 Annika De Sousa Linhares1
Annika De Sousa Linhares1 Peter Steinberger
Peter Steinberger- 1Division of Immune Receptors and T Cell Activation, Medical University of Vienna, Vienna, Austria
- 2Division of Clinical and Experimental Immunology, Center for Pathophysiology, Infectiology, and Immunology, Institute of Immunology, Medical University of Vienna, Vienna, Austria
Antibodies that block T cell inhibition via the immune checkpoints CTLA-4 and PD-1 have revolutionized cancer therapy during the last 15 years. T cells express additional inhibitory surface receptors that are considered to have potential as targets in cancer immunotherapy. Antibodies against LAG-3 and TIM-3 are currently clinically tested to evaluate their effectiveness in patients suffering from advanced solid tumors or hematologic malignancies. In addition, blockade of the inhibitory BTLA receptors on human T cells may have potential to unleash T cells to effectively combat cancer cells. Much research on these immune checkpoints has focused on mouse models. The analysis of animals that lack individual inhibitory receptors has shed some light on the role of these molecules in regulating T cells, but also immune responses in general. There are current intensive efforts to gauge the efficacy of antibodies targeting these molecules called immune checkpoint inhibitors alone or in different combinations in preclinical models of cancer. Differences between mouse and human immunology warrant studies on human immune cells to appreciate the potential of individual pathways in enhancing T cell responses. Results from clinical studies are not only highlighting the great benefit of immune checkpoint inhibitors for treating cancer but also yield precious information on their role in regulating T cells and other cells of the immune system. However, despite the clinical relevance of CTLA-4 and PD-1 and the high potential of the emerging immune checkpoints, there are still substantial gaps in our understanding of the biology of these molecules, which might prevent the full realization of their therapeutic potential. This review addresses PD-1, CTLA-4, BTLA, LAG-3, and TIM-3, which are considered major inhibitory immune checkpoints expressed on T cells. It provides summaries of our current conception of the role of these molecules in regulating T cell responses, and discussions about major ambiguities and gaps in our knowledge. We emphasize that each of these molecules harbors unique properties that set it apart from the others. Their distinct functional profiles should be taken into account in therapeutic strategies that aim to exploit these pathways to enhance immune responses to combat cancer.
Introduction
Although T cells can recognize tumor antigens, they depend on therapeutic intervention to effectively combat malignant cells in cancer patients. While many attempts with antigen-based therapies failed, antigen-independent strategies that enhance T cell responses by blocking inhibitory pathways have been shown to be effective in a significant proportion of treated patients. This therapeutic success is achieved by antibodies often referred to as immune checkpoint inhibitors (ICIs) (1). CTLA-4 was the first immune checkpoint that was targeted to enhance T cell responses in patients suffering from melanoma (2). Antibodies interfering with PD-1 mediated inhibition of T cells and potentially other immune cells were introduced a few years later and have had the greatest success so far (3–6). Inhibitory immune checkpoints help maintaining tolerance and consequently a broad spectrum of side effects–immune-related adverse events (IRAEs)–are observed in treated patients (7). Moreover, monotherapy with ICIs that are currently in use is only beneficial in a subset of cancer patients and frequently leads to acquired resistance (8–10). Consequently, there have been many attempts to evaluate the efficacy of combining PD-1 blockers with conventional cancer treatments (chemotherapy, radiation, surgery) or targeted therapies. Co-administration of PD-1 and CTLA-4 antibodies to patients with melanoma was shown to increase therapeutic efficacy, whereas adverse events were only moderately increased as compared to CTLA-4 blockade alone (11). It is possible that blocking other inhibitory receptors might also augment the therapeutic benefit of PD-1 blockade. Antibodies targeting BTLA, TIM-3, and LAG-3 are promising candidates to boost T cell responses alone or in combination with ICIs disrupting PD-1 mediated inhibition.
Consequently, there is great interest to understand the biology of these inhibitory receptors, which, like CTLA-4, are clearly distinct from PD-1. The original concept of inhibitory receptors was shaped by a group of receptors described on NK cells over 20 years ago (12, 13). These molecules were shown to contain inhibitory motifs (ITIMs), which upon engagement by their ligands, counteract activating signaling processes mediated by ITAM-containing receptors (14). These classical inhibitory receptors exert their function by recruiting SH2-containing phosphatases SHP-1 and SHP-2, which dephosphorylate signaling molecules, thereby directly interfering with activating signaling processes. It was later established that receptors can exert inhibitory functions independent of ITIM motifs. Therefore, inhibitory receptors are now defined by their function rather than by the presence of an ITIM motif in their cytoplasmic domain (13, 15). It is quite clear that BTLA, LAG-3, TIM-3, and CTLA-4 deviate from the classical inhibitory receptors described above. They also differ considerably from PD-1, which can be regarded as the prototypic T cell-expressed immune checkpoint that induces inhibitory intracellular signaling upon engagement with its non-signaling ligands that are preferentially expressed on antigen-presenting cells (APC) and tumor cells.
Here, we want to illustrate that each of these inhibitory receptors has unique properties and we want to draw attention to important open questions regarding BTLA, LAG-3, TIM-3 and CTLA-4 (Figure 1). Distinct features of each immune checkpoint should be accounted for when developing strategies to exploit these pathways therapeutically and unresolved issues and controversies need to be addressed to better validate their potential as targets in cancer immunotherapy.
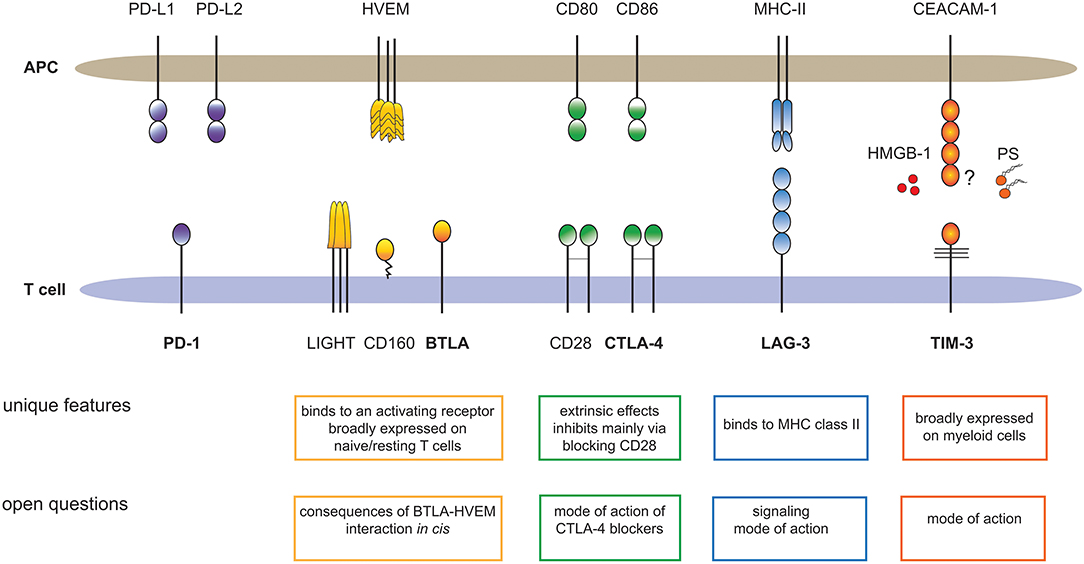
Figure 1. Major T cell expressed immune checkpoints and their ligands. The cartoon summarizes unique features of BTLA, CTLA-4, LAG-3, and TIM-3, which sets these receptors apart from PD-1, the primary immune checkpoint on T cells. In addition, important open questions regarding these pathways are outlined. HMGB-1, high-mobility group box 1; PS, Phosphatidylserine.
Programmed Cell Death Protein-1 (PD-1)
Programmed cell death-1 (PD-1) is a type 1 transmembrane receptor that belongs to the immunoglobulin superfamily (Ig-SF). Its cytoplasmic domain contains two inhibitory motifs: an immunoreceptor tyrosine-based inhibition motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM) (Figure 2). Following TCR-ligation, the phosphatase SHP-2 associates with the intracellular domain of PD-1 via these motifs (16). However, PD-1 ligation is required for inhibition, suggesting that PD-1 must co-localize with the TCR-CD3 complex or CD28 to exert its function (17). Whereas earlier work has suggested that strong CD28 costimulation can override PD-1 costimulation (18), two recent studies reported that CD28 is a major target of PD-1 signaling (19, 20). The B7-family members programmed cell death ligand-1 (PD-L1) and−2 (PD-L2) are ligands for PD-1. PD-L2 expression is mainly restricted to professional APCs such as dendritic cells (DCs) and macrophages, whereas PD-L1 is broadly expressed on cells of the hematopoietic lineage including activated T cells (21). Inflammatory stimuli induce PD-L1 expression and this ligand is also expressed in a wide variety of non-hematopoietic tissues and importantly in many different types of tumor cells (22, 23). PD-1 is a potent negative regulator of T cell activation and studies on PD-1−/− mice highlighted an essential role of PD-1 in maintaining tolerance and preventing autoimmunity. Mice deficient in PD-1 develop features of a lupus-like disease and autoimmunity is promoted in NOD and MLR mice (24, 25). The interaction of PD-1 with its ligands promotes tolerance and dampens T cell immunity at several levels. PD-1 helps to maintain central tolerance by regulating positive and negative selection (26). It critically contributes to peripheral tolerance, e.g., by promoting Treg induction, expression of PD-ligands on resting DCs and upregulation of PD-L1 on host tissues and endothelial cells during inflammation (27–29). However, PD-1 also limits productive T cell immunity against pathogens and tumor cells (30). PD-1 is induced upon T cell activation, and PD-ligands are constitutively expressed on APCs such as DCs. Consequently, PD-1 is broadly engaged on T cells responding to their cognate antigens. Importantly, PD-1 gains importance on T cells that are exposed to persistent antigenic challenge through antigens derived from chronic viruses or tumor cells. Such T cells enter a state of functional impairment that is often described as exhaustion (31). It was shown that PD-1 is constitutively expressed on mouse, macaque and human CD8 T cells specific for LCMV and HIV antigens, respectively (32–34). Importantly, blockade of PD-1 signaling reverts the functional impairment of exhausted T cells in both models. Signs of exhaustion are frequently observed in tumor resident T cells (35) and their capability to combat tumor cells is frequently impaired by the presence of PD-L1 on their targets. Moreover, blocking PD-1 or PD-L1 was demonstrated to enhance anti-tumor responses in murine models of cancer (36–38). Taken together, these findings provided a rationale for targeting PD-1 to enhance anti-tumor responses in humans. Several antibodies blocking PD-1 signaling by either binding to PD-1 or to PD-L1 have shown clinical efficacy in solid tumors and hematological malignancies such as melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), head, and neck squamous cell carcinoma, cervical cancer, uterine cancer, breast cancer, Merkel cell carcinoma, Hodgkin's lymphoma, diffuse large B cell lymphoma, and follicular lymphoma (39). Although these antibodies represent a great advance in cancer treatment, there is a great variation in patient response to PD-1 blockade with a significant proportion not responding. Consequently, there are intense efforts underway to combine PD-1 blockers with conventional therapies or targeting of other inhibitory receptors to further increase the response rate in cancer patients.

Figure 2. Intracellular domains of human immune checkpoints. The amino acid sequences of the cytoplasmic domains of human PD-1, BTLA, CTLA-4, LAG-3, and TIM-3 are shown. Binding motifs for signaling molecules are indicated.
B and T Lymphocyte Attenuator (BTLA)
B and T lymphocyte attenuator (BTLA) is a type I transmembrane receptor belonging to the Ig-superfamily. It bears similarities to PD-1; its extracellular domain has an IgV-like fold and its cytoplasmic domain also harbors an ITIM and an ITSM motif, two classical inhibitory motifs (Figure 2). Engagement of BTLA was reported to lead to the recruitment of the SH2-domain containing phosphatases SHP-1 and SHP-2, which subsequently mediate the inhibitory effects of this receptor (40, 41). However, BTLA, which has a long cytoplasmic tail of 111 amino acids can also engage activating signaling pathways via a putative Grb-2 binding motif located upstream of ITIM/ITSM sequences (42) (Figure 2). As implied by its name, BTLA is preferentially expressed on B and T cells, but it is also present on innate immune cells such as monocytes and DCs (43). The only known ligand expressed in human cells is the Herpes virus entry mediator (HVEM), a member of the tumor necrosis factor receptor superfamily (TNFR-SF) (44). HVEM is an activating receptor that also interacts with members of the TNF-SF, LIGHT, and LT-α (43). In addition, HVEM is a binding partner of CD160, which is also a member of the Ig-SF (45). The role of CD160 in T cell activation processes is controversial because both activating and inhibitory effects have been reported (45–47). CD160 is mainly expressed as a GPI-linked molecule and it is currently unclear how this receptor engages the intracellular signaling machinery of T cells.
In mice, BTLA deficiency is associated with hyper-reactive B and T cells and enhanced susceptibility to autoimmunity (43). Interestingly, HVEM-deficiency results in a similar phenotype, indicating that inhibitory BTLA signaling might play a dominant role in the HVEM network (48). Several reports have found that BTLA blockers can enhance human T cell responses when used alone or in combination with antibodies against PD-1 (49–52). Work by Derré and colleagues demonstrated that although BTLA is down-regulated during activation and differentiation, this receptor is prominently expressed on human T cells in the tumor microenvironment and can function to inhibit tumor-specific T cells (53). However, recent studies indicate that the role of BTLA in tumor-resident T cells is complex, as engagement by its ligand HVEM inhibits proliferation and cytokine production but promotes survival of tumor-infiltrating lymphocytes (TILs) (54). Signaling mediated via PI3K recruitment to the Grb-2 binding motif of BTLA has been implicated in these activating effects (54, 55).
Unique Features of BTLA
The intracellular domain of BTLA bears classical inhibitory motifs and it is well established that BTLA mainly functions as a negative regulator of lymphocytes. However, two striking features set BTLA apart from other inhibitory immune checkpoints expressed on T cells. One peculiarity is that it has a ligand that functions as activating receptor. Therefore, the interaction of BTLA with the TNFR-family member HVEM not only generates inhibitory signals in BTLA expressing cells but also stimulatory signals in the cells that express HVEM. Therefore, both BTLA and HVEM have a dual role as a ligand and receptor when they interact with each other (43, 56, 57). It seems counter-intuitive that upon engagement of an inhibitory receptor also an activating signal is generated, and currently the significance of this phenomenon is not understood.
Another unique feature of BTLA is that it is prominently expressed on naïve T cells and tentatively down-regulated upon activation and differentiation (58). This is in stark contrast to other inhibitory immune checkpoints, which are largely absent on naïve cells. PD-1, CTLA-4, or LAG-3 expression is associated with activation and persistent stimulation, which is consistent with a role of these molecules in limiting and terminating immune reactions. Currently the significance of the unusual expression pattern of BTLA expression is not clear.
Important Open Questions About BTLA
Elegant work by Cheung and colleagues has shown that BTLA and HVEM interact with each other on cells co-expressing these molecules (59). The majority of human T cells harbor HVEM, and thus BTLA and its ligand HVEM are extensively co-expressed in these cells. It has not yet been addressed whether such in cis engagement of BTLA and HVEM during the activation of T cells results in signaling by either of these molecules. However, there is evidence that in cis engagement of HVEM prevents the interaction of this receptor with ligands in trans, thereby precluding HVEM signaling (59). To date, no studies have addressed whether engagement of BTLA by co-expressed HVEM also attenuates BTLA signaling by interfering with interaction with HVEM in trans. The majority of co-inhibitory receptors expressed on T cells is tightly regulated and can only be detected on the surface of activated or “exhausted” T cells. In contrast, BTLA is broadly expressed on T cells and it is tempting to speculate that its function is controlled by co-expression of HVEM. Future studies should test this hypothesis and aim to reveal the interrelationship of BTLA and HVEM on T cells. Eventually, these studies might help to gauge the potential of BTLA as a target of tumor immunotherapy and to devise immune checkpoint inhibitors that optimally target this pathway.
Cytotoxic T Lymphocyte Antigen-4 (CTLA-4)
Cytotoxic T cell lymphocyte antigen-4 (CTLA-4) is a type-1 transmembrane protein harboring a IgV-like Ig-domain. Conventional T cells express CTLA-4 upon activation, whereas Tregs express it constitutively (60). CTLA-4 resides in intracellular vesicles and is quickly exported to the surface upon activation. Importantly, CD28, the primary costimulatory receptor, and CTLA-4 share their extracellular ligands CD80 and CD86, but CTLA-4 binds both molecules with higher affinity. Two papers on CTLA-4-deficient mice published in 1995 clearly established that CTLA-4 functions as a negative regulator of T cell responses. These studies demonstrated that mice lacking CTLA-4 suffer from autoimmune phenomena and immune dysregulation which results in early death (61, 62).
The 36 amino acid cytoplasmic tail of CTLA-4 is highly conserved and interaction with several intracellular signaling molecules has been reported (63). Interestingly, CTLA-4 and its activating counterpart CD28 have common intracellular binding partners including the p85 subunit of PI3K and the phosphatase PP2A (63). In Tregs, the protein kinase C-η (PKC-η) associates with CTLA-4 and signaling via the CTLA-4–PKC-η axis was found to be required for contact-dependent suppression (64). Arguably, the best-established relationship of a binding motif within the cytoplasmic tail of CTLA-4 with a function is the YVKM motif that interacts with the clathrin adaptor complex AP-2, thereby promoting internalization and localization of CTLA-4 in intracellular vesicles (Figure 2) (65, 66). For surface expression of CTLA-4 a molecular complex comprised of TRIM, LAX and Rab8 is formed, which shuttle CTLA-4 from the trans-Golgi networt to the surface (67). It has been suggested that the function of the cytoplasmic domain of CTLA-4 is to control the turnover and cellular location of this molecule rather than transmitting inhibitory signals (68).
Unique Features of CTLA-4
Initial research focused on the contribution of the cytoplasmic tail of CTLA-4 to T cell inhibition. These studies revealed that several intracellular signaling molecules can interact with motifs contained in the intracellular domain of CTLA-4. In addition to “classical” effects like recruitment of enzymes that counteract TCR mediated downstream signaling processes (69, 70), it was found that CTLA-4 mediates a reversal of the “stop-signal” initiated upon cognate T cell-APC interaction and thereby prevents efficient cytokine production and proliferation (71). Although these mechanisms contribute to T cell inhibition, there is increasing evidence that CTLA-4 exhibits inhibitory functions that are independent of its intracellular moiety (72–74). Therefore, one unique property of CTLA-4 is that “extrinsic effects,” specifically its capacity to interfere with CD28 costimulation, critically contribute to its function as an attenuator of T cell immunity. Two major mechanisms have been demonstrated in this context. First, CTLA-4, which has a higher affinity for CD80 and CD86 than CD28, binds these ligands and thereby prevents CD28 costimulation (73, 75, 76). More recently, it was shown that CTLA-4 depletes B7 molecules on APC by literally ripping out these costimulatory ligands, a process termed trans-endocytosis (77). However, it is currently not clear to which extend this process contributes to the extrinsic function of CTLA-4. Results of a study were a transgene encoding tail-less CTLA-4 and full length CTLA-4 was introduced into CTLA-4 deficient mice only mice expressing the full length molecule were completely healthy, whereas expression of a tail-less molecule only partly restored immune function. This indicated that both intrinsic and extrinsic effects contribute to maintenance of immune homeostastis by CTLA-4 (78). An important function of CTLA-4 on conventional T cells and Tregs may be the regulation of activating signals via the primary costimulatory receptor CD28. Indeed, induction of autoimmune disease in CTLA-4−/− mice is only observed when in vivo CD28 costimulation is in place (79).
Important Open Questions About CTLA-4
As outlined above CTLA-4 has been implicated to mediate T cell inhibition by numerous quite distinct mechanisms. Although there is mounting evidence that signaling-independent processes have a major role, the contribution of individual mechanisms is a matter of ongoing debate. Tregs, which have a variety of mechanisms to inhibit immune responses, are characterized by constitutive and high CTLA-4 expression. Studies in mouse tumor models showing that CTLA-4 antibodies can function by depleting intratumoral Tregs via Fc-receptor dependent mechanisms have received much attention (80–82). Recent work by Romano and colleagues demonstrated that patients responding to ipilimumab have higher frequencies of non-classical monocytes and that ipilimumab can mediate killing of CTLA-4high cells by these cells (83). In addition, there is evidence that in melanoma patients response to ipilimumab was associated with the CD16a-V158F high affinity polymorphism (84). Taken together, these results suggest that ipilimumab, which is an IgG1 antibody that is fully capable of interacting with Fc-receptors, may mediate killing of Tregs in vivo. However, more investigations are required to substantiate that Treg depletion is a major mechanism of ipilimumab action in cancer patients. Such studies might also help determine potential of strategies aiming at Treg depletion in cancer therapy.
Lymphocyte Activation Gene-3 (LAG-3)
Lymphocyte activation gene-3 is a type 1 transmembrane protein that has significant homology to CD4 and was first described by Triebel and colleagues in 1990 (85). Expression of LAG-3 has been described on activated T cells, B cells, and NK cells but also on plasmacytoid DCs (85–87). Its extracellular part contains four Ig-like domains and shares high structural homology to CD4. Like CD4, LAG-3 binds to MHC class II molecules, albeit with much higher affinity (88). LAG-3 is heavily glycosylated and interacts with the lectins galectin 3 and the cell surface resident liver sinusoidal endothelial lectin (LSECtin), which is a member of the DC-SIGN family (89, 90).
The 54 amino acid cytoplasmic tail of LAG-3 is devoid of classical motifs involved in the recruitment of inhibitory phosphatases. Instead, it contains a potential serine phosphorylation motif (S454), an unusual sequence consisting of glutamic acid and proline dipeptide motifs (EP motif) and a highly conserved KIEELE motif (Figure 2). A protein termed LAG-3-associated protein (LAP) was shown to bind to the repeated EP motif of LAG-3 but functional effects of this interaction were not studied (91). Follow-up studies on this finding are lacking. The role of the cytoplasmic tail in the function of LAG-3 has to date only been addressed in a singular study by Workman and colleagues published more than 15 years ago (92). The authors expressed wild type and mutated variants of LAG-3 in a murine hen egg lysozyme-specific LAG-3-negative CD4+ T cell hybridoma line. They found that wildtype, but not tailless, LAG-3 inhibited IL-2 production in response to antigen. Moreover, the authors reported that LAG-3 inhibition depended on its ligation to MHC class II as well as on the presence of CD4, since a CD4-deficient subline was not inhibited (92). Different LAG-3 mutants were tested and it was found that the KIEELE motif was required, whereas S454 and the EP motif were dispensable for LAG-3 function (92). Collectively, these data suggest that although the presence of CD4 is required for LAG-3 inhibition, this receptor does not simply function by interfering with MHC class II–CD4 interaction, since the intracellular motifs of LAG-3 are required for inhibition. Several studies have shown that LAG-3 functions as an intrinsic negative regulator of CD4+ but also CD8+ T cells (92–95). In addition, LAG-3 is constitutively expressed on Tregs and can contribute to Treg mediated inhibition (96, 97). Interaction of Treg expressed LAG-3 with MHC class II molecules was shown to induce inhibitory signaling pathways in DCs (98). A number of studies in murine tumor models have provided a rationale for LAG-3 blockade to limit tumor growth. It was shown that LAG-3 antibodies alone or in combination with PD-1 blockers curtailed growth of malignant cells and promoted tumor clearance (99–102). Several antibodies targeting LAG-3, including the bispecific agent MGD013 that simultaneously binds LAG-3 and PD-1, are in clinical development. Most of these aim at enhancing T cell responses, but a depleting antibody that should function by killing activated effector memory T cells, thus reducing unwanted T cell responses, is also being developed (103, 104). In addition, IMP321, a LAG-3 immunoglobulin fusion protein that exerts immune potentiating functions by activating APCs via MHC class II molecules, is being tested in several clinical trials (103, 105).
Unique Features of LAG-3
A striking feature of LAG-3 is that it ligates to MHC class II molecules rather than to a generic co-inhibitory ligand. Related to this, LAG-3 has a large extracellular domain compared to other T cell expressed co-inhibitory molecules like PD-1, BTLA, and CTLA-4. In addition, LAG-3 has an unusual cytoplasmic tail containing motifs that are not found in other co-inhibitory receptors. Therefore, inhibitory mechanisms of LAG-3 are likely to be unique and clearly distinct from those exerted by other immune checkpoints; potential extrinsic effects will affect the antigen-specific signals rather than costimulatory signals (signal 1 rather than signal 2) and intrinsic effects will engage unique pathways that are not used by other inhibitory receptors.
Important Open Questions About LAG-3
The mode of action of LAG-3 mediated inhibition is currently incompletely understood. LAP binds to the EP motifs of LAG-3 but the consequences of this interaction are not known (91). The KIEELE domain is highly conserved and was described to be required for the inhibitory function of LAG-3. However, there is a complete lack of data showing how the intracellular signaling machinery of T cells connects with this motif to counteract activating T cell signaling processes. Workman and colleagues have described that LAG-3 inhibits CD4-dependent, but not CD4-independent T cell function (92). Thus, it is possible that the role of the intracellular domain of LAG-3 is to promote the extrinsic effects of LAG-3 for instance by ensuring optimal spatial orientation of LAG-3 in the immunological synapse. However, direct inhibition of CD8+ T cells by LAG-3 has also been described and distinct mechanisms have to be involved for such a function of LAG-3 (93–95). We have found that blocking LAG-3 alone or in combination with PD-1 on T cells stimulated with allogeneic DC or virus antigens had limited efficacy (49, 50). In general, there are scarce data describing a robust effect of LAG-3 on human T cell responses in vitro. Establishing stimulation conditions for primary human T cells where LAG-3 blockade exerts a strong and reproducible effect would be valuable to further our understanding of LAG-3 function and aid the development of improved therapeutic strategies targeting this immune checkpoint in T cells.
Another important issue is the consequence of MHC class II engagement by LAG-3. The LAG-3 fusion protein IMP321 shows adjuvant properties and enhances immunogenicity of tumor vaccines (105). Induction of DC maturation via engagement of MHC class II has been proposed as a mechanism underlying this effect (106, 107). Interestingly, binding of MHC class II molecules on a CD4 T cell clone by a LAG-3 fusion protein inhibited proliferation and cytokine production upon stimulation with antigen (108). Moreover, Tregs were shown to inhibit DC maturation via LAG-3 (98). Thus it appears that engagement of MHC class II molecules by membrane-bound or soluble LAG-3 can transduce either activating or inhibitory signals and dissecting the mechanisms behind this functional dichotomy will certainly help to understand the complex pathways used by LAG-3 to regulate immune responses. LAG-3 is also released from CD4 T cells after activation but it is not known whether this has a role in immune regulation (109).
T Cell Immunoglobulin and Mucin-Domain Containing Protein-3 (TIM-3)
T cell immunoglobulin and mucin-domain containing protein-3 (TIM-3) is a member of the TIM family, which has two additional members in humans: TIM-1 and TIM-4. TIM-molecules are type I transmembrane proteins that contain an N-terminal IgV-like domain and a mucin domain (110). TIM-3 is constitutively expressed on innate immune cells such as monocytes/macrophages, DCs, mast cells, and mature NK cells, whereas on T cells its expression is associated with activated and terminally differentiated states (110–113). Several ligands have been proposed for TIM-3. Like all TIMs, it binds phosphatidylserine (PtdSer), yet compared to TIM-1 and TIM-4, its capacity to interact with these molecules appears to be considerably lower (110, 114). TIM-3 also binds to high-mobility group box 1 (HMGB-1), a damage-associated molecular pattern protein that is released from stressed innate immune cells and can interact with different molecules including nucleic acids and lipopolysaccharide (LPS) (115). Based on intracellular binding experiments Galectin-9 (Gal-9) was reported to serve as a binding partner for TIM-3 (116). The galectins are a family of beta-galactoside-binding proteins and Gal-9 was also implicated in binding 4-1BB, CD40, CD44, and Dectin-1 (117–120). We performed a series of experiments that produced no evidence for a specific interaction of human or mouse TIM-3 with Gal-9 (121). CEACAM-1, a co-inhibitory molecule expressed on T cells that functions as a self-ligand, was reported as another ligand for TIM-3 (122). An interaction between TIM-3 and CEACAM-1 on cell surfaces was not shown in this study. Instead, co-precipitation experiments were performed and the crystal structure of a heterodimer of the V-domains of human CEACAM-1 and human TIM-3 was published (122). The heterodimer models have since been withdrawn and further work is required to establish an interaction between CEACAM-1 and TIM-3 (123).
Human TIM-3 has a cytoplasmic tail of 71 amino acids that lacks classical activating or inhibitory signaling motifs like ITAMs or ITIMs (Figure 2). However, several studies report that the cytoplasmic domain of TIM-3 can mediate intracellular signaling in T cells and myeloid cells. Two tyrosines (Y256 and Y263 in human TIM-3) whose phosphorylation enables interaction with SH2 domain containing molecules appear to have a significant role in this process. Intracellular signaling proteins that have been reported to interact with TIM-3 include p85 of PI3K, PLC-γ, ZAP-70, Lck, and SLP-76 (124). Rangachari et al. found that HLA-B-associated transcript 3 (Bat3) associates with the cytoplasmic domain of TIM-3, thereby preventing T cell dysfunction and exhaustion (125). Recent work by Avery and colleagues showed that TIM-3 promotes Akt/mTOR signaling and is essential for optimal effector T cell responses (126).
An autoimmune phenotype of mice lacking TIM-3 was not described but consistent for an inhibitory role of TIM-3 in immunity these animals were found to be refractory to tolerance induction (127). Gorman et al. however found that TIM-3 knockout mice had reduced magnitudes of both primary and secondary CD8 T cell responses. They showed that this effect was cell intrinsic, suggesting that TIM-3 can mediate a stimulatory effect on CD8 T cell responses (128).
Unique Features of TIM-3
Unlike the other immune checkpoints described in this review, TIM-3 is constitutively expressed on several cell types of the myeloid lineage. TIM-3 acts as a receptor for ligands like HMGB-1 and phosphatidylserine, which is consistent with a molecule that is primarily expressed on innate immune cells. CTLA-4, PD-1, LAG-3, and BTLA interact with cell surface molecules preferentially expressed on professional APCs, whereas APC-expressed membrane-bound ligands for TIM-3 have not been reported. Although TIM-3 is present on activated and exhausted T cells, a recent study reported that TIM-3-positive cells in breast cancer cell samples were of myeloid rather than T cell origin (129). Thus, therapeutic approaches targeting TIM-3 are likely to have a strong impact on APCs such as macrophages and DCs.
Important Open Questions About TIM-3
TIM-3 is expressed in many immune cells and activating as well as inhibitory functions have been ascribed to this receptor. Phosphatidylserine, HMGB-1, Galectin-9, and CEACAM-1 were proposed as binding partners for this molecule, but it is currently not clear whether all of these molecules act as bona fide TIM-3 ligands. In many studies, TIM-3 function was not linked to a specific TIM-3 ligand, and Galectin-9 and CEACAM-1 can regulate T cells independent of TIM-3 (120, 130–133).
Several reports found that antibodies against human TIM-3 enhance T cells responses alone or in combination with PD-1 blockers and thus provide a rationale to explore strategies to enhance anti-cancer immunity by targeting TIM-3 (49, 50, 113, 134, 135). TIM-3 antibodies could directly act on T cells or indirectly by potentiating APC functions, which in turn could enhance T cell responses. In this context, it should be noted that TIM-3 antibodies were shown to induce activating signals in human DCs (5, 111). Gain of function studies on TIM-3 in human T cell lines have yielded conflicting results; while one group obtained results that point to an activating role of TIM-3 (124), others have observed effects that are consistent with an inhibitory role of TIM-3 (136). T cell reporter systems based on the human T cell line Jurkat are powerful tools to assess mechanisms of co-inhibition and to test immune checkpoint inhibitors. Although such reductionist assay systems for evaluating antibodies against PD-1, CTLA-4, BTLA, and LAG-3 are commercially available and have been described in the literature (72, 137–140), a validated test system for antibodies targeting TIM-3 has not yet been described to our knowledge. A recent report by Sabins and colleagues demonstrated that a TIM-3 antibody that was used in several studies to target human TIM-3 could function as an agonist and promoted CD8 T cell differentiation through activation of mTORC1 (141). Thus, it will be necessary to address whether functionally active antibodies to human TIM-3 act as agonists or antagonists to understand the role of TIM-3 in human T cell responses.
General Open Questions and Outlook
Exhaustion and Immune Checkpoints
It is generally accepted that persistent stimulation with an antigen can result in a state of functional impairment referred to as exhaustion in T cells specific for virus and tumor antigens. A landmark paper by Blackburn and colleagues showed that exhausted T cells can upregulate several co-inhibitory receptors (142). Subsequently, it was shown that Melan-A-specific T cells in patients with melanoma resemble exhausted T cells in chronic infections (143). Importantly, several studies have demonstrated that PD-1 antagonists can revert dysfunction in exhausted T cells (32, 33, 144, 145). Based on these findings, immune checkpoint receptors have been phenotypically and functionally linked to T cell exhaustion (146, 147). Consequently, it is often inferred that immune checkpoint inhibitors mainly function to reinvigorate exhausted T cells. Although inhibitory receptors are involved in T cell exhaustion, it is important to emphasize that the expression of immune checkpoint inhibitors on T cells is by no means limited to exhausted populations (147, 148). In addition, the presence of a particular inhibitory receptor on exhausted T cells neither proves that the receptor is the cause of their state nor that it critically contributes to their functional impairment (13). A better understanding on the relationship of inhibitory immune checkpoints and their role in exhaustion is highly desired and will help to understand the potential but also the limitations of ICIs in targeting exhausted tumor specific T cells.
Tregs and Inhibitory Immune Checkpoints
Tregs and T cell-expressed inhibitory immune checkpoints play important roles in maintaining peripheral tolerance. However, they can both limit protective immunity against pathogens and tumor cells. As summarized in a recent review addressing the immune checkpoint inhibitors in Tregs, these cells constitutively express immune checkpoints like CTLA-4 but also PD-1, BTLA, LAG-3, and TIM-3, and upregulate inhibitory receptors during activation and at tumor sites (1). Although there is ample evidence for a role of co-inhibitory receptors in Treg function (96, 149, 150), many aspects of the interrelation between these two pillars of tolerance are incompletely understood. Specifically, it is not clear how immune checkpoints that inhibit T cells by downregulating intracellular signaling pathways function in Tregs: is engagement of such receptors on Tregs mainly attenuating or enhancing their regulatory function? If the former would be true, ICI-therapy would potentiate Treg function, which could result in reduced efficacy of such regimens. Whereas in the latter case, immune checkpoint inhibitors might exert their beneficial function at least in part by targeting Tregs.
Emerging Immune Checkpoints
There are several additional co-inhibitory pathways that are implicated in limiting T cell responses and thus might have potential in cancer immunotherapy. One such protein is TIGIT (T cell immunoreceptor with Ig and ITIM domains), which bears similarities to CTLA-4 as it shares binding partners with an activating receptor (CD226), which binds these ligands with lower affinity (151–154).
V-domain Ig suppressor of T cell activation (VISTA), also known as B7-H5, PD-1H, and Gi24, is expressed on T cells, myeloid cells, and NK-cells (155). VISTA, an orphan receptor on T cells, can also function as a ligand for an unknown receptor on T cells (156–158). Studies in mice and murine cells indicate that VISTA has an inhibitory role in immunity. VISTA was knocked out in mice by two independent approaches and both showed signs of enhanced immune activity and autoimmunity albeit to different degrees (159, 160). In addition, there are several studies in mice that suggest that blocking VISTA might enhance tumor immunity (155, 156, 158, 161, 162). In addition several studies show VISTA expression in tumors and treatment with ipilimumab was found to upregulate VISTA in patients with prostate cancer (162–165). Therefore, antibodies blocking VISTA on T cells as well as the interaction of APC-expressed VISTA with its unknown receptor expressed on T cells may have potential in cancer immunotherapy since they could enhance T cell responses by disrupting two inhibitory signaling pathways in T cells.
Surprisingly few studies have addressed the role of VISTA in human T cells and myeloid cells. Lines et al. reported that a VISTA immunoglobulin fusion protein blocks T cell activation and promotes the generation of human Tregs (166). By contrast Baraj and colleagues found that overexpression of VISTA on human monocytes promoted their activation and subsequently enhanced T cell responses (167). To date there is a lack of information not only regarding receptors and ligands on T cells and APC, respectively, that mediate the proposed effects of VISTA but also on downstream signaling events induced upon VISTA engagement. Despite, this and based on promising result in mice, a clinical trial with a monoclonal antibody to VISTA was initiated (NCT02671955).
B7-H7, also known as HERV-H LTR associating 2 (HHLA2), is a member of the extended B7 family and inhibits proliferation and cytokine production of human CD4 and CD8 T cells (168). B7-H7 is expressed on human APCs such as monocytes or B cells, but it is also widely expressed in non-hematopoietic tissues and cancers (168, 169). CD28H, also known as TMIGD2, was shown to function as a binding partner for HHLA2 (170). HHLA2 was designated as B7-H5 in this publication and it thus should be stressed that HHLA2 is distinct from VISTA, which has also been referred to as B7-H5. The interaction of TMIGD2 with HHLA2 has since been confirmed by an independent study (169). Interestingly, engagement of TMIGD2 was found to costimulate cytokine production and proliferation in human T cells (170). Thus, it is possible that HHLA2 interacts with another yet-unidentified inhibitory receptor on T cells. Interestingly, neither HHLA2 nor TMIGD2 are expressed in mice and rats (168, 170).
In addition, there are orphan ligand molecules including B7-H3 (CD276), B7-H4 (also known as B7S1, B7x or VTCN1), and ILDR2 that have been reported to inhibit T cell responses (171–177). The identification of receptors for orphan ligands like B7-H3 will be mandatory to target pathways involving these molecules to enhance T cell responses (178). B7-H3 is broadly expressed in cancer cells and B7-H3 antibodies targeting B7-H3+ tumors are currently being tested in several clinical trials (179). It is currently not known whether these B7-H3 antibodies interfere with T cell inhibitory effects of this molecule.
The Future: Novel Immune Checkpoint Inhibitors and Beyond
The arrival of ICIs has dramatically changed the therapeutic landscape of cancer. Despite the enormous success of these immunotherapies, it is becoming increasingly clear that combining ICIs with a second drug may have superior potential to combat cancer. Currently, numerous clinical trials testing combinations of established immune checkpoint inhibitors (PD-1 and PD-L1 antibodies) with conventional treatments (chemotherapy, radiation, or targeted therapy) are carried out and are likely to result in improved treatment modalities for many different types of cancer (180). It is noteworthy to mentioned that ICIs are not the only mean to target inhibitory receptors. Taylor et al. have recently shown that glycogen synthase kinase 3 (GSK-3) has a key role in the regulation of PD-1 expression in CD8+T cells (181). Follow up work by the same group has demonstrated that GSK-inhibitors are as effective in enhancing anti-tumor responses in preclinical models as PD-1 and PD-L1 antibodies (182).
Promising clinical data were obtained upon co-administration of antibodies targeting PD-1 and CTLA-4 (11, 183). These two immune checkpoints might mediate anti-tumor effects through distinct non-redundant mechanisms (184, 185). The CTLA-4 antibody ipilimumab acts early during T cell activation and mainly exerts extrinsic effects by outcompeting the primary costimulatory receptor CD28. It promotes the expansion of Th1-like CD4 T cells and potentially the deletion of tumor-resident Tregs. In contrast, PD-1 blockade mainly acts intrinsically on tumor-infiltrating exhausted-like CD8 T cells (184). These cells expand but maintain PD-1 expression indicating that PD-1 blockade does not reprogram them into a non-exhausted state, which is consistent with a epigenetic regulation of exhaustion (184). In addition, these results suggests that despite their exhausted like phenotype these cells are capable to expert potent anti-tumor activity following PD-1 blockade. Since PD-L1 is frequently expressed on tumor cells, PD-1 blockade can have a dual role in the tumor microenvironment–expansion of effector cells and also promoting anti-tumor effector functions.
The successful co-targeting of PD-1 and CTLA-4 and encouraging results obtained in preclinical models that combined PD-1 antibodies with other immune checkpoint inhibitors has fostered strategies to combine immune checkpoint inhibitors to enhance anti-tumor responses in patients with cancer. Several clinical trials where PD-1 antibodies are tested in combination with antibodies targeting TIM-3 and LAG-3 are ongoing (https://clinicaltrials.gov). Distinct properties of PD-1 versus TIM-3 and LAG-3 might result in synergistic effects of such combinations.
Adoptive therapy with T cells genetically engineered to express chimeric antigen receptors (CARs) or TCRs specific for tumor antigens is able to induce impressive anti-tumor responses. However, the upregulation of inhibitory receptors in genetically engineered T cells following transfer reduces their efficacy (186, 187). Multiple clinical trials investigating combinations of CAR T cells with antibodies to PD-1 or PD-L1 are ongoing (188). Engineering CAR T cells or TCR-transgenic T cells that are refractory to inhibition by immune checkpoints represents a promising future avenue to specifically protect engineered tumor-specific T cells against functional impairment through inhibitory pathways such as PD-1/PD-L1. This could be achieved by silencing or knocking out inhibitory receptors but also by co-introducing genes encoding PD-1/PD-L1 antibodies or so-called chimeric switch receptors (188–190).
The triumph of immune checkpoint inhibitors has underpinned that T cells have the potential to efficiently fight tumor cells, but in the majority of cases can only do so upon therapeutic intervention. Immune checkpoint blockade is effective but associated with severe side effects since it interferes with vital mechanisms of peripheral tolerance. Recent work by the Schreiber group has identified T cells that are reactivated upon immune checkpoint inhibitor treatment and mediate tumor rejection in a mouse model. The authors went on to show that tumor rejection can also be achieved by specifically boosting these T cells by peptide vaccination (191). The identification of antigens that are recognized by T cells in patients responding to immune checkpoint therapy might thus offer possibilities to target these antigens by vaccination or introduction of TCR-transgenic autologous T cells. Such approaches may increase the specificity of tumor targeting, thereby potentially enhancing therapy effects while reducing autoimmune toxicity.
Specificity is a hallmark of adaptive immunity and it seems paradoxical that immune checkpoint inhibition, which is an antigen-independent approach, has had the most spectacular success in cancer immunotherapy to date. Recent technological progress has facilitated the identification of mutations, which give rise to neoantigens in the tumors of individual cancer patients (192, 193). Studies in mouse models have demonstrated that vaccination with neoantigens can result in tumor control (191, 194). Strategies that combine patient-tailored approaches aimed at enhancing immune responses to individual neoantigens (e.g., by synthetic vaccines, oncolytic viruses or tumor radiation therapy but also adoptive therapy with in vitro expanded neoantigen-specific T cells) and interference with inhibitory pathways might represent particular promising avenues to improve anti-cancer immunotherapy (195, 196).
Author Contributions
AD, JL, KG-P, and PS wrote the manuscript. AD and JL prepared figures.
Funding
This work was supported by the Austrian Science Fund FWF (DK W 1248-B30). The funder had no role in study design, deceision to publish or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Daniel Speiser critically read the manuscript. We wish to thank him for his invaluable comments and suggestions.
References
1. Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors in cancer therapy: a focus on T-regulatory cells. Immunol Cell Biol. (2018) 96:21–33. doi: 10.1111/imcb.1003
2. Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv Immunol. (2006) 90:297–339. doi: 10.1016/S0065-2776(06)90008-X
3. Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. (2015) 33:4015–22. doi: 10.1200/JCO.2015.62.3397
4. Iwai Y, Hamanishi J, Chamoto K, Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. (2017) 24:26. doi: 10.1186/s12929-017-0329-9
5. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. (2016) 374:2542–52. doi: 10.1056/NEJMoa1603702
6. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
7. Marrone KA, Ying W, Naidoo J. Immune-related adverse events from immune checkpoint inhibitors. Clin Pharmacol Ther. (2016) 100:242–51. doi: 10.1002/cpt.394
8. Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity (2016) 44:1255–69. doi: 10.1016/j.immuni.2016.06.001
9. Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer (2018) 118:9–16. doi: 10.1038/bjc.2017.434
10. Gide TN, Wilmott JS, Scolyer RA, Long GV. Primary and acquired resistance to immune checkpoint inhibitors in metastatic melanoma. Clin Cancer Res. (2018) 24:1260–70. doi: 10.1158/1078-0432.CCR-17-2267
11. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
12. Lanier LL. NK cell receptors. Annu Rev Immunol. (1998) 16:359–93. doi: 10.1146/annurev.immunol.16.1.359
13. Fuertes Marraco SA, Neubert NJ, Verdeil G, Speiser DE. Inhibitory receptors beyond T cell exhaustion. Front. Immunol. (2015) 6:310. doi: 10.3389/fimmu.2015.00310
14. Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. (1999) 17:875–904. doi: 10.1146/annurev.immunol.17.1.875
15. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. J Immunol. (2012) 188:2957–65. doi: 10.4049/jimmunol.1100038
16. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
17. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. (2004) 173:945–54.
18. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34.
19. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
20. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science (2017) 355:1423–27. doi: 10.1126/science.aaf0683
21. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
22. Selenko-Gebauer N, Majdic O, Szekeres A, Hofler G, Guthann E, Korthauer U, et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol. (2003) 170:3637–44.
23. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. (2016) 9:5023–39. doi: 10.2147/OTT.S105862
24. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51.
25. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science (2001) 291:319–22. doi: 10.1126/science.291.5502.319
26. Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med. (2000) 191:891–8.
27. Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. (2005) 6:280–6. doi: 10.1038/ni1165
28. Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. (2003) 33:3117–26. doi: 10.1002/eji.200324270
29. Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. (2006) 203:883–95. doi: 10.1084/jem.20051776
30. Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. (2007) 8:239–45. doi: 10.1038/ni1443
32. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature (2006) 439:682–7. doi: 10.1038/nature04444
33. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature (2006) 443:350–4. doi: 10.1038/nature05115
34. Petrovas C, Price DA, Mattapallil J, Ambrozak DR, Geldmacher C, Cecchinato V, et al. SIV-specific CD8+ T cells express high levels of PD1 and cytokines but have impaired proliferative capacity in acute and chronic SIVmac251 infection. Blood (2007) 110:928–36. doi: 10.1182/blood-2007-01-069112
35. Baitsch L, Legat A, Barba L, Fuertes Marraco SA, Rivals JP, Baumgaertner P, et al. Extended co-expression of inhibitory receptors by human CD8 T-cells depending on differentiation, antigen-specificity and anatomical localization. PLoS ONE (2012) 7:e30852. doi: 10.1371/journal.pone.0030852
36. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
37. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
38. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. (2004) 64:1140–5.
39. Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. (2016) 7:550. doi: 10.3389/fimmu.2016.00550
40. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. (2003) 312:1236–43.
41. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. (2003) 4:670–9. doi: 10.1038/ni944
42. Gavrieli M, Murphy KM. Association of Grb-2 and PI3K p85 with phosphotyrosile peptides derived from BTLA. Biochem Biophys Res Commun. (2006) 345:1440–5. doi: 10.1016/j.bbrc.2006.05.036
43. Murphy TL, Murphy KM. Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol. (2010) 28:389–411. doi: 10.1146/annurev-immunol-030409-101202
44. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. (2005) 6:90–8. doi: 10.1038/ni1144
45. Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. (2008) 9:176–85. doi: 10.1038/ni1554
46. Peretz Y, He Z, Shi Y, Yassine-Diab B, Goulet JP, Bordi R, et al. CD160 and PD-1 Co-Expression on HIV-Specific CD8 T Cells Defines a Subset with Advanced Dysfunction. PLoS Pathog. (2012) 8:e1002840. doi: 10.1371/journal.ppat.1002840
47. El-Far M, Pellerin C, Pilote L, Fortin JF, Lessard IA, Peretz Y, et al. CD160 isoforms and regulation of CD4 and CD8 T-cell responses. J Trans Med. (2014) 12:217. doi: 10.1186/s12967-014-0217-y
48. Wang Y, Subudhi SK, Anders RA, Lo J, Sun Y, Blink S, et al. The role of herpesvirus entry mediator as a negative regulator of T cell-mediated responses. J Clin Invest. (2005) 115:711–7. doi: 10.1172/JCI22982
49. Stecher C, Battin C, Leitner J, Zettl M, Grabmeier-Pfistershammer K, Holler C, et al. PD-1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol. (2017) 8:572. doi: 10.3389/fimmu.2017.00572
50. Grabmeier-Pfistershammer K, Stecher C, Zettl M, Rosskopf S, Rieger A, Zlabinger GJ, et al. Antibodies targeting BTLA or TIM-3 enhance HIV-1 specific T cell responses in combination with PD-1 blockade. Clin Immunol. (2017) 183:167–73. doi: 10.1016/j.clim.2017.09.002
51. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. (2012) 72:887–96. doi: 10.1158/0008-5472.CAN-11-2637
52. Hobo W, Norde WJ, Schaap N, Fredrix H, Maas F, Schellens K, et al. B and T lymphocyte attenuator mediates inhibition of tumor-reactive CD8+ T cells in patients after allogeneic stem cell transplantation. J Immunol. (2012) 189:39–49. doi: 10.4049/jimmunol.1102807
53. Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. (2010) 120:157–67. doi: 10.1172/JCI40070
54. Haymaker CL, Wu RC, Ritthipichai K, Bernatchez C, Forget MA, Chen JQ, et al. BTLA marks a less-differentiated tumor-infiltrating lymphocyte subset in melanoma with enhanced survival properties. Oncoimmunology (2015) 4:e1014246. doi: 10.1080/2162402X.2015.1014246
55. Ritthipichai K, Haymaker CL, Martinez M, Aschenbrenner A, Yi X, Zhang M, et al. Multifaceted role of BTLA in the control of CD8(+) T-cell fate after antigen encounter. Clin Cancer Res. (2017) 23:6151–64. doi: 10.1158/1078-0432.CCR-16-1217
56. Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci USA. (2009) 106:6244–9. doi: 10.1073/pnas.0902115106
57. Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev. (2009) 229:244–58. doi: 10.1111/j.1600-065X.2009.00783.x
58. Serriari NE, Gondois-Rey F, Guillaume Y, Remmerswaal EB, Pastor S, Messal N, et al. B and T lymphocyte attenuator is highly expressed on CMV-specific T cells during infection and regulates their function. J Immunol. (2010) 185:3140–8. doi: 10.4049/jimmunol.0902487
59. Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol. (2009) 183:7286–96. doi: 10.4049/jimmunol.0902490
60. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131:58–67. doi: 10.1182/blood-2017-06-741033
61. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270:985–8.
62. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3:541–7.
63. Rudd CE, Schneider H. Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling. Nat Rev Immunol. (2003) 3:544–56. doi: 10.1038/nri1131
64. Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat Immunol. (2014) 15:465–72. doi: 10.1038/ni.2866
65. Schneider H, Martin M, Agarraberes FA, Yin L, Rapoport I, Kirchhausen T, et al. Cytolytic T lymphocyte-associated antigen-4 and the TCR zeta/CD3 complex, but not CD28, interact with clathrin adaptor complexes AP-1 and AP-2. J Immunol. (1999) 163:1868–79.
66. Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, et al. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity (1997) 6:583–9.
67. Banton MC, Inder KL, Valk E, Rudd CE, Schneider H. Rab8 binding to immune cell-specific adaptor LAX facilitates formation of trans-Golgi network-proximal CTLA-4 vesicles for surface expression. Mol Cell Biol. (2014) 34:1486–99. doi: 10.1128/MCB.01331-13
68. Walker LS. Treg and CTLA-4: two intertwining pathways to immune tolerance. J Autoimmun. (2013) 45:49–57. doi: 10.1016/j.jaut.2013.06.006
69. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation by CTLA-4. Science (1998) 282:2263–6.
70. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. (2001) 1:220–8. doi: 10.1038/35105024
71. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, et al. Reversal of the TCR stop signal by CTLA-4. Science (2006) 313:1972–5. doi: 10.1126/science.1131078
72. Jutz S, Hennig A, Paster W, Asrak O, Dijanovic D, Kellner F, et al. A cellular platform for the evaluation of immune checkpoint molecules. Oncotarget (2017) 8:64892–906. doi: 10.18632/oncotarget.17615
73. Corse E, Allison JP. Cutting edge: CTLA-4 on effector T cells inhibits in trans. J Immunol. (2012) 189:1123–7. doi: 10.4049/jimmunol.1200695
74. Walker LS, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. (2011) 11:852–63. doi: 10.1038/nri3108
75. Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, et al. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J Immunol. (2006) 177:4376–83.
76. Sojka DK, Hughson A, Fowell DJ. CTLA-4 is required by CD4+CD25+ Treg to control CD4+ T-cell lymphopenia-induced proliferation. Eur J Immunol. (2009) 39:1544–51. doi: 10.1002/eji.200838603
77. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
78. Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural analysis of CTLA-4 function in vivo. J Immunol. (2000) 164:5319–27.
79. Tai X, Van Laethem F, Sharpe AH, Singer A. Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci USA. (2007) 104:13756–61. doi: 10.1073/pnas.0706509104
80. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
81. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. (2013) 210:1685–93. doi: 10.1084/jem.20130573
82. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326-6066.CIR-13-0013
83. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
84. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell (2018) 33:649–63 e4. doi: 10.1016/j.ccell.2018.02.010
85. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405.
86. Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur J Immunol. (2005) 35:2081–8. doi: 10.1002/eji.200526090
87. Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol. (2009) 182:1885–91. doi: 10.4049/jimmunol.0800185
88. Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, et al. Characterization of the lymphocyte activation gene 3-encoded protein. a new ligand for human leukocyte antigen class II antigens. J Exp Med. (1992) 176:327–37.
89. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
90. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. (2014) 74:3418–28. doi: 10.1158/0008-5472.CAN-13-2690
91. Iouzalen N, Andreae S, Hannier S, Triebel F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. Eur J Immunol. (2001) 31:2885–91. doi: 10.1002/1521-4141(2001010)31:10<2885::AID-IMMU2885>3.0.CO;2-2
92. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol. (2002) 169:5392–5.
93. Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology (2005) 115:170–8. doi: 10.1111/j.1365-2567.2005.02145.x
94. Cook KD, Whitmire JK. LAG-3 confers a competitive disadvantage upon antiviral CD8+ T cell responses. J Immunol. (2016) 197:119–27. doi: 10.4049/jimmunol.1401594
95. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. (2007) 117:3383–92. doi: 10.1172/JCI31184
96. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
97. Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA, et al. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE (2014) 9:e109080. doi: 10.1371/journal.pone.0109080
98. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J Immunol. (2008) 180:5916–26.
99. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
100. Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol. (2013) 190:4899–909. doi: 10.4049/jimmunol.1300271
101. Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget (2015) 6:27359–77. doi: 10.18632/oncotarget.4751
102. Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood (2018) 131:1617–21. doi: 10.1182/blood-2017-06-792267
103. Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. (2017) 276:80–96. doi: 10.1111/imr.12519
104. He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, et al. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. (2016) 107:1193–7. doi: 10.1111/cas.12986
105. Sierro S, Romero P, Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets (2011) 15:91–101. doi: 10.1517/14712598.2011.540563
106. Andreae S, Buisson S, Triebel F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood (2003) 102:2130–7. doi: 10.1182/blood-2003-01-0273
107. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. (2015) 15:45–56. doi: 10.1038/nri3790
108. Huard B, Prigent P, Pages F, Bruniquel D, Triebel F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur J Immunol. (1996) 26:1180–6. doi: 10.1002/eji.1830260533
109. Annunziato F, Manetti R, Tomasevic I, Guidizi MG, Biagiotti R, Gianno V, et al. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. FASEB J. (1996) 10:769–76.
110. Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. (2010) 235:172–89. doi: 10.1111/j.0105-2896.2010.00903.x
111. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science (2007) 318:1141–3. doi: 10.1126/science.1148536
112. Ocana-Guzman R, Torre-Bouscoulet L, Sada-Ovalle I. TIM-3 regulates distinct functions in macrophages. Front Immunol. (2016) 7:229. doi: 10.3389/fimmu.2016.00229
113. Jones RB, Ndhlovu LC, Barbour JD, Sheth PM, Jha AR, Long BR, et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J Exp Med. (2008) 205:2763–79. doi: 10.1084/jem.20081398
114. DeKruyff RH, Bu X, Ballesteros A, Santiago C, Chim YL, Lee HH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. (2010) 184:1918–30. doi: 10.4049/jimmunol.0903059
115. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. (2012) 13:832–42. doi: 10.1038/ni.2376
116. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. (2005) 6:1245–52. doi: 10.1038/ni1271
117. Madireddi S, Eun SY, Lee SW, Nemcovicova I, Mehta AK, Zajonc DM, et al. Galectin-9 controls the therapeutic activity of 4-1BB-targeting antibodies. J Exp Med. (2014) 211:1433–48. doi: 10.1084/jem.20132687
118. Vaitaitis GM, Wagner DH Jr. Galectin-9 controls CD40 signaling through a Tim-3 independent mechanism and redirects the cytokine profile of pathogenic T cells in autoimmunity. PLoS ONE (2012) 7:e38708. doi: 10.1371/journal.pone.0038708
119. Katoh S, Ishii N, Nobumoto A, Takeshita K, Dai SY, Shinonaga R, et al. Galectin-9 inhibits CD44-hyaluronan interaction and suppresses a murine model of allergic asthma. Am J Respir Crit Care Med. (2007) 176:27–35. doi: 10.1164/rccm.200608-1243OC
120. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med. (2017) 23:556–67. doi: 10.1038/nm.4314
121. Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog. (2013) 9:e1003253. doi: 10.1371/journal.ppat.1003253
122. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi: 10.1038/nature13848
123. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2016) 536:359. doi: 10.1038/nature17421
124. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
125. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3-mediated cell death and exhaustion. Nat Med. (2012) 18:1394–400. doi: 10.1038/nm.2871
126. Avery L, Filderman J, Szymczak-Workman AL, Kane LP. Tim-3 co-stimulation promotes short-lived effector T cells, restricts memory precursors, and is dispensable for T cell exhaustion. Proc Natl Acad Sci USA. (2018) 115:2455–60. doi: 10.1073/pnas.1712107115
127. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. (2003) 4:1102–10. doi: 10.1038/ni988
128. Gorman JV, Starbeck-Miller G, Pham NL, Traver GL, Rothman PB, Harty JT, et al. Tim-3 directly enhances CD8 T cell responses to acute Listeria monocytogenes infection. J Immunol. (2014) 192:3133–42. doi: 10.4049/jimmunol.1302290
129. de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM-3 Regulates CD103(+) Dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell (2018) 33:60–74 e6. doi: 10.1016/j.ccell.2017.11.019
130. Su EW, Bi S, Kane LP. Galectin-9 regulates T helper cell function independently of Tim-3. Glycobiology (2011) 21:1258–65. doi: 10.1093/glycob/cwq214
131. Oomizu S, Arikawa T, Niki T, Kadowaki T, Ueno M, Nishi N, et al. Galectin-9 suppresses Th17 cell development in an IL-2-dependent but Tim-3-independent manner. Clin Immunol. (2012) 143:51–8. doi: 10.1016/j.clim.2012.01.004
132. Chen CJ, Shively JE. The cell-cell adhesion molecule carcinoembryonic antigen-related cellular adhesion molecule 1 inhibits IL-2 production and proliferation in human T cells by association with Src homology protein-1 and down-regulates IL-2 receptor. J Immunol. (2004) 172:3544–52.
133. Li Y, Shively JE. CEACAM1 regulates Fas-mediated apoptosis in Jurkat T-cells via its interaction with beta-catenin. Exp Cell Res. (2013) 319:1061–72. doi: 10.1016/j.yexcr.2013.02.020
134. McMahan RH, Golden-Mason L, Nishimura MI, McMahon BJ, Kemper M, Allen TM, et al. Tim-3 expression on PD-1+ HCV-specific human CTLs is associated with viral persistence, and its blockade restores hepatocyte-directed in vitro cytotoxicity. J Clin Invest. (2010) 120:4546–57. doi: 10.1172/JCI43127
135. Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. (2009) 83:9122–30. doi: 10.1128/JVI.00639-09
136. Tomkowicz B, Walsh E, Cotty A, Verona R, Sabins N, Kaplan F, et al. TIM-3 Suppresses Anti-CD3/CD28-induced TCR activation and IL-2 expression through the NFAT signaling pathway. PLoS ONE (2015) 10:e0140694. doi: 10.1371/journal.pone.0140694
137. Bhagwat B, Cherwinski H, Sathe M, Seghezzi W, McClanahan TK, de Waal Malefyt R, et al. Establishment of engineered cell-based assays mediating LAG3 and PD1 immune suppression enables potency measurement of blocking antibodies and assessment of signal transduction. J Immunol Methods (2018) 456:7–14. doi: 10.1016/j.jim.2018.02.003
138. Wang L, Yu C, Yang Y, Gao K, Wang J. Development of a robust reporter gene assay to measure the bioactivity of anti-PD-1/anti-PD-L1 therapeutic antibodies. J Pharm Biomed Anal. (2017) 145:447–53. doi: 10.1016/j.jpba.2017.05.011
139. Rosskopf S, Jutz S, Neunkirchner A, Candia MR, Jahn-Schmid B, Bohle B, et al. Creation of an engineered APC system to explore and optimize the presentation of immunodominant peptides of major allergens. Sci Rep. (2016) 6:31580. doi: 10.1038/srep31580
140. Jutz S, Leitner J, Schmetterer K, Doel-Perez I, Majdic O, Grabmeier-Pfistershammer K, et al. Assessment of costimulation and coinhibition in a triple parameter T cell reporter line: simultaneous measurement of NF-kappaB, NFAT and AP-1. J Immunol Methods (2016) 430:10–20. doi: 10.1016/j.jim.2016.01.007
141. Sabins NC, Chornoguz O, Leander K, Kaplan F, Carter R, Kinder M, et al. TIM-3 engagement promotes effector memory T cell differentiation of human antigen-specific CD8 T cells by activating mTORC1. J Immunol. (2017) 199:4091–102. doi: 10.4049/jimmunol.1701030
142. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. (2009) 10:29–37. doi: 10.1038/ni.1679
143. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
144. Lee J, Ahn E, Kissick HT, Ahmed R. Reinvigorating exhausted T cells by blockade of the PD-1 pathway. For Immunopathol Dis Therap. (2015) 6:7–17. doi: 10.1615/ForumImmunDisTher.2015014188
145. Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. (2006) 12:1198–202. doi: 10.1038/nm1482
146. Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, et al. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. (2010) 6:e1000947. doi: 10.1371/journal.ppat.1000947
147. Haymaker C, Wu R, Bernatchez C, Radvanyi L. PD-1 and BTLA and CD8(+) T-cell “exhaustion” in cancer: “Exercising” an alternative viewpoint. Oncoimmunology (2012) 1:735–38. doi: 10.4161/onci.20823
148. Legat A, Speiser DE, Pircher H, Zehn D, Fuertes Marraco SA. Inhibitory receptor expression depends more dominantly on differentiation and activation than “Exhaustion” of human CD8 T cells. Front Immunol. (2013) 4:455. doi: 10.3389/fimmu.2013.00455
149. Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. (2000) 192:303–10.
150. Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T. Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci USA. (2016) 113:8490–5. doi: 10.1073/pnas.1608873113
151. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.12518
152. Lozano E, Dominguez-Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. (2012) 188:3869–75. doi: 10.4049/jimmunol.1103627
153. Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol. (2011) 186:1338–42. doi: 10.4049/jimmunol.1003081
154. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
155. Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R, et al. Immunoregulatory functions of VISTA. Immunol Rev. (2017) 276:66–79. doi: 10.1111/imr.12525
156. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J Clin Invest. (2014) 124:1966–75. doi: 10.1172/JCI74589
157. Flies DB, Wang S, Xu H, Chen L. Cutting edge: a monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J Immunol. (2011) 187:1537–41. doi: 10.4049/jimmunol.1100660
158. Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med. (2011) 208:577–92. doi: 10.1084/jem.20100619
159. Wang L, Le Mercier I, Putra J, Chen W, Liu J, Schenk AD, et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc Natl Acad Sci USA. (2014) 111:14846–51. doi: 10.1073/pnas.1407447111
160. Yoon KW, Byun S, Kwon E, Hwang SY, Chu K, Hiraki M, et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science (2015) 349:1261669. doi: 10.1126/science.1261669
161. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. (2014) 74:1933–44. doi: 10.1158/0008-5472.CAN-13-1506
162. Kondo Y, Ohno T, Nishii N, Harada K, Yagita H, Azuma M. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. (2016) 57:54–60. doi: 10.1016/j.oraloncology.2016.04.005
163. Kuklinski LF, Yan S, Li Z, Fisher JL, Cheng C, Noelle RJ, et al. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol Immunother. (2018) 67:1113–21. doi: 10.1007/s00262-018-2169-1
164. Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. (2017) 23:551–55. doi: 10.1038/nm.4308
165. Villarroel-Espindola F, Yu X, Datar I, Mani N, Sanmamed M, Velcheti V, et al. Spatially resolved and quantitative analysis of VISTA/PD-1H as a novel immunotherapy target in human non-small cell lung cancer. Clin Cancer Res. (2018) 24:1562–73. doi: 10.1158/1078-0432.CCR-17-2542
166. Lines JL, Sempere LF, Broughton T, Wang L, Noelle R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol Res. (2014) 2:510–7. doi: 10.1158/2326-6066.CIR-14-0072
167. Bharaj P, Chahar HS, Alozie OK, Rodarte L, Bansal A, Goepfert PA, et al. Characterization of programmed death-1 homologue-1 (PD-1H) expression and function in normal and HIV infected individuals. PLoS ONE (2014) 9:e109103. doi: 10.1371/journal.pone.0109103
168. Zhao R, Chinai JM, Buhl S, Scandiuzzi L, Ray A, Jeon H, et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T-cell function. Proc Natl Acad Sci USA. (2013) 110:9879–84. doi: 10.1073/pnas.1303524110
169. Janakiram M, Chinai JM, Fineberg S, Fiser A, Montagna C, Medavarapu R, et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin Cancer Res. (2015) 21:2359–66. doi: 10.1158/1078-0432.CCR-14-1495
170. Zhu Y, Yao S, Iliopoulou BP, Han X, Augustine MM, Xu H, et al. B7-H5 costimulates human T cells via CD28H. Nat Commun. (2013) 4:2043. doi: 10.1038/ncomms3043
171. Leitner J, Klauser C, Pickl WF, Stockl J, Majdic O, Bardet AF, et al. B7-H3 is a potent inhibitor of human T-cell activation: no evidence for B7-H3 and TREML2 interaction. Eur J Immunol. (2009) 39:1754–64. doi: 10.1002/eji.200839028
172. Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity (2018) 48:773–86.e5. doi: 10.1016/j.immuni.2018.03.018
173. Steinberger P, Majdic O, Derdak SV, Pfistershammer K, Kirchberger S, Klauser C, et al. Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four Ig-like domains. J Immunol. (2004) 172:2352–9.
174. Hecht I, Toporik A, Podojil JR, Vaknin I, Cojocaru G, Oren A, et al. ILDR2 is a novel B7-like protein that negatively regulates T cell responses. J Immunol. (2018) 200:2025–37. doi: 10.4049/jimmunol.1700325
175. Prasad DV, Nguyen T, Li Z, Yang Y, Duong J, Wang Y, et al. Murine B7-H3 is a negative regulator of T cells. J Immunol. (2004) 173:2500–6.
176. Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity (2003) 18:849–61.
177. Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci USA. (2003) 100:10388–92. doi: 10.1073/pnas.1434299100
178. Steinberger P. B7-H3 ameliorates GVHD. Blood (2015) 125:3219–21. doi: 10.1182/blood-2015-04-638304
179. Janakiram M, Shah UA, Liu W, Zhao A, Schoenberg MP, Zang X. The third group of the B7-CD28 immune checkpoint family: HHLA2, TMIGD2, B7x, and B7-H3. Immunol Rev. (2017) 276:26–39. doi: 10.1111/imr.12521
180. Hughes PE, Caenepeel S, Wu LC. Targeted therapy and checkpoint immunotherapy combinations for the treatment of cancer. Trends Immunol. (2016) 37:462–76. doi: 10.1016/j.it.2016.04.010
181. Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE. Glycogen synthase kinase 3 inactivation drives T-bet-mediated downregulation of Co-receptor PD-1 to enhance CD8(+) cytolytic T cell responses. Immunity (2016) 44:274–86. doi: 10.1016/j.immuni.2016.01.018
182. Taylor A, Rothstein D, Rudd CE. Small-molecule inhibition of PD-1 transcription is an effective alternative to antibody blockade in cancer therapy. Cancer Res. (2018) 78:706–17. doi: 10.1158/0008-5472.CAN-17-0491
183. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. (2015) 33:1974–82. doi: 10.1200/JCO.2014.59.4358
184. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell (2017) 170:1120–33 e17. doi: 10.1016/j.cell.2017.07.024
185. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science (2018) 359:1350–55. doi: 10.1126/science.aar4060
186. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
187. Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O'Brien S, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res. (2016) 22:436–47. doi: 10.1158/1078-0432.CCR-15-1070
188. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-Ts): combination or built-in CAR-T. Int J Mol Sci. (2018) 19: E340. doi: 10.3390/ijms19020340
189. Ankri C, Shamalov K, Horovitz-Fried M, Mauer S, Cohen CJ. Human T cells engineered to express a programmed death 1/28 costimulatory retargeting molecule display enhanced antitumor activity. J Immunol. (2013) 191:4121–9. doi: 10.4049/jimmunol.1203085
190. Rosskopf S, Leitner J, Paster W, Morton LT, Hagedoorn RS, Steinberger P, et al. A Jurkat 76 based triple parameter reporter system to evaluate TCR functions and adoptive T cell strategies. Oncotarget (2018) 9:17608–19. doi: 10.18632/oncotarget.24807
191. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515:577–81. doi: 10.1038/nature13988
192. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. (2013) 19:747–52. doi: 10.1038/nm.3161
193. Duan F, Duitama J, Al Seesi S, Ayres CM, Corcelli SA, Pawashe AP, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med. (2014) 211:2231–48. doi: 10.1084/jem.20141308
194. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. (2012) 72:1081–91. doi: 10.1158/0008-5472.CAN-11-3722
195. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
Keywords: cancer immunotherapy, inhibitory receptors, immune checkpoints, PD-1, BTLA, LAG-3, CTLA-4
Citation: De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K and Steinberger P (2018) Not All Immune Checkpoints Are Created Equal. Front. Immunol. 9:1909. doi: 10.3389/fimmu.2018.01909
Received: 30 May 2018; Accepted: 02 August 2018;
Published: 31 August 2018.
Edited by:
Alexandre M. Carmo, i3S, Instituto de Investigação e Inovação em Saúde, PortugalReviewed by:
Christopher E. Rudd, Université de Montréal, CanadaLewis Zhichang Shi, Case Western Reserve University, United States
Copyright © 2018 De Sousa Linhares, Leitner, Grabmeier-Pfistershammer and Steinberger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Steinberger, cGV0ZXIuc3RlaW5iZXJnZXJAbWVkdW5pd2llbi5hYy5hdA==