 Charles J. Kroger
Charles J. Kroger Matthew Clark
Matthew Clark Qi Ke
Qi Ke Roland M. Tisch
Roland M. Tisch- 1Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
- 2Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, United States
Type 1 diabetes (T1D) is an autoimmune disease that is generally considered to be T cell-driven. Accordingly, most strategies of immunotherapy for T1D prevention and treatment in the clinic have targeted the T cell compartment. To date, however, immunotherapy has had only limited clinical success. Although certain immunotherapies have promoted a protective effect, efficacy is often short-term and acquired immunity may be impacted. This has led to the consideration of combining different approaches with the goal of achieving a synergistic therapeutic response. In this review, we will discuss the status of various T1D therapeutic strategies tested in the clinic, as well as possible combinatorial approaches to restore β cell tolerance.
Introduction
Type 1 diabetes (T1D) is an autoimmune disease marked by the dysfunction and/or destruction of the insulin-producing β cells found in the pancreatic islets of Langerhans (1–4). T1D most often arises in children but also develops in adults. To compensate for impaired β cell function, T1D individuals require daily insulin therapy. Despite insulin administration, however, establishing optimal glycemic control long-term is often problematic resulting in a number of complications, including retinopathy, nephropathy, vasculopathy, and neuropathy. This morbidity coupled with an increasing incidence of T1D world-wide, underscores an urgent need for effective immunotherapies for T1D prevention and treatment.
The goal of an immunotherapy is threefold: (1) selectively suppress ongoing autoimmunity, (2) reestablish self-tolerance long-term, and (3) preserve acquired immunity. This review will focus on the different strategies of immunotherapy being tested experimentally and in the clinic to suppress β cell autoimmunity, and prevent clinical onset and/or treat T1D.
Immunopathology of T1D
The loss of β cell tolerance involves both genetic and ill-defined environmental factors (5–16). A strong genetic association with specific human leukocyte antigen haplotypes, coupled with several variants of genes expressed by β cells, T cells, and other immune effectors underscore the complexity of the autoimmune response of T1D (17–23). β cell autoimmunity is typically viewed as a chronic inflammatory response characterized by the progressive infiltration of the pancreatic islets with various immune effectors (24). In the nonobese diabetic (NOD) mouse, a spontaneous model of T1D, islet infiltration is initiated at an early age by macrophages and dendritic cells (DC), CD4+ and CD8+ T cells, and B cells. This insulitis initially exhibits benign properties with little effect on β cell viability or function. However, at a late preclinical stage, an ill-defined qualitative change occurs within the islet infiltrate, which promotes efficient β cell destruction leading to the onset of overt diabetes. In NOD mice and other rodent models of T1D, pathogenic β cell-specific CD4+ and CD8+ effector T cells (Teff) are essential drivers of autoimmunity. Diabetogenic CD4+ and CD8+ T cells target several β cell autoantigens and related peptide epitopes, including proinsulin/insulin, glutamic acid decarboxylase 65 (GAD65), chromogranin A (ChgA), zinc transporter 8 (ZnT8), glucose-6-phosphatase catalytic subunit-related protein (IGRP), and hybrid insulin peptides (HIPs) among others (25–34). It is thought that early in the disease process only few β cell autoantigens and epitopes are targeted by T cells. However, over time additional epitopes within a given autoantigen as well as new autoantigens are recognized. Furthermore, chronic inflammation negatively affects β cells in part by generating neo-autoantigens, such as HIPs, via post-translational modification events (32–39). These neo-autoantigens further diversify the diabetogenic response. Together this epitope spread effectively amplifies the β cell-specific T cell response (28, 40–42).
Analyses of human cadaveric T1D pancreases have also demonstrated islet infiltrates consisting of CD8+ T cells and macrophages, and to a lesser extent CD4+ T cells, and B cells (29, 31, 43–52). However, T1D pancreases have been reported that lack T cell infiltrates suggesting that the immunopathology of human T1D is heterogeneous (53, 54). The prevalence of T cell-independent subsets of T1D is unclear, and thought to be primarily associated with adult T1D onset. On the other hand, evidence indicates that the rapid and severe T1D that develops in children and adolescents is T cell-mediated (44). For instance, recent reports show that childhood onset is marked by a broader and more aggressive β cell-specific T cell response compared to adult T1D (29, 31, 43–52, 55–57). Multiple β cell autoantigens are recognized by human CD4+ and CD8+ T cells found in peripheral blood, as well as the islets of T1D subjects; many of which are also targeted in the NOD mouse diabetogenic response (e.g., insulin, GAD65, IGRP, and ZnT8) (4, 25, 28, 57).
Pathogenic β cell-specific CD4+ and CD8+ Teff in NOD and human T1D typically exhibit a type 1 or T helper 1 (Th1) phenotype marked by IFNγ production (47, 58, 59). IL-17-producing CD4+ Th17 cells have also been implicated in mediating β cell destruction (60–62). Differentiation and expansion of pathogenic Teff are in part attributed to aberrant peripheral immunoregulation (63–68). An impaired pool of thymic-derived FoxP3-expressing immunoregulatory T cells (Foxp3+Treg) has been linked to T1D (68–70). In general, Foxp3+Treg play an essential role in maintaining peripheral self-tolerance through cytokine and contact-dependent mechanisms of suppression (71). Decreased survival of islet-resident Foxp3+Treg is thought to be a key factor in promoting the progression from benign to pathogenic insulitis in NOD mice (69). Failure to maintain islet Foxp3+Treg numbers in NOD mice is due to insufficient local levels of IL-2, a critical cytokine needed for Foxp3+Treg survival, fitness, and function (69, 72–74). FOXP3+Treg from T1D subjects have defective IL-2 receptor (R) signaling which limits fitness and function of FOXP3+Treg (66, 75). Additionally, production of the proinflammatory cytokine IL-21, which is critical for T1D development, can inhibit IL-2 expression by T cells which negatively impacts Foxp3+Treg viability and function (76). Human T1D is also marked by deficiencies in non-FoxP3-expressing adaptive (a) Treg. For example, the frequency of β cell-specific IL-10-secreting Tr1 cells is reduced in T1D versus healthy subjects (77–79). In both NOD and human T1D, Teff exhibit a reduced sensitivity to Treg-mediated suppression, which further permits expansion of the diabetogenic Teff pool (63, 64).
Dysregulation among antigen-presenting cells (APC), such as DC, macrophages, and B cells, has also been reported to contribute to T1D (80–85). Although detection of autoantibodies is a key indicator of β cell autoimmunity, B cells are thought to play a critical role in the development of T1D by functioning primarily as an APC (86–88). APC exhibiting proinflammatory properties also skew differentiation of naïve β cell-specific T cells toward pathogenic Teff, as well as amplify islet inflammation and β cell destruction. For instance cytokines, such as IFNγ, TNFα, and IL-1β secreted by islet APC are cytotoxic to β cells in vitro (89). The culmination of the adaptive and innate effector immune response within the islets results in β cell destruction/dysfunction and elevated blood glucose levels (Figure 1).
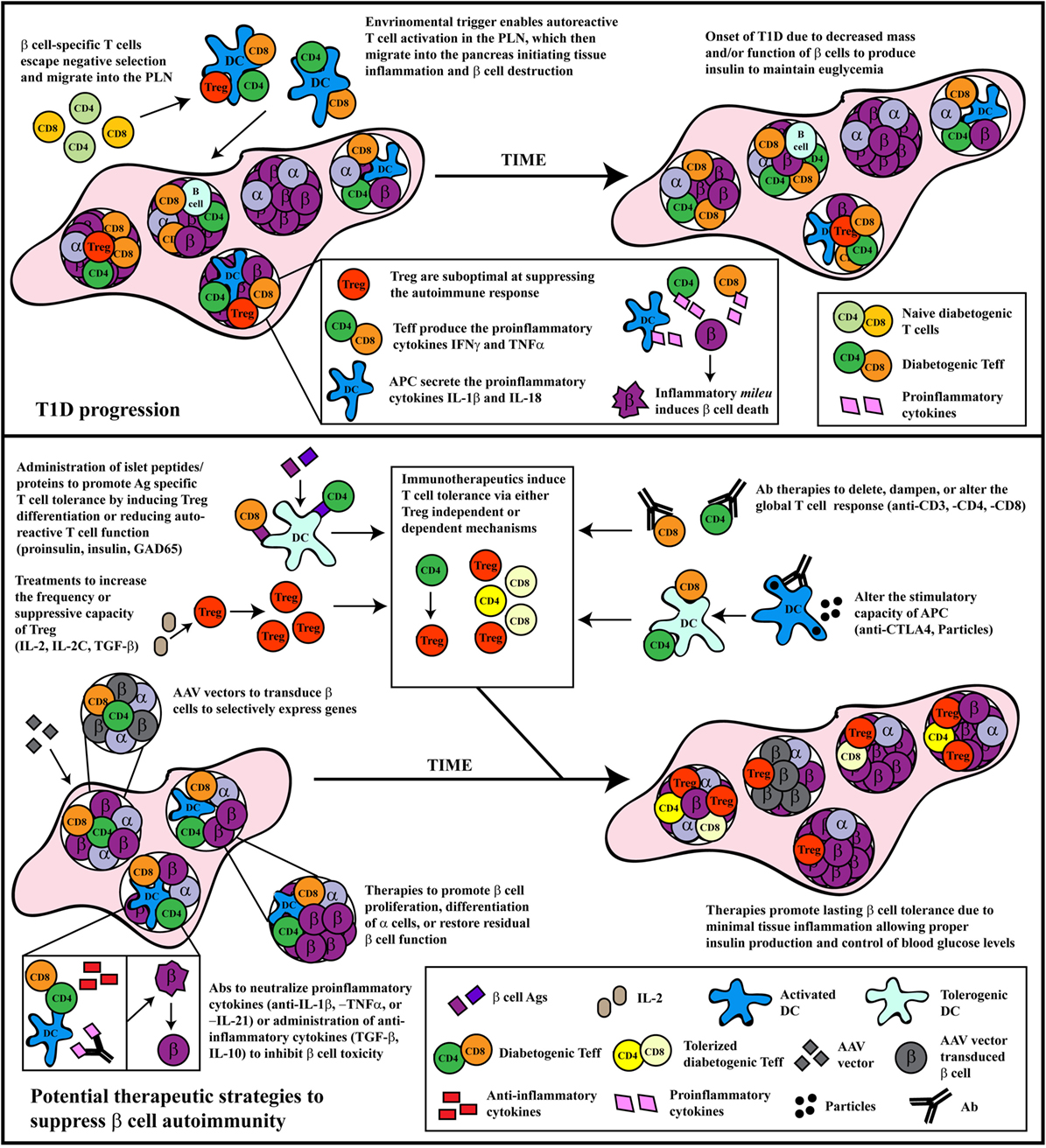
Figure 1. The progression and treatment of β cell autoimmunity. Top panel: In general, overt diabetes results from the gradual loss of functional insulin-producing β cells due to the inflammatory environment driven by infiltrating self-reactive T cells and antigen-presenting cells (APC). Although β cell-specific T cell clones are detected in both healthy and type 1 diabetes (T1D) susceptible individuals, a number of factors promote T1D development in the latter population. Decreased efficiency of negative selection in the thymus allows for the increased escape of β cell-specific T cell clones into the periphery. In the periphery, β cell-specific T cells are stimulated in the pancreatic lymph nodes (PLN) by APC derived from the islets, leading to effector T cells (Teff) differentiation due to suboptimal Foxp3+Treg suppression. These pathogenic Teff then infiltrate the islets and drive inflammation leading to reduced β cell function and/or survival. Bottom panel: Several distinct therapeutic interventions have been tested for the prevention or treatment of T1D. The major approaches have included: administration of β cell autoantigens, expansion of Foxp3+Treg, Ab therapies that alter T cell responses, therapeutic reagents that tolerize APC function, neutralization of proinflammatory molecules, or treatments that expand or enhance β cell function and/or survival. Some of these approaches have had clinical, albeit limited, success. Future therapeutic interventions should look toward refinement in the specificity of these treatments, and the development of combinatorial therapies targeting multiple arms of the immune system, as T1D is a multi-pronged autoimmune disease.
Aberrant peripheral immunoregulation has been the focus of most T1D immunotherapies. Different strategies have attempted to selectively suppress the pathogenic proinflammatory events that affect β cells, while reestablishing protective immunoregulation that persists long-term without altering acquired immunity (Figure 1). Achieving these goals in the clinic, however, has proven to be highly challenging.
Immunotherapy of T1D
The progression of T1D affords three windows of therapeutic intervention to alter disease outcome (90, 91). The first is during the prodromal stage of T1D, which may persist for a number of years. At-risk individuals with ongoing β cell autoimmunity, identified by serum autoantibodies specific for various β cell autoantigens, are candidates for prevention of clinical onset of T1D (90–92). The second window of intervention is at the time of clinical onset. The goal here is to suppress β cell autoimmunity, rescue residual β cell mass, and ideally reverse clinical diabetes. Early studies provided proof of principle that sufficient β cell mass exists at the time of diagnosis to reverse diabetes (93, 94). Administration of the immunosuppressive drug cyclosporine A (CsA) induces remission in new onset children. However, CsA results in severe kidney toxicity, and once therapy is halted, patients develop recurrent diabetes (95, 96). The third window of intervention is in long-standing T1D patients to protect residual β cell function. Evidence indicates that after T1D onset low but sustained C-peptide levels are observed (93, 94). Therefore, maintenance of a small amount of functional β mass may aid in the control of glycemia as well as limit morbidity.
Two general approaches of immunotherapy have been tested experimentally and in the clinic for the prevention and treatment of T1D. The first approach entails manipulating the autoimmune response independent of β cell specificity. Typically, antibodies (Ab) targeting specific immune effectors or effector molecules have been employed for this approach. The second approach makes use of β cell antigen-specific strategies. Here, β cell autoantigens or corresponding peptides are administered under various conditions. In the following sections, we will discuss different strategies of immunotherapy that are included in these two general approaches, highlighting related strengths and weaknesses (Figure 1).
Antigen-Independent Immunotherapies
Ab and cytokines have been administered to modulate or block the function of immune effector cells and/or molecules. The general approach is advantageous, since pathogenic effectors can be targeted en masse, often resulting in rapid and robust outcomes in experimental settings. However, the limited specificity of these therapeutics can lead to unwanted systemic effects impacting acquired immunity for instance. Nevertheless, a sizable body of work has demonstrated clinical efficacy for certain Ab therapies, whereas promising early results are being seen with cytokine- and cell-based strategies.
Antibody-Based Therapies
Arguably, the most effective strategy to clinically alter T1D progression has been to target diabetogenic Teff with anti-CD3 monoclonal Ab (mAb) (97, 98). In new onset NOD mice anti-CD3 mAb therapy reverses diabetes in part by driving apoptosis of islet-infiltrating Teff. Engulfment of the apoptotic T cells induces TGF-β secretion by APC that promotes differentiation of Foxp3+Treg. Once established this pool of induced (i) Foxp3+Treg mediate long-lasting β cell tolerance (99–101). In addition, peripheral Teff exhibit reduced IFNγ production, and increased markers of exhaustion and anergy (102). Interestingly, anti-CD3 mAb therapy given at a preclinical stage fails to prevent diabetes onset in NOD mice (98, 103). This indicates that a given immunotherapy may be effective only at particular stages of T1D progression.
For clinical trials, humanized anti-CD3 mAb hOKT3γ1 (Ala-Ala) (teplizumab) or CCHAglyCD3 (otelixizumab) have been engineered to prevent binding to Fc receptor, and minimize proinflammatory cytokine release by APC (97, 100, 104–112). Anti-CD3 mAb therapy administered to new onset T1D patients reduces the rate of loss of β cell function in the majority of individuals (113). The mechanism of protection is ill-defined, although expansion of CD8+ Treg and CD8+ Teff exhibiting an exhausted phenotype has been reported (109). Despite this efficacy, diabetes reversal is not achieved, and protection of residual β cell mass is short-term, lasting up to 4 years in some individuals (113). Furthermore, anti-CD3 mAb binding can activate T cells resulting in cytokine release and unwanted inflammation, albeit transient. Moreover, the transient depletion of T cells systemically by anti-CD3 mAb is linked to recurrent viral infections (100, 104). Nevertheless, these findings provide direct evidence that progression of β cell autoimmunity can be modulated by targeting T cells. However, further refinement of the approach is required to enhance β cell protection while minimizing effects on acquired immunity. This may in part be achieved by using anti-CD3 mAb in combination with other therapeutics (114–116). For instance, treatment of NOD mice with anti-CD3 mAb and the β cell trophic growth factor prolactin, increases both diabetes reversal and β cell proliferation relative to anti-CD3 mAb alone (116).
The aim of Ab-based combinatorial strategies is to establish a synergistic or additive effect to enhance both efficacy and safety. An increased tolerogenic response may permit reduced dosing and, therefore, minimize unwanted complications with a given therapeutic. One combinatorial strategy being tested in the clinic is the application of antithymocyte globulin (ATG) and granulocyte colony stimulating factor (G-CSF). Low dose ATG induces apoptosis and transiently depletes T cells, whereas G-CSF promotes mobilization of aTreg and induction of tolerogenic DC. In preclinical studies, ATG alone induces ~30% remission in newly diabetic NOD mice, whereas the combination of ATG and G-CSF increases diabetes reversal >twofold (117). Initial phase I clinical results for low dose ATG plus G-CSF therapy given to recent onset T1D patients indicate that β cell function can be maintained up to 2 years in a group of responders (118). This protective effect correlates with quantitative and qualitative changes in the conventional T cell pool, coupled with an increase in the frequency of FOXP3+Treg (118).
Although most T1D immunotherapies have targeted T cells, success has also been demonstrated by altering the B cell response in various autoimmune diseases including T1D, both in mice and humans (88, 119, 120). As previously mentioned, serum autoantibodies indicate B cells also target β cell antigens, including GAD65 and insulin, which serve as a biomarker for individuals with an increased risk for developing T1D (90–92). NOD mice lacking B cells are protected from insulitis and diabetes onset (86, 87). Several molecules, such as CD20, CD22, BAFF, and BCMA have been targeted to modulate the B cell pool (121–125). In different murine models of T1D, anti-CD20 mAb suppresses the progression of β cell autoimmunity as well as reverses diabetes onset, by promoting Foxp3+Treg and regulatory B cell populations (121, 122). In recent onset T1D patients, anti-CD20 mAb (Rituximab), which has also been used in the treatment of rheumatoid arthritis (RA), transiently depleted B cells and prolonged β cell function (126, 127). However, this protective effect was only transient, diminishing after 1 year, suggesting brief deletion of B cells alone is not sufficient to restore β cell tolerance.
The depleting effects of a given Ab make dosing problematic, as well as limit the frequency of treatments. On the other hand, nondepleting (ND) Ab provide a strategy to modulate the function of Teff and other immune cells without systemic cell depletion. Human IgG4 is intrinsically ND due to a decreased capacity to fix complement and bind to Fc receptor, and, therefore, is well suited for this purpose. Other IgG isotypes can be engineered accordingly to establish a ND property. A striking example of altering the activity of β cell-specific Teff is observed with the application of ND anti-CD4 and -CD8α Ab. A short-course of ND anti-CD4 and -CD8α Ab induces remission in the majority of new onset diabetic NOD mice without affecting systemic T cell numbers (128–131). Induction of remission is due to Ab-mediated crosslinking of the CD4 and CD8 molecules, which reduces TCR signaling as well as upregulates additional signaling pathways (128, 131–133). The result is suppressed expression of proinflammatory cytokines by Teff combined with the induction of a promigratory phenotype that drives pancreatic egress of Teff (128, 133).
ND Ab and immunoglobulin (Ig) fusion molecules can also serve to block the function of key surface effector molecules. For instance, CTLA-4-Ig (Abatacept)-mediated blockade of the CD80 and CD86 co-stimulatory molecules inhibits APC function and alters the Teff pool in recent onset T1D patients. Abatacept therapy administered monthly for 24 months extends β cell function up to 30 months (134, 135). Examination of peripheral blood shows a decrease in the central memory CD4+ T cell population with a concomitant increase in naïve T cells in Abatacept-treated individuals, supporting the notion that Teff expansion is limited (135).
Inhibiting cytotoxic pathways with ND blocking Ab offer another option to protect β cell viability. NOD mice deficient in Fas lack islet-infiltrating effector cells and fail to develop T1D (136, 137). Ab blockade of FasL between 2 and 4 weeks of age prevents T1D in NOD mice. Interestingly, FasL blockade results in the presence of protective IL-10-producing B cells in the pancreas of NOD mice after treatment (138). These results suggest the targeting the Fas–FasL pathway may be a viable therapeutic avenue for the treatment of clinical T1D.
In addition to directly targeting immune effector cells, Ab or Ig-fusion molecules have been used to neutralize secreted effector molecules, such as proinflammatory cytokines. Neutralization of TNFα with several antagonists including mAb (infliximab, adalimumab) and a recombinant TNFα receptor fusion protein, etanercept, have been widely used to treat the autoimmunity driving RA (139). Although TNFα is directly cytotoxic to β cells, its role in the pathogenesis of T1D is controversial. In NOD mice, for instance, TNFα accelerates the progression of β cell autoimmunity when given at a young but not older age (140, 141). In a 24-week clinical study, anti-TNFα therapy in human T1D patients was seen to reduce levels of glycated hemoglobin A1C (HbA1c), a marker indicative of blood glucose values over a 3-month period, as well as increase C-peptide levels suggesting preservation of β cell function (142). In a case report, treatment with infliximab was also found to reduce HbA1c levels, increase insulin secretion, and alleviate insulin resistance in a T1D patient also diagnosed with Crohn’s disease (143). Anti-TNFα therapy, however, fails to prevent the development of T1D in at-risk patients, and may in fact accelerate disease progression (144). These results further underscore how efficacy (or lack of) is dependent on the stage of disease progression at which a given therapeutic is administered.
IL-21 is an appealing cytokine to target and suppress β cell autoimmunity. IL-21 is chiefly produced by T follicular helper cells (Tfh) and Th17 cells, and has diverse roles that include regulating B cell and CD8+ Teff function (145–147). The emerging role for both Tfh and Th17 cells in the progression of T1D, and the protective phenotype seen in NOD mice deficient of IL-21 suggests that neutralizing human IL-21 is a promising approach (60, 148–152). Indeed, neutralization of IL-21 with an IL-21R-Ig prevents diabetes in NOD mice (153).
Cytokine-Based Immunotherapies
Administration of cytokines offers an approach to reestablish peripheral immunoregulation and β cell tolerance. One such strategy currently being investigated is administration of low dose IL-2 (65, 154–156). Conventional CD4+ and CD8+ T cells but not Foxp3+Treg are producers of IL-2, which plays a central role in driving both pro- and anti-inflammatory responses (157, 158). IL-2 for instance is needed for conventional T cell activation and expansion, and IL-2 drives proinflammatory responses by other immune effectors (158). IL-2 also plays an essential role in Foxp3+ Treg survival, expansion, and suppressor function (159–161). Constitutive expression of the high affinity IL-2R (CD25) allows Foxp3+Treg to out-compete conventional T cells for limiting amounts of IL-2. The latter provides rationale for using low dose IL-2 to preferentially affect Foxp3+Treg versus conventional T cells (154). Indeed, low dose IL-2 has been effective in the clinic for the treatment of graft-versus-host disease, and systemic vasculitis (162, 163). In NOD mice, low-dose IL-2 prevents clinical onset and reverses diabetes via an expanded pool of Foxp3+ Treg in the islets and draining pancreatic lymph nodes (69, 164, 165). Similarly, low-dose IL-2 in combination with rapamycin in recent onset T1D patients increases the frequency of Foxp3+Treg in blood (166). However, these patients also exhibit an accelerated rate of β cell loss (166), suggesting an enhanced pathogenic response, and highlighting the key problem of administering a cytokine with pleiotropic effects (167, 168).
Different strategies are being developed to enhance the efficacy of IL-2 (and other cytokines), while avoiding unwanted systemic effects (169, 170). One approach is to promote selective binding of IL-2 to Foxp3+Treg via IL-2-Ab complexes (IL-2C) (170–172). Targeting particular epitopes on IL-2 with anti-IL-2 Ab can favor binding to the high affinity IL-2R constitutively expressed by Foxp3+Treg (173). Administration of IL-2C readily expands Foxp3+Treg in mice and prevents autoimmunity (170–172). While promising, polyclonal expansion of Foxp3+Treg by IL-2C may compromise protective immunity against pathogens.
An additional strategy to minimize the systemic effects of IL-2 while expanding β cell-specific Foxp3+Treg is to target cytokine expression to β cells in vivo. Adeno-associated virus (AAV) vectors have been used to deliver and target expression of an insulin promoter-driven IL-2 transgene (AAViP-IL2) to β cells in vivo (72–74, 174). In general, AAV vectors are appealing for in vivo gene delivery due to limited immunogenicity, lack of integration into the genome, and efficient transduction of non-proliferating cells (175). Notably, treatment of NOD mice at a late preclinical T1D stage with AAViP-IL2 results in the expansion of islet-resident Foxp3+Treg, suppression of β cell-specific Teff, and prevention of diabetes onset (72–74). AAV vectors can be further exploited by co-delivering genes encoding other anti-inflammatory cytokines (e.g., IL-10, TGFβ1, and IL-35) and/or pro-survival proteins to enhance both the tolerogenic effect and maintenance of β cell mass in vivo (72, 174, 176–179).
Foxp3+Treg-Mediated Therapy
An alternative strategy to manipulate the Foxp3+Treg population in vivo is to transfer Foxp3+Treg that have been expanded in vitro (180–182). The approach is effective in preventing diabetes onset in NOD mice (183). However, in vitro expansion of a homogeneous and stable pool of Foxp3+Treg, particularly when starting from relatively few cells, has been technically difficult. Expansion protocols are being devised using drugs such as rapamycin to prevent outgrowth of Teff, as well as DNA methyltransferase and histone deacetylase inhibitors to enhance Foxp3+Treg stability (184, 185). Despite these hurdles, clinical studies have shown Foxp3+Treg therapy is well tolerated and therapeutic efficacy is observed for various diseases (180, 181). Furthermore, a phase I trial has demonstrated that in vitro-expanded Foxp3+Treg persist long-term and exhibit a stable phenotype when transferred back into T1D subjects (180). Notably, loss of C peptide is also limited in some patients. These results provide justification for additional clinical studies to directly assess the efficacy of Foxp3+Treg transfer for T1D treatment.
Microbiome Interventions in T1D
In recent years, the importance of the gut microbiota in maintaining the normal function of the immune system has emerged (186). Reports have documented that changes in gut microbiota can markedly influence T1D development, and that differences in the colonizing microflora contributes to variations in T1D onset among NOD mouse colonies (187, 188). Toll-like receptors (TLRs) play a crucial role in the innate immune system’s recognition of common bacterial and viral components. TLR signaling triggers APC maturation and production of proinflammatory mediators (189). One of the major TLR signaling adaptor molecules driving APC maturation is MyD88, which activates several downstream signaling pathways (190). NOD mice lacking MyD88 expression and housed under normal specific-pathogen-free conditions exhibit a reduced T1D incidence. In contrast, MyD88-deficient NOD mice housed in germ-free conditions develop robust diabetes (16). Interestingly sex differences in the gut microbiota also influence T1D susceptibility. For example, young female NOD mice receiving gut microbiota from male NOD mice exhibit a reduced T1D incidence (191). These results demonstrate that the gut microbiota can have a protective role against autoimmunity, and specifically T1D. Furthermore, differences in the biodiversity of the gut microbiome are seen between T1D patients and healthy individuals (192–196). Similarly, the microbiome within the gut of T1D infants fails to diversify during development in comparison to healthy individuals, potentially due to hyperglycemia (197). Here, changes in the production of microbial metabolites such as short-chain fatty acids found in the colonic lumen and peripheral blood are thought to impact immunoregulatory networks. Indeed, a recent study showed that diets that enhance production of acetate and butyrate by the gut microbiome, effectively prevent diabetes in NOD mice (198). Increased acetate is seen to reduce β cell-specific Teff, whereas elevated butyrate expands Foxp3+Treg. Together these results indicate an association between modified gut microbiota and compromised self-tolerance. Manipulating the gut microbiota may prove to be an effective adjuvant therapy to limit the progression of β cell autoimmunity in at-risk individuals.
Antigen-Dependent Immunotherapies
The goal of antigen-based therapies is to selectively tolerize the autoreactive Teff pool and induce or expand autoantigen-specific Treg (199–202). The approach is appealing, since β cell antigen vaccination is expected to have no effect on acquired immunity. Teff are tolerized via whole antigen or peptide by various mechanisms, including: (i) clonal anergy, (ii) clonal deletion, or (iii) exhaustion. This has classically been achieved by administration of high doses of soluble antigen or peptide. On the other hand, antigen vaccination has been used to induce differentiation of naïve β cell-specific T cells into aTreg, including iFoxp3+Treg. Depending on the conditions of antigen vaccination, distinct subsets of aTreg can be induced. Expanding the aTreg pool is advantageous, since local cytokine-mediated suppression is independent of Teff antigen-specificity; this bystander-mediated suppression also can downregulate the activity of proinflammatory APC. Success of either tolerizing Teff or inducing/expanding Treg is dictated by multiple factors including dose, the frequency, and route of antigen vaccination. Also important is the context in which the antigen is delivered. Antigen administration in the context of an adjuvant, microparticles, or DC can significantly alter the nature of the T cell response. The most critical factor determining efficacy, however, is the identity of the autoantigen (or peptide) used for vaccination. To effectively block the progression of β cell autoimmunity, prevalent, or immunodominant Teff clonotypes need to be targeted. Ongoing efforts defining the specificity of islet-resident Teff are expected to identify immunodominant Teff clonotypes (31). Furthermore, promising antigen-based immunotherapies are exploiting advancements in targeted drug delivery systems to effectively tolerize Teff and promote Treg-mediated suppression.
β Cell-Autoantigen Vaccination
Early clinical studies have highlighted the difficulty in establishing a protective, β cell-specific T cell response via autoantigen vaccination (203). The Diabetes Prevention Trial-type 1 tested parenteral insulin delivery via intravenous (i.v.), subcutaneous (s.c.), and oral routes in at-risk individuals (202, 204–206). No significant difference, however, was detected in the frequency and onset of diabetes in the treatment and control cohorts. Similarly, phase II and III studies of s.c. vaccination of aluminum hydroxide (alum)-formulated GAD65 to new onset T1D patients had no marked effect on C-peptide secretion over time (207, 208). Alum as an adjuvant is included due to a potent capacity to induce IL-4-secreting Th2 cells, which in turn can block the differentiation of pathogenic IFNγ-producing Teff (209, 210).
Additional studies have tested insulin and proinsulin peptides to restore β cell tolerance in human T1D patients. An altered-peptide ligand of the insulin B chain 9–23 epitope administered s.c. to recent onset T1D subjects reduces peptide-specific IFNγ-secreting Teff, while elevating Th2-like aTreg (211, 212). Nevertheless, no clinical benefit was detected, suggesting that altering insulin B chain-specific Teff reactivity alone is insufficient to induce robust β cell tolerance. Clinical trials assessing intradermal vaccination of a natural peptide derived from proinsulin (C19-A3) have also been carried out (213, 214). Vaccination with low (30ug) versus high (300 µg) doses of C19-A3 increases the frequency of peptide-specific CD4+ Tr1 cells in long-standing T1D subjects (213). A similar increase in CD4+ Tr1 cells is detected in newly diagnosed T1D subjects vaccinated with C19-A3 (214). Notably, FOXP3+ Treg are also increased, and evidence suggests that residual β cell function is preserved in some individuals (214). Treatment with a pool of peptides derived from additional β cell autoantigens may promote a more robust tolerogenic effect on Teff, as well as enhance induction and/or expansion of the aTreg pool.
Strategies to Enhance the Efficacy of β Cell-Autoantigen Vaccination
As noted above the context of autoantigen vaccination is critical for determining the nature and magnitude of the induced T cell response, and in turn therapeutic efficacy. Various strategies are being developed to better “tailor” the antigen-specific T cell response. One such approach has been the application of plasmid DNA (pDNA) vaccination. Injection of soluble pDNA or via gene gun delivered pDNA-coated particles results in significant levels of transgene expression lasting several weeks in vivo (215, 216). Co-vaccination of pDNA encoding β cell autoantigens and anti-inflammatory cytokines both induces protective β cell-specific aTreg, and prevents diabetes onset in NOD mice (174, 217–220). In a T1D clinical trial, patients received intramuscular injections of pDNA encoding proinsulin over a 12-week period. C-peptide levels were maintained up to 3 months after treatment, which corresponded with a reduced frequency of proinsulin-specific CD8+ T cells in peripheral blood (221). Notably, the frequency of CD8+ T cells specific for foreign antigen remained unperturbed, illustrating a key strength of β cell-autoantigen vaccination.
An additional approach to enhance the efficacy of β cell-autoantigen vaccines is the use of small molecule particles (222, 223). Typically, injected β cell antigens and other soluble reagents have a short half-life in vivo, which can be greatly enhanced by encapsulation into a particle. Particles are generated using different compounds, including poly(lactide-co-glycolide) and acetalated dextran (222, 224). The particle size and composition can be readily manipulated to determine cellular uptake and cargo release in vivo (225). Some studies have included cell-specific Ab or ligands to target particles to a desired cell population, and avoid off-target effects (222, 226–228). However, most treatments utilize the natural phagocytic function of DC and macrophages to engulf the injected particles. In this way, a tolerogenic APC phenotype can also be established in vivo by encapsulating various immunomodulatory agents such as the aryl hydrocarbon receptor ligand 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester, rapamycin, dexamethasone, cytokines, and/or antisense oligodeoxynucleotides (222, 223, 229–232). These APC upon presenting the loaded antigen more readily tolerize Teff and/or induce aTreg (229–232).
As opposed to delivering a cargo to APC, nanoparticles have also been used as surrogate APC (233, 234). Here the surface of nanoparticles is complexed with MHC class II (MHCII) molecules tethered to a β cell-derived peptide (234). These MHCII-peptide-coated nanoparticles promote differentiation of CD4+ Teff into Tr1 cells that are readily expanded, and in turn drive diabetes remission in NOD mice (234). Another approach has exploited the tolerogenic properties of apoptotic cells. Uptake of apoptotic bodies by immature DC and macrophages blocks subsequent APC maturation and promotes upregulation of TGFβ1 (235). Accordingly, i.v. injection of 1-ethyl-3-(3 dimethylaminopropyl) carbodiimide (ECDI) fixed splenocytes loaded with autoantigen suppresses experimental autoimmune encephalomyelitis, a murine model of multiple sclerosis (MS) (236). Currently, this approach is being tested in clinical trials in which MS patients receive an i.v. transfusion of ECDI-fixed peripheral blood mononuclear cells loaded with MS-relevant peptides recognized by CD4+ T cells (237, 238).
Summary
Type 1 diabetes is a complex disease driven by pathogenic adaptive and innate responses leading to the dysfunction and destruction of β cells (1–4). Although disease immunopathology is heterogeneous, T cells are considered to be the key mediators of β cell destruction/dysfunction for the majority of cases of human T1D (57). Consequently, T cells have been the focus of most strategies of immunotherapy. Suppression of pathogenic β cell-specific T cell reactivity in the islets long-term, while preserving acquired immunity is the ultimate goal for a given immunotherapy (Figure 1). Achieving this goal in the clinic, however, has been elusive. To date, the most effective strategies have been broadly acting on the majority if not all T cells although protection has also been demonstrated by targeting B cells (88, 100, 119, 120, 239). Efficacy, however, is short-lived. Furthermore, systemic depletion and unwanted effects on protective immunity seen with anti-CD3 mAb therapy for instance, limit dosing and repeated application for efforts to extend β cell protection. On the other hand, antigen-based immunotherapy, such as β cell protein/peptide or particle vaccination, must contend with what is likely to be a high degree of diversity in the β cell-specific T cell response among T1D individuals and T1D subsets (199–201). Therefore, the ability to tolerize a sufficient number of disease-driving self-reactive T cell clones residing in the islets will be challenging. The most effective approach may reside in combining strategies functioning via distinct mechanisms. ND Ab specific for CD4 and CD8, for example, can be used to purge pathogenic Teff residing in the islets (128, 133). This would then be followed by an antigen-based strategy in which the need to establish a sufficiently sized pool of aTreg becomes less stringent. A novel combinatorial approach recently described utilizes IL-2C and MHC class II-peptide tetramers. This combination expands the frequency of Foxp3+Treg in a peptide-specific manner that results in prevention of diabetes in NOD mice (240).
Further studies defining common biomarkers of T1D will be critical in designing treatment protocols for individual patients, as well as for monitoring the effects of a given immunotherapy. Current advancements in large-scale genomic and proteomic analyses are making identification of these types of markers a reality, in addition to “matching” at risk or T1D patients with the most appropriate immunotherapy.
Author Contributions
CJK, MC, QK, and RMT contributed to the preparation of the review article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by National Institutes of Health grants R01DK100256 and R01DK1035486 (RMT) and T32AI007273 (MC).
References
1. Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev (1994) 15(4):516–42. doi:10.1210/edrv-15-4-516
2. Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell (1996) 85(3):291–7. doi:10.1016/S0092-8674(00)81106-X
3. Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol (2004) 552:306–10.
4. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi:10.1146/annurev.immunol.23.021704.115643
5. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi:10.1038/ng.381
6. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what’s next? Diabetes (2010) 59(7):1561–71. doi:10.2337/db10-0076
7. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(1):a007732. doi:10.1101/cshperspect.a007732
8. Onengut-Gumuscu S, Chen WM, Burren O, Cooper NJ, Quinlan AR, Mychaleckyj JC, et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat Genet (2015) 47(4):381–6. doi:10.1038/ng.3245
9. Op de Beeck A, Eizirik DL. Viral infections in type 1 diabetes mellitus – why the beta cells? Nat Rev Endocrinol (2016) 12(5):263–73. doi:10.1038/nrendo.2016.30
10. Laitinen OH, Honkanen H, Pakkanen O, Oikarinen S, Hankaniemi MM, Huhtala H, et al. Coxsackievirus B1 is associated with induction of beta-cell autoimmunity that portends type 1 diabetes. Diabetes (2014) 63(2):446–55. doi:10.2337/db13-0619
11. Oikarinen S, Tauriainen S, Hober D, Lucas B, Vazeou A, Sioofy-Khojine A, et al. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes (2014) 63(2):655–62. doi:10.2337/db13-0620
12. Atkinson MA. The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(11). doi:10.1101/cshperspect.a007641
13. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers (2017) 3:17016. doi:10.1038/nrdp.2017.16
14. Knip M, Siljander H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat Rev Endocrinol (2016) 12(3):154–67. doi:10.1038/nrendo.2015.218
15. Vaarala O, Atkinson MA, Neu J. The “perfect storm” for type 1 diabetes: the complex interplay between intestinal microbiota, gut permeability, and mucosal immunity. Diabetes (2008) 57(10):2555–62. doi:10.2337/db08-0331
16. Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature (2008) 455(7216):1109–13. doi:10.1038/nature07336
17. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. HL-A antigens and diabetes mellitus. Lancet (1974) 2(7885):864–6. doi:10.1016/S0140-6736(74)91201-X
18. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36(4):337–8. doi:10.1038/ng1323
19. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JM, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes (2004) 53(11):3020–3. doi:10.2337/diabetes.53.11.3020
20. Nistico L, Buzzetti R, Pritchard LE, Van der Auwera B, Giovannini C, Bosi E, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Hum Mol Genet (1996) 5(7):1075–80. doi:10.1093/hmg/5.7.1075
21. Ueda H, Howson JM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423(6939):506–11. doi:10.1038/nature01621
22. Field LL, Larsen Z, Pociot F, Nerup J, Tobias R, Bonnevie-Nielsen V. Evidence for a locus (IDDM16) in the immunoglobulin heavy chain region on chromosome 14q32.3 producing susceptibility to type 1 diabetes. Genes Immun (2002) 3(6):338–44. doi:10.1038/sj.gene.6363857
23. Morahan G, Huang D, Ymer SI, Cancilla MR, Stephen K, Dabadghao P, et al. Linkage disequilibrium of a type 1 diabetes susceptibility locus with a regulatory IL12B allele. Nat Genet (2001) 27(2):218–21. doi:10.1038/84872
24. Clark M, Kroger CJ, Tisch RM. Type 1 diabetes: a chronic anti-self-inflammatory response. Front Immunol (2017) 8:1898. doi:10.3389/fimmu.2017.01898
25. Mallone R, Brezar V, Boitard C. T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol (2011) 2011:513210. doi:10.1155/2011/513210
26. Enee E, Kratzer R, Arnoux JB, Barilleau E, Hamel Y, Marchi C, et al. ZnT8 is a major CD8+ T cell-recognized autoantigen in pediatric type 1 diabetes. Diabetes (2012) 61(7):1779–84. doi:10.2337/db12-0071
27. Han S, Donelan W, Wang H, Reeves W, Yang LJ. Novel autoantigens in type 1 diabetes. Am J Transl Res (2013) 5(4):379–92.
28. Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med (2012) 2(4):a007781. doi:10.1101/cshperspect.a007781
29. Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, et al. Islet-derived CD4 T cells targeting proinsulin in human autoimmune diabetes. Diabetes (2017) 66(3):722–34. doi:10.2337/db16-1025
30. Delong T, Baker RL, He J, Barbour G, Bradley B, Haskins K. Diabetogenic T-cell clones recognize an altered peptide of chromogranin A. Diabetes (2012) 61(12):3239–46. doi:10.2337/db12-0112
31. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi:10.1038/nm.4203
32. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi:10.1126/science.aad2791
33. Baker RL, Jamison BL, Wiles TA, Lindsay RS, Barbour G, Bradley B, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes (2018) 67(8):db180200. doi:10.2337/db18-0200
34. Wiles TA, Delong T, Baker RL, Bradley B, Barbour G, Powell RL, et al. An insulin-IAPP hybrid peptide is an endogenous antigen for CD4 T cells in the non-obese diabetic mouse. J Autoimmun (2017) 78:11–8. doi:10.1016/j.jaut.2016.10.007
35. Roep BO, Kracht MJ, van Lummel M, Zaldumbide A. A roadmap of the generation of neoantigens as targets of the immune system in type 1 diabetes. Curr Opin Immunol (2016) 43:67–73. doi:10.1016/j.coi.2016.09.007
36. Kracht MJ, van Lummel M, Nikolic T, Joosten AM, Laban S, van der Slik AR, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med (2017) 23(4):501–7. doi:10.1038/nm.4289
37. Phelps EA, Cianciaruso C, Michael IP, Pasquier M, Kanaani J, Nano R, et al. Aberrant accumulation of the diabetes autoantigen GAD65 in Golgi membranes in conditions of ER stress and autoimmunity. Diabetes (2016) 65(9):2686–99. doi:10.2337/db16-0180
38. Eizirik DL, Sammeth M, Bouckenooghe T, Bottu G, Sisino G, Igoillo-Esteve M, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet (2012) 8(3):e1002552. doi:10.1371/journal.pgen.1002552
39. Ortis F, Naamane N, Flamez D, Ladriere L, Moore F, Cunha DA, et al. Cytokines interleukin-1beta and tumor necrosis factor-alpha regulate different transcriptional and alternative splicing networks in primary beta-cells. Diabetes (2010) 59(2):358–74. doi:10.2337/db09-1159
40. Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol (2001) 166(8):5265–70. doi:10.4049/jimmunol.166.8.5265
41. You S, Chatenoud L. Proinsulin: a unique autoantigen triggering autoimmune diabetes. J Clin Invest (2006) 116(12):3108–10. doi:10.1172/JCI30760
42. von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol (2007) 7(12):988–94. doi:10.1038/nri2192
43. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi:10.1111/j.1365-2249.2008.03860.x
44. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127(8):2881–91. doi:10.1172/JCI94549
45. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1):51–60. doi:10.1084/jem.20111187
46. Atkinson MA, von Herrath M, Powers AC, Clare-Salzler M. Current concepts on the pathogenesis of type 1 diabetes – considerations for attempts to prevent and reverse the disease. Diabetes Care (2015) 38(6):979–88. doi:10.2337/dc15-0144
47. Arif S, Gibson VB, Nguyen V, Bingley PJ, Todd JA, Guy C, et al. Beta-cell specific T-lymphocyte response has a distinct inflammatory phenotype in children with type 1 diabetes compared with adults. Diabet Med (2017) 34(3):419–25. doi:10.1111/dme.13153
48. Heninger AK, Eugster A, Kuehn D, Buettner F, Kuhn M, Lindner A, et al. A divergent population of autoantigen-responsive CD4+ T cells in infants prior to beta cell autoimmunity. Sci Transl Med (2017) 9(378):eaaf8848. doi:10.1126/scitranslmed.aaf8848
49. Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol (2011) 33(1):9–21. doi:10.1007/s00281-010-0205-0
50. Coppieters KT, Roep BO, von Herrath MG. Beta cells under attack: toward a better understanding of type 1 diabetes immunopathology. Semin Immunopathol (2011) 33(1):1–7. doi:10.1007/s00281-010-0236-6
51. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and beta-cell mass in the natural history of type 1 diabetes. Diabetes (2016) 65(3):719–31. doi:10.2337/db15-0779
52. Campbell-Thompson M. Organ donor specimens: what can they tell us about type 1 diabetes? Pediatr Diabetes (2015) 16(5):320–30. doi:10.1111/pedi.12286
53. In’t Veld P. Insulitis in human type 1 diabetes: the quest for an elusive lesion. Islets (2011) 3(4):131–8. doi:10.4161/isl.3.4.15728
54. Skog O, Korsgren S, Melhus A, Korsgren O. Revisiting the notion of type 1 diabetes being a T-cell-mediated autoimmune disease. Curr Opin Endocrinol Diabetes Obes (2013) 20(2):118–23. doi:10.1097/MED.0b013e32835edb89
55. Atkinson MA, Gianani R. The pancreas in human type 1 diabetes: providing new answers to age-old questions. Curr Opin Endocrinol Diabetes Obes (2009) 16(4):279–85. doi:10.1097/MED.0b013e32832e06ba
56. Kent SC, Mannering SI, Michels AW, Babon JAB. Deciphering the pathogenesis of human type 1 diabetes (T1D) by interrogating T cells from the “scene of the crime”. Curr Diab Rep (2017) 17(10):95. doi:10.1007/s11892-017-0915-y
57. Gomez-Tourino I, Arif S, Eichmann M, Peakman M. T cells in type 1 diabetes: instructors, regulators and effectors: a comprehensive review. J Autoimmun (2016) 66:7–16. doi:10.1016/j.jaut.2015.08.012
58. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest (2004) 113(3):451–63. doi:10.1172/JCI19585
59. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol (2016) 183(1):16–29. doi:10.1111/cei.12672
60. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes (2009) 58(6):1302–11. doi:10.2337/db08-1113
61. Marwaha AK, Crome SQ, Panagiotopoulos C, Berg KB, Qin H, Ouyang Q, et al. Cutting edge: increased IL-17-secreting T cells in children with new-onset type 1 diabetes. J Immunol (2010) 185(7):3814–8. doi:10.4049/jimmunol.1001860
62. Ferraro A, Socci C, Stabilini A, Valle A, Monti P, Piemonti L, et al. Expansion of Th17 cells and functional defects in T regulatory cells are key features of the pancreatic lymph nodes in patients with type 1 diabetes. Diabetes (2011) 60(11):2903–13. doi:10.2337/db11-0090
63. Schneider A, Rieck M, Sanda S, Pihoker C, Greenbaum C, Buckner JH. The effector T cells of diabetic subjects are resistant to regulation via CD4+ FOXP3+ regulatory T cells. J Immunol (2008) 181(10):7350–5. doi:10.4049/jimmunol.181.10.7350
64. D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, et al. The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci U S A (2008) 105(50):19857–62. doi:10.1073/pnas.0810713105
65. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes (2012) 61(1):14–22. doi:10.2337/db11-1213
66. Garg G, Tyler JR, Yang JH, Cutler AJ, Downes K, Pekalski M, et al. Type 1 diabetes-associated IL2RA variation lowers IL-2 signaling and contributes to diminished CD4+CD25+ regulatory T cell function. J Immunol (2012) 188(9):4644–53. doi:10.4049/jimmunol.1100272
67. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes (2005) 54(5):1407–14. doi:10.2337/diabetes.54.5.1407
68. Lindley S, Dayan CM, Bishop A, Roep BO, Peakman M, Tree TI. Defective suppressor function in CD4(+)CD25(+) T-cells from patients with type 1 diabetes. Diabetes (2005) 54(1):92–9. doi:10.2337/diabetes.54.1.92
69. Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, et al. Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity (2008) 28(5):687–97. doi:10.1016/j.immuni.2008.03.016
70. Hull CM, Peakman M, Tree TIM. Regulatory T cell dysfunction in type 1 diabetes: what’s broken and how can we fix it? Diabetologia (2017) 60(10):1839–50. doi:10.1007/s00125-017-4377-1
71. Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med (2007) 13(3):108–16. doi:10.1016/j.molmed.2007.01.003
72. Goudy KS, Johnson MC, Garland A, Li C, Samulski RJ, Wang B, et al. Inducible adeno-associated virus-mediated IL-2 gene therapy prevents autoimmune diabetes. J Immunol (2011) 186(6):3779–86. doi:10.4049/jimmunol.1001422
73. Johnson MC, Garland AL, Nicolson SC, Li C, Samulski RJ, Wang B, et al. Beta-cell-specific IL-2 therapy increases islet Foxp3+Treg and suppresses type 1 diabetes in NOD mice. Diabetes (2013) 62(11):3775–84. doi:10.2337/db13-0669
74. Flores RR, Zhou L, Robbins PD. Expression of IL-2 in beta cells by AAV8 gene transfer in pre-diabetic NOD mice prevents diabetes through activation of FoxP3-positive regulatory T cells. Gene Ther (2014) 21(8):715–22. doi:10.1038/gt.2014.45
75. Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, et al. Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes (2010) 59(2):407–15. doi:10.2337/db09-0694
76. Attridge K, Wang CJ, Wardzinski L, Kenefeck R, Chamberlain JL, Manzotti C, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood (2012) 119(20):4656–64. doi:10.1182/blood-2011-10-388546
77. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes (2014) 63(11):3835–45. doi:10.2337/db14-0365
78. Sanda S, Roep BO, von Herrath M. Islet antigen specific IL-10+ immune responses but not CD4+CD25+FoxP3+ cells at diagnosis predict glycemic control in type 1 diabetes. Clin Immunol (2008) 127(2):138–43. doi:10.1016/j.clim.2007.12.003
79. Petrich de Marquesini LG, Fu J, Connor KJ, Bishop AJ, McLintock NE, Pope C, et al. IFN-gamma and IL-10 islet-antigen-specific T cell responses in autoantibody-negative first-degree relatives of patients with type 1 diabetes. Diabetologia (2010) 53(7):1451–60. doi:10.1007/s00125-010-1739-3
80. Jin Y, Chen X, Podolsky R, Hopkins D, Makala LH, Muir A, et al. APC dysfunction is correlated with defective suppression of T cell proliferation in human type 1 diabetes. Clin Immunol (2009) 130(3):272–9. doi:10.1016/j.clim.2008.10.005
81. Serreze DV, Gaskins HR, Leiter EH. Defects in the differentiation and function of antigen presenting cells in NOD/Lt mice. J Immunol (1993) 150(6):2534–43.
82. Katz JD, Ondr JK, Opoka RJ, Garcia Z, Janssen EM. Cutting edge: merocytic dendritic cells break T cell tolerance to beta cell antigens in nonobese diabetic mouse diabetes. J Immunol (2010) 185(4):1999–2003. doi:10.4049/jimmunol.1001398
83. Poligone B, Weaver DJ Jr, Sen P, Baldwin AS Jr, Tisch R. Elevated NF-kappaB activation in nonobese diabetic mouse dendritic cells results in enhanced APC function. J Immunol (2002) 168(1):188–96. doi:10.4049/jimmunol.168.1.188
84. Liu J, Beller DI. Distinct pathways for NF-kappa B regulation are associated with aberrant macrophage IL-12 production in lupus- and diabetes-prone mouse strains. J Immunol (2003) 170(9):4489–96. doi:10.4049/jimmunol.170.9.4489
85. Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia (2007) 50(3):596–601. doi:10.1007/s00125-006-0569-9
86. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med (1996) 184(5):2049–53. doi:10.1084/jem.184.5.2049
87. Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, Naji A. B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes (1997) 46(6):941–6. doi:10.2337/diabetes.46.6.941
88. Marino E, Silveira PA, Stolp J, Grey ST. B cell-directed therapies in type 1 diabetes. Trends Immunol (2011) 32(6):287–94. doi:10.1016/j.it.2011.03.006
89. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes (2005) 54(Suppl 2):S97–107. doi:10.2337/diabetes.54.suppl_2.S97
90. Simmons KM, Michels AW. Type 1 diabetes: a predictable disease. World J Diabetes (2015) 6(3):380–90. doi:10.4239/wjd.v6.i3.380
91. Regnell SE, Lernmark A. Early prediction of autoimmune (type 1) diabetes. Diabetologia (2017) 60(8):1370–81. doi:10.1007/s00125-017-4308-1
92. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA (2013) 309(23):2473–9. doi:10.1001/jama.2013.6285
93. Chen C, Cohrs CM, Stertmann J, Bozsak R, Speier S. Human beta cell mass and function in diabetes: recent advances in knowledge and technologies to understand disease pathogenesis. Mol Metab (2017) 6(9):943–57. doi:10.1016/j.molmet.2017.06.019
94. Shields BM, McDonald TJ, Oram R, Hill A, Hudson M, Leete P, et al. C-peptide decline in type 1 diabetes has two phases: an initial exponential fall and a subsequent stable phase. Diabetes Care (2018) 41(7):1486–92. doi:10.2337/dc18-0465
95. Bougneres PF, Landais P, Boisson C, Carel JC, Frament N, Boitard C, et al. Limited duration of remission of insulin dependency in children with recent overt type I diabetes treated with low-dose cyclosporin. Diabetes (1990) 39(10):1264–72. doi:10.2337/diab.39.10.1264
96. Stiller CR, Dupre J, Gent M, Jenner MR, Keown PA, Laupacis A, et al. Effects of cyclosporine immunosuppression in insulin-dependent diabetes mellitus of recent onset. Science (1984) 223(4643):1362–7. doi:10.1126/science.6367043
97. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346(22):1692–8. doi:10.1056/NEJMoa012864
98. Shoda LK, Young DL, Ramanujan S, Whiting CC, Atkinson MA, Bluestone JA, et al. A comprehensive review of interventions in the NOD mouse and implications for translation. Immunity (2005) 23(2):115–26. doi:10.1016/j.immuni.2005.08.002
99. Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med (2008) 14(5):528–35. doi:10.1038/nm1749
100. Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol (2007) 7(8):622–32. doi:10.1038/nri2134
101. Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9(9):1202–8. doi:10.1038/nm924
102. Wallberg M, Recino A, Phillips J, Howie D, Vienne M, Paluch C, et al. Anti-CD3 treatment up-regulates programmed cell death protein-1 expression on activated effector T cells and severely impairs their inflammatory capacity. Immunology (2017) 151(2):248–60. doi:10.1111/imm.12729
103. Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol (1997) 158(6):2947–54.
104. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352(25):2598–608. doi:10.1056/NEJMoa043980
105. Herold KC, Gitelman S, Greenbaum C, Puck J, Hagopian W, Gottlieb P, et al. Treatment of patients with new onset Type 1 diabetes with a single course of anti-CD3 mAb Teplizumab preserves insulin production for up to 5 years. Clin Immunol (2009) 132(2):166–73. doi:10.1016/j.clim.2009.04.007
106. Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes (2013) 62(11):3766–74. doi:10.2337/db13-0345
107. Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes (2005) 54(6):1763–9. doi:10.2337/diabetes.54.6.1763
108. Herold KC, Gitelman SE, Willi SM, Gottlieb PA, Waldron-Lynch F, Devine L, et al. Teplizumab treatment may improve C-peptide responses in participants with type 1 diabetes after the new-onset period: a randomised controlled trial. Diabetologia (2013) 56(2):391–400. doi:10.1007/s00125-012-2753-4
109. Long SA, Thorpe J, DeBerg HA, Gersuk V, Eddy J, Harris KM, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol (2016) 1(5):eaai7793. doi:10.1126/sciimmunol.aai7793
110. Long SA, Thorpe J, Herold KC, Ehlers M, Sanda S, Lim N, et al. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol (2017) 319:3–9. doi:10.1016/j.cellimm.2017.07.007
111. Tooley JE, Vudattu N, Choi J, Cotsapas C, Devine L, Raddassi K, et al. Changes in T-cell subsets identify responders to FcR-nonbinding anti-CD3 mAb (teplizumab) in patients with type 1 diabetes. Eur J Immunol (2016) 46(1):230–41. doi:10.1002/eji.201545708
112. Vudattu NK, Herold KC. Treatment of new onset type 1 diabetes with teplizumab: successes and pitfalls in development. Expert Opin Biol Ther (2014) 14(3):377–85. doi:10.1517/14712598.2014.881797
113. Daifotis AG, Koenig S, Chatenoud L, Herold KC. Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol (2013) 149(3):268–78. doi:10.1016/j.clim.2013.05.001
114. Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: evidence of improved immune regulation. Diabetes (2012) 61(1):145–54. doi:10.2337/db11-1033
115. Sherry NA, Chen W, Kushner JA, Glandt M, Tang Q, Tsai S, et al. Exendin-4 improves reversal of diabetes in NOD mice treated with anti-CD3 monoclonal antibody by enhancing recovery of beta-cells. Endocrinology (2007) 148(11):5136–44. doi:10.1210/en.2007-0358
116. Hyslop CM, Tsai S, Shrivastava V, Santamaria P, Huang C. Prolactin as an adjunct for type 1 diabetes immunotherapy. Endocrinology (2016) 157(1):150–65. doi:10.1210/en.2015-1549
117. Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, et al. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes (2009) 58(10):2277–84. doi:10.2337/db09-0557
118. Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Perry DJ, Schultz AR, et al. Antithymocyte globulin plus G-CSF combination therapy leads to sustained immunomodulatory and metabolic effects in a subset of responders with established type 1 diabetes. Diabetes (2016) 65(12):3765–75. doi:10.2337/db16-0823
119. Hamad AR, Ahmed R, Donner T, Fousteri G. B cell-targeted immunotherapy for type 1 diabetes: what can make it work? Discov Med (2016) 21(115):213–9.
120. Musette P, Bouaziz JD. B cell modulation strategies in autoimmune diseases: new concepts. Front Immunol (2018) 9:622. doi:10.3389/fimmu.2018.00622
121. Hu CY, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, et al. Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J Clin Invest (2007) 117(12):3857–67. doi:10.1172/JCI32405
122. Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol (2008) 180(5):2863–75. doi:10.4049/jimmunol.180.5.2863
123. Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes (2008) 57(11):3013–24. doi:10.2337/db08-0420
124. Zekavat G, Rostami SY, Badkerhanian A, Parsons RF, Koeberlein B, Yu M, et al. In vivo BLyS/BAFF neutralization ameliorates islet-directed autoimmunity in nonobese diabetic mice. J Immunol (2008) 181(11):8133–44. doi:10.4049/jimmunol.181.11.8133
125. Marino E, Villanueva J, Walters S, Liuwantara D, Mackay F, Grey ST. CD4(+)CD25(+) T-cells control autoimmunity in the absence of B-cells. Diabetes (2009) 58(7):1568–77. doi:10.2337/db08-1504
126. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med (2009) 361(22):2143–52. doi:10.1056/NEJMoa0904452
127. Cohen MD, Keystone E. Rituximab for rheumatoid arthritis. Rheumatol Ther (2015) 2(2):99–111. doi:10.1007/s40744-015-0016-9
128. Yi Z, Diz R, Martin AJ, Morillon YM, Kline DE, Li L, et al. Long-term remission of diabetes in NOD mice is induced by nondepleting anti-CD4 and anti-CD8 antibodies. Diabetes (2012) 61(11):2871–80. doi:10.2337/db12-0098
129. Phillips JM, Parish NM, Raine T, Bland C, Sawyer Y, De La Pena H, et al. Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabet Stud (2009) 6(2):97–103. doi:10.1900/RDS.2009.6.97
130. Hutchings P, O’Reilly L, Parish NM, Waldmann H, Cooke A. The use of a non-depleting anti-CD4 monoclonal antibody to re-establish tolerance to beta cells in NOD mice. Eur J Immunol (1992) 22(7):1913–8. doi:10.1002/eji.1830220735
131. Phillips JM, Harach SZ, Parish NM, Fehervari Z, Haskins K, Cooke A. Nondepleting anti-CD4 has an immediate action on diabetogenic effector cells, halting their destruction of pancreatic beta cells. J Immunol (2000) 165(4):1949–55. doi:10.4049/jimmunol.165.4.1949
132. Pullar CE, Morris PJ, Wood KJ. Altered proximal T-cell receptor signalling events in mouse CD4+ T cells in the presence of anti-CD4 monoclonal antibodies: evidence for reduced phosphorylation of Zap-70 and LAT. Scand J Immunol (2003) 57(4):333–41. doi:10.1046/j.1365-3083.2003.01241.x
133. Martin AJ, Clark M, Gojanovich G, Manzoor F, Miller K, Kline DE, et al. Anti-coreceptor therapy drives selective T cell egress by suppressing inflammation-dependent chemotactic cues. JCI Insight (2016) 1(17):e87636. doi:10.1172/jci.insight.87636
134. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet (2011) 378(9789):412–9. doi:10.1016/S0140-6736(11)60886-6
135. Orban T, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care (2014) 37(4):1069–75. doi:10.2337/dc13-0604
136. Itoh N, Imagawa A, Hanafusa T, Waguri M, Yamamoto K, Iwahashi H, et al. Requirement of Fas for the development of autoimmune diabetes in nonobese diabetic mice. J Exp Med (1997) 186(4):613–8. doi:10.1084/jem.186.4.613
137. Chervonsky AV, Wang Y, Wong FS, Visintin I, Flavell RA, Janeway CA Jr, et al. The role of Fas in autoimmune diabetes. Cell (1997) 89(1):17–24. doi:10.1016/S0092-8674(00)80178-6
138. Xiao Z, Mohamood AS, Uddin S, Gutfreund R, Nakata C, Marshall A, et al. Inhibition of Fas ligand in NOD mice unmasks a protective role for IL-10 against insulitis development. Am J Pathol (2011) 179(2):725–32. doi:10.1016/j.ajpath.2011.04.016
139. Kalden JR, Schulze-Koops H. Immunogenicity and loss of response to TNF inhibitors: implications for rheumatoid arthritis treatment. Nat Rev Rheumatol (2017) 13(12):707–18. doi:10.1038/nrrheum.2017.187
140. Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, et al. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J Exp Med (1994) 180(3):995–1004. doi:10.1084/jem.180.3.995
141. Chee J, Angstetra E, Mariana L, Graham KL, Carrington EM, Bluethmann H, et al. TNF receptor 1 deficiency increases regulatory T cell function in nonobese diabetic mice. J Immunol (2011) 187(4):1702–12. doi:10.4049/jimmunol.1100511
142. Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S, et al. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care (2009) 32(7):1244–9. doi:10.2337/dc09-0054
143. Timper K, Hruz P, Beglinger C, Donath MY. Infliximab in the treatment of Crohn disease and type 1 diabetes. Diabetes Care (2013) 36(7):e90–1. doi:10.2337/dc13-0199
144. Tack CJ, Kleijwegt FS, Van Riel PL, Roep BO. Development of type 1 diabetes in a patient treated with anti-TNF-alpha therapy for active rheumatoid arthritis. Diabetologia (2009) 52(7):1442–4. doi:10.1007/s00125-009-1381-0
145. Tian Y, Cox MA, Kahan SM, Ingram JT, Bakshi RK, Zajac AJ. A context-dependent role for IL-21 in modulating the differentiation, distribution, and abundance of effector and memory CD8 T cell subsets. J Immunol (2016) 196(5):2153–66. doi:10.4049/jimmunol.1401236
146. Tian Y, Zajac AJ. IL-21 and T cell differentiation: consider the context. Trends Immunol (2016) 37(8):557–68. doi:10.1016/j.it.2016.06.001
147. Beringer A, Noack M, Miossec P. IL-17 in chronic inflammation: from discovery to targeting. Trends Mol Med (2016) 22(3):230–41. doi:10.1016/j.molmed.2016.01.001
148. Maurer MF, Garrigues U, Jaspers SR, Meengs B, Rixon MW, Stevens BL, et al. Generation and characterization of human anti-human IL-21 neutralizing monoclonal antibodies. MAbs (2012) 4(1):69–83. doi:10.4161/mabs.4.1.18713
149. Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, et al. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes (2009) 58(5):1144–55. doi:10.2337/db08-0882
150. Kuriya G, Uchida T, Akazawa S, Kobayashi M, Nakamura K, Satoh T, et al. Double deficiency in IL-17 and IFN-gamma signalling significantly suppresses the development of diabetes in the NOD mouse. Diabetologia (2013) 56(8):1773–80. doi:10.1007/s00125-013-2935-8
151. Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci U S A (2008) 105(37):14028–33. doi:10.1073/pnas.0804358105
152. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi:10.1172/JCI76238
153. McGuire HM, Walters S, Vogelzang A, Lee CM, Webster KE, Sprent J, et al. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes (2011) 60(3):867–75. doi:10.2337/db10-1157
154. Pham MN, von Herrath MG, Vela JL. Antigen-specific regulatory T cells and low dose of IL-2 in treatment of type 1 diabetes. Front Immunol (2015) 6:651. doi:10.3389/fimmu.2015.00651
155. Long SA, Buckner JH, Greenbaum CJ. IL-2 therapy in type 1 diabetes: “trials” and tribulations. Clin Immunol (2013) 149(3):324–31. doi:10.1016/j.clim.2013.02.005
156. Dwyer CJ, Ward NC, Pugliese A, Malek TR. Promoting immune regulation in type 1 diabetes using low-dose interleukin-2. Curr Diab Rep (2016) 16(6):46. doi:10.1007/s11892-016-0739-1
157. Bayer AL, Pugliese A, Malek TR. The IL-2/IL-2R system: from basic science to therapeutic applications to enhance immune regulation. Immunol Res (2013) 57(1–3):197–209. doi:10.1007/s12026-013-8452-5
158. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi:10.1038/nri3156
159. Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med (2005) 201(5):723–35. doi:10.1084/jem.20041982
160. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity (2009) 30(2):204–17. doi:10.1016/j.immuni.2008.11.014
161. Barron L, Dooms H, Hoyer KK, Kuswanto W, Hofmann J, O’Gorman WE, et al. Cutting edge: mechanisms of IL-2-dependent maintenance of functional regulatory T cells. J Immunol (2010) 185(11):6426–30. doi:10.4049/jimmunol.0903940
162. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med (2011) 365(22):2055–66. doi:10.1056/NEJMoa1108188
163. Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med (2011) 365(22):2067–77. doi:10.1056/NEJMoa1105143
164. Grinberg-Bleyer Y, Baeyens A, You S, Elhage R, Fourcade G, Gregoire S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med (2010) 207(9):1871–8. doi:10.1084/jem.20100209
165. Rabinovitch A, Suarez-Pinzon WL, Shapiro AM, Rajotte RV, Power R. Combination therapy with sirolimus and interleukin-2 prevents spontaneous and recurrent autoimmune diabetes in NOD mice. Diabetes (2002) 51(3):638–45. doi:10.2337/diabetes.51.3.638
166. Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes (2012) 61(9):2340–8. doi:10.2337/db12-0049
167. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun (2015) 58:48–58. doi:10.1016/j.jaut.2015.01.001
168. Hartemann A, Bensimon G, Payan CA, Jacqueminet S, Bourron O, Nicolas N, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol (2013) 1(4):295–305. doi:10.1016/S2213-8587(13)70113-X
169. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science (2018) 359(6379):1037–42. doi:10.1126/science.aar3246
170. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311(5769):1924–7. doi:10.1126/science.1122927
171. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206(4):751–60. doi:10.1084/jem.20082824
172. Manirarora JN, Wei CH. Combination therapy using IL-2/IL-2 monoclonal antibody complexes, rapamycin, and islet autoantigen peptides increases regulatory T cell frequency and protects against spontaneous and induced type 1 diabetes in nonobese diabetic mice. J Immunol (2015) 195(11):5203–14. doi:10.4049/jimmunol.1402540
173. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity (2015) 42(5):815–25. doi:10.1016/j.immuni.2015.04.015
174. Johnson MC, Wang B, Tisch R. Genetic vaccination for re-establishing T-cell tolerance in type 1 diabetes. Hum Vaccin (2011) 7(1):27–36. doi:10.4161/hv.7.1.12848
175. Grieger JC, Samulski RJ. Adeno-associated virus vectorology, manufacturing, and clinical applications. Methods Enzymol (2012) 507:229–54. doi:10.1016/B978-0-12-386509-0.00012-0
176. Manzoor F, Johnson MC, Li C, Samulski RJ, Wang B, Tisch R. beta-cell-specific IL-35 therapy suppresses ongoing autoimmune diabetes in NOD mice. Eur J Immunol (2017) 47(1):144–54. doi:10.1002/eji.201646493
177. Li C, Zhang L, Chen Y, Lin X, Li T. Protective role of adenovirus vector-mediated interleukin-10 gene therapy on endogenous islet beta-cells in recent-onset type 1 diabetes in NOD mice. Exp Ther Med (2016) 11(5):1625–32. doi:10.3892/etm.2016.3169
178. Mallol C, Casana E, Jimenez V, Casellas A, Haurigot V, Jambrina C, et al. AAV-mediated pancreatic overexpression of Igf1 counteracts progression to autoimmune diabetes in mice. Mol Metab (2017) 6(7):664–80. doi:10.1016/j.molmet.2017.05.007
179. Song S, Goudy K, Campbell-Thompson M, Wasserfall C, Scott-Jorgensen M, Wang J, et al. Recombinant adeno-associated virus-mediated alpha-1 antitrypsin gene therapy prevents type I diabetes in NOD mice. Gene Ther (2004) 11(2):181–6. doi:10.1038/sj.gt.3302156
180. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi:10.1126/scitranslmed.aad4134
181. Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin Ther Targets (2015) 19(8):1091–103. doi:10.1517/14728222.2015.1037282
182. Gitelman SE, Bluestone JA. Regulatory T cell therapy for type 1 diabetes: may the force be with you. J Autoimmun (2016) 71:78–87. doi:10.1016/j.jaut.2016.03.011
183. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi:10.1084/jem.20040139
184. Putnam AL, Brusko TM, Lee MR, Liu W, Szot GL, Ghosh T, et al. Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes (2009) 58(3):652–62. doi:10.2337/db08-1168
185. Miyara M, Chader D, Burlion A, Goldstein J, Sterlin D, Norol F, et al. Combination of IL-2, rapamycin, DNA methyltransferase and histone deacetylase inhibitors for the expansion of human regulatory T cells. Oncotarget (2017) 8(62):104733–44. doi:10.18632/oncotarget.10914
186. Lin L, Zhang J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol (2017) 18(1):2. doi:10.1186/s12865-016-0187-3
187. Alkanani AK, Hara N, Gottlieb PA, Ir D, Robertson CE, Wagner BD, et al. Alterations in intestinal microbiota correlate with susceptibility to type 1 diabetes. Diabetes (2015) 64(10):3510–20. doi:10.2337/db14-1847
188. Dunne JL, Triplett EW, Gevers D, Xavier R, Insel R, Danska J, et al. The intestinal microbiome in type 1 diabetes. Clin Exp Immunol (2014) 177(1):30–7. doi:10.1111/cei.12321
189. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi:10.1016/j.cell.2006.02.015
190. Troutman TD, Bazan JF, Pasare C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell Cycle (2012) 11(19):3559–67. doi:10.4161/cc.21572
191. Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science (2013) 339(6123):1084–8. doi:10.1126/science.1233521
192. Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, et al. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case-control study. BMC Med (2013) 11:46. doi:10.1186/1741-7015-11-46
193. Tai N, Wong FS, Wen L. The role of gut microbiota in the development of type 1, type 2 diabetes mellitus and obesity. Rev Endocr Metab Disord (2015) 16(1):55–65. doi:10.1007/s11154-015-9309-0
194. de Goffau MC, Luopajarvi K, Knip M, Ilonen J, Ruohtula T, Harkonen T, et al. Fecal microbiota composition differs between children with beta-cell autoimmunity and those without. Diabetes (2013) 62(4):1238–44. doi:10.2337/db12-0526
195. Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe (2015) 17(2):260–73. doi:10.1016/j.chom.2015.01.001
196. Endesfelder D, Zu Castell W, Ardissone A, Davis-Richardson AG, Achenbach P, Hagen M, et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes (2014) 63(6):2006–14. doi:10.2337/db13-1676
197. Giongo A, Gano KA, Crabb DB, Mukherjee N, Novelo LL, Casella G, et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J (2011) 5(1):82–91. doi:10.1038/ismej.2010.92
198. Marino E, Richards JL, McLeod KH, Stanley D, Yap YA, Knight J, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol (2017) 18(5):552–62. doi:10.1038/ni.3713
199. Clemente-Casares X, Tsai S, Huang C, Santamaria P. Antigen-specific therapeutic approaches in Type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(2):a007773. doi:10.1101/cshperspect.a007773
200. Miller SD, Turley DM, Podojil JR. Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease. Nat Rev Immunol (2007) 7(9):665–77. doi:10.1038/nri2153
201. Smith EL, Peakman M. Peptide immunotherapy for type 1 diabetes-clinical advances. Front Immunol (2018) 9:392. doi:10.3389/fimmu.2018.00392
202. Pozzilli P, Gisella Cavallo M. Oral insulin and the induction of tolerance in man: reality or fantasy? Diabetes Metab Res Rev (2000) 16(5):306–7. doi:10.1002/1520-7560(200009/10)16:5<306::AID-DMRR150>3.0.CO;2-J
203. Skyler JS. Type 1 diabetes TrialNet Study G. Update on worldwide efforts to prevent type 1 diabetes. Ann N Y Acad Sci (2008) 1150:190–6. doi:10.1196/annals.1447.055
204. Skyler JS, Krischer JP, Wolfsdorf J, Cowie C, Palmer JP, Greenbaum C, et al. Effects of oral insulin in relatives of patients with type 1 diabetes: the diabetes prevention trial – type 1. Diabetes Care (2005) 28(5):1068–76. doi:10.2337/diacare.28.5.1068
205. Pozzilli P, Pitocco D, Visalli N, Cavallo MG, Buzzetti R, Crino A, et al. No effect of oral insulin on residual beta-cell function in recent-onset type I diabetes (the IMDIAB VII). IMDIAB Group. Diabetologia (2000) 43(8):1000–4. doi:10.1007/s001250051482
206. Chaillous L, Lefevre H, Thivolet C, Boitard C, Lahlou N, Atlan-Gepner C, et al. Oral insulin administration and residual beta-cell function in recent-onset type 1 diabetes: a multicentre randomised controlled trial. Diabete Insuline Orale group. Lancet (2000) 356(9229):545–9. doi:10.1016/S0140-6736(00)02579-4
207. Wherrett DK, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Antigen-based therapy with glutamic acid decarboxylase (GAD) vaccine in patients with recent-onset type 1 diabetes: a randomised double-blind trial. Lancet (2011) 378(9788):319–27. doi:10.1016/S0140-6736(11)60895-7
208. Ludvigsson J, Krisky D, Casas R, Battelino T, Castano L, Greening J, et al. GAD65 antigen therapy in recently diagnosed type 1 diabetes mellitus. N Engl J Med (2012) 366(5):433–42. doi:10.1056/NEJMoa1107096
209. Awate S, Babiuk LA, Mutwiri G. Mechanisms of action of adjuvants. Front Immunol (2013) 4:114. doi:10.3389/fimmu.2013.00114
210. Axelsson S, Hjorth M, Akerman L, Ludvigsson J, Casas R. Early induction of GAD(65)-reactive Th2 response in type 1 diabetic children treated with alum-formulated GAD(65). Diabetes Metab Res Rev (2010) 26(7):559–68. doi:10.1002/dmrr.1126
211. Alleva DG, Gaur A, Jin L, Wegmann D, Gottlieb PA, Pahuja A, et al. Immunological characterization and therapeutic activity of an altered-peptide ligand, NBI-6024, based on the immunodominant type 1 diabetes autoantigen insulin B-chain (9-23) peptide. Diabetes (2002) 51(7):2126–34. doi:10.2337/diabetes.51.7.2126
212. Walter M, Philotheou A, Bonnici F, Ziegler AG, Jimenez R, Group NBIS. No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care (2009) 32(11):2036–40. doi:10.2337/dc09-0449
213. Thrower SL, James L, Hall W, Green KM, Arif S, Allen JS, et al. Proinsulin peptide immunotherapy in type 1 diabetes: report of a first-in-man Phase I safety study. Clin Exp Immunol (2009) 155(2):156–65. doi:10.1111/j.1365-2249.2008.03814.x
214. Alhadj Ali M, Liu YF, Arif S, Tatovic D, Shariff H, Gibson VB, et al. Metabolic and immune effects of immunotherapy with proinsulin peptide in human new-onset type 1 diabetes. Sci Transl Med (2017) 9(402):eaaf7779. doi:10.1126/scitranslmed.aaf7779
215. Donnelly JJ, Wahren B, Liu MA. DNA vaccines: progress and challenges. J Immunol (2005) 175(2):633–9. doi:10.4049/jimmunol.175.2.633
216. Abdulhaqq SA, Weiner DB. DNA vaccines: developing new strategies to enhance immune responses. Immunol Res (2008) 42(1–3):219–32. doi:10.1007/s12026-008-8076-3
217. Goudy KS, Wang B, Tisch R. Gene gun-mediated DNA vaccination enhances antigen-specific immunotherapy at a late preclinical stage of type 1 diabetes in nonobese diabetic mice. Clin Immunol (2008) 129(1):49–57. doi:10.1016/j.clim.2008.06.001
218. Tisch R, Wang B, Weaver DJ, Liu B, Bui T, Arthos J, et al. Antigen-specific mediated suppression of beta cell autoimmunity by plasmid DNA vaccination. J Immunol (2001) 166(3):2122–32. doi:10.4049/jimmunol.166.3.2122
219. Seifarth C, Pop S, Liu B, Wong CP, Tisch R. More stringent conditions of plasmid DNA vaccination are required to protect grafted versus endogenous islets in nonobese diabetic mice. J Immunol (2003) 171(1):469–76. doi:10.4049/jimmunol.171.1.469
220. Weaver DJ Jr, Liu B, Tisch R. Plasmid DNAs encoding insulin and glutamic acid decarboxylase 65 have distinct effects on the progression of autoimmune diabetes in nonobese diabetic mice. J Immunol (2001) 167(1):586–92. doi:10.4049/jimmunol.167.1.586
221. Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Harrison LC, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci Transl Med (2013) 5(191):191ra82. doi:10.1126/scitranslmed.3006103
222. Keselowsky BG, Xia CQ, Clare-Salzler M. Multifunctional dendritic cell-targeting polymeric microparticles: engineering new vaccines for type 1 diabetes. Hum Vaccin (2011) 7(1):37–44. doi:10.4161/hv.7.1.12916
223. Lewis JS, Allen RP. An introduction to biomaterial-based strategies for curbing autoimmunity. Exp Biol Med (Maywood) (2016) 241(10):1107–15. doi:10.1177/1535370216650294
224. Bachelder EM, Pino EN, Ainslie KM. Acetalated dextran: a tunable and acid-labile biopolymer with facile synthesis and a range of applications. Chem Rev (2017) 117(3):1915–26. doi:10.1021/acs.chemrev.6b00532
225. Chen N, Collier MA, Gallovic MD, Collins GC, Sanchez CC, Fernandes EQ, et al. Degradation of acetalated dextran can be broadly tuned based on cyclic acetal coverage and molecular weight. Int J Pharm (2016) 512(1):147–57. doi:10.1016/j.ijpharm.2016.08.031
226. Wattendorf U, Coullerez G, Voros J, Textor M, Merkle HP. Mannose-based molecular patterns on stealth microspheres for receptor-specific targeting of human antigen-presenting cells. Langmuir (2008) 24(20):11790–802. doi:10.1021/la801085d
227. Kwon YJ, James E, Shastri N, Frechet JM. In vivo targeting of dendritic cells for activation of cellular immunity using vaccine carriers based on pH-responsive microparticles. Proc Natl Acad Sci U S A (2005) 102(51):18264–8. doi:10.1073/pnas.0509541102
228. Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, et al. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release (2010) 144(2):118–26. doi:10.1016/j.jconrel.2010.02.013
229. Machen J, Harnaha J, Lakomy R, Styche A, Trucco M, Giannoukakis N. Antisense oligonucleotides down-regulating costimulation confer diabetes-preventive properties to nonobese diabetic mouse dendritic cells. J Immunol (2004) 173(7):4331–41. doi:10.4049/jimmunol.173.7.4331
230. Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, et al. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes (2008) 57(6):1544–55. doi:10.2337/db07-0507
231. Engman C, Wen Y, Meng WS, Bottino R, Trucco M, Giannoukakis N. Generation of antigen-specific Foxp3+ regulatory T-cells in vivo following administration of diabetes-reversing tolerogenic microspheres does not require provision of antigen in the formulation. Clin Immunol (2015) 160(1):103–23. doi:10.1016/j.clim.2015.03.004
232. Yeste A, Takenaka MC, Mascanfroni ID, Nadeau M, Kenison JE, Patel B, et al. Tolerogenic nanoparticles inhibit T cell-mediated autoimmunity through SOCS2. Sci Signal (2016) 9(433):ra61. doi:10.1126/scisignal.aad0612
233. Tsai S, Shameli A, Yamanouchi J, Clemente-Casares X, Wang J, Serra P, et al. Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity (2010) 32(4):568–80. doi:10.1016/j.immuni.2010.03.015
234. Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature (2016) 530(7591):434–40. doi:10.1038/nature16962
235. Trahtemberg U, Mevorach D. Apoptotic cells induced signaling for immune homeostasis in macrophages and dendritic cells. Front Immunol (2017) 8:1356. doi:10.3389/fimmu.2017.01356
236. Vanderlugt CL, Neville KL, Nikcevich KM, Eagar TN, Bluestone JA, Miller SD. Pathologic role and temporal appearance of newly emerging autoepitopes in relapsing experimental autoimmune encephalomyelitis. J Immunol (2000) 164(2):670–8. doi:10.4049/jimmunol.164.2.670
237. Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med (2013) 5(188):188ra75. doi:10.1126/scitranslmed.3006168
238. Peschl P, Reindl M, Schanda K, Sospedra M, Martin R, Lutterotti A. Antibody responses following induction of antigen-specific tolerance with antigen-coupled cells. Mult Scler (2015) 21(5):651–5. doi:10.1177/1352458514549405
239. Kaufman A, Herold KC. Anti-CD3 mAbs for treatment of type 1 diabetes. Diabetes Metab Res Rev (2009) 25(4):302–6. doi:10.1002/dmrr.933
Keywords: autoimmunity, diabetes, immunoregulation, immunotherapy
Citation: Kroger CJ, Clark M, Ke Q and Tisch RM (2018) Therapies to Suppress β Cell Autoimmunity in Type 1 Diabetes. Front. Immunol. 9:1891. doi: 10.3389/fimmu.2018.01891
Received: 14 June 2018; Accepted: 31 July 2018;
Published: 16 August 2018
Edited by:
Gustavo Javier Martinez, Rosalind Franklin University of Medicine and Science, United StatesReviewed by:
Abdel Rahim A. Hamad, Johns Hopkins University, United StatesMaja Wallberg, University of Cambridge, United Kingdom
Rachel Friedman, National Jewish Health, United States
Copyright: © 2018 Kroger, Clark, Ke and Tisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland M. Tisch, cm10aXNjaEBtZWQudW5jLmVkdQ==