 Duong Thi Bich Thuan1
Duong Thi Bich Thuan1 Hatem Zayed2
Hatem Zayed2 Ali H. Eid2,3,4
Ali H. Eid2,3,4 Haissam Abou-Saleh4
Haissam Abou-Saleh4 Gheyath K. Nasrallah2,5
Gheyath K. Nasrallah2,5 Arduino A. Mangoni6*
Arduino A. Mangoni6* Gianfranco Pintus2,5*
Gianfranco Pintus2,5*- 1Department of Biochemistry, Hue University of Medicine and Pharmacy, University of Hue, Hue, Vietnam
- 2Department of Biomedical Sciences, College of Health Sciences, Qatar University, Doha, Qatar
- 3Department of Pharmacology and Toxicology, Faculty of Medicine, American University of Beirut, Beirut, Lebanon
- 4Department of Biological and Environmental Sciences, College of Arts and Sciences, Qatar University, Doha, Qatar
- 5Biomedical Research Center, Qatar University, Doha, Qatar
- 6Department of Clinical Pharmacology, College of Medicine and Public Health, Flinders Medical Centre, Flinders University, Adelaide, SA, Australia
Systemic sclerosis (SSc), an autoimmune disease that is associated with a number of genetic and environmental risk factors, is characterized by progressive fibrosis and microvasculature damage in the skin, lungs, heart, digestive system, kidneys, muscles, joints, and nervous system. These abnormalities are associated with altered secretion of growth factor and profibrotic cytokines, such as transforming growth factor-beta (TGF-β), interleukin-4 (IL-4), platelet-derived growth factor (PDGF), and connective-tissue growth factor (CTGF). Among the cellular responses to this proinflammatory environment, the endothelial cells phenotypic conversion into activated myofibroblasts, a process known as endothelial to mesenchymal transition (EndMT), has been postulated. Reactive oxygen species (ROS) might play a key role in SSs-associated fibrosis and vascular damage by mediating and/or activating TGF-β-induced EndMT, a phenomenon that has been observed in other disease models. In this review, we identified and critically appraised published studies investigating associations ROS and EndMT and the presence of EndMT in SSc, highlighting a potential link between oxidative stress and EndMT in this condition.
Introduction
Systemic sclerosis or scleroderma (SSc) is a complex multisystem autoimmune disease characterized by progressive fibrosis of the skin and visceral organs and significant vascular alterations (1). The pathogenesis of SSc remains unclear, particularly the mechanisms involved in the development of vascular lesions (2). Oxidative stress-mediated-vascular dysfunction and Endothelial-to-Mesenchymal Transition (EndMT) are likely to play a role in SSc-mediated vascular damage (3). Several studies have shown increased production of reactive oxygen species (ROS), altered redox state, and excessive extracellular matrix (ECM) deposition in organs and tissues of SSc patients (2). Although the primary cellular effector of diseases-associated fibrotic conditions remains to be identified (4) some potential sources have been proposed.
In addition to Epithelial-to-Mesenchymal Transition (EMT), which takes place in vivo in the lung (5–8), as well as in a variety of other fibrotic processes (9–13) including SSc (14, 15), the potential involvement of EndMT in SSc has also been suggested. EndMT accounts for the increased fibroproliferative vasculopathy and fibrosis in several diseases (16) and is considered a novel mechanism for the generation of activated myofibroblasts in SSc (17–20). On the other hand, increased ROS generation has been reported to mediate TGF-β-induced EndMT in several conditions including atherosclerosis, Fuchs endothelial corneal dystrophy, and diabetic nephropathy (21–23). TGF-β-mediated ROS generation also promotes cardiac fibroblast differentiation into myofibroblasts, which accounts for the increased production of ECM proteins such as type I and III collagen and the initiation of α-smooth muscle actin expression (α-SMA) during the EndMT process (24). Noteworthy, although not specifically in SSc, the regulation of TGF-β signaling by mitochondrial-derived ROS has also been reported in lung fibrosis (25, 26).
In this review, we summarize the most relevant research regarding the correlation between oxidative stress and EndMT, and their role in SSc-associated vascular damage and remodeling. Readers interested in a more comprehensive discussion concerning the mechanisms involved in the onset and progression of the fibrotic process can refer to other recent excellent reviews (27–30).
Oxidative Stress and SSc
The term ROS indicates oxygen-containing free radicals harboring one or more unpaired electrons in the atom or the outer molecular orbitals (31). Unpaired electrons make free radicals highly reactive. Among them, the superoxide radical (), hydrogen peroxide (H2O2), hydroxyl radical (·OH−), hypochlorous acid (HOCl) and peroxynitrite (ONOO−) are key oxidative molecules within the ROS family (32). In this regard, oxidative stress reflects an imbalance between the generation of ROS and the biological system's ability to counteract or detoxify their harmful effects. Therefore, when present in excessive concentrations, ROS cause oxidative stress and cellular damage, potentially leading to cell transformation and/or cell death (33).
ROS and oxidative stress are considered to play a key role in the onset and progression of SSc through several processes, such as ischemia-reperfusion injury (34–36). In addition, ligand-mediated receptor activation by cytokines and growth factors can also increase ROS generation (2, 35, 37). For example, TGF-β is a profibrotic cytokine that plays a key role in the ligand-mediated receptor process that triggers the onset and progression of SSc (2, 37–41). Other putative factors involved in the pathogenesis of SSc include the platelet-derived growth factors (PDGF), vascular endothelial growth factor (VEGF), connective tissue growth factor (CTGF), angiotensin II, interleukin 3, interleukin 6, tumor necrosis factor-alpha (TNF-α), nerve growth factor, and fibroblast growth factor (FGF). These factors can generate ROS in vascular smooth muscle cells, cardiac, lung and skin fibroblasts by activating signaling pathways coupled to nicotinamide adenine dinucleotide phosphate oxidase (NOX) family members (42). Cytokines can also modulate ROS generation by influencing the cellular concentrations of NOX at both RNA and protein levels, as well as by improving both stability and NOX translocation to the cell membrane by stimulating the phosphorylation of NOX complex's components (42). For instance, in rat aortic smooth muscle cells, production of ROS by IL-1β is mediated by NOX4 (43). Although IL1-β induces its expression (44), NOX1 does not appear to be involved in IL1-β-induced ROS production (43). In addition, TGF-β has been shown to induce NOX4 expression as well as ROS production in human arterial smooth muscle cells (45). IFN-γ, via the JAK/STAT pathway (46), and TNF-α, likely via the NFκB pathway (47), also induce expression of NOX1 and NOX4 in human aortic smooth muscle cells (48). Indeed, NOX inhibitors appear to attenuate the effect of TNF-α in vascular smooth muscle cells, supporting the hypothesis that this cytokine elicits its effects via NOX activity (49). The signaling pathways involved in cytokines-induced ROS generation in fibrosis and SSc are quite complex and depend on specific cytokines, NOX isoforms and target cells. For a more detailed information regarding this aspect we refer the readers to this excellent review (42) Furthermore, besides cytokines-induced ROS generation, the increased concentrations of superoxide from different cells, including fibroblasts and monocytes, can contribute per se to ROS elevation in SSc (2, 50–54).
About 90% of patients with SSc suffer from Raynaud's phenomenon, a condition where the cold-induced constriction of dermal arterioles is excessively augmented and results in vasospasm and skin color change. Patients with Raynaud's phenomenon secondary to underlying diseases typically present with more severe manifestations such as ulcer, scar, or gangrene (55, 56). Although the detailed molecular pathology of the Raynaud's phenomenon, and its association with SSc, is not clearly understood, both oxidative and non-oxidative pathways appear to be involved (35, 56–58). The systemic increase of ROS concentrations in SSc is likely to be an important factor for the worsening of the Raynaud's phenomenon. In this context, the concentrations of 8-isoprostane, a biomarker of oxidative stress, antioxidant deficiency and lipid peroxidation, have been shown to correlate with the extent of vascular lesions in Raynaud's phenomenon and the severity of fibrosis in patients with SSc (59–61). The free radical nitric oxide (NO), released by the endothelial cells, plays an essential role in the homeostatic control of vascular tone and blood pressure as well as in preventing thrombosis and cell damage. However, during the reperfusion phase in the Raynaud's phenomenon, free radicals and NO lead to peroxynitrite formation, which precedes oxidative vascular damage and endothelial apoptosis. Therefore, in this specific situation, NO further aggravates vascular damage (35, 62).
A growing number of in vitro and in vivo studies have demonstrated the direct role of ROS in the pathogenesis of SSc (61, 63, 64). Grygiel-Gorniak and Puszczewicz et al. skin and visceral fibroblasts from SSc patients spontaneously produce large amounts of ROS that initiate collagen synthesis (35). Indeed, fibroblasts from SSc patients have higher baseline NOX-inhibitable intracellular ROS concentrations (65) when compared to fibroblasts from healthy donors (65). This phenomenon appears to be triggered by the stimulation of the PDGF receptor and further maintained through ROS-ERK1/2 signals mediated by Ha-Ras (66). It is important to emphasize, however, that normal fibroblasts can also respond to stimulation by different cytokines with a NOX-dependent increase in intracellular ROS concentrations (65).
A preliminary study by Boin et al. (67) showed a significant increase in intracellular ROS concentrations in human pulmonary artery smooth muscle cells (HPASMCs) after treatment with sera from patients with SSc and pulmonary artery hypertension (PAH). NOX2ds-tat (gp91ds-tat), a specific inhibitor of NOX2, prevented the PAH-SSc sera -induced ROS generation, suggesting the mechanistic involvement of NOX2 in this phenomenon (67). Exposure of HPASMCs to SSc-PAH sera also resulted in a progressive increase of the Collagen promoter activity. Similarly, this effect was prevented by NOX2ds-tat treatment, suggesting that the collagen synthesis activation in HPAMSCs is driven by SSc-related PAH sera through NADPH oxidase-dependent ROS generation (67). Moreover, in human dermal fibroblasts, NOX inhibition caused a significant reduction in the expression of fibronectin, collagen type I and alpha-smooth muscle actin (68). Similarly, the selective NOX1/NOX4 inhibitor, GKT-137831, abolished TGF-β-induced expression of the profibrotic genes CCN2 and alpha-SMA (41). Importantly, GKT-137831 was also able to reduce collagen gel contraction as well as expression of alpha-SMA and CCN2 protein overexpression in fibroblasts isolated from dermal lesions in SSc patients (41). A NOX4-derived increase of ROS has also been reported to be involved in the vascular smooth muscle cells contractile to synthetic phenotype switch elicited by agonistic anti-PDGF receptor autoantibodies from SSc patients (69) Taken together, these findings indicate that the NOX system, in addition to ROS production, mediates the activation of collagen synthesis and profibrotic genes.
In vivo studies in BALB/c mice injected with HOCl daily for 6 weeks (a murine model of SSc) showed the induction of chronic oxidative stress and the concomitant development of cutaneous and lung fibrosis. Furthermore, HOCl-treated mice overexpressed α smooth muscle actin (α-SMA), a marker of myofibroblast activation. These processes were mediated by the ROS-activated intracellular signaling pathways ADAM17/Notch and Ras-ERK (70–72). Blockade of the Ras-ERK pathway by propylthiouracil (PTU) or simvastatin prevented both cutaneous and lung fibrosis (71, 72).
Some studies reported that, in SSc, the excessive production of ROS activates fibroblasts through the binding of stimulatory serum autoantibodies to the PDGFR (73). In this regard, it has been reported that sera from mice and patients with SSc induce fibroblast proliferation and H2O2 production by endothelial cells, phenomena that appear to be mediated by oxidized auto-antigens such as the oxidized DNA topoisomerase (74). Noteworthy, the same paper indicated that the nature of ROS, as well as the induction of different antibodies, appear to dictate the form of SSc in both mice and humans. BALB/c SCID mice treated with peroxynitrites developed skin fibrosis and serum anti centromere protein autoantibodies (anti-CENP-B), as reported in patients with limited cutaneous scleroderma, while mice treated with hypochlorite or hydroxyl radicals developed skin and lung fibrosis and DNA topoisomerase I autoantibodies, as reported in patients with diffuse cutaneous scleroderma (74). While pro-oxidants may cause an increase in autoantibodies, other studies failed to demonstrate a significant association between autoantibodies in endothelial cells and fibroblasts and serum-induced ROS or cell proliferation. Despite this uncertainty, there is good evidence supporting the role played by autoantibodies in SSc, particularly anti-endothelial cell antibodies (AECA), in the development of pulmonary fibrosis (35, 75). Oxidative stress may either directly activate ROS-induced differentiation of fibroblasts into myofibroblasts (70), disrupt the balance between protease and protease inhibitors, or both. TGF-β upregulates the expression of extracellular matrix proteins including collagens, but also suppresses protein degradation through enhancing the activities of protease inhibitors such as plasmin activator inhibitor 1 (PAI-1) and tissue inhibitors of metalloproteinases (TIMPs) (38). This protease-antiprotease imbalance is likely to represent a critical factor in the development of SSc lung fibrosis (76). Furthermore, although not specifically demonstrated in the SSc-associated fibrotic process, a mitochondrial-generated increase of ROS has been found to induce lung fibrosis (25–27). Effect of ROS in the development of fibroproliferative vasculopathy and fibrosis in SSc (Figure 1).
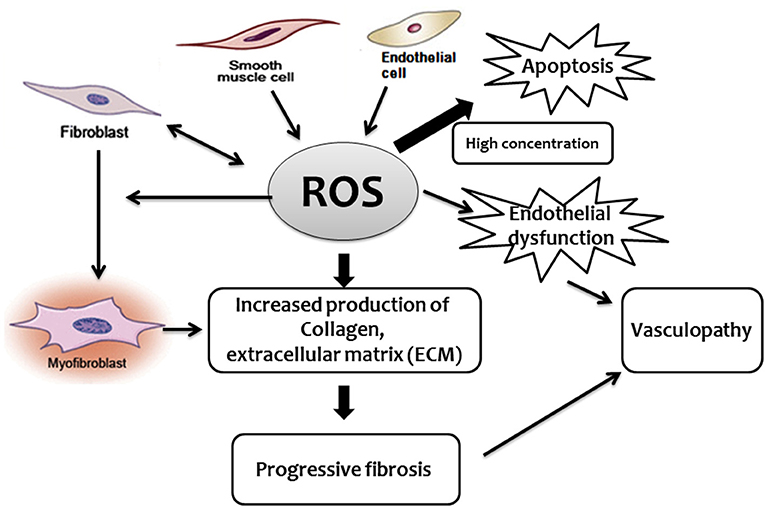
Figure 1. Schematic diagram describing the central role played by ROS in the development of fibroproliferative vasculopathy and fibrosis in SSc.
Endothelial-to-Mesenchymal Transition and SSc
The pathogenesis of SSc involves several complex mechanisms associated with (i) microvascular fibroproliferative lesions, (ii) innate and adaptive immune system abnormalities and uncontrolled accumulation of collagen, as well as (iii) other extracellular matrix compartments produced by fibroblasts and activated myofibroblasts in the skin and other organs (17, 77). Physiologically, myofibroblasts die through apoptosis and/or the transition to a quiescent/senescent state in the late stages of wound healing. However, the persistence of activated myofibroblasts contributes to progressive fibrogenesis (78, 79) and favors the onset and progression of interstitial and perivascular fibrosis in the lungs, heart, kidneys and other organs, which accounts for the high mortality of SSc patients (80).
Myofibroblast activation in SSc has been demonstrated in pericytes and smooth muscle cells (SMCs) from vessel walls, resident fibroblasts, and bone marrow-derived fibroblasts (17, 81, 82). Although the ontogenesis of myofibroblasts in fibrotic conditions remains an area of active research (4, 83–86), an increasing number of studies indicated EMT as a potential source of activated fibroblasts by which epithelial cells transform into myofibroblasts (5–8, 14, 15). In this regard, some authors suggest that diseases-associated fibrotic processes may be the result of injury-elicited cellular stress responses such as senescence or apoptosis (28, 87–91), These processes, under yet unknown conditions, could also promote tissue repair by activating and/or recruiting resident progenitor cells (92) especially in the lungs (93–95). However, this hypothesis does not exclude the coexistence of other injury-activated processes such as the cellular transdifferentiation of endothelial cells to profibrotic activated myofibroblasts during EndMT, a phenomenon that has been reported also in vivo (19, 96–102). The latter might provide a plausible explanation for the excessive secretion of extracellular matrix proteins that takes place in this pathological condition (103). EndMT is considered a distinct form of EMT since vascular endothelial cells share several similar characteristics and molecular mechanisms with epithelial cells in generating fibroblasts and myofibroblasts (104, 105). EndMT contributes to the development of cardiac, pulmonary, renal, liver, and intestinal fibrosis, and idiopathic portal hypertension in SSc (7, 12, 13, 17, 18, 97, 98, 106–109). EndMT has also been reported in vivo in pulmonary hypertension, a process that is closely associated to SSc (101, 102, 110). It is also important to mention that, despite its role in many pathologies, EndMT may be beneficial during angiogenic sprouting, as it allows cells to lose intercellular junctions and delaminate from the parent vessel (111).
During the conversion of endothelium into mesenchyme elicited by TGF-β or Notch ligands, endothelial cells undergo morphological alterations and loss of characteristic cell-surface markers, acquiring mesenchymal, fibroblast-like properties such as spindle-shaped morphology, migratory capacity, invasiveness, and enhanced resistance to apoptosis (82, 112). During EndMT, the structure of vessel-lining is disrupted due to resident endothelial cells disaggregating from the organized cells layer in the vessel walls and invading the surrounding tissue (112–114). Cell-surface markers such as vascular endothelial cadherin (VE-Cadherin), CD31 (platelet endothelial cell adhesion molecule-1, PECAM 1), and von Willebrand factor (vWF) are gradually replaced by markers such as fibroblast-specific protein-1 (FSP-1), α-SMA, vimentin and type I and type III collagen (12, 18, 114).
Several studies have demonstrated the presence of transitional EndMT cells in the pulmonary vasculature of patients with SSc-PAH, indicating their possible contribution to vascular remodeling and fibrosis (18). For instance, the coexpression of cell surface markers specific for both endothelial and mesenchymal/fibroblastic cells, such as Willebrand factor (vWF) and α-SMA, has been reported in an experimental murine model of PAH and in pulmonary endothelium samples of SSc-PAH patients (110). Similar findings have been reported in lung tissue from patients suffering from SSc-associated interstitial lung disease (19). Cells in the intermediate stages of EndoMT have also been found in dermal vessels from patients with SSc and in two types of SSc animal models, bleomycin-induced SSc and urokinase-type plasminogen activator receptor (uPAR)-deficient mouse model (115). When compared to normal skin microvascular endothelial cells from healthy donors, cells from SSc patients displayed a spindle-shaped figure along with co-expression of both endothelial (CD31 and VE-cadherin) and myofibroblast markers (α-SMA, S100A4, type I collagen). Moreover, exposure of healthy donor-derived microvascular endothelial cells to either SSc sera or TGFβ1 triggered the transition to a myofibroblast-like morphology, contractile phenotype, downregulation of endothelial markers and induction of mesenchymal markers (115). Similarly, exposure of pulmonary artery endothelial cells to a mixture of proinflammatory cytokines such as IL-1β, TNF-α, and TGF-β abolished their “cobblestone” structure and prompted a spindle-like appearance along with induction of mesenchymal markers (110). Induced endothelial-mesenchymal transition (I-EndMT) cells exhibit an increased secretion of proinflammatory proteins and collagen type I. In addition, the presence of I-EndMT cells in the cellular barriers leads to a significant increase in paracellular and transcellular permeability, an early sign of vascular dysfunction in SSc (76, 110).
Vascular abnormalities in SSc patients have been reported to affect the structure and function of several organs and systems, such as the kidney, lung, skin, heart, gut, penis and large vessels (17). Therefore, the mechanistic contribution of EndMT to the pathogenesis of SSc vasculopathy is postulated to involve a synergistic and complex activation of a large variety of endothelial cell types from different body districts exposed to multiple local biological mediators, particularly TGF-β, but also IL-1β, TNF-α, PDGF, VEGF, and endothelin-1 (ET-1) (17, 18, 77, 78, 112, 114, 116–119). Indeed, cytokines-mediated receptor activation induces the expression of endothelial cell adhesion molecules such as ICAM, VCAM-1, and E-selectin (120, 121), which promote both recruitment and activation of chronic inflammatory cells, such as T- and B-lymphocytes and profibrotic macrophages, in the perivascular tissue and in the parenchymal organs interstitium. Recruited chronic inflammatory cells secrete transforming TGF-β, CTGF, and other profibrotic growth factors which along with endothelial cell released mediators, such as endothelin-1, enhance the fibroproliferative vasculopathy characteristic of the disease and potentially activate EndMT (17, 122, 123). In this regard, the interaction between proinflammatory stimuli and endothelial cells has been demonstrated by a recent paper reporting the upregulation of ET-1 and TGF-β in human microvascular endothelial cells induced by IF-γ, (118). Nevertheless, the precise molecular mechanisms by which inflammatory cytokines, growth factors, and signaling pathways mediate EndMT are not completely elucidated and necessitate further investigation.
Using a transgenic mice model, a recent study demonstrated that the endothelial cell-specific activation of TGF-β1 downstream signaling pathway induces EndMT in lung vessels (20). An elegant study using co-culture of microvascular endothelial cells and fibroblasts isolated from the skin of SSc patients demonstrated the ability of SSc-fibroblasts to induce endothelial cells EndMT transition. Consistent with this finding, normal fibroblasts treated with TGF-β and ET-1 were able to promote the same effect when co-cultured with microvascular endothelial cells (124). TGF-β2 has also been reported to mediate Interferon-γ-induced EndMT in human dermal microvascular endothelial cells (118). Protein kinase C has also been reported to be involved in TG-induced EndMT of mouse pulmonary endothelial cells in vitro (125).
All TGF-β isoforms have been reported able to elicit EndMT although the precise role of each isoform appears to differ between species and requires further studies to be clearly elucidated. While TGF-β 1 appears to be the main isoform involved in fibrosis-associated EndMT processes, TGF-β 2 seems to be primarily involved in EndMT associated to embryonic heart development (126–128). Nevertheless, also TGF-β 2 has been recently reported to be involved in EndMT associated with some pathological process such as cardiac hypertrophy and renal fibrosis (129, 130). In this regard, Maleszewska et al. (131) showed that TGF-β can either promote EndMT per se or synergize with TNF-α and IL-1β to induce the transdifferentiation of endothelial cells toward the profibrotic activated myofibroblasts phenotype. The authors showed that, in the framework of an inflammatory co-stimulation, TGF-β 2 is more potent that TGF-β1 in inducing EndMT, suggesting that TGF-β 2 may be the primary EndMT trigger, while IL-1 is necessary for the efficient induction of EndMT, but is not essential for its maintenance (131). The finding that endothelial cells in proinflammatory environment respond differently to TGF1 and TGF2 suggests that, similar to what reported in the embryonic development, TGF2 might play a major role in EndMT associated to pathological inflammation. In this regard, Good et al. (110) showed that a combination of TGF-β with IL-1β and TNF-α induced EndMT in pulmonary artery endothelial cells. Notably, the withdrawal of IL-1β, TNF-α, and TGF-β after 6 days failed to revert this process, suggesting that the phenotypic change might be permanent (104, 110). This observation was in agreement with previous studies in cells exposed to activated Ras and TGF-β treatment (107). Recently, Cipriani et al. (119) reported that Macitentan, an Endothelin-1 Receptor Antagonist, blocks both Endothelin-1- and TGF-β-induced EndMT in microvascular endothelial cells isolated from healthy donors and SSc patients (119). Similar results, using both Bosentan and Macitentan, were also obtained by Corallo et al. (124) in the fibroblast and microvascular endothelial cells coculture model previously discussed (124). Other studies reported that some anti-fibrotic mediators could dedifferentiate established myofibroblasts. These findings suggest the possibility that EndMT is a reversible process, providing a new intriguing therapeutic target for fibrotic diseases (81, 132).
TGF-β plays a critical role in other signaling pathways involved in EndMT, such as the Smad-dependent and Smad-independent pathways (Figure 2). Smad proteins have been shown to bind directly to the Snail gene promoter and regulate its transcription (117). Snail-1 is a zinc-finger transcription factor that forms a complex with Smad3/Smad4, and by acting as transcriptional repressor, plays a crucial role in the TGF-β-induced mesenchymal transdifferentiation of embryonic stem cell-derived ECs. The active Smad3/Smad4/Snail-1 complex is a potent inhibitor of E-cadherin expression by directly integrating into specific sequences within the gene promoter and blocking its transcription. In addition to E-cadherin inhibition, Snail-1 precedes transcriptional events that lead to the expression of a mesenchymal-cell-specific phenotype (17, 18).
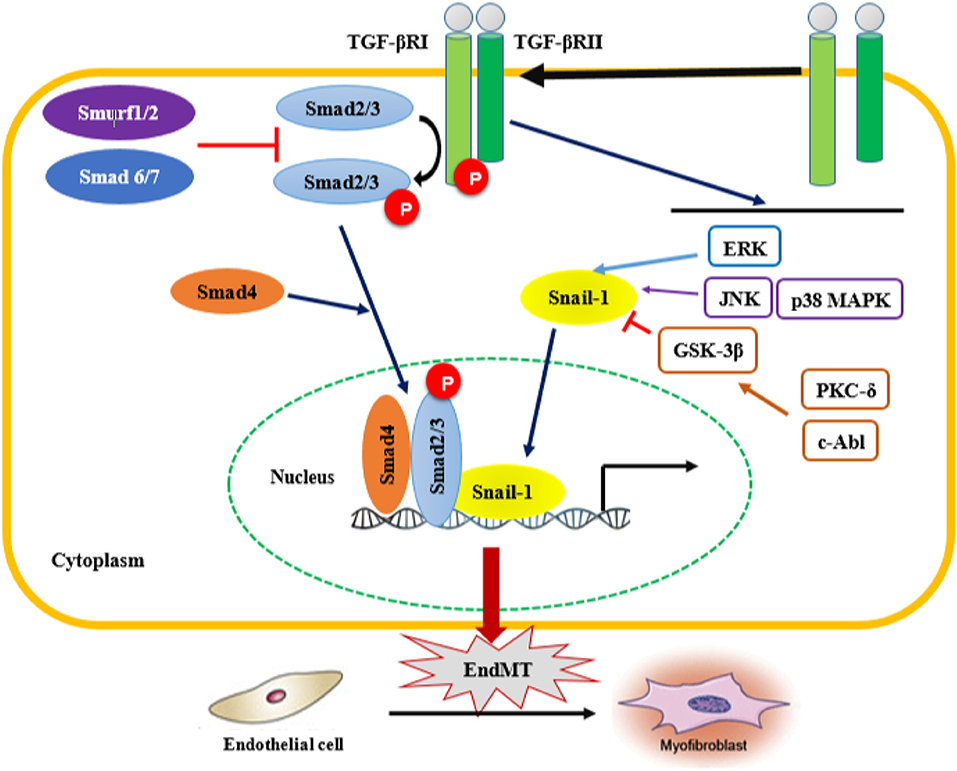
Figure 2. Smad-dependent and Smad-independent pathways of TGF-β signaling associated with EndMT. Transforming growth fator-β (TGF-β) signaling activates the downstream signal transduction cascades, Smad and non-Smad pathways. TGF-β binds the TGF-β type II receptor (TGF-βRII), which recruits and activates the type I TGF-β receptor. TGF-βRI in turn phosphorylates Smad2/3, which forms a complex with Smad 4. In addition, TGF-β activates Smad-independent pathways. Activation of Smad-independent TGF-β pathway causes phosphorylation of GSK-3β mediated by PKC-δ and c-Abl. Phosphorylation of GSK-3β causes its own inhibition which then allows Snail-1 to enter the nucleus. TGF-β/Smad-dependent and Smad-independent pathways upregulate the transcription of TGF-β target genes such as α-smooth muscle actin (α-SMA), fibronectin and type I collagen, as well as the transcription factor Snail-1 involved in EndMT. EndMT leads to the transdifferentation of ECs into mesenchymal cells, which subsequently transform into myofibroblasts, therefore contributing to the progression of fibrotic diseases. ERK, extracellular signal-regulated kinase; JNK, jun N-terminal kinase; p38 MAPK, p38 mitogen-activated protein kinases; PKC-δ, protein kinase C δ; c-Abl, c-Abl protein kinase; GSK-3β, Glycogen synthase kinase 3β.
In the Smad-independent pathway of TGF-β signaling, there is an involvement of important kinases such as c-Abl protein kinase (c-Abl), protein kinase Cδ (PKC-δ), RhoA and glycogen synthase kinase 3β (GSK-3β). The function of RhoA in actin and microtubule cytoskeleton organization is well-established, and as such contributes to the structural/phenotypic changes observed during EndMT (133). On the other hand, activation the of Smad-independent TGF-β pathway causes GSK-3β inhibition allowing Snail-1 to enter the nucleus. Indeed, phosphorylation of GSK-3β at the specific Ser9 residue causes its own inhibition, which in turn induces Snail-1 up-regulation and promotes its subsequent translocation into the nucleus. This process increases the nuclear accumulation of Snail-1, which consequently drives the expression of mesenchymal cell-specific markers such as α-SMA and type I collagen, while reducing the expression of VE-cadherin harbored in endothelial cells. Inhibitors of c-Abl and PKC-δ, such as imatinib and rottlerin, counteract the phosphorylation of GSK-3β, which allows GSK-3β to phosphorylate Snail-1, targeting it for proteasomal degradation and ultimately abolishing the transdifferentiation of ECs into myofibroblasts. This intervention could be a therapeutic strategy to counteract the acquisition of the myofibroblastic phenotype during EndMT (125). TGF-β2-downstream signals mediated by MEK, PI3K, and p38MAPK pathways are also essential for ECs undergoing EndMT transition (117). In addition, regulation of EndMT by Wnt (134), NOTCH (135), and Caveolin-1 signaling (136, 137) has been observed prior to the TGF-β-induced endothelial-mesenchymal transition. ET-1-mediated TGF-β1-induced EndMT has been also confirmed in skin and lungs in vivo in an animal model of TGF-β1-induced tissue fibrosis (138). Finally, other cytokines and growth factors such as PDGF, VEGF, ET-1, CTGF, and some MicroRNAs (miRNAs) might also be involved in the endothelial-mesenchymal transition (17, 18).
miRNAs are small non-coding RNAs containing about 22 nucleotides, which are post-transcriptional repressors of gene function (139). miRNAs have been recently reported to play important roles in SSc pathogenesis as well as in the EndMT process (140, 141). However, further studies are required to determine whether the SSc-associated profibrotic or antifibrotic effects of specific miRNAs are mediated by EndoMT. Recent studies reported the involvement of miRNAs 125b and 126 in the development of EndoMT (142–144) and the interaction between TFG β and several miRNAs in modulating EndMT. For instance, miRNA21 partially mediated TGF-β-induced EndoMT (145, 146). miR-155 is higly overexpressed during the EndoMT process and potently inhibits TGF-β induced EndoMT through a mechanism involving RhoA signaling (147). The overexpression of miR-148b increased EC migration, proliferation, and angiogenesis, whereas its inhibition promoted TGF-β-induced EndoMT (148). On the other hand, other studies indicated some miRNAs as positive modulators of TGF-β-induced EndoMT. For instance, the constitutively expression of miR-31 positively regulates TGF-β-induced EndMT in cultured endothelial cells (149). Interestingly, the overexpression of miR-130a, which is upregulated in monocrotaline-induced the Pulmonary arterial hypertension (PAH) mouse model, can elicit the expression of α-smooth muscle actin, a critical component in EndMT transition and fibrogenesis (150). Since PAH is a widely SSc-associated condition, this paper my pave the way for further experimentations in this direction. Many other miRNAs have also been reported to be involved in EndoMT modulation including let-7 (151, 152) and miRNA 29s (153). Given the pivotal regulatory effects exerted by miRNAs on the multitude of signaling pathways involved in the pathophysiology of multiple diseases, we expect that the knowledge regarding the putative contribution of miRNA to the EndMT process in conditions such as SSc will rapidly expand providing valuable information both to unravel the EndoMT mechanistic processes and to identify potential therapeutic targets for fibrotic disorders in general.
Reactive Oxygen Species and Endothelial-to-Mesenchymal Transition
ROS have been proposed as key mediators of EMT in renal tubular epithelial cells, human epithelial keratinocytes, and lung epithelial cells (32, 105, 154–156). An emerging issue is whether there is a similar tendency of endothelial cells exposed to oxidative stress to form transitional EndMT cells. Although EMT and EndMT share several similarities, in terms of signaling pathways and outcome, the two processes need to be differentiated due to the various origin, functions, and microenvironment of epithelial and endothelial cells (157, 158). Whether EndMT is a reversible biological process, similar to EMT (157), is an intriguing question that deserves further investigations.
ROS Activate/Mediate TGF-β-Dependent Signaling Pathways in EndMT
TGF-β is a multifunctional protein, including three isoforms (TGF-β1, TGF-β2, and TGF-β3), which regulates several important physiological processes such as cell proliferation, differentiation, apoptosis, adhesion, and migration (38, 159). However, a critical, potentially vicious, cycle of TGF-beta, and ROS interaction exists. For instance, TGF-β stimulates the generation of ROS in various cells while ROS can activate TGF-β and mediate its effects. Moreover, TGF-β elevates ROS production via NOX4, mitochondria, or microsomes, and ROS, in turn, can induce TGF-β gene expression and activate its signaling through oxidizing latency association protein (LAP) or activating MMPs which promotes LAP release (38). It is well known that TGF-β is synthesized as a non-active pro-form combining with LAP to create a latent complex. The ROS-oxidable redox center present in LAP can trigger a conformational change resulting in the release of TGF-β1 (160). Hence, under the stimulation of ROS, TGF-β is activated and increasingly expressed.
It is well documented that TGF-β induces EndMT in cardiac, pulmonary, renal, intestinal, and skin tissues (12, 106, 107, 161, 162). In this context, NADPH oxidases and ROS play a pivotal role in mediating TGF-β induced fibrotic responses via Smad2/Smad 3 activation (159). Of note, NOX4-dependent generation of H2O2 is required for TGF-β1-induced myofibroblast differentiation and ECM production (163, 164). A recent study by Montorfano et al. (32) used human umbilical vein endothelial cells (HUVECs) to investigate the role of ROS as well as the underlying mechanism in the conversion of ECs into myofibroblasts (32). The authors demonstrated that oxidative stress is a crucial factor in generating the EndMT phenotypic conversion of ECs via TGF-β1 and TGF-β2-dependent pathway and that the ALK5/Smad3/NF-κB intracellular pathway mediated the observed phenomenon. This study supports the hypothesis of an interaction between ROS, TGF-β, and EndMT. Indeed, increased ROS prompted the expression and secretion of TGF-β1/2, with consequent activation of its downstream signals, while silencing of TGF-β1/2 abolished the oxidative stress-induced conversion (32).
Another recent study by Echeverría et al. (165) provided evidence that lipopolysaccharide-induced ROS could lead to an EndMT-like process through an ALK5 activity-dependent mechanism (165). In line with this observation, Toshio et al. (109) showed that endotoxemia-derived oxidative stress promotes TGF-β-mediated EndMT in pulmonary vascular endothelial cells (109). This evidence was strongly supported by another study showing that the expression of TGF-β1 and TGF-β2 is crucial for the development of endotoxin-induced endothelial fibrosis (166). The increase in endotoxin-induced TGF-β1 and TGF-β2 expression required the activation of NOX and the subsequent generation of ROS (166). Collectively, these data suggest that oxidative stress mediates the EndMT process induced by TGF-β.
Oxidative Stress and EndMT in SSc: Is There a Link?
Despite the fact that the relationships between ROS and SSc, and between EndMT and SSc, have been extensively investigated, relatively few studies have investigated the effects of oxidative stress on EndMT in SSc. Xu et al. (167) showed that chronic oxidative stress mediates EndMT in a murine model of SSc (167). The authors isolated microfibrils from the skin of tight skin (Tsk+/−) mice, which showed abnormal big fibrillin-1. Culturing ECs with this abnormal extracellular matrix led to morphological and functional cellular changes, and increased the concentrations of 4-HNE, a well-known fission product of polyunsaturated fatty acid oxidation. Transdifferentiation from ECs to mesenchymal cells with the increased presence of FSP-1 and Twist (a transcription factor implicated in the endothelial cell to fibroblast transition), along with the decreased expression of VE-cadherin, were also noted (167). Furthermore, the abnormal big fibrillin expression was associated with oxidative stress (reduced nitric-oxide-to-superoxide anion ratio) suggesting changes in the intracellular redox state involved in the observed transition. Interestingly, chronic mice pretreatment with D-4F, a peptide binding with high affinity to oxidized lipids, attenuated or abolished EndMT, indicating that oxidized lipids might play a central role in other chronic conditions where oxidative stress promotes endothelial-mesenchymal transition (167). However, the authors did not dissect the role of TGF-β although, as mentioned above, TGF-β is known to be a crucial player in the initiation of EndMT in various diseases (17, 18). Of note, ECM including fibrillin functions as a reservoir for TGF-β and other growth factors to control mesenchymal differentiation (168). The possibility of an interaction between TGF-β and oxidative stress prior to EndMT is intriguing and requires further research.
It is speculated that the local availability of tetrahydrobiopterin (BH4) contributes to endothelial physiology (169), and that its insufficiency might be involved in the endothelial dysfunction observed in SSc (170). Decreased concentration of BH4 induces eNOS uncoupling, leading to superoxide, rather than NO, production, which in turn induces a state of oxidative stress (171). Although not able to reverse blood oxidative stress markers, the acute administration of BH4 has been shown to improve endothelial function in patients with SSc, without affecting their blood pressure (170). However, in this paper the authors did not investigate whether endothelial, rather than systemic, oxidative stress was ameliorated. Indeed, it may be possible that the concentration of BH4 used was sufficient to improve endothelial function but not appropriate to ameliorate the oxidative insult seen in blood samples of SSc patients. More importantly, the patients only received BH4 for 5 h. This period might be too short to influence systemic oxidative parameters. In fact, more data support the involvement of BH4 in SSc-associated endothelial dysfunction. For instance, circulating concentrations of BH4 have been found to be lower in plasma and pulmonary arteries of patients with IPF, and in rats with bleomycin-induced pulmonary fibrosis, when compared to their healthy counterparts (172). Notably, TGβ1 and ET-1 were able to induce EndMT in human pulmonary artery endothelial cells by decreasing BH4 and eNOS expression. Finally, treatment with sepiapterin, a BH4 precursor, blunted bleomycin-induced pulmonary fibrosis, ameliorated vascular remodeling in vivo by increasing plasma BH4 and vascular eNOS expression in rats and counteracted TGβ1- and ET-1-induced EndMT in human pulmonary artery endothelial cells in vitro (172).
In models of SSc-like bleomycin (BLM)-induced fibrosis, BLM-induced expression of collagen (I and III) synthesis mediated by ROS (173). Antioxidants such as NAC were found to attenuate BLM-induced lung fibrosis in mice and rats (174, 175) as well as collagen expression (173). The above data are consistent with earlier findings showing higher ROS production in type II alveolar epithelial cells and lung phagocytes in a rat model of BLM-induced fibrosis (176). Although the above-mentioned studies did not explore the involvement of EndoMT in the observed SSc-associated fibrotic process induced by ROS, recent evidence supports the role of ROS in promoting EndoMT in association with other pathological fibrotic conditions in the kidney (22, 177, 178). Furthermore, antioxidants have been shown to reduce EndMT of vascular endothelial cells (177). Indeed, suppressing oxidative stress has been shown to reduce in vivo EndMT in glomerular endothelial cells (22).
The BLM-induced fibrosis model has also been used in other studies (107, 179–181). Notably, Qi et al. (181) demonstrated that EndMT in the BLM-induced scleroderma mouse model is inhibited by geniposide, a constituent of the Chinese herbal compound Zhizi. According to Chinese herbal medicine, its “bitterness and coldness” properties are appropriate to attenuate various inflammatory conditions, including the early inflammation stage of SSc (181). The study proved the ability of Geniposide to block BLM-mediated EndMT not only in vivo (mouse model) but also in vitro (HUVECs). Further investigation of the EndMT-inhibiting effects exerted by Geniposide showed its ability to down-regulate key transcription factors involved in EndMT (Slug, Snail, Twist). Although the above-mentioned study does not report the involvement of oxidative stress, it is important to emphasize the well-known prooxidant effect of BLM which can damage surrounding cells, resulting in fibroblast activation (182). Interestingly, among all the pharmacological properties of Geniposide, its antioxidant activity was highlighted as protective in preventing cells from undergoing oxidative damage via MAP kinase pathway (183). Of note, ROS and EndMT interact through TGF-β-dependent and -independent pathways, as previously discussed. Based on the reported data, one could hypothesize that Geniposide might act as an anti-oxidant and anti-inflammatory factor, promoting an inhibitory effect on BLM-induced EndMT in the SSc mouse model. Although this hypothesis requires further investigations, a recent study demonstrated that salvianolic acid A (SSA), a natural polyphenol antioxidant, prevented EndMT both in vitro and in vivo by inhibiting oxidative stress (184). In vitro, EndMT was induced in human pulmonary arterial endothelial cells by TFG- β treatment, while in vivo EndMT was studied in a rat model of monocrotaline-induced pulmonary hypertension. SSA, by reducing ROS concentrations, was able to counteract EndMT-associated cell functions, signaling, and proteins both in vitro and in the lung of rats with pulmonary hypertension (184).
Concluding Remarks and Future Direction
SSc represents a public health and economic burden with a high rate of mortality and morbidity. ROS play a critical role in the pathogenesis of SSc. Both oxidative stress and EndMT are involved in the onset and progression of the fibroproliferative vasculopathy and fibrotic process in SSc. TGF-β is involved in the fibrotic process through EndMT, where ROS mediate and/or activate TGF-β to induce EndMT. Although numerous studies demonstrated the potential involvement of both ROS and EndMT in the development of fibrosis, only a few studies investigated the potential relationship between ROS and EndMT, and their involvement in the pathogenesis of SSc. The potential role of oxidative stress in inducing EndMT directly, or through the TGF-β-dependent pathway, is an emerging area of investigation that needs to be addressed. Elucidating the mechanism of SSc pathogenesis associated with ROS will contribute to the identification of therapeutic strategies to alleviate the costs and health burden of this disease.
Author Contributions
DT and GP ideated the review. DT wrote the first draft. HZ, AE, HA-S, GN, AM, and GP contributed to the editing and review of the different versions. GP and AM performed the final editing and GP submitted the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This review article constitutes original research work that was part of Dr. Thuan's Doctoral Thesis (185). This research was supported by grants from the Qatar National Research Fund (UREP20-051-3-012) and the Qatar University (QUCG-CHS-2018/2019-1). The article processing charge (APC) for the publication of this article was funded by the (Qatar National Library).
Abbreviations
4-HNE, 4-hydroxynonenal, a common byproduct of lipid peroxidation during oxidative stress; ADAM17/NOTCH, Disintergrin and metalloproteinase domain-containing protein 17 involved in the activation of the Notch signaling pathway; AECA, Anti-endothelial cell antibodies; ALK5, Activin receptor-like kinase 5; BH4, Tetrahydrobiopterin; BLM, Bleomycin; c-Abl, c-Abl protein kinase; CTGF, Connective tissue growth factor; EMT, Epithelial-to-Mesenchymal Transition; EndMT, Endothelial-to-Mesenchymal Transition; ERK, Extracellular signal-regulated kinases; ET-1, Endothelial-1; FSP-1, Fibroblast specific protein-1; GSK-3β, Glycogen synthase kinase 3β; GTPase, Guanosine triphosphate intracellular signaling; HOCl, Hypochlorous acid; HPASMCs, Human pulmonary artery smooth muscle cells; I-EndMT, Induced EndMT; IL-1β, Interleukin-1β; miRNAs, MicroRNAs; MMP, Matrix metalloproteinase-1; NAC, N-Acetyl-Cysteine; NADPH oxidase (NOX), Nicotinamide adenine dinucleotide phosphate oxidase; NF-κB, Nuclear factor kappa-light-chain-enhancer of activated B cells; NO, Nitric oxide; , Superoxide radical; OH, Hydroxyl radical; ONOO−, Peroxynitrite; p38MAPK, P38 Mitogen-activated protein kinases; PAH, Pulmonary artery hypertension; PAI-1, Plasmin activator inhibitor 1; PDGF, Platelet derived growth factors; PDGFR, Platelet derived growth factor receptor; PECAM 1, Platelet endothelial cell adhesion molecule-1; PI3K, Phosphoinositide 3-kinase; PKC-δ, Protein kinase Cδ; PTU, Propylthiouracil; Ras, A small GTP-binding protein; ROS, Reactive oxygen species; SSc, Systemic sclerosis; TGF-β, Transforming growth factor-β; TIMPs, Tissue inhibitors of metalloproteinases; TNF-α, Tumor necrosis factor-alpha; VE-Cadherin, Vascular endothelial cadherin; VEGF, Vascular endothelial growth factor; vWF, Von Willebrand factor; α-SMA, α-smooth muscle actin.
References
1. Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. (2009) 360:1989–2003. doi: 10.1056/NEJMra0806188
2. Gabrielli A, Svegliati S, Moroncini G, Amico D. New insights into the role of oxidative stress in scleroderma fibrosis. Open Rheumatol J. (2012) 6:87–95. doi: 10.2174/1874312901206010087
3. Mostmans Y, Cutolo M, Giddelo C, Decuman S, Melsens K, Declercq H, et al. The role of endothelial cells in the vasculopathy of systemic sclerosis: a systematic review. Autoimmun Rev. (2017) 16:774–86. doi: 10.1016/j.autrev.2017.05.024
4. Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. (2011) 108:E1475–83. doi: 10.1073/pnas.1117988108
5. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
6. Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. (2013) 32:231–44. doi: 10.1038/emboj.2012.336
7. Sohal SS. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig. (2017) 55:104–13. doi: 10.1016/j.resinv.2016.11.006
8. Han Q, Lin L, Zhao B, Wang N, Liu X. Inhibition of mTOR ameliorates bleomycin-induced pulmonary fibrosis by regulating epithelial-mesenchymal transition. Biochem Biophys Res Commun. (2018) 500:839–45. doi: 10.1016/j.bbrc.2018.04.148
9. Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. (2007) 117:3810–20. doi: 10.1172/JCI30487
10. Chapman HA. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu Rev Physiol. (2011) 73:413–35. doi: 10.1146/annurev-physiol-012110-142225
11. Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation (2012) 125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352
12. He J, Xu Y, Koya D, Kanasaki K. Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin Exp Nephrol. (2013) 17:488–97. doi: 10.1007/s10157-013-0781-0
13. Sterzer V, Alsamman M, Bretag T, Scholten D. EMT in liver fibrosis. Curr Pathobiol Rep. (2014) 2:201–7. doi: 10.1007/s40139-014-0056-9
14. Nakamura M, Tokura Y. Expression of SNAI1 and TWIST1 in the eccrine glands of patients with systemic sclerosis: possible involvement of epithelial-mesenchymal transition in the pathogenesis. Br J Dermatol. (2011) 164:204–5. doi: 10.1111/j.1365-2133.2010.10021.x
15. Nikitorowicz-Buniak J, Denton CP, Abraham D, Stratton R. Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased tgfbeta signaling within lesional scleroderma skin. PLoS ONE (2015) 10:e0134092. doi: 10.1371/journal.pone.0134092
16. Piera-Velazquez S, Mendoza FA, Jimenez SA. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of human fibrotic diseases. J Clin Med. (2016) 5:45. doi: 10.3390/jcm5040045
17. Jimenez SA. Role of endothelial to mesenchymal transition in the pathogenesis of the vascular alterations in systemic sclerosis. ISRN Rheumatol. (2013) 2013:835948. doi: 10.1155/2013/835948
18. Jimenez SA, Piera-Velazquez S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. (2016) 51:26–36. doi: 10.1016/j.matbio.2016.01.012
19. Mendoza FA, Piera-Velazquez S, Farber JL, Feghali-Bostwick C, Jimenez SA. Endothelial cells expressing endothelial and mesenchymal cell gene products in lung tissue from patients with systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. (2016) 68:210–7. doi: 10.1002/art.39421
20. Wermuth PJ, Carney KR, Mendoza FA, Piera-Velazquez S, Jimenez SA. Endothelial cell-specific activation of transforming growth factor-beta signaling in mice induces cutaneous, visceral, and microvascular fibrosis. Lab Invest. (2017) 97:806–18. doi: 10.1038/labinvest.2017.23
21. Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman K-R, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. (2016) 7:11853. doi: 10.1038/ncomms11853
22. Ma Z, Zhu L, Liu Y, Wang Z, Yang Y, Chen L, et al. Lovastatin alleviates endothelial-to-mesenchymal transition in glomeruli via suppression of oxidative stress and TGF-beta1 signaling. Front Pharmacol. (2017) 8:473. doi: 10.3389/fphar.2017.00473
23. Katikireddy KR, White TL, Miyajima T, Vasanth S, Raoof D, Chen Y, et al. NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy. Free Radic Biol Med. (2018) 116:19–30. doi: 10.1016/j.freeradbiomed.2017.12.036
24. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. (2005) 97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D
25. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. (2013) 288:770–7. doi: 10.1074/jbc.M112.431973
26. Rangarajan S, Bernard K, Thannickal VJ. Mitochondrial dysfunction in pulmonary fibrosis. Ann Am Thorac Soc. (2017) 14:S383–8. doi: 10.1513/AnnalsATS.201705-370AW
27. Gonzalez-Gonzalez FJ, Chandel NS, Jain M, Budinger GS. Reactive oxygen species as signaling molecules in the development of lung fibrosis. Transl. Res. (2017) 190:61–8. doi: 10.1016/j.trsl.2017.09.005
28. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
29. Zhang X, Hu M, Lyu X, Li C, Thannickal VJ, Sanders YY. DNA methylation regulated gene expression in organ fibrosis. Biochim Biophys Acta (2017) 1863:2389–97. doi: 10.1016/j.bbadis.2017.05.010
30. Kim KK, Sheppard D, Chapman HA. TGF-beta1 signaling and tissue fibrosis. Cold Spring Harb Perspect Biol. (2018) 10:a022293. doi: 10.1101/cshperspect.a022293
31. Murrell DF. A radical proposal for the pathogenesis of scleroderma. J Am Acad Dermatol. (1993) 28:78–85. doi: 10.1016/0190-9622(93)70014-K
32. Montorfano I, Becerra A, Cerro R, Echeverría C, Sáez E, Morales MG, et al. Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab Investig. (2014). 94:1068–82. doi: 10.1038/labinvest.2014.100
33. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. (2012) 5:9–19. doi: 10.1097/WOX.0b013e3182439613
34. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheumat. (2013) 65:1953–62. doi: 10.1002/art.37988
35. Grygiel-Gorniak B, Puszczewicz M. Oxidative damage and antioxidative therapy in systemic sclerosis. Mediators Inflamm. (2014) 389582:8. doi: 10.1155/2014/389582
36. Shroff A, Mamalis A, Jagdeo J. Oxidative stress and skin fibrosis. Curr Pathobiol Rep. (2014) 2:257–67. doi: 10.1007/s40139-014-0062-y
37. Dumoitier N, Lofek S, Mouthon L. Pathophysiology of systemic sclerosis: state of the art in 2014. La Presse Médicale (2014) 43:e267–78. doi: 10.1016/j.lpm.2014.08.001
38. Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. (2010) 48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026
39. Piera-Velazquez S, Jimenez SA. Role of oxidative stress and reactive oxygen radicals in the pathogenesis of systemic sclerosis. In: Studies on Arthritis and Joint Disorders. New York, NY: Humana Press; Springer (2013). p. 183–97. doi: 10.1007/978-1-4614-6166-1_10
40. Akamata K, Wei J, Bhattacharyya M, Cheresh P, Bonner MY, Arbiser JL, et al. SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis. Oncotarget (2016) 7:69321–36. doi: 10.18632/oncotarget.12504
41. Murphy-Marshman H, Quensel K, Shi-Wen X, Barnfield R, Kelly J, Peidl A, et al. Antioxidants and NOX1/NOX4 inhibition blocks TGFbeta1-induced CCN2 and alpha-SMA expression in dermal and gingival fibroblasts. PLoS ONE (2017) 12:e0186740. doi: 10.1371/journal.pone.0186740
42. Svegliati S, Spadoni T, Moroncini G, Gabrielli A. NADPH oxidase, oxidative stress and fibrosis in systemic sclerosis. Free Radic Biol Med. (2018). doi: 10.1016/j.freeradbiomed.2018.04.554. [Epub ahead of print].
43. Ginnan R, Jourd'heuil FL, Guikema B, Simons M, Singer HA, Jourd'heuil D. NADPH oxidase 4 is required for interleukin-1β-mediated activation of protein kinase Cδ and downstream activation of c-jun N-terminal kinase signaling in smooth muscle. Free Radic Biol Med. (2013) 54:125–34. doi: 10.1016/j.freeradbiomed.2012.09.026
44. Aguado A, Fischer T, Rodriguez C, Manea A, Martinez-Gonzalez J, Touyz RM, et al. Hu antigen R is required for NOX-1 but not NOX-4 regulation by inflammatory stimuli in vascular smooth muscle cells. J Hypertens. (2016) 34:253–65. doi: 10.1097/HJH.0000000000000801
45. Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L661–73. doi: 10.1152/ajplung.00269.2005
46. Manea A, Tanase LI, Raicu M, Simionescu M. Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH oxidase in human aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. (2010) 30:105–12. doi: 10.1161/ATVBAHA.109.193896
47. Manea A, Tanase LI, Raicu M, Simionescu M. Transcriptional regulation of NADPH oxidase isoforms, Nox1 and Nox4, by nuclear factor-kappaB in human aortic smooth muscle cells. Biochem Biophys Res Commun. (2010) 396:901–7. doi: 10.1016/j.bbrc.2010.05.019
48. Moe KT, Aulia S, Jiang F, Chua YL, Koh TH, Wong MC, et al. Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-alpha in human aortic smooth muscle and embryonic kidney cells. J Cell Mol Med. (2006) 10:231–9. doi: 10.1111/j.1582-4934.2006.tb00304.x
49. St Hilaire C, Koupenova M, Carroll SH, Smith BD, Ravid K. TNF-alpha upregulates the A2B adenosine receptor gene: the role of NAD(P)H oxidase 4. Biochem Biophys Res Commun. (2008) 375:292–6. doi: 10.1016/j.bbrc.2008.07.059
50. Lee S, Lee S, Sharma K. The pathogenesis of fibrosis and renal disease in scleroderma: recent insights from glomerulosclerosis. Curr Rheumatol Rep. (2004) 6:141–8. doi: 10.1007/s11926-004-0059-3
51. Dooley A, Shi-Wen X, Aden N, Tranah T, Desai N, Denton CP, et al. Modulation of collagen type I, fibronectin and dermal fibroblast function and activity, in systemic sclerosis by the antioxidant epigallocatechin-3-gallate. Rheumatology (2010) 49:2024–36. doi: 10.1093/rheumatology/keq208
52. Bielecki M, Kowal K, Lapinska A, Chwiesko-Minarowska S, Chyczewski L, Kowal-Bielecka O. Peripheral blood mononuclear cells from patients with systemic sclerosis spontaneously secrete increased amounts of vascular endothelial growth factor (VEGF) already in the early stage of the disease. Adv Med Sci. (2011) 56:255–63. doi: 10.2478/v10039-011-0025-z
53. Stawski L, Han R, Bujor AM, Trojanowska M. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther. (2012) 14:R194. doi: 10.1186/ar4028
54. Spadoni T, Svegliati Baroni S, Amico D, Albani L, Moroncini G, Avvedimento EV, et al. A reactive oxygen species-mediated loop maintains increased expression of NADPH oxidases 2 and 4 in skin fibroblasts from patients with systemic sclerosis. Arthritis Rheumatol. (2015) 67:1611–22. doi: 10.1002/art.39084
55. Herrick AL, Worthington J. Genetic epidemiology: systemic sclerosis. Arthritis Res. (2002) 4:165–8. doi: 10.1186/ar402
56. Fardoun MM, Nassif J, Issa K, Baydoun E, Eid AH. Raynaud's phenomenon: a brief review of the underlying mechanisms. Front Pharmacol. (2016) 7:438. doi: 10.3389/fphar.2016.00438
57. Eid AH, Maiti K, Mitra S, Chotani MA, Flavahan S, Bailey SR, et al. Estrogen increases smooth muscle expression of alpha2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am J Physiol Heart Circ Physiol. (2007) 293:H1955–61. doi: 10.1152/ajpheart.00306.2007
58. Eid AH, Chotani MA, Mitra S, Miller TJ, Flavahan NA. Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle alpha2C-adrenoceptors. Am J Physiol Heart Circ Physiol. (2008) 295:H266–72. doi: 10.1152/ajpheart.00084.2008
59. Stein CM, Tanner SB, Awad JA, Roberts LJ II, Morrow JD. Evidence of free radical-mediated injury (isoprostane overproduction) in scleroderma. Arthritis Rheum. (1996) 39:1146–50. doi: 10.1002/art.1780390711
60. Montuschi P, Toni GC, Paredi P, Pantelidis P, Du Bois RM, Kharitonov SA, et al. 8-Isoprostane as a biomarker of oxidative stress in interstitial lung diseases. Am J Respir Crit Care Med. (1998) 158:1524–7. doi: 10.1164/ajrccm.158.5.9803102
61. Ogawa F, Shimizu K, Muroi E, Hara T, Hasegawa M, Takehara K, et al. Serum levels of 8-isoprostane, a marker of oxidative stress, are elevated in patients with systemic sclerosis. Rheumatology (2006) 45:815–8. doi: 10.1093/rheumatology/kel012
62. Cerinic MM, Kahaleh M. Beauty and the beast. The nitric oxide paradox in systemic sclerosis. Rheumatology (2002) 41:843–7. doi: 10.1093/rheumatology/41.8.843
63. Herrick AL, Matucci Cerinic M. The emerging problem of oxidative stress and the role of antioxidants in systemic sclerosis. Clin Exp Rheumatol. (2001) 19:4–8.
64. Servettaz A, Guilpain P, Goulvestre C, Chereau C, Hercend C, Nicco C, et al. Radical oxygen species production induced by advanced oxidation protein products predicts clinical evolution and response to treatment in systemic sclerosis. Ann Rheum Dis. (2007) 66:1202–9. doi: 10.1136/ard.2006.067504
65. Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, et al. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. (2001) 44:2653–64. doi: 10.1002/1529-0131(200111)44:11<2653::AID-ART445>3.0.CO;2-1
66. Svegliati S, Cancello R, Sambo P, Luchetti M, Paroncini P, Orlandini G, et al. Platelet-derived growth factor and reactive oxygen species (ROS) regulate Ras protein levels in primary human fibroblasts via ERK1/2. Amplification of ROS and Ras in systemic sclerosis fibroblasts. J Biol Chem. (2005) 280:36474–82. doi: 10.1074/jbc.M502851200
67. Boin F, Erre GL, Posadino AM, Cossu A, Giordo R, Spinetti G, et al. Oxidative stress-dependent activation of collagen synthesis is induced in human pulmonary smooth muscle cells by sera from patients with scleroderma-associated pulmonary hypertension. Orphanet J Rare Dis. (2014) 9:123. doi: 10.1186/s13023-014-0123-7
68. Dosoki H, Stegemann A, Taha M, Schnittler H, Luger TA, Schroder K, et al. Targeting of NADPH oxidase in vitro and in vivo suppresses fibroblast activation and experimental skin fibrosis. Exp Dermatol. (2017) 26:73–81. doi: 10.1111/exd.13180
69. Svegliati S, Amico D, Spadoni T, Fischetti C, Finke D, Moroncini G, et al. Agonistic anti-PDGF receptor autoantibodies from patients with systemic sclerosis impact human pulmonary artery smooth muscle cells function in vitro. Front Immunol. (2017) 8:75. doi: 10.3389/fimmu.2017.00075
70. Kavian N, Servettaz A, Mongaret C, Wang A, Nicco C, Chéreau C, et al. Targeting ADAM-17/notch signaling abrogates the development of systemic sclerosis in a murine model. Arthritis Rheumat. (2010) 62:3477–87. doi: 10.1002/art.27626
71. Bagnato G, Bitto A, Irrera N, Pizzino G, Sangari D, Cinquegrani M, et al. Propylthiouracil prevents cutaneous and pulmonary fibrosis in the reactive oxygen species murine model of systemic sclerosis. Arthritis Res Ther. (2013) 15:R120. doi: 10.1186/ar4300
72. Bagnato G, Bitto A, Pizzino G, Irrera N, Sangari D, Cinquegrani M, et al. Simvastatin attenuates the development of pulmonary and cutaneous fibrosis in a murine model of systemic sclerosis. (2013). Rheumatology 52:1377–86. doi: 10.1093/rheumatology/ket144
73. Baroni SS, Santillo M, Bevilacqua F, Luchetti M, Spadoni T, Mancini M, et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med. (2006) 354:2667–76. doi: 10.1056/NEJMoa052955
74. Servettaz A, Goulvestre C, Kavian N, Nicco C, Guilpain P, Chereau C, et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J Immunol (2009) 182:5855–64. doi: 10.4049/jimmunol.0803705
75. Ihn H, Sato S, Fujimoto M, Igarashi A, Yazawa N, Kubo M, et al. Characterization of autoantibodies to endothelial cells in systemic sclerosis (SSc): association with pulmonary fibrosis. Clin Exp Immunol. (2000) 119:203–9. doi: 10.1046/j.1365-2249.2000.01115.x
76. Gabrielli A, Svegliati S, Moroncini G, Pomponio G, Santillo M, Avvedimento EV. Oxidative stress and the pathogenesis of scleroderma: the Murrell's hypothesis revisited. Semin Immunopathol. (2008) 30:329–37. doi: 10.1007/s00281-008-0125-4
77. Pattanaik D, Brown M, Postlethwaite BC, Postlethwaite AE. Pathogenesis of systemic sclerosis. Front Immunol. (2015) 6:272. doi: 10.3389/fimmu.2015.00272
78. Mun JH, Kim YM, Kim BS, Kim JH, Kim MB, Ko HC. Simvastatin inhibits transforming growth factor-β1-induced expression of type I collagen, CTGF, and α-SMA in keloid fibroblasts. Wound Repair Regener. (2014) 22:125–33. doi: 10.1111/wrr.12136
79. Siani A, Tirelli N. Myofibroblast differentiation: main features, biomedical relevance, and the role of reactive oxygen species. Antioxidants Redox Signal. (2014) 21:768–85. doi: 10.1089/ars.2013.5724
80. Bhattacharyya S, Wei J, Varga J. Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol. (2012) 8:42–54. doi: 10.1038/nrrheum.2011.149
81. Yang X, Chen B, Liu T, Chen X. Reversal of myofibroblast differentiation: a review. Eur J Pharmacol. (2014) 734:83–90. doi: 10.1016/j.ejphar.2014.04.007
82. Hua-Huy T, Dinh-Xuan AT. Cellular and molecular mechanisms in the pathophysiology of systemic sclerosis. Pathol Biol. (2015) 63:61–8. doi: 10.1016/j.patbio.2015.03.003
83. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. (2007) 170:1807–16. doi: 10.2353/ajpath.2007.070112
84. Mcanulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. (2007) 39:666–71. doi: 10.1016/j.biocel.2006.11.005
85. Kis K, Liu X, Hagood JS. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev Mol Med. (2011) 13:e27. doi: 10.1017/S1462399411001967
86. Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast—implications for kidney fibrosis. Nat Rev Nephrol. (2015) 11:233. doi: 10.1038/nrneph.2014.246
87. Thannickal VJ, Horowitz JC. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc Am Thorac Soc. (2006) 3:350–6. doi: 10.1513/pats.200601-001TK
88. Johnson A, Dipietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J. (2013) 27:3893–901. doi: 10.1096/fj.12-214189
89. Hecker L, Logsdon NJ, Kurundkar D, Kurundkar A, Bernard K, Hock T, et al. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci Transl Med. (2014) 6:231ra247. doi: 10.1126/scitranslmed.3008182
90. Kim SJ, Cheresh P, Eren M, Jablonski RP, Yeldandi A, Ridge KM, et al. Klotho, an antiaging molecule, attenuates oxidant-induced alveolar epithelial cell mtDNA damage and apoptosis. Am J Physiol Lung Cell Mol Physiol. (2017) 313:L16–26. doi: 10.1152/ajplung.00063.2017
91. Xie J, Chen Y, Hu C, Pan Q, Wang B, Li X, et al. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget (2017) 8:57981–90. doi: 10.18632/oncotarget.19853
92. Donati G, Rognoni E, Hiratsuka T, Liakath-Ali K, Hoste E, Kar G, et al. Wounding induces dedifferentiation of epidermal Gata6(+) cells and acquisition of stem cell properties. Nat Cell Biol. (2017) 19:603–13. doi: 10.1038/ncb3532
93. Lynch TJ, Anderson PJ, Rotti PG, Tyler SR, Crooke AK, Choi SH, et al. Submucosal gland myoepithelial cells are reserve stem cells that can regenerate mouse tracheal epithelium. Cell Stem Cell (2018) 22:653–667.e5. doi: 10.1016/j.stem.2018.04.007
94. Tata A, Kobayashi Y, Chow RD, Tran J, Desai A, Massri AJ, et al. Myoepithelial cells of submucosal glands can function as reserve stem cells to regenerate airways after injury. Cell Stem Cell (2018) 22:668–683.e6. doi: 10.1016/j.stem.2018.03.018
95. Zacharias WJ, Frank DB, Zepp JA, Morley MP, Alkhaleel FA, Kong J, et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature (2018) 555:251–5. doi: 10.1038/nature25786
96. Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Pechoux C, Bogaard HJ, et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation (2015) 131:1006–18. doi: 10.1161/CIRCULATIONAHA.114.008750
97. Suzuki T, Tada Y, Gladson S, Nishimura R, Shimomura I, Karasawa S, et al. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir Res. (2017) 18:177. doi: 10.1186/s12931-017-0660-4
98. Fang S, Guo H, Cheng Y, Zhou Z, Zhang W, Han B, et al. circHECTD1 promotes the silica-induced pulmonary endothelial-mesenchymal transition via HECTD1. Cell Death Dis. (2018) 9:396. doi: 10.1038/s41419-018-0432-1
99. Lv Z, Wang Y, Liu YJ, Mao YF, Dong WW, Ding ZN, et al. NLRP3 inflammasome activation contributes to mechanical stretch-induced endothelial-mesenchymal transition and pulmonary fibrosis. Crit Care Med. (2018) 46:e49–58. doi: 10.1097/CCM.0000000000002799
100. Mammoto T, Muyleart M, Konduri GG, Mammoto A. Twist1 in hypoxia-induced pulmonary hypertension through transforming growth factor-β-Smad signaling. Am J Respir Cell Mol Biol. (2018) 58:194–207. doi: 10.1165/rcmb.2016-0323OC
101. Suzuki T, Carrier EJ, Talati MH, Rathinasabapathy A, Chen X, Nishimura R, et al. Isolation and characterization of endothelial-to-mesenchymal transition cells in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L118–26. doi: 10.1152/ajplung.00296.2017
102. Tang H, Babicheva A, Mcdermott KM, Gu Y, Ayon RJ, Song S, et al. Endothelial HIF-2alpha contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. (2018) 314:L256–75. doi: 10.1152/ajplung.00096.2017
103. Tomos IP, Tzouvelekis A, Aidinis V, Manali ED, Bouros E, Bouros D, et al. Extracellular matrix remodeling in idiopathic pulmonary fibrosis. It is the 'bed' that counts and not 'the sleepers.' Expert Rev Respir Med. (2017) 11:299–309. doi: 10.1080/17476348.2017.1300533
104. Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. (2011) 179:1074–80. doi: 10.1016/j.ajpath.2011.06.001
105. Lee K, Nelson CM. New insights into the regulation of epithelial-mesenchymal transition and tissue fibrosis. Int Rev Cell Mol Biol. (2012) 294:171–221. doi: 10.1016/B978-0-12-394305-7.00004-5
106. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, Mcmullen JR, Gustafsson E, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. (2007) 13:952–61. doi: 10.1038/nm1613
107. Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, et al. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. (2010) 43:161–72. doi: 10.1165/rcmb.2009-0031OC
108. Sato Y, Nakanuma Y. Role of endothelial-mesenchymal transition in idiopathic portal hypertension. Histol Histopathol. (2013) 28:145–54. doi: 10.14670/HH-28.145
109. Toshio S, Rintaro N, Takashi U, Ayumi S, Takeshi K, Koichiro T. Oxydative stress-induced endothelial-to-mesenchymal transition in septic pulmonary vascular endothelial cells. In: D42. Cytoskeleton and Inflammation in the Pulmonary Circulation. Colorado, AZ: American Thoracic Society (2015). A5940.
110. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, et al. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary artery hypertension. Am J Pathol. (2015). 185:1850–8. doi: 10.1016/j.ajpath.2015.03.019
111. Welch-Reardon KM, Wu N, Hughes CC. A role for partial endothelial-mesenchymal transitions in angiogenesis? Arterioscler Thromb Vasc Biol. (2015) 35:303–8. doi: 10.1161/ATVBAHA.114.303220
112. Van Meeteren LA, Ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tiss Res. (2012) 347:177–86. doi: 10.1007/s00441-011-1222-6
113. Lin F, Wang N, Zhang T-C. The role of endothelial–mesenchymal transition in development and pathological process. IUBMB Life (2012) 64:717–23. doi: 10.1002/iub.1059
114. Piera-Velazquez S, Jimenez SA. Molecular mechanisms of endothelial to mesenchymal cell transition (EndoMT) in experimentally induced fibrotic diseases. Fibrogenesis Tissue Repair (2012) 6.
115. Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. (2017) 76:924–34. doi: 10.1136/annrheumdis-2016-210229
116. Goumans M-J, Liu Z, Ten Dijke P. TGF-β signaling in vascular biology and dysfunction. Cell Res. (2009) 19:116–27. doi: 10.1038/cr.2008.326
117. Medici D, Potenta S, Kalluri R. Transforming growth factor-beta2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J. (2011) 437:515–20. doi: 10.1042/BJ20101500
118. Chrobak I, Lenna S, Stawski L, Trojanowska M. Interferon-gamma promotes vascular remodeling in human microvascular endothelial cells by upregulating endothelin (ET)-1 and transforming growth factor (TGF) beta2. J Cell Physiol. (2013) 228:1774–83. doi: 10.1002/jcp.24337
119. Cipriani P, Di Benedetto P, Ruscitti P, Capece D, Zazzeroni F, Liakouli V, et al. The endothelial-mesenchymal transition in systemic sclerosis is induced by endothelin-1 and transforming growth factor-β and may be blocked by macitentan, a dual endothelin-1 receptor antagonist. J Rheumatol. (2015) 42:1808–16. doi: 10.3899/jrheum.150088
120. Andersen GN, Caidahl K, Kazzam E, Petersson A-S, Waldenström A, Mincheva-Nilsson L, et al. Correlation between increased nitric oxide production and markers of endothelial activation in systemic sclerosis: findings with the soluble adhesion molecules E-selectin, intercellular adhesion molecule 1, and vascular adhesion molecule 1. Arthritis Rheumat. (2000) 43:1085–93. doi: 10.1002/1529-0131(200005)43:5<1085::AID-ANR19>3.0.CO;2-7
121. Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S. Soluble adhesion molecules (sVCAM-1, sE-selectin), vascular endothelial growth factor (VEGF) and endothelin-1 in patients with systemic sclerosis: relationship to organ systemic involvement. Clin Rheumatol. (2005) 24:111–6. doi: 10.1007/s10067-004-0987-3
122. Trojanowska M. Cellular and molecular aspects of vascular dysfunction in systemic sclerosis. Nat Rev Rheumatol. (2010) 6:453–60. doi: 10.1038/nrrheum.2010.102
123. Giordano N, Papakostas P, Pecetti G, Nuti R. Cytokine modulation by endothelin-1 and possible therapeutic implications in systemic sclerosis. J Biol Regul Homeost Agents (2011) 25:487–92.
124. Corallo C, Cutolo M, Kahaleh B, Pecetti G, Montella A, Chirico C, et al. Bosentan and macitentan prevent the endothelial-to-mesenchymal transition (EndoMT) in systemic sclerosis: in vitro study. Arthritis Res Ther. (2016) 18:228. doi: 10.1186/s13075-016-1122-y
125. Li Z, Jimenez SA. Protein kinase Cdelta and c-Abl kinase are required for transforming growth factor beta induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. (2011) 63:2473–83. doi: 10.1002/art.30317
126. Camenisch TD, Molin DG, Person A, Runyan RB, Gittenberger-De Groot AC, Mcdonald JA, et al. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis. Dev Biol. (2002) 248:170–81. doi: 10.1006/dbio.2002.0731
127. Tavares ALP, Mercado-Pimentel ME, Runyan RB, Kitten GT. TGFβ-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dynamics (2006) 235:1589–98. doi: 10.1002/dvdy.20771
128. Mercado-Pimentel ME, Runyan RB. Multiple transforming growth factor-β isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs (2007) 185:146–56. doi: 10.1159/000101315
129. Wang B, Koh P, Winbanks C, Coughlan MT, Mcclelland A, Watson A, et al. miR-200a prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes (2010) 60:280–7. doi: 10.2337/db10-0892
130. Zhao M, Fajardo G, Urashima T, Spin JM, Poorfarahani S, Rajagopalan V, et al. Cardiac pressure overload hypertrophy is differentially regulated by β-adrenergic receptor subtypes. Am J Physiol Heart Circul Physiol. (2011) 301:H1461–70. doi: 10.1152/ajpheart.00453.2010
131. Maleszewska M, Moonen J-RA, Huijkman N, Van De Sluis B, Krenning G, Harmsen MC. IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology (2013) 218:443–54. doi: 10.1016/j.imbio.2012.05.026
132. Garrison G, Huang SK, Okunishi K, Scott JP, Kumar Penke LR, Scruggs AM, et al. Reversal of myofibroblast differentiation by prostaglandin e2. Am J Respir Cell Mol Biol. (2013) 48:550–8. doi: 10.1165/rcmb.2012-0262OC
133. Mihira H, Suzuki HI, Akatsu Y, Yoshimatsu Y, Igarashi T, Miyazono K, et al. TGF-beta-induced mesenchymal transition of MS-1 endothelial cells requires Smad-dependent cooperative activation of Rho signals and MRTF-A. J Biochem. (2012) 151:145–56. doi: 10.1093/jb/mvr121
134. Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. (2012) 64:2734–45. doi: 10.1002/art.34424
135. Noseda M, Mclean G, Niessen K, Chang L, Pollet I, Montpetit R, et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circulation Res. (2004) 94:910–7. doi: 10.1161/01.RES.0000124300.76171.C9
136. Del Galdo F, Lisanti MP, Jimenez SA. Caveolin-1, transforming growth factor-beta receptor internalization, and the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. (2008) 20:713–9. doi: 10.1097/BOR.0b013e3283103d27
137. Del Galdo F, Sotgia F, De Almeida CJ, Jasmin JF, Musick M, Lisanti MP, et al. Decreased expression of caveolin 1 in patients with systemic sclerosis: crucial role in the pathogenesis of tissue fibrosis. Arthritis Rheum. (2008) 58:2854–65. doi: 10.1002/art.23791
138. Wermuth PJ, Li Z, Mendoza FA, Jimenez SA. Stimulation of transforming growth factor-β1-induced endothelial-to-mesenchymal transition and tissue fibrosis by endothelin-1 (ET-1): a novel profibrotic effect of ET-1. PLoS ONE (2016) 11:e0161988. doi: 10.1371/journal.pone.0161988
139. Nelson P, Kiriakidou M, Sharma A, Maniataki E, Mourelatos Z. The microRNA world: small is mighty. Trends Biochem Sci. (2003) 28:534–40. doi: 10.1016/j.tibs.2003.08.005
140. Miao CG, Xiong YY, Yu H, Zhang XL, Qin MS, Song TW, et al. Critical roles of microRNAs in the pathogenesis of systemic sclerosis: new advances, challenges and potential directions. Int Immunopharmacol. (2015) 28:626–33. doi: 10.1016/j.intimp.2015.07.042
141. Kim J. MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. BMB Rep. (2018) 51:65–72. doi: 10.5483/BMBRep.2018.51.2.011
142. Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE. Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal. (2012) 24:1031–6. doi: 10.1016/j.cellsig.2011.12.024
143. Zhang J, Zhang Z, Zhang DY, Zhu J, Zhang T, Wang C. microRNA 126 inhibits the transition of endothelial progenitor cells to mesenchymal cells via the PIK3R2-PI3K/Akt signalling pathway. PLoS ONE (2013) 8:e83294. doi: 10.1371/journal.pone.0083294
144. Nagpal V, Rai R, Place AT, Murphy SB, Verma SK, Ghosh AK, et al. MiR-125b is critical for fibroblast-to-myofibroblast transition and cardiac fibrosis. Circulation (2016) 133:291–301. doi: 10.1161/CIRCULATIONAHA.116.022627
145. Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park D-H, Thum T. Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arteriosclerosis Thrombosis Vasc Biol. (2012) 32:361–9. doi: 10.1161/ATVBAHA.111.234286
146. Guo Y, Li P, Bledsoe G, Yang Z-R, Chao L, Chao J. Kallistatin inhibits TGF-β-induced endothelial–mesenchymal transition by differential regulation of microRNA-21 and eNOS expression. Exp Cell Res. (2015) 337:103–10. doi: 10.1016/j.yexcr.2015.06.021
147. Kumar S, Kim CW, Simmons RD, Jo H. Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler Thromb Vasc Biol. (2014) 34:2206–16. doi: 10.1161/ATVBAHA.114.303425
148. Miscianinov V, Martello A, Rose L, Parish E, Cathcart B, Mitic T, et al. MicroRNA-148b targets the TGF-beta pathway to regulate angiogenesis and endothelial-to-mesenchymal transition during skin wound healing. Mol Ther. (2018). 26:1996–2007. doi: 10.1016/j.ymthe.2018.05.002
149. Katsura A, Suzuki HI, Ueno T, Mihira H, Yamazaki T, Yasuda T, et al. Micro RNA-31 is a positive modulator of endothelial–mesenchymal transition and associated secretory phenotype induced by TGF-β. Genes Cells (2016) 21:99–116. doi: 10.1111/gtc.12323
150. Li L, Kim I-K, Chiasson V, Chatterjee P, Gupta S. NF-κB mediated miR-130a modulation in lung microvascular cell remodeling: implication in pulmonary hypertension. Exp Cell Res. (2017) 359:235–42. doi: 10.1016/j.yexcr.2017.07.024
151. Chen P-Y, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. (2012) 2:1684–96. doi: 10.1016/j.celrep.2012.10.021
152. Nagai T, Kanasaki M, Srivastava SP, Nakamura Y, Ishigaki Y, Kitada M, et al. N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. BioMed Res Int. (2014) 2014:696475. doi: 10.1155/2014/696475
153. Kanasaki K, Shi S, Kanasaki M, He J, Nagai T, Nakamura Y, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes (2014). 63:2120–31. doi: 10.2337/db13-1029
154. Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh S-T, et al. Role of reactive oxygen species in TGF-β1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. (2005) 16:667–75. doi: 10.1681/ASN.2004050425
155. Fukawa T, Kajiya H, Ozeki S, Ikebe T, Okabe K. Reactive oxygen species stimulates epithelial mesenchymal transition in normal human epidermal keratinocytes via TGF-beta secretion. Exp Cell Res. (2012) 318:1926–32. doi: 10.1016/j.yexcr.2012.05.023
156. Ding S-Z, Yang Y-X, Li X-L, Michelli-Rivera A, Han S-Y, Wang L, et al. Epithelial–mesenchymal transition during oncogenic transformation induced by hexavalent chromium involves reactive oxygen species-dependent mechanism in lung epithelial cells. Toxicol Appl. Pharmacol. (2013) 269:61–71. doi: 10.1016/j.taap.2013.03.006
157. Arciniegas E, Frid MG, Douglas IS, Stenmark KR. Perspectives on endothelial-to-mesenchymal transition: potential contribution to vascular remodeling in chronic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2007) 293:L1–8. doi: 10.1152/ajplung.00378.2006
158. Cho JG, Lee A, Chang W, Lee MS, Kim J. Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front Immunol. (2018) 9:294. doi: 10.3389/fimmu.2018.00294
159. Jiang F, Liu G-S, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. (2014) 2:267–72. doi: 10.1016/j.redox.2014.01.012
160. Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, et al. Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiation Res. (2006) 166:839–48. doi: 10.1667/RR0695.1
161. Chaudhuri V, Zhou L, Karasek M. Inflammatory cytokines induce the transformation of human dermal microvascular endothelial cells into myofibroblasts: a potential role in skin fibrogenesis. J Cutaneous Pathol. (2007) 34:146–53. doi: 10.1111/j.1600-0560.2006.00584.x
162. Rieder F, Kessler SP, West GA, Bhilocha S, De La Motte C, Sadler TM, et al. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. (2011) 179:2660–73. doi: 10.1016/j.ajpath.2011.07.042
163. Hecker L, Vittal R, Jones T, Jagirdar R, Luckhardt TR, Horowitz JC, et al. NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat Med. (2009) 15:1077–81. doi: 10.1038/nm.2005
164. Jarman ER, Khambata VS, Cope C, Jones P, Roger J, Ye LY, et al. An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am J Respir Cell Mol Biol. (2013) 50:158–69. doi: 10.1165/rcmb.2013-0174OC
165. Echeverría C, Montorfano I, Sarmiento D, Becerra A, Nunez-Villena F, Figueroa XF, et al. Lipopolysaccharide induces a fibrotic-like phenotype in endothelial cells. J Cell Mol Med. (2013) 17:800–14. doi: 10.1111/jcmm.12066
166. Echeverría C, Montorfano I, Tapia P, Riedel C, Cabello-Verrugio C, Simon F. Endotoxin-induced endothelial fibrosis is dependent on expression of transforming growth factors beta1 and beta2. Infect Immun. (2014) 82:3678–86. doi: 10.1128/IAI.02158-14
167. Xu H, Zaidi M, Struve J, Jones DW, Krolikowski JG, Nandedkar S, et al. Abnormal fibrillin-1 expression and chronic oxidative stress mediate endothelial mesenchymal transition in a murine model of systemic sclerosis. Am J Physiol Cell Physiol. (2011) 300:15. doi: 10.1152/ajpcell.00123.2010
168. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Investig. (2007) 117:557–67. doi: 10.1172/JCI31139
169. Cosentino F, Luscher TF. Tetrahydrobiopterin and endothelial function. Eur Heart J. (1998) 19(Suppl G), G3–8.
170. Machin DR, Clifton HL, Richardson RS, Wray DW, Donato AJ, Frech TM. Acute oral tetrahydrobiopterin administration ameliorates endothelial dysfunction in systemic sclerosis. Clin Exp Rheumatol. (2017) 35(Suppl 106):167–72.
171. Wever RM, Van Dam T, Van Rijn HJ, De Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. (1997) 237:340–4. doi: 10.1006/bbrc.1997.7069
172. Almudever P, Milara J, De Diego A, Serrano-Mollar A, Xaubet A, Perez-Vizcaino F, et al. Role of tetrahydrobiopterin in pulmonary vascular remodelling associated with pulmonary fibrosis. Thorax (2013) 68:938–48. doi: 10.1136/thoraxjnl-2013-203408
173. Kokot A, Sindrilaru A, Schiller M, Sunderkotter C, Kerkhoff C, Eckes B, et al. Alpha-melanocyte-stimulating hormone suppresses bleomycin-induced collagen synthesis and reduces tissue fibrosis in a mouse model of scleroderma: melanocortin peptides as a novel treatment strategy for scleroderma? Arthritis Rheum. (2009) 60:592–603. doi: 10.1002/art.24228
174. Hagiwara SI, Ishii Y, Kitamura S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med. (2000) 162:225–31. doi: 10.1164/ajrccm.162.1.9903129
175. Serrano-Mollar A, Closa D, Prats N, Blesa S, Martinez-Losa M, Cortijo J, et al. In vivo antioxidant treatment protects against bleomycin-induced lung damage in rats. Br J Pharmacol. (2003) 138:1037–48. doi: 10.1038/sj.bjp.0705138
176. Inghilleri S, Morbini P, Oggionni T, Barni S, Fenoglio C. In situ assessment of oxidant and nitrogenic stress in bleomycin pulmonary fibrosis. Histochem Cell Biol. (2006) 125:661–9. doi: 10.1007/s00418-005-0116-7
177. Kang DH. Hyperuricemia and progression of chronic kidney disease: role of phenotype transition of renal tubular and endothelial cells. Contrib Nephrol. (2018) 192:48–55. doi: 10.1159/000484278
178. Zhao XP, Chang SY, Liao MC, Lo CS, Chenier I, Luo H, et al. Hedgehog interacting protein promotes fibrosis and apoptosis in glomerular endothelial cells in murine diabetes. Sci Rep. (2018) 8:5958. doi: 10.1038/s41598-018-24220-6
179. Taniguchi T, Asano Y, Akamata K, Noda S, Takahashi T, Ichimura Y, et al. Fibrosis, vascular activation, and immune abnormalities resembling systemic sclerosis in bleomycin-treated Fli-1-haploinsufficient mice. Arthritis Rheumatol. (2015) 67:517–26. doi: 10.1002/art.38948
180. Toyama T, Asano Y, Akamata K, Noda S, Taniguchi T, Takahashi T, et al. Tamibarotene ameliorates bleomycin-induced dermal fibrosis by modulating phenotypes of fibroblasts, endothelial cells, and immune cells. J Invest Dermatol. (2016) 136:387–98. doi: 10.1016/j.jid.2015.10.058
181. Qi Q, Mao Y, Tian Y, Zhu K, Cha X, Wu M, et al. Geniposide inhibited endothelial-mesenchymal transition via the mTOR signaling pathway in a bleomycin-induced scleroderma mouse model. Am J Transl Res. (2017) 9:1025–36.
182. Batteux F, Kavian N, Servettaz A. New insights on chemically induced animal models of systemic sclerosis. Curr Opin Rheumatol. (2011) 23:511–8. doi: 10.1097/BOR.0b013e32834b1606
183. Liu J, Yin F, Zheng X, Jing J, Hu Y. Geniposide, a novel agonist for GLP-1 receptor, prevents PC12 cells from oxidative damage via MAP kinase pathway. Neurochem Int. (2007) 51:361–9. doi: 10.1016/j.neuint.2007.04.021
Keywords: Endothelial-to-Mesenchymal Transition, oxidative stress, reactive oxygen species, scleroderma, systemic sclerosis
Citation: Thuan DTB, Zayed H, Eid AH, Abou-Saleh H, Nasrallah GK, Mangoni AA and Pintus G (2018) A Potential Link Between Oxidative Stress and Endothelial-to-Mesenchymal Transition in Systemic Sclerosis. Front. Immunol. 9:1985. doi: 10.3389/fimmu.2018.01985
Received: 13 April 2018; Accepted: 13 August 2018;
Published: 19 September 2018.
Edited by:
Philippe Guilpain, Université De Montpellier, FranceReviewed by:
Mauricio Rojas, University of Pittsburgh, United StatesOrnella Parolini, Università Cattolica del Sacro Cuore, Italy
Copyright © 2018 Thuan, Zayed, Eid, Abou-Saleh, Nasrallah, Mangoni and Pintus. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arduino A. Mangoni, YXJkdWluby5tYW5nb25pQGZsaW5kZXJzLmVkdS5hdQ==
Gianfranco Pintus, Z3BpbnR1c0BxdS5lZHUucWE=