 Jessica C. Hargarten
Jessica C. Hargarten Peter R. Williamson
Peter R. Williamson- Laboratory of Clinical Immunology and Microbiology (LCIM), National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, United States
Autoimmune diseases are a significant cause of debilitation and mortality globally and are in need of cost-effective therapeutics. Autophagy is a cellular pathway that facilitates immune modulation involved in both pathogen control and autoimmunity. Regulation is multifactorial and includes a number of epigenetic pathways which can involve modification of DNA-binding histones to induce autophagy-related mRNA synthesis or microRNA and decapping-associated mRNA degradation which results in autophagy suppression. Appreciation of epigenetic-based pathways involved in autophagy and autoimmunity may facilitate application of a burgeoning group of epigenetic pharmaceuticals to these important diseases.
Introduction
Autoimmunity-related diseases are a common cause of debilitation globally. The search for treatments is an active area of research, in no part due to the fact that three of the top six best-selling prescription drugs in 2015 were for the control of autoimmune disorders. In addition to the widespread prevalence of these diseases, the compelling economic benefit of these agents is borne out by a recent study showing that maintenance of even a mild degree of inflammation in patients resulted in comparative employee productivity to that of unaffected employees (1).
FDA approved epigenetic drugs include the histone deacetylase inhibitors romidepsin (cutaneous and peripheral T-cell lymphomas), belonostat (refractory peripheral T-cell lymphoma), panobinostat (refractory multiple myeloma), and vorinostat (refractory T-cell lymphoma) as well as a number of histone acetyltransferase inhibitors such as azacitidine and decitabine (both for chronic myelomonocytic leukemia and myelodysplastic syndrome) (2). As development progresses, it is likely that pharmaceutical epigenetic therapies will be adapted to other diseases including autoinflammatory diseases and small molecule inhibitors, such as these, may prove cost-effective. While this could have tremendous implications for patients with these diseases, it is important to identify regulatory pathways inherent to epigenetic regulation including autophagy to minimize side effects that are unexpected only because of ignorance of a relevant pathway(s). “On target” treatment toxicity is common. For example, the increased risk of Aspergillus infections in patients taking the B-cell-directed Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib (3), due to an unexpected role of these inhibitors in a TLR9-BTK-calcineurin-nuclear factor of activated T-cells pathway in innate immunity to the fungus (4). Thus, a thorough understanding of the impact of epigenetic pathways may be key to avoiding unexpected toxicities of these agents.
The Balance of Autophagy During Infection and Autoimmunity
Autophagy was first described in yeast as a mechanism of intracellular recycling during nutrient stress (5). During cellular stress, specific autophagy-related proteins (designated Atg) orchestrate the sequestration of cytosolic materials to be recycled into a double-membraned structure, called the autophagosome (Figure 1D). Recently, the role of autophagy in mammalian immune modulation has been demonstrated in both innate and adaptive immunity (6, 7). Autophagy plays a direct role in eliminating invading pathogens by phagocytic processes (8), as well as MAP1LC3-associated phagocytosis (LAP) and sequestosome-like receptor recruitment (9). Autophagy also limits excessive inflammation during pathogen control by: removing residual microbial debris, known to activate the inflammasome pathway; digesting dysfunctional mitochondria, which typically mediate production of reactive oxygen species (ROS); or through direct removal of inflammasome complexes (10). These residual “mop up operations” of autophagy can also be induced by secondary “danger” signals (11) typically mediated by the mTOR pathway that harkens back to the role of this pathway in the yeast nutrient response. In adaptive immunity, autophagy also facilitates effective major histocompatibility complex presentation for T-cell activation (12), serving to control pathogens and remove inflammatory microbial products. Indeed, the importance of autophagy has been recognized by the pathogens themselves in that many utilize host autophagy to protect themselves against killing and support survival within host cells. The fungus Cryptococcus neoformans, which causes lethal meningoencephalitis (13), as well as certain bacterial pathogens, such as Mycobacterium tuberculosis (Mtb), have co-opted autophagic vesicles to conceal their intracellular residence and prevent lysosomal fusion and microbial killing (14, 15).
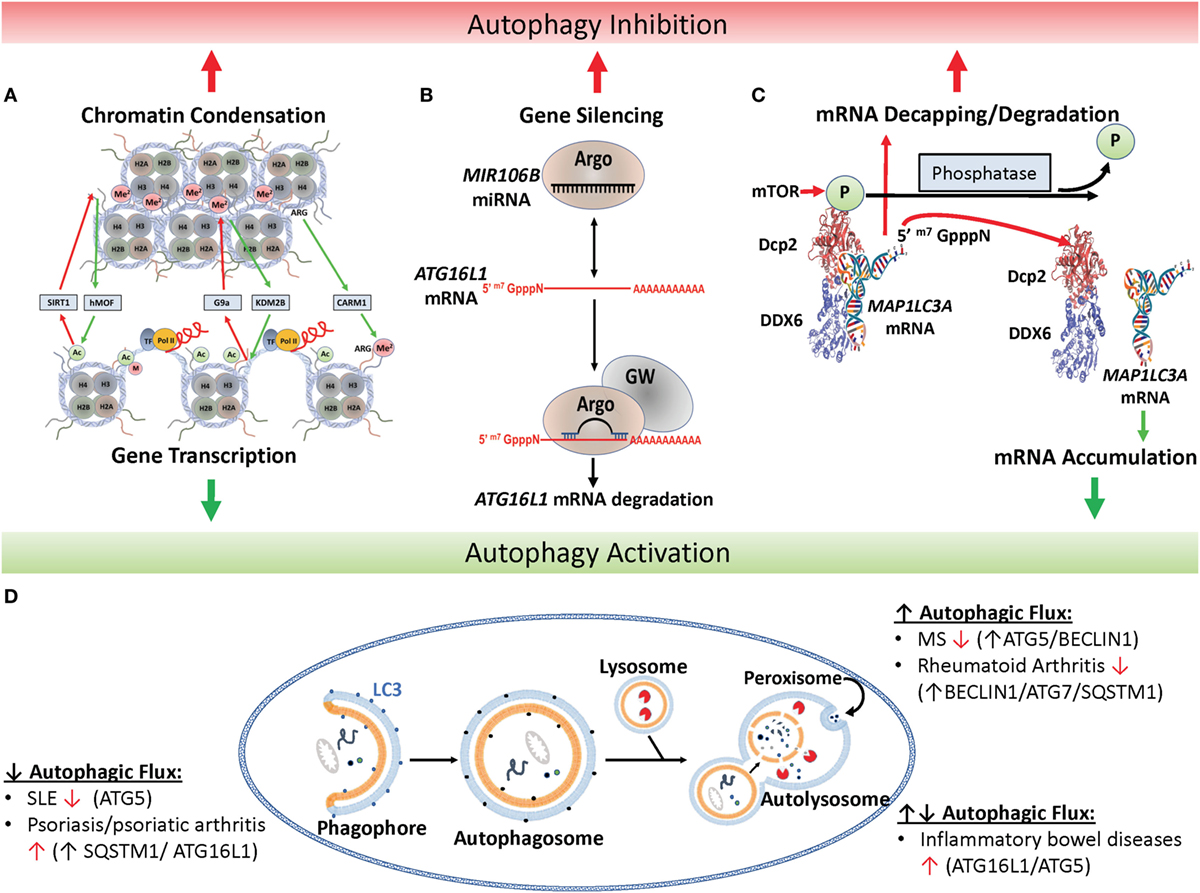
Figure 1. Epigenetic regulation of autophagy. (A) Histone marks facilitate either chromatin condensation (top panel) or an open matrix which facilitates transcription (lower panel). Repressors include the histone deacetylase SIRT1, the H3 histone methyltransferase G9a and activators include the H3 histone acetyltransferase hMof, the H3 demethylase KMS2B and the H3 arginine methyltransferase co-activator-associated arginine methyltransferase 1 (CARM1). (B) Canonical microRNAs (miRNAs), such as MIR106B, is recruited to Argonaut members (ARG) which recruits target mRNAs, such as the autophagy-related ATG16L1 mRNA, for degradation and gene silencing in concert with GW-motif proteins (GW). (C) Mechanism of mTOR-dependent decapping/degradation. mTOR-dependent phosphorylation of the decapping protein DCP2 facilitates recruitment of target mRNA molecules for decapping followed by degradation. Conversely, low mTOR activities in the presence of phosphatases result in dissociation of the decapping complex from the mRNA target with resultant accumulation of target transcripts, illustrated here with the autophagy-associated transcription, MAP1LC3A. [Model of putative mammalian DCP2-DDX6 interaction with MAP1LC3A mRNA adapted from Ref. (16).] (D) Illustration of autophagy and autoimmune diseases associated with alterations in autophagic flux. In autophagy, protein aggregates, misfolded proteins, and pathogens are recruited to the phagophore and then enclosed by a double-membrane vesicle to form the autophagosome. Following lysosome fusion with the autophagosome, proteinaceous material undergoes degradation in the autolysosome. Red arrow found next to autoimmune disease names indicates whether disease improves (↑) or declines (↓) following pharmacological inhibition of autophagy (17).
Although a direct link between autophagy, autoimmunity, and infectious disease is still under investigation, monogenic primary immune deficiencies in humans highlight the growing evidence for their interconnection. Activated PI3K-delta syndrome (APDS), for example, whose dysregulation results in immune-mediated cytopenias treatable by the PI3K inhibitor leniolisib (18)—has been associated with higher risk for developing autoimmune diseases (19). Similarly, chronic granulomatous disease (CGD), resulting from defects in the NADPH oxidase complex is not only associated with reduced ROS production, multiple recurrent infections (20), and chronic inflammation in patients—whose inflammatory colitis can be successfully treated with the IL-1 receptor antagonist, anakinra (21), but also autoimmunity (22). Interestingly, the increased IL-1β production in CGD was linked to a reduction in autophagy that also resulted in defects in phagocyte killing of internalized bacteria and fungi (23), demonstrating a link between autoimmunity, autophagy, and infectious disease.
Therapeutic interventions against autoimmune diseases are also strongly associated with susceptibility to infection. This is exemplified in patients undergoing treatment for multiple sclerosis (MS) who are at an increased risk of life-threatening Histoplasma capsulatum infections with the use of TNFα inhibitors, such as infliximab and etanercept (24), or increased risk of CNS infections with the very late antigen 4 (VLA-4) inhibitor, natalizumab, used for minimized autoimmune inflammation (25, 26). A number of “off-target” epigenetic side effects have also been described that associate autophagy and epigenetics. For example, the psychotropic drug lithium acts to downregulate HDAC1 translation, leading to decrease in histone deacetylation and upregulation of autophagy (27). Clearly, an appreciation of regulatory pathways related to autophagy and immunity will be useful to anticipate side effects of epigenetic modifying pharmaceuticals.
mRNA Transcript Synthesis: Role of Histone Modification
The field of epigenetics has been a slowly evolving and often controversial concept in genetics. Indeed, some of the first epigenetic molecular work was published in 1964 by Allfey et al. who proposed a role for histone modifications in gene regulation. However, the field progressed slowly until ignited by the synthesis of histone epigenetic studies by Strahl and Allis (28) and Turner (29). Since that time, epigenetic studies have identified a number of covalent histone post-translational modifications, including acetylation, methylation, phosphorylation (30, 31), ADP-ribosylation (32), ubiquitination (33), SUMOylation (30), citrullination (34), glycosylation (35), hydroxylation (36), and isomerization (37, 38). Prominent among these are acetylation and methylation with a number of these histone modifications related directly to the regulation of autophagy and will therefore be the focus of this review. However, since the field of epigenetic regulation of autoimmunity is still in its infancy, many areas remain to be elucidated.
Histone post-translational modifications control gene expression by a number of mechanisms including altering the electrostatic associations between nucleosomes, modulating interactions between nucleosomes and DNA, interfering with transcription factor binding to promoter/enhancer regions, or recruiting either activating or repressing protein complexes to the specific histone modification (39). Typical modifications occur at the epsilon amino group of lysine sidechains within the polypeptide and serve to reduce the electrostatic charge of histones by acetylation and methylation or reverse this by phosphorylation. Some of the best-known modifications affecting autophagy are exhibited in Figure 1A. For example, the histone acetyltransferase hMOF/KAT8 acts to add an acetyl group to H4K16 facilitating chromatin decondensation, which sterically allows transcriptional machinery and enhancers access to DNA facilitating expression of autophagy-related genes (40). Conversely, overexpression of the NAD-dependent histone deacetylase sirtuin 1 (SIRT1) antagonizes H4K16 acetylation reducing basal levels of autophagy, which can be inhibited by the drug valproic acid (41). However, the relationship is complicated by a feedback loop whereby SIRT1 acts on non-histone targets in an mTOR-dependent fashion to induce autophagy, which subsequently inhibits hMOF/KAT8 activity (40).
SIRT1 may also play a critical role in regulating the immune system by modulating the activity of essential transcriptional regulators. Specifically, SIRT1 deacetylates RAR-related orphan receptor gamma promoting its transcriptional activity and Th17 cell differentiation (40). In thymic epithelial cells, SIRT1 is an essential regulator of AIRE-mediated expression of tissue-restricted antigens, a critical step for immunological self-tolerance (42, 43). Interestingly, polymorphisms in SIRT1 are associated with autoimmune thyroiditis and high titers of anti-thyroid antibodies (44), suggesting a link between epigenetic regulators and autoimmunity. Immune consequences for overlapping regulation of autophagy and immunity can be seen with other related histone deacetylases. SIRT6 potentiates autophagy activation through effects on autophagy-related genes (ATG12, ATG3, and ATG7), as well as the well-known Crohn’s colitis-associated autophagy gene, IRGM (45, 46). Broad spectrum deacetylases, such as those found within the HDAC family—exemplified by HDAC4’s ability to deacetylate H3K9, 14, 18, and 23 and H4K5, 8, 12, and 16—are well known for their role in cancer biology prompting development of the HDAC inhibitors described above and in Table 1 (47). But they also show promise for the treatment of autoimmune diseases, as HDAC4 inhibitors have been shown to alleviate vascular inflammation resulting from activation of autophagy (48). Similarly, HDAC6 has been shown to modulate ATG6 (BECN1) and ATG7 and the inhibitor tubastatin A was found to potentiate autophagy with inhibition of the pro-inflammatory cytokine, IL-6 (49). These experiences suggest that further studies may identify epigenetic pathways relevant to HDAC inhibition that may prove useful in autoinflammatory disorders.
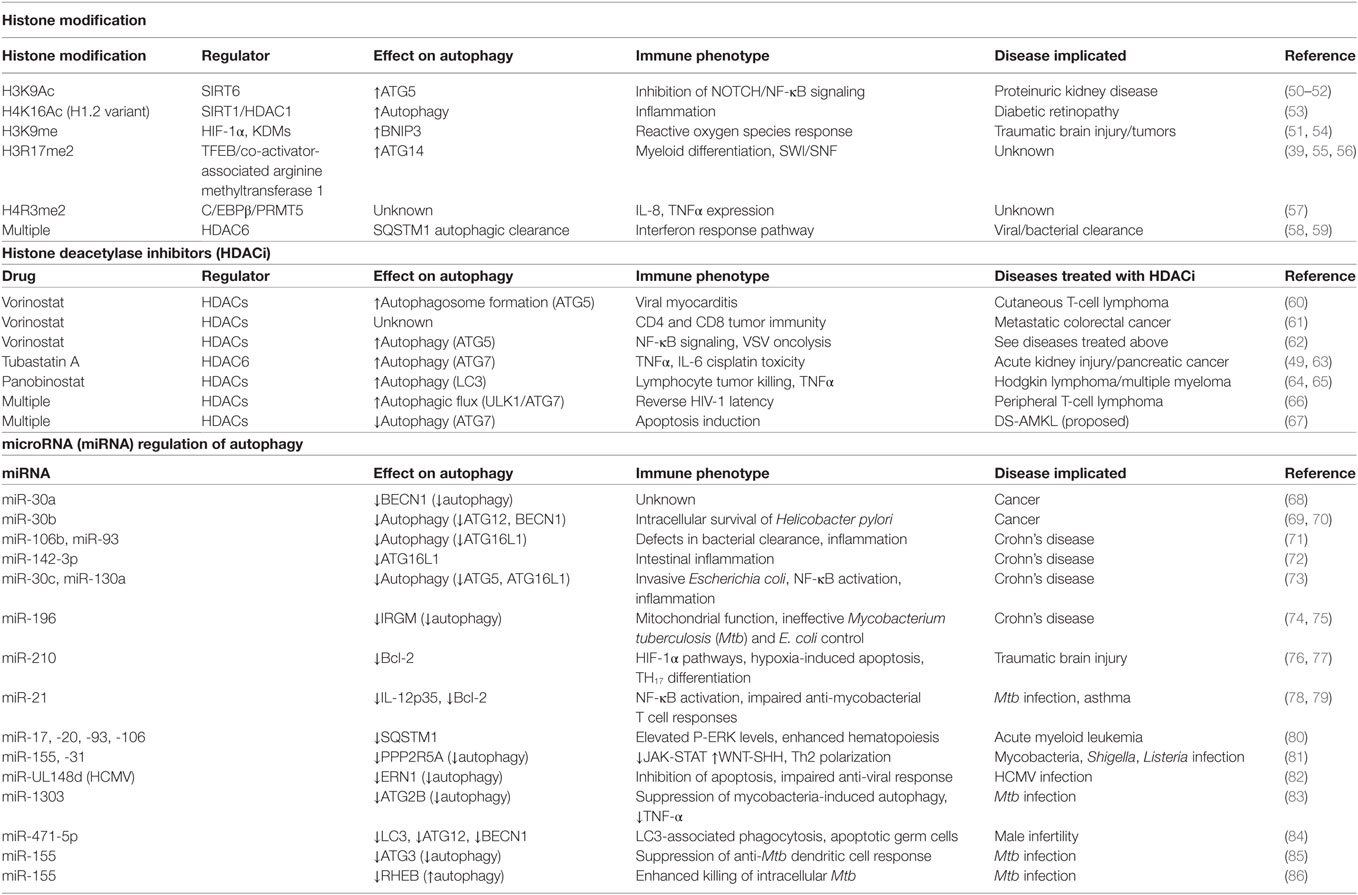
Table 1. Epigenetic regulators associated with autophagy and immunity.
A second set of histone modifiers are those involved in H3K9 methylation by the euchromatic histone-lysine N-methyltransferase 2 (G9a/EHMT2) and GLP/EHMT1 methyl transferases which form heteromeric complexes via their Su(var)3-9-Enhancer of zeste-trithorax (SET) domains (87). These lysine methyltransferases transfer one to three methyl groups from S-adenosyl-l-methionine to the target lysine e-amino group, causing a similar charge disruption as acetylation described above. In addition, H3K9me3 moieties (marks) recruit histone mark “readers” such as heterochromatin protein 1 (HP1) whose unique chromodomain mediates protein binding, heterochromatin formation (a tight lattice of DNA bound to histones), and transcriptional repression (Figure 1A, top panel). The methyltransferase activities of G9A/EHMT2 and the polycomb-repressive complex member, enhancer of zeste homolog 2 (EZH2), converge onto H3K27, whose methylation is associated with derepression of mTOR (88) and autophagy repression (89). Pharmacological inhibition of G9a/EHMT2 with BIX01294 results in induction of autophagy demonstrated by increased LC3B-positive autophagic vesicles (90), but autoimmunity was not studied in this context. Long non-coding RNAs (lncRNA) add an additional layer of regulation to this pathway that might hint at a connect to autoimmunity. Specifically, the lncRNA HOTAIR acts as a scaffold to recruitment the histone methyltransferase EZH2 to target genes, facilitating H3K27me-mediated gene repression (91). Aberrant expression of HOTAIR is associated with various cancers (92) and MS (93).
Interestingly, the hypoxia-inducible transcription factor (HIF1α)—recently shown to impact T-cell differentiation (94) and B-cell-related autoimmune disease (95)—was also found to regulate the H3K9 lysine (K)-specific demethylase, KDM2B, as well as the related demethylase KDM1A. This epigenetic regulation leads to activation of autophagy as well as mTOR, NF-κB, and TGF-β pathways important in T-cell adaptive signaling (96). In addition, HP1 located at H3K9me3 marked histones can recruit DNA methylases, such as DNA (cytosine-5)-methyltransferase 1 (DNMT1), which provide more permanent heterochromatin formation. DNA methylation carried out by DNMT1 have been implicated in susceptibility to endogenous retrovirus-induced systemic lupus erythematosus (97) related to increased antigen processing of hypomethylated DNA (98) or altered gene expression of inflammatory genes directly, including IL-17 (99).
An unusual histone modifying enzyme associated with autophagy is the arginine-specific H3R17 methyltransferase, co-activator-associated arginine methyltransferase 1 (CARM1), which collaborates with the transcription factor, TFEB, to enhance histone methylation allowing access of transcription factors in the AMP-SKP-CARM1 signaling cascade to autophagy-related genes (55). Previous studies have also shown a role for CARM1 as a promoter-specific regulator of NF-κB signaling (100), important for a number of innate and adaptive immune responses, demonstrating the intimate relationship between autophagy activation and autoimmunity.
mRNA Transcript Degradation: Role of microRNAs (miRNAs)
The complexity of miRNAs has been quite daunting, but its complexity is important to the programmatic modulation of autophagy and its effect on inflammation. miRNAs are the shortest of the non-coding RNAs at approximately 22 nucleotides in length. Most miRNAs are considered “canonical” in that they undergo primary miRNA processing in the nucleus by the “microprocessor” complex, which contains the RNase III enzyme Drosha and the dsRNA-binding protein Dgcr8, and further maturation in the cytoplasm by the ribonuclease DICER. The resulting ~22 nucleotide duplex is loaded into the Argonaute-containing RNA-induced silencing complex (RISC), which then recruits mature mRNAs for degradation (Figure 1B). Interestingly, the complete structure of RISC is still unresolved and reports of size and component variability suggest complex adaptability to induction conditions or the passengers in question. In addition to “canonical” miRNAs, various non-canonical miRNAs appear to remain dependent on DICER, but are processed independent of Drosha or Dgcr8 (101). The best known of these are mirtrons, which are processed through a unique intronic splicing mechanism and have been recently associated with the pathophysiology of IgA deficiency via regulation of immunoglobulin heavy constant alpha 1 (IGHA1) and IGHA2 (102). It is likely that this intricate web of miRNA regulation furnishes an important modulating capacity for the immune system to optimize survival against a range of pathogenic organisms under strong evolutionary pressure. But when inappropriately triggered may result in the “off-target” consequence of autoimmunity.
One of the first miRNAs identified to play a role in immunity was the global regulator miR-155. Prominently associated with IFN-γ expression and germinal center function (103–105), it was not determined until much later that some of the direct regulatory targets of miR-155 were autophagy-related (86). One of the best-known targets of miRNA-regulated autophagy is the immunity-related GTPase family M protein (IRGM) clinically associated with inflammatory bowel disease (106, 107). The canonical miRNA miR-196 targets and regulates IRGM whose levels confer either autophagic protection or cell death in target cells, implicated in both defense against the intracellular pathogen Mtb and damaging inflammation caused by Crohn’s disease. Such studies are an important demonstration of the importance of the exquisite immunological balance necessary to provide both microbiological protection and avoid autoimmune pathology. Pertinent to the importance of epigenetic mechanisms of regulation, clinical genetic studies identified a disease variant that was originally felt to be dispensable due to a lack of effects on either IRGM protein sequence or splice site selection, but later found to result in downregulation of an IRGM protective variant, but not a risk-associated allele due to a miRNA-based alteration in IRGM regulation (74).
Another prominent target of miRNA degradation is the autophagy-associated gene autophagy-related 16 like 1 (ATG16L1). Typically, ATG16L1 interacts with ATG12-ATG5 facilitating the phosphatidylethanolamine lipidation of the vesicular shaping protein, MAP1LC3A, and elongation of the nascent autophagosomal membrane (108, 109). In one study, levels of MIR106B were found to be elevated in the intestinal epithelium of patients with active Crohn’s disease along with decreased levels of ATG16L1 transcripts (Figure 1B) (71). This was found in human cell lines to be associated with defects in autophagy-dependent eradication of intracellular bacteria. Studies in the same year identified a role for MIR142-3p in the same target gene (72) and others also found that Crohn’s disease-associated adherent Escherichia coli were able to modulate MIR30C and 130A to effect changes in ATG16L1 transcripts as well as the autophagy-conjugation gene, ATG5 (73). Interestingly, ATG16L1 has recently been found to play a role in NOD2 inflammasome activation (110) and is associated with inflammatory bowel disease (111). Recently, ATG16L1 gene polymorphisms were found to be associated with necrotizing enterocolitis in premature infants, again stressing the potential importance of this regulatory pathway to autoimmunity (112).
mRNA Transcript Degradation: Role of Regulated mRNA Decapping/Degradation
A more recently identified mechanism of post-transcriptional regulation implicated a well-characterized mRNA decay pathway, characterized as a housekeeping function to remove RNA in yeast (113) and mammalian systems (114). In this process, mRNA undergoes a reversible poly-A tail deadenylation followed by an irreversible 5′-decapping by the decapping enzyme DCP2 and subsequent XRN1-exonuclease mediated degradation of the RNA in the 5′–3′ direction. More recently, studies in the yeasts Saccharomyces cerevisiae as well as C. neoformans identified an RNA-binding protein, ATP-dependent RNA helicase Dhh1/Vad1, as an RNA chaperone that binds and recruits targeted autophagy-related mRNA to the decapping complex resulting in suppression of autophagic flux (Figures 1C,D). Regulation of this process by mTOR was demonstrated by a specific mTOR-dependent phosphorylation of the DCP2 protein in humans, without which mRNA recruitment and decapping was prevented. These studies were extended to patients with monogenic dominant-activating mutations in a PI3K p110δ subunit who were further characterized by increased mTOR activity and autoimmune-associated leukopenia (115). Increased mTOR activity in these patients resulted in accelerated decapping and degradation of relevant autophagy mRNA transcripts with resultant decreased autophagy activity. Further studies, prompted by the recent finding of a role for autophagy in modulation of inflammasome-related IL-1β levels (116) demonstrated that the reduced autophagy activity in these patients resulted in elevated levels of IL-1β, suggesting an etiology of the patient’s autoimmunity. Conversely, knockdown of DDX6 by siRNA was successful in pseudonormalization of IL-1β levels, suggesting both a pathway for rapamycin-treatment of this disorder and new targets for pharmacological intervention against autophagy-related IL1β-associated autoimmunity.
A Question of Balance and the Role of Transcriptional “Futile Cycles”
As suggested by the parallel pathways described in Figure 1, epigenetic mechanisms have the ability to modulate each pathway’s activity. This coupling of mRNA synthesis with mRNA degradation is exemplative of transcriptional “futile cycles” first describe in yeast (117). Futile cycles were first described in energy metabolism with the classic example concerning gluconeogenesis, where regulated inhibition of a degradative phosphatase resulted in a rapid accumulation of fructose 1,6-bisphosphate required for de novo glucose synthesis during the “fight or flight” response (118, 119). These cycles were termed futile, because it was not yet understood why energy would be exerted to simultaneously synthesize and degrade a required cellular precursor. However, maintenance of basal levels of synthesis (metabolic intermediate or mRNA) even during periods of disuse allows more rapid induction of synthetic enzymes without the need to start from zero. It also allows rapid adaptation to newly required steady states by simultaneous modulations in both synthesis and degradation. The concept is well suited for immune mechanisms, demonstrated by the induction of inflammasome activation by TLR4, accompanied by the simultaneous induction of autophagy to degrade IL-1β, resulting in mechanisms to optimize pathogen control and yet avoid autoimmunity (116).
While relationships between epigenetic regulation, autophagy, and immunity are just now being elucidated, the study of HDAC inhibitors in cancer demonstrates some of the complexity of epigenetic manipulations. For example, studies of breast cancer carcinogenesis in the presence of the chemopreventative DNMT1 inhibitor, 3,6-dihydroxyflavone (3,6-DHF), demonstrated reduced DNA methylation with resulting activation of autophagy, as well as epigenetic activation of the MIR21 promoter, resulting in an unexpected induction of the NOTCH-1 pathway (120). Applying some of the known pathways in Figure 1 may help to anticipate some side effects. For example, mTOR-dependent signaling of SIRT1-dependent H3K9 acetylation would be expected to increase autophagy activity (121). However, high mTOR activities would be expected to phosphorylate the S249 amino acid of DCP2, resulting in increased autophagy-related transcript suppression, which modulates the effect of autophagy on IL-1β and autoimmunity with potential reductions in pathogen clearance. However, without experimental probing of these relationships, it is difficult to discern which effects would predominate under a given condition of autoimmunity or infection. Clearly, the study of the role of epigenetic networks in autophagy and autoimmunity is in its infancy, but is critical to the application of a developing repertoire of epigenetic pharmaceuticals to autoimmunity as well as for the anticipation of their potential side effects.
Author Contributions
PW and JH participated in writing and preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by funding from the NIAID intramural program, AI001123 and AI001124.
References
1. Allen H, Bunn WB III, Naim AB. The self-reported health and productivity burden of autoimmune disorders at a major self-insured employer. J Occup Environ Med (2012) 54(9):1049–63. doi:10.1097/JOM.0b013e3182602447
2. U.S. Food and Drug Administration. (2018). Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (Accessed: April 3, 2018).
3. Lionakis MS, Dunleavy K, Roschewski M, Widemann BC, Butman JA, Schmitz R, et al. Inhibition of B cell receptor signaling by ibrutinib in primary CNS lymphoma. Cancer Cell (2017) 31(6):833–43.e5. doi:10.1016/j.ccell.2017.04.012
4. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-dependent activation of a TLR9-BTK-calcineurin-NFAT pathway co-ordinates innate immunity to Aspergillus fumigatus. EMBO Mol Med (2015) 7(3):240–58. doi:10.15252/emmm.201404556
5. Wen X, Klionsky DJ. An overview of macroautophagy in yeast. J Mol Biol (2016) 428(9 Pt A):1681–99. doi:10.1016/j.jmb.2016.02.021
6. Deretic V, Klionsky DJ. Autophagy and inflammation: a special review issue. Autophagy (2018) 14(2):179–80. doi:10.1080/15548627.2017.1412229
7. Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: consequences for human disease. Autophagy (2016) 12(2):245–60. doi:10.1080/15548627.2015.1071759
8. Kwon DH, Song HK. A structural view of xenophagy, a battle between host and microbes. Mol Cells (2018) 41(1):27–34. doi:10.14348/molcells.2018.2274
9. Heckmann BL, Boada-Romero E, Cunha LD, Magne J, Green DR. LC3-associated phagocytosis and inflammation. J Mol Biol (2017) 429(23):3561–76. doi:10.1016/j.jmb.2017.08.012
10. Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev (2018) 281(1):62–73. doi:10.1111/imr.12613
11. Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol (2001) 13(1):114–9. doi:10.1016/S0952-7915(00)00191-6
12. Crotzer VL, Blum JS. Autophagy and its role in MHC-mediated antigen presentation. J Immunol (2009) 182(6):3335–41. doi:10.4049/jimmunol.0803458
13. Hu G, Hacham M, Waterman SR, Panepinto J, Shin S, Liu X, et al. PI3K signaling of autophagy is required for starvation tolerance and virulence of Cryptococcus neoformans. J Clin Invest (2008) 118(3):1186–97. doi:10.1172/JCI32053
14. Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell (2004) 119(6):753–66. doi:10.1016/j.cell.2004.11.038
15. Qin QM, Luo J, Lin X, Pei J, Li L, Ficht TA, et al. Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog (2011) 7(6):e1002078. doi:10.1371/journal.ppat.1002078
16. Hu G, McQuiston T, Bernard A, Park YD, Qiu J, Vural A, et al. A conserved mechanism of TOR-dependent RCK-mediated mRNA degradation regulates autophagy. Nat Cell Biol (2015) 17(7):930–42. doi:10.1038/ncb3189
17. Wu DJ, Adamopoulos IE. Autophagy and autoimmunity. Clin Immunol (2017) 176:55–62. doi:10.1016/j.clim.2017.01.007
18. Rao VKet al. Effective “activated PI3Kdelta syndrome”-targeted therapy with the PI3Kdelta inhibitor leniolisib. Blood (2017) 130(21):2307–16. doi:10.1182/blood-2017-08-801191
19. Zhang KJH, Husami A, Marsh R, Jordan MB. Identification of a phosphoinositide 3-kinase (PI-3K) p110δ (PIK3CD) deficient individual. J Clin Immunol (2013) (33):673–4.
20. Marciano BE, Holland SM. Primary immunodeficiency diseases: current and emerging therapeutics. Front Immunol (2017) 8:937. doi:10.3389/fimmu.2017.00937
21. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci U S A (2014) 111(9):3526–31. doi:10.1073/pnas.1322831111
22. De Ravin SS, Naumann N, Cowen EW, Friend J, Hilligoss D, Marquesen M, et al. Chronic granulomatous disease as a risk factor for autoimmune disease. J Allergy Clin Immunol (2008) 122(6):1097–103. doi:10.1016/j.jaci.2008.07.050
23. van de Veerdonk FL, Dinarello CA. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy (2014) 10(6):1141–2. doi:10.4161/auto.28638
24. Lee JH, Slifman NR, Gershon SK, Edwards ET, Schwieterman WD, Siegel JN, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum (2002) 46(10):2565–70. doi:10.1002/art.10583
25. Wijburg MT, Kleerekooper I, Lissenberg-Witte BI, de Vos M, Warnke C, Uitdehaag BMJ, et al. Association of progressive multifocal leukoencephalopathy lesion volume with JC virus polymerase chain reaction results in cerebrospinal fluid of natalizumab-treated patients with multiple sclerosis. JAMA Neurol (2018) 75(7):827–33. doi:10.1001/jamaneurol.2018.0094
26. Gundacker ND, Jordan SJ, Jones BA, Drwiega JC, Pappas PG. Acute cryptococcal immune reconstitution inflammatory syndrome in a patient on natalizumab. Open Forum Infect Dis (2016) 3(1):ofw038. doi:10.1093/ofid/ofw038
27. Wu S, Zheng SD, Huang HL, Yan LC, Yin XF, Xu HN, et al. Lithium down-regulates histone deacetylase 1 (HDAC1) and induces degradation of mutant huntingtin. J Biol Chem (2013) 288(49):35500–10. doi:10.1074/jbc.M113.479865
28. Strahl BD, Allis CD. The language of covalent histone modifications. Nature (2000) 403(6765):41–5. doi:10.1038/47412
29. Turner BM. Histone acetylation and an epigenetic code. Bioessays (2000) 22(9):836–45. doi:10.1002/1521-1878(200009)22:9<836::AID-BIES9>3.0.CO;2-X
30. Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci U S A (2003) 100(23):13225–30. doi:10.1073/pnas.1735528100
31. Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics (2012) 7(10):1098–108. doi:10.4161/epi.21975
32. Hottiger MO. Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu Rev Biochem (2015) 84:227–63. doi:10.1146/annurev-biochem-060614-034506
33. Zhang Y. Transcriptional regulation by histone ubiquitination and deubiquitination. Genes Dev (2003) 17(22):2733–40. doi:10.1101/gad.1156403
34. Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, et al. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature (2014) 507(7490):104–8. doi:10.1038/nature12942
35. Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, et al. GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature (2011) 480(7378):557–60. doi:10.1038/nature10656
36. Tsukada Y. Hydroxylation mediates chromatin demethylation. J Biochem (2012) 151(3):229–46. doi:10.1093/jb/mvs003
37. Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell (2006) 126(5):905–16. doi:10.1016/j.cell.2006.07.026
38. Howe FS, Boubriak I, Sale MJ, Nair A, Clynes D, Grijzenhout A, et al. Lysine acetylation controls local protein conformation by influencing proline isomerization. Mol Cell (2014) 55(5):733–44. doi:10.1016/j.molcel.2014.07.004
39. Shin HR, Kim H, Kim KI, Baek SH. Epigenetic and transcriptional regulation of autophagy. Autophagy (2016) 12(11):2248–9. doi:10.1080/15548627.2016.1214780
40. Füllgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature (2013) 500(7463):468–71. doi:10.1038/nature12313
41. Hajji N, Wallenborg K, Vlachos P, Füllgrabe J, Hermanson O, Joseph B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene (2010) 29(15):2192–204. doi:10.1038/onc.2009.505
42. Lim HWet al. SIRT1 deacetylates RORgammat and enhances Th17 cell generation. J Exp Med (2015) 212(5):607–17. doi:10.1084/jem.2013237805062015c
43. Chuprin A, Avin A, Goldfarb Y, Herzig Y, Levi B, Jacob A, et al. The deacetylase Sirt1 is an essential regulator of Aire-mediated induction of central immunological tolerance. Nat Immunol (2015) 16(7):737–45. doi:10.1038/ni.3194
44. Sarumaru M, Watanabe M, Inoue N, Hisamoto Y, Morita E, Arakawa Y, et al. Association between functional SIRT1 polymorphisms and the clinical characteristics of patients with autoimmune thyroid disease. Autoimmunity (2016) 49(5):329–37. doi:10.3109/08916934.2015.1134506
45. Garcia-Peterson LM, Ndiaye MA, Singh CK, Chhabra G, Huang W, Ahmad N. SIRT6 histone deacetylase functions as a potential oncogene in human melanoma. Genes Cancer (2017) 8(9–10):701–12. doi:10.18632/genesandcancer.153
46. Cleynen I, González JR, Figueroa C, Franke A, McGovern D, Bortlík M, et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: results from the IBDchip European Project. Gut (2013) 62(11):1556–65. doi:10.1136/gutjnl-2011-300777
47. Wang Z, Qin G, Zhao TC. HDAC4: mechanism of regulation and biological functions. Epigenomics (2014) 6(1):139–50. doi:10.2217/epi.13.73
48. Yang D, Xiao C, Long F, Su Z, Jia W, Qin M, et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc Res (2018) 114(7):1016–28. doi:10.1093/cvr/cvy051
49. Tang J, Shi Y, Liu N, Xu L, Zang X, Li P, et al. Blockade of histone deacetylase 6 protects against cisplatin-induced acute kidney injury. Clin Sci (Lond) (2018) 132(3):339–59. doi:10.1042/CS20171417
50. Liu M, Liang K, Zhen J, Zhou M, Wang X, Wang Z, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun (2017) 8(1):413. doi:10.1038/s41467-017-00498-4
51. Huber TB, Edelstein CL, Hartleben B, Inoki K, Jiang M, Koya D, et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy (2012) 8(7):1009–31. doi:10.4161/auto.19821
52. Sanchez-Fidalgo S, Villegas I, Sanchez-Hidalgo M, de la Lastra CA. Sirtuin modulators: mechanisms and potential clinical implications. Curr Med Chem (2012) 19(15):2414–41. doi:10.2174/092986712800269272
53. Wang W, Wang Q, Wan D, Sun Y, Wang L, Chen H, et al. Histone HIST1H1C/H1.2 regulates autophagy in the development of diabetic retinopathy. Autophagy (2017) 13(5):941–54. doi:10.1080/15548627.2017.1293768
54. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol (2015) 35(5):615–21. doi:10.1007/s10571-015-0166-x
55. Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature (2016) 534(7608):553–7. doi:10.1038/nature18014
56. Kowenz-Leutz E, Pless O, Dittmar G, Knoblich M, Leutz A. Crosstalk between C/EBPbeta phosphorylation, arginine methylation, and SWI/SNF/mediator implies an indexing transcription factor code. EMBO J (2010) 29(6):1105–15. doi:10.1038/emboj.2010.3
57. Tsutsui T, Fukasawa R, Shinmyouzu K, Nakagawa R, Tobe K, Tanaka A, et al. Mediator complex recruits epigenetic regulators via its two cyclin-dependent kinase subunits to repress transcription of immune response genes. J Biol Chem (2013) 288(29):20955–65. doi:10.1074/jbc.M113.486746
58. Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J Biol Chem (2015) 290(3):1485–95. doi:10.1074/jbc.M114.593871
59. Moreno-Gonzalo O, Ramírez-Huesca M, Blas-Rus N, Cibrián D, Saiz ML, Jorge I, et al. HDAC6 controls innate immune and autophagy responses to TLR-mediated signalling by the intracellular bacteria Listeria monocytogenes. PLoS Pathog (2017) 13(12):e1006799. doi:10.1371/journal.ppat.1006799
60. Zhou L, He X, Gao B, Xiong S. Inhibition of histone deacetylase activity aggravates coxsackievirus B3-induced myocarditis by promoting viral replication and myocardial apoptosis. J Virol (2015) 89(20):10512–23. doi:10.1128/JVI.01028-15
61. Patel S, Hurez V, Nawrocki ST, Goros M, Michalek J, Sarantopoulos J, et al. Vorinostat and hydroxychloroquine improve immunity and inhibit autophagy in metastatic colorectal cancer. Oncotarget (2016) 7(37):59087–97. doi:10.18632/oncotarget.10824
62. Shulak L, Beljanski V, Chiang C, Dutta SM, Van Grevenynghe J, Belgnaoui SM, et al. Histone deacetylase inhibitors potentiate vesicular stomatitis virus oncolysis in prostate cancer cells by modulating NF-kappaB-dependent autophagy. J Virol (2014) 88(5):2927–40. doi:10.1128/JVI.03406-13
63. Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PLoS One (2012) 7(12):e52095. doi:10.1371/journal.pone.0052095
64. Klein JM, Henke A, Sauer M, Bessler M, Reiners KS, Engert A, et al. The histone deacetylase inhibitor LBH589 (panobinostat) modulates the crosstalk of lymphocytes with Hodgkin lymphoma cell lines. PLoS One (2013) 8(11):e79502. doi:10.1371/journal.pone.0079502
65. Laubach JP, Moreau P, San-Miguel JF, Richardson PG. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res (2015) 21(21):4767–73. doi:10.1158/1078-0432.CCR-15-0530
66. Campbell GR, Bruckman RS, Chu YL, Spector SA. Autophagy induction by histone deacetylase inhibitors inhibits HIV type 1. J Biol Chem (2015) 290(8):5028–40. doi:10.1074/jbc.M114.605428
67. Stankov MV, El Khatib M, Kumar Thakur B, Heitmann K, Panayotova-Dimitrova D, Schoening J, et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia (2014) 28(3):577–88. doi:10.1038/leu.2013.264
68. Zhu H, Wu H, Liu X, Li B, Chen Y, Ren X, et al. Regulation of autophagy by a beclin 1-targeted microRNA, miR-30a, in cancer cells. Autophagy (2009) 5(6):816–23. doi:10.4161/auto.9064
69. Tang B, Li N, Gu J, Zhuang Y, Li Q, Wang HG, et al. Compromised autophagy by miR30B benefits the intracellular survival of Helicobacter pylori. Autophagy (2012) 8(7):1045–57. doi:10.4161/auto.20159
70. Jin L, Li Y, He T, Hu J, Liu J, Chen M, et al. Identification of miR30b as an oncogene in renal cell carcinoma. Mol Med Rep (2017) 15(4):1837–46. doi:10.3892/mmr.2017.6197
71. Lu C, Chen J, Xu HG, Zhou X, He Q, Li YL, et al. miR106B and miR93 prevent removal of bacteria from epithelial cells by disrupting ATG16L1-mediated autophagy. Gastroenterology (2014) 146(1):188–99. doi:10.1053/j.gastro.2013.09.006
72. Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, Boone DL, et al. Human autophagy gene ATG16L1 is post-transcriptionally regulated by miR142-3p. Autophagy (2014) 10(3):468–79. doi:10.4161/auto.27553
73. Nguyen HT, Dalmasso G, Müller S, Carrière J, Seibold F, Darfeuille-Michaud A. Crohn’s disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology (2014) 146(2):508–19. doi:10.1053/j.gastro.2013.10.021
74. Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat Genet (2011) 43(3):242–5. doi:10.1038/ng.762
75. Singh SB, Ornatowski W, Vergne I, Naylor J, Delgado M, Roberts E, et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol (2010) 12(12):1154–65. doi:10.1038/ncb2119
76. Chio CC, Lin JW, Cheng HA, Chiu WT, Wang YH, Wang JJ, et al. MicroRNA-210 targets antiapoptotic Bcl-2 expression and mediates hypoxia-induced apoptosis of neuroblastoma cells. Arch Toxicol (2013) 87(3):459–68. doi:10.1007/s00204-012-0965-5
77. Wang H, Flach H, Onizawa M, Wei L, McManus MT, Weiss A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat Immunol (2014) 15(4):393–401. doi:10.1038/ni.2846
78. Wu Z, Lu H, Sheng J, Li L. Inductive microRNA-21 impairs anti-mycobacterial responses by targeting IL-12 and Bcl-2. FEBS Lett (2012) 586(16):2459–67. doi:10.1016/j.febslet.2012.06.004
79. Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol (2011) 8(5):706–13. doi:10.4161/rna.8.5.16154
80. Meenhuis A, van Veelen PA, de Looper H, van Boxtel N, van den Berge IJ, Sun SM, et al. miR-17/20/93/106 promote hematopoietic cell expansion by targeting sequestosome 1-regulated pathways in mice. Blood (2011) 118(4):916–25. doi:10.1182/blood-2011-02-336487
81. Holla S, Kurowska-Stolarska M, Bayry J, Balaji KN. Selective inhibition of IFNG-induced autophagy by miR155- and miR31-responsive WNT5A and SHH signaling. Autophagy (2014) 10(2):311–30. doi:10.4161/auto.27225
82. Babu SG, Pandeya A, Verma N, Shukla N, Kumar RV, Saxena S. Role of HCMV miR-UL70-3p and miR-UL148D in overcoming the cellular apoptosis. Mol Cell Biochem (2014) 393(1–2):89–98. doi:10.1007/s11010-014-2049-8
83. Au KY, Pong JC, Ling WL, Li JC. miR-1303 regulates mycobacteria induced autophagy by targeting Atg2B. PLoS One (2016) 11(1):e0146770. doi:10.1371/journal.pone.0146770
84. Panneerdoss S, Viswanadhapalli S, Abdelfattah N, Onyeagucha BC, Timilsina S, Mohammad TA, et al. Cross-talk between miR-471-5p and autophagy component proteins regulates LC3-associated phagocytosis (LAP) of apoptotic germ cells. Nat Commun (2017) 8(1):598. doi:10.1038/s41467-017-00590-9
85. Etna MP, Sinigaglia A, Grassi A, Giacomini E, Romagnoli A, Pardini M, et al. Mycobacterium tuberculosis-induced miR-155 subverts autophagy by targeting ATG3 in human dendritic cells. PLoS Pathog (2018) 14(1):e1006790. doi:10.1371/journal.ppat.1006790
86. Wang J, Yang K, Zhou L, Minhaowu, Wu Y, Zhu M, et al. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog (2013) 9(10):e1003697. doi:10.1371/journal.ppat.1003697
87. Shinkai Y, Tachibana M. H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev (2011) 25(8):781–8. doi:10.1101/gad.2027411
88. Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy (2015) 11(12):2309–22. doi:10.1080/15548627.2015.1117734
89. Katona BW, Liu Y, Ma A, Jin J, Hua X. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol Ther (2014) 15(12):1677–87. doi:10.4161/15384047.2014.972776
90. Ke XX, Zhang D, Zhu S, Xia Q, Xiang Z, Cui H. Inhibition of H3K9 methyltransferase G9a repressed cell proliferation and induced autophagy in neuroblastoma cells. PLoS One (2014) 9(9):e106962. doi:10.1371/journal.pone.0106962
91. Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A (2009) 106(28):11667–72. doi:10.1073/pnas.0904715106
92. Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature (2010) 464(7291):1071–6. doi:10.1038/nature08975
93. Pahlevan Kakhki M, Nikravesh A, Shirvani Farsani Z, Sahraian MA, Behmanesh M. HOTAIR but not ANRIL long non-coding RNA contributes to the pathogenesis of multiple sclerosis. Immunology (2018) 153(4):479–87. doi:10.1111/imm.12850
94. Tao JH, Barbi J, Pan F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A review in the theme: cellular responses to hypoxia. Am J Physiol Cell Physiol (2015) 309(9):C580–9. doi:10.1152/ajpcell.00204.2015
95. Meng Xet al. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat Commun (2018) 9(1):251. doi:10.1038/s41467-017-02683-x
96. Salminen A, Kaarniranta K, Kauppinen A. Hypoxia-inducible histone lysine demethylases: impact on the aging process and age-related diseases. Aging Dis (2016) 7(2):180–200. doi:10.14336/AD.2015.0929
97. Ogasawara H, Okada M, Kaneko H, Hishikawa T, Sekigawa I, Hashimoto H. Possible role of DNA hypomethylation in the induction of SLE: relationship to the transcription of human endogenous retroviruses. Clin Exp Rheumatol (2003) 21(6):733–8.
98. Januchowski R, Prokop J, Jagodzinski PP. Role of epigenetic DNA alterations in the pathogenesis of systemic lupus erythematosus. J Appl Genet (2004) 45(2):237–48.
99. Rauen Tet al. cAMP-responsive element modulator (CREM)alpha protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem (2011) 286(50):43437–46. doi:10.1074/jbc.M111.299313
100. Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, et al. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J (2005) 24(1):85–96. doi:10.1038/sj.emboj.7600500
101. Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature (2010) 465(7298):584–9. doi:10.1038/nature09092
102. Chitnis N, Clark PM, Kamoun M, Stolle C, Brad Johnson F, Monos DS. An expanded role for HLA genes: HLA-B encodes a microRNA that regulates IgA and other immune response transcripts. Front Immunol (2017) 8:583. doi:10.3389/fimmu.2017.00583
103. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al. Requirement of bic/microRNA-155 for normal immune function. Science (2007) 316(5824):608–11. doi:10.1126/science.1139253
104. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A (2007) 104(5):1604–9. doi:10.1073/pnas.0610731104
105. Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science (2007) 316(5824):604–8. doi:10.1126/science.1141229
106. Fisher SA, Tremelling M, Anderson CA, Gwilliam R, Bumpstead S, Prescott NJ, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet (2008) 40(6):710–2. doi:10.1038/ng.145
107. Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR. Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun (2008) 9(6):561–5. doi:10.1038/gene.2008.49
108. Boada-Romero E, Letek M, Fleischer A, Pallauf K, Ramón-Barros C, Pimentel-Muiños FX. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J (2013) 32(4):566–82. doi:10.1038/emboj.2013.8
109. Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell (2014) 55(2):238–52. doi:10.1016/j.molcel.2014.05.021
110. Negroni A, Colantoni E, Vitali R, Palone F, Pierdomenico M, Costanzo M, et al. NOD2 induces autophagy to control AIEC bacteria infectiveness in intestinal epithelial cells. Inflamm Res (2016) 65(10):803–13. doi:10.1007/s00011-016-0964-8
111. Al Nabhani Z, Dietrich G, Hugot JP, Barreau F. Nod2: the intestinal gate keeper. PLoS Pathog (2017) 13(3):e1006177. doi:10.1371/journal.ppat.1006177
112. Sampath V, Bhandari V, Berger J, Merchant D, Zhang L, Ladd M, et al. A functional ATG16L1 (T300A) variant is associated with necrotizing enterocolitis in premature infants. Pediatr Res (2017) 81(4):582–8. doi:10.1038/pr.2016.260
113. Nissan T, Rajyaguru P, She M, Song H, Parker R. Decapping activators in Saccharomyces cerevisiae act by multiple mechanisms. Mol Cell (2010) 39(5):773–83. doi:10.1016/j.molcel.2010.08.025
114. Grudzien-Nogalska E, Kiledjian M. New insights into decapping enzymes and selective mRNA decay. Wiley Interdiscip Rev RNA (2017) 8(1). doi:10.1002/wrna.1379
115. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110delta result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi:10.1038/ni.2771
116. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol (2012) 13(3):255–63. doi:10.1038/ni.2215
117. Park YD, Williamson PR. ‘Popping the clutch’: novel mechanisms regulating sexual development in Cryptococcus neoformans. Mycopathologia (2012) 173(5–6):359–66. doi:10.1007/s11046-011-9464-0
118. Shulman GI, Ladenson PW, Wolfe MH, Ridgway EC, Wolfe RR. Substrate cycling between gluconeogenesis and glycolysis in euthyroid, hypothyroid, and hyperthyroid man. J Clin Invest (1985) 76(2):757–64. doi:10.1172/JCI112032
119. Suh SH, Paik IY, Jacobs K. Regulation of blood glucose homeostasis during prolonged exercise. Mol Cells (2007) 23(3):272–9.
120. Peng X, Chang H, Gu Y, Chen J, Yi L, Xie Q, et al. 3,6-Dihydroxyflavone suppresses breast carcinogenesis by epigenetically regulating miR-34a and miR-21. Cancer Prev Res (Phila) (2015) 8(6):509–17. doi:10.1158/1940-6207.CAPR-14-0357
121. García-Rodríguez JL, Barbier-Torres L, Fernández-Álvarez S, Gutiérrez-de Juan V, Monte MJ, Halilbasic E, et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology (2014) 59(5):1972–83. doi:10.1002/hep.26971
Keywords: histone, epigenetics, autoimmunity, autophagy, mRNA degradation, histone deacetylase inhibitors, miRNA
Citation: Hargarten JC and Williamson PR (2018) Epigenetic Regulation of Autophagy: A Path to the Control of Autoimmunity. Front. Immunol. 9:1864. doi: 10.3389/fimmu.2018.01864
Received: 07 April 2018; Accepted: 27 July 2018;
Published: 14 August 2018
Edited by:
Xu-jie Zhou, Peking University First Hospital, ChinaReviewed by:
Jennifer Martinez, National Institute of Environmental Health Sciences (NIEHS), United StatesRaman Chandrasekar, Kansas State University, United States
Suowen Xu, University of Rochester, United States
Copyright: © 2018 Hargarten and Williamson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter R. Williamson, d2lsbGlhbXNvbnByQG1haWwubmloLmdvdg==