Biochem Biophys Commun (1974) 58(3):749–55. doi:10.1016/S0006-291X(74)80481-X
43. Mitra S, Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochim Biophys Acta (2006) 1762(8):732–41. doi:10.1016/j.bbadis.2006.06.011
44. Wang X, Luo F, Zhao H. Paraquat-induced reactive oxygen species inhibit neutrophil apoptosis via a p38 MAPK/NF-kB-IL-6/TNF-α positive-feedback circuit. PLoS One (2014) 9(4):e93837. doi:10.1371/journal.pone.0093837
45. Fu H, Karlsson J, Bylund J, Movitz C, Karlsson A, Dahlgren C. Ligand recognition and activation of formyl peptide receptors in neutrophils. J Leukoc Biol (2006) 79(2):247–56. doi:10.1189/jlb.0905498
46. Schepetkin IA, Kirpotina LN, Khlebnikov AI, Quinn MT. High-throughput screening for small-molecule activators of neutrophils: identification of novel N-formyl peptide receptor agonists. Mol Pharm (2007) 71:1061–74. doi:10.1124/mol.106.033100
47. Bürli RW, Xu H, Zou X, Muller K, Golden J, Frohn M, et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorg Med Chem Lett (2006) 16:3713–8. doi:10.1016/j.bmcl.2006.04.068
48. Sogawa Y, Shimizugawa A, Ohyama T, Maeda H, Hirahara K. The pyrazolone originally reported to be a formyl peptide receptor (FPR) 2/ALX-selective agonist is instead an FPR1 and FPR2/ALX dual agonist. J Pharmacol Sci (2009) 111(3):317–21. doi:10.1254/jphs.09196SC
49. Forsman H, Kalderén C, Nordin A, Nordling E, Jensen AJ, Dahlgren C. Stable formyl peptide receptor agonists that activate the neutrophil NADPH-oxidase identified through screening of a compound library. Biochem Pharmacol (2011) 81:402–11. doi:10.1016/j.bcp.2010.11.005
50. Nanamori M, Cheng X, Mei J, Sang H, Xuan Y, Zhou C, et al. A novel nonpeptide ligand for formyl peptide receptor-like 1. Mol Pharmacol (2004) 66:1213–22. doi:10.1124/mol.104.004309
51. Zhou C, Zhang S, Nanamori M, Zhang Y, Liu Q, Li N, et al. Pharmacological characterization of a novel nonpeptide antagonist for formyl peptide receptor like 1. Mol Pharmacol (2007) 72(4):976–83. doi:10.1124/mol.107.037564
52. Olofsson P, Holmberg J, Tordsson J, Lu S, Akerström B, Holmdahl R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat Genet (2003) 33:25–32. doi:10.1038/ng1058
53. Wallner KF, Olofsson JP, Hultquist MK, Pelcman BH. Quinolinone derivatives for use in the treatment of an autoimmune disease and/or an inflammatory disease. WIPO Patent (2011) WO2012127214A1.
54. Li YM, Broome JD. Arsenic targets tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res (1999) 59:776–80.
55. Yang CH, Kuo ML, Chen JC, Chen YC. Arsenic trioxide sensitivity is associated with low level of glutathione in cancer cells. Br J Cancer (1999) 81(5):796–9. doi:10.1038/sj.bjc.6690766
56. Binet F, Cavalli H, Moisan É, Girard D. Arsenic trioxide (AT) is a novel human neutrophil pro-apoptotic agent: effects of catalase on AT-induced apoptosis, degradation of cytoskeletal proteins and de novo protein synthesis. Br J Haematol (2005) 132:349–58. doi:10.1111/j.1365-2141.2005.05866.x
57. Ogino T, Packer L, Maguire JJ. Neutrophil antioxidant capacity during the respiratory burst: loss of glutathione induced by chloramines. Free Radic Biol Med (1997) 23(3):445–52. doi:10.1016/S0891-5849(97)00115-9
58. Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, Nombela-Arrieta C, et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity (2015) 42:159–71. doi:10.1016/j.immuni.2014.12.017
59. Zhu H, Kwak H-J, Liu P, Bajrami B, Xu Y, Park S-Y, et al. Reactive oxygen species-producing myeloid cells act as a bone marrow niche for sterile inflammation-induced granulopoiesis. J Immunol (2017) 198:2854–64. doi:10.4049/jimmunol.1602006
60. Kinnula VL, Soini Y, Kvist-Mäkelä K, Savolainen ER, Koistinen P. Antioxidant defense mechanism in human neutrophils. Antioxid Redox Signal (2002) 4(1):27–34. doi:10.1089/152308602753625825
61. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife (2017) 6:e24437. doi:10.7554/eLife.24437
62. Kajitani N, Kobuchi H, Fujita H, Yano H, Fujiwara T, Yasuda T, et al. Mechanism of A23187-induced apoptosis in HL-60 cells: dependency on mitochondrial permeability transition, but not on NADPH oxidase. Biosci Biotechnol Biochem (2007) 71(11):2701–11. doi:10.1271/bbb.70304
63. Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochim Biophys Acta (2009) 1787(11):1395–401. doi:10.1016/j.bbabio.2009.06.009
64. Hokin MR, Brown DF. Inhibition by gamma-hexachlorocyclohexane of acetylcholine-stimulated phosphatidylinositol (PI) synthesis in cerebral cortex slices and of phosphatidic acid-inositol transferase in cerebral cortex particular fractions. J Neurochem (1969) 16:475. doi:10.1111/j.1471-4159.1969.tb06846.x
65. Holian A, Marchiarullo MA, Stickle DF. Gamma-hexachlorocyclohexane activation in alveolar macrophage phosphatidylinositol cycle, calcium mobilization of O2- production. FEBS Lett (1984) 176(1):151–4. doi:10.1016/0014-5793(84)80930-8
66. English D, Schell M, Siakotos A, Gabig TG. Reversible activation of the neutrophil superoxide generating system by hexachlorocyclohexane: correlation with effects on a subcellular superoxide-generating fraction. J Immunol (1986) 137:283–90.
67. Paschke A, Schüürmann G. Concentration dependence of the octanol/water partition coefficients of the hexachlorocyclohexane isomers at 25°C. Chem Eng Technol (2000) 23(8):666–70. doi:10.1002/1521-4125(200008)23:8<666::AID-CEAT666>3.0.CO;2-5
68. Parries GS, Hokin-Neaverson M. Inhibition of phosphatidylinositol synthase and other membrane-associated enzymes by stereoisomers of hexachlorocyclohexane. J Biol Chem (1985) 260(5):2687–93.
69. Matono R, Mlyano K, Klyohara T, Sumimoto H. Arachidonic acid induces direct interaction of the p67phox-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. J Biol Chem (2014) 289(36):24874–84. doi:10.1074/jbc.M114.581785
70. Cocco T, De Paola M, Papa S, Lorusso M. Arachidonic acid interaction with the mitochondrial electron transport chain promotes reactive oxygen species generation. Free Radic Biol Med (1999) 27(1–2):51–9. doi:10.1016/S0891-5849(99)00034-9
72. Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol (2005) 70:177–88. doi:10.1016/j.bcp.2005.03.033
73. Pérez-Ortiz JM, Tranque P, Burgos M, Vaquero CF, Llopis J. Glitazones induce astroglioma cell death by releasing reactive oxygen species from mitochondria: modulation of cytotoxicity by nitric oxide. Mol Pharm (2007) 72(2):407–17. doi:10.1124/mol.106.032458
74. Fernandez-Boyanapalli RF, Frasch SC, Thomas SM, Malcolm KC, Nicks M, Harbeck RJ, et al. Pioglitazone restores phagocyte mitochondrial oxidants and bactericidal capacity in chronic granulomatous disease. J Allergy Clin Immunol (2015) 135(2):517–27.e12. doi:10.1016/j.jaci.2014.10.034
75. García-Ruiz I, Solís-Muños P, Fernández-Moreira D, Muñoz-Yagüe T, Solís-Herruzo JA. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol (2013) 11:88. doi:10.1186/1741-7007-11-88
76. Zmijewski JW, Lorne E, Zhao X, Tsuruda Y, Sha Y, Liu G, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med (2008) 178:168–79. doi:10.1164/rccm.200710-1602OC
77. Kienhöfer D, Hahn J, Stoof J, Csepregi JZ, Reinwald C, Urbonaviciute V, et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight (2017) 2(10):e92920. doi:10.1172/jci.insight.92920
78. Kikushima K, Kita S, Higuchi H. A non-invasive imaging for the in vivo tracking of high-speed vesicle transport in mouse neutrophils. Sci Rep (2013) 3:1913. doi:10.1038/srep01913
79. Daley JM, Thomay AA, Connolly MD, Reicher JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol (2008) 83(1):64–70. doi:10.1189/jlb.0407247
80. Locke LW, Chordia MD, Zhang Y, Kundu B, Kennedy D, Landseadel J, et al. A novel neutrophil-specific PET imaging agent: cFLFLFK-PEG-64Cu. J Nucl Med (2009) 50:790–7. doi:10.2967/jnumed.108.056127
81. Xiao L, Zhang Y, Berr SS, Chordia MD, Pramoonjago P, Pu L, et al. A novel near-infrared fluorescence imaging probe for in vivo neutrophil tracking. Mol Imaging (2012) 11(5):372–82. doi:10.2310/7290.2011.00054
82. Albertine KH, Gee MH. In vivo labeling of neutrophils using a fluorescence cell linker. J Leuk Biol (1996) 59:631–8. doi:10.1002/jlb.59.5.631
83. Oh DJ, Lee GM, Francis K, Palsson BO. Phototoxicity of the fluorescent membrane dyes PKH2 and PKH26 on the human hematopoietic KG1a progenitor cell line. Cytometry (1999) 36:312–8. doi:10.1002/(SICI)1097-0320(19990801)36:4<312::AID-CYTO5>3.0.CO;2-V
84. Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J Med Chem (2012) 55(2):924–34. doi:10.1021/jm2014937
85. Marzenell P, Hagen H, Sellner L, Zenz T, Grinyte R, Pavlov V, et al. Aminoferrocene-based prodrugs and their effects on human normal and cancer as well as bacterial cells. J Med Chem (2013) 56(17):6935–44. doi:10.1021/jm400754c
86. Schikora M, Reznikov A, Chaykovskaya L, Sachinska O, Polyakova L, Mokhir A. Activity of aminoferrocene-based prodrugs against prostate cancer. Bioorg Med Chem Lett (2015) 25(17):3447–50. doi:10.1016/j.bmcl.2015.07.013
87. Daum S, Chekhun V, Todor I, Lukianova N, Shvets Y, Sellner L, et al. Improved synthesis of N-benzylaminoferrocene-based prodrugs and evaluation of their toxicity and antileukemic activity. J Med Chem (2015) 58(4):2015–24. doi:10.1021/jm5019548
88. Kinski E, Marzenell P, Hofer W, Hagen H, Raskatov JA, Knaup KX, et al. 4-Azidobenzyl ferrocenylcarbamate as an anticancer prodrug activated at reductive conditions. J Inorg Biochem (2016) 160:218–24. doi:10.1016/j.jinorgbio.2016.02.023
89. Reshetnikov V, Daum S, Mokhir A. Cancer specific, intracellular, reductive activation of anticancer Pt(IV)-prodrugs. Chem Eur J (2017) 23(24):5678–81. doi:10.1002/chem.201701192
90. Daum S, Reshetnikov V, Sisa M, Dumych T, Lootsik MD, Bilyy R, et al. Lysosome-targeting amplifiers of reactive oxygen species as anticancer prodrugs. Angew Chem Int Ed Engl (2017) 56(49):15545–9. doi:10.1002/anie.201706585
91. Daum S, Babiy S, Konovalova H, Hofer W, Shtemenko A, Shtemenko N, et al. Tuning the structure of aminoferrocene-based anticancer prodrugs to prevent their aggregation in aqueous solution. J Inorg Biochem (2018) 178:9–17. doi:10.1016/j.jinorgbio.2017.08.038
92. Kasparkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, et al. Design of ultrasensitive probes for human neutrophil esterase through hybrid combinatorial substrate library profiling. Proc Natl Acad Sci U S A (2014) 111(7):2518–23. doi:10.1073/pnas.1318548111
Keywords: aminoferrocenes, autoimmune disease, chronic granulomatous disease, inflammation, NADPH oxidase 2, neutrophils, reactive oxygen species, therapy
Citation: Reshetnikov V, Hahn J, Maueröder C, Czegley C, Munoz LE, Herrmann M, Hoffmann MH and Mokhir A (2018) Chemical Tools for Targeted Amplification of Reactive Oxygen Species in Neutrophils. Front. Immunol. 9:1827. doi: 10.3389/fimmu.2018.01827
Received: 18 April 2018; Accepted: 24 July 2018;
Published: 13 August 2018
Edited by:
Gabor Csanyi, Augusta University, United StatesReviewed by:
Angelo A. Manfredi, Università Vita-Salute San Raffaele, ItalyJohn Hancock, University of the West of England, United Kingdom
Copyright: © 2018 Reshetnikov, Hahn, Maueröder, Czegley, Munoz, Herrmann, Hoffmann and Mokhir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus H. Hoffmann, bWFya3VzLmhvZmZtYW5uQHVrLWVybGFuZ2VuLmRl;
Andriy Mokhir, YW5kcml5Lm1va2hpckBmYXUuZGU=