 Arim Lee1
Arim Lee1 Sang Ho Choi
Sang Ho Choi Tae Sung Kim
Tae Sung Kim- 1Department of Life Sciences, College of Life Sciences and Biotechnology, Korea University, Seoul, South Korea
- 2Institute of Convergence Science, Korea University, Seoul, South Korea
- 3National Research Laboratory of Molecular Microbiology and Toxicology, Department of Agricultural Biotechnology, Seoul National University, Seoul, South Korea
T helper type 17 (Th17) cells are a subset of pro-inflammatory T helper cells that mediate host defense and pathological inflammation. We have previously reported that host dendritic cells (DCs) infected with Vibrio vulnificus induce Th17 responses through the production of several pro-inflammatory cytokines, including interleukin (IL)-1β and IL-6. V. vulnificus produces RTX toxin (RtxA), an important virulence factor that determines successful pathophysiology. In this study, we investigated the involvement of RtxA from V. vulnificus in Th17 cell induction through the activation and maturation of DCs. The increased expression of the DC surface marker CD40 caused by V. vulnificus wild-type infection was reduced by rtxA gene mutation in V. vulnificus. The mRNA and protein levels of Th17 polarization-related cytokines also decreased in V. vulnificus rtxA mutant-infected DCs. In addition, the co-culture of Th cells and DCs infected with rtxA mutant V. vulnificus resulted in reduction in DC-mediated Th17 responses. Th17 cell responses in the small intestinal lamina propria decreased in mice inoculated with V. vulnificus rtxA mutant as compared to those inoculated with the wild-type strain. These decreases in DC maturation, Th17-polarizing cytokine secretion, and Th17 responses attributed to rtxA mutation were restored following infection with the rtxA revertant strain. Furthermore, the mutation in the hlyU gene encoding the activator of rtxA1 gene reproduced the results observed with rtxA mutation. Taken together, V. vulnificus, by means of RtxA, induces inflammatory Th17 responses, which may be associated with adaptive responses of the host against V. vulnificus infection.
Introduction
Vibrio vulnificus is a halophilic estuarine bacterium that causes disease by infection of wounds or ingestion of contaminated seafood (1). V. vulnificus is the causative agent of several diseases, including necrotizing fasciitis, gastroenteritis, and primary septicemia.
The pathogenicity of V. vulnificus is associated with several virulence factors that include lipopolysaccharide (LPS) (2), capsular polysaccharide (CPS) (3), elastolytic protease (VvpE) (4), hemolysin (VvhA) (5), peroxiredoxin (6), and RTX toxin (RtxA) (7). Of these, the VvhA and RtxA cytotoxins may be the most important virulence factors.
Recent studies have elucidated the adaptive immune response against V. vulnificus infection (8, 9). Previously we demonstrated that V. vulnificus infection induces Th17 responses via maturation and activation of dendritic cells (DCs). In addition, V. vulnificus infection following oral ingestion results in the induction of Th17 cell response in the small intestinal lamina propria (8). Furthermore, V. vulnificus infection induces Th1 and T follicular helper (Tfh) cells and VvhA is involved in these responses. (9). However, the specific virulence factor of V. vulnificus necessary for the induction of Th17 cells is unclear.
RtxA is a member of the multifunctional-autoprocessing repeats in toxin, a subgroup of RTX toxin family with tandem nonapeptide repeats near the C-terminal region (10). RtxA is exported via the modified type I secretion system (11). Several studies have evaluated the cytotoxic and cytopathic effects of RtxA, and reported that RtxA was related to the in vivo growth of bacteria (12), host cell necrosis (12), apoptosis (13), inflammasome activation (14), actin aggregation (15), phagocytosis inhibition (16), and the production of reactive oxygen species (17). The role of RTX toxin in pathogenesis has been investigated in V. vulnificus and other bacterial species. However, its effect on host adaptive immune responses against V. vulnificus infection remains unclear. Therefore, we investigated whether RtxA influences Th17 cell responses following V. vulnificus infection.
We found that the rtxA mutant of V. vulnificus induced lower levels of DC maturation and activation than wild-type (WT) V. vulnificus. Furthermore, the ability of V. vulnificus to induce Th17 cell responses became diminished following mutation of the rtxA gene, consistent with the observation of reduced expression and secretion of Th17 cell-polarizing cytokines. Involvement of RtxA in Th17 cell induction was confirmed through the recovery of the decreased Th17-related responses following infection with an rtxA revertant. Furthermore, the mutation of the hlyU gene, an anti-repressor of rtxA gene, resulted in defective Th17 cell responses. Taken together, our results suggest that V. vulnificus induces Th17 cell responses in vitro and in vivo through RtxA.
Materials and Methods
Mice
All animals used in this study were purchased from Orient Bio Inc. (Seoul, Korea). Seven- to 11-week-old female C57BL/6 mice were used for experiments. All mice were capable of accessing a standard laboratory chow diet (cat. no. 1314; Altromin Spezialfutter GmbH & Co. KG, Lage, Nordrhein-Westfalen, Germany) and water. The animals were housed in an SPF facility under a strict light cycle (lights on at 07:00 a.m. and off at 07:00 p.m.) at 22 ± 1°C and 52.5 ± 2.5% relative humidity, and all animal experiments were ethically performed in accordance with the guidelines of the Korea University Institutional Animal Care and Use Committee (Seoul, Korea; approval no. KUIACUC-2016-170, 2017-113).
Bacterial Strains, Plasmids, and Culture Conditions
All V. vulnificus strains and plasmids used in this study are listed in Table 1. Unless otherwise noted, V. vulnificus strains were cultured in Luria-Bertani (LB) medium supplemented with 2.0% (wt/vol) sodium chloride (NaCl; LBS) at 30°C. For the infection experiments, the bacteria were cultured overnight, followed by their incubation in fresh LBS medium at 30°C. The culture was diluted to ~1.8 × 108 CFU/mL in LBS and centrifuged at 2,420 × g for 3 min at room temperature. The cells were resuspended in antibiotic-free growth medium before infection of DCs or in phosphate-buffered saline (PBS) before oral administration into mice.

Table 1. Bacterial strains and plasmids used in this study.
Construction of the rtxA Mutant and Revertant Strain
To inactivate rtxA gene in vitro, the region from 15- to 1,068-bp downstream of the translational initiation codon of rtxA was deleted using the polymerase chain reaction (PCR)-mediated linker-scanning mutation method, as previously described (7, 25). In brief, Briefly, pairs of primers RTXA01-F and -R (for amplification of the 5′ amplicon) or RTXA02-F and -R (for amplification of the 3′ amplicon) were designed and used (Table 2). The resulting rtxA mutant was amplified by PCR using the mixture of both amplicons as the template and RTXA01-F and RTXA02-R as primers. The 1.2-kb DNA fragment carrying nptI encoding for aminoglycoside 3′-phosphotransferae and conferring resistance to kanamycin (26) was inserted into a unique BamHI site present within the ΔrtxA in pKK1621. The ΔrtxA::nptI cartridge from the resulting construction (pKK1625) was liberated and ligated with SpeI-SphI-digested pDM4 (24) to generate pKK1628 (Table 1). Escherichia coli S17-1 λpir, tra strain (22) containing pKK1628 was used as a conjugal donor to V. vulnificus MO6-24/O to generate the rtxA mutant KK1604 (Table 1). The conjugation and isolation of the transconjugants were conducted using a previously described method (27).
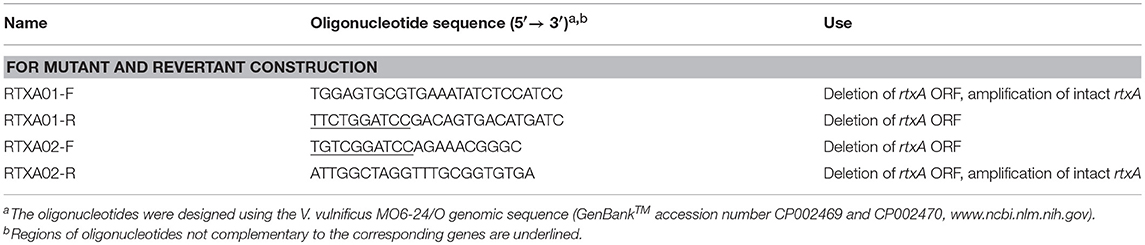
Table 2. Oligonucleotides used in this study.
To complement the rtxA mutation, rtxA revertant was constructed as previously described (16). In brief, the region containing the intact sequences of rtxA was amplified by PCR using RTXA01-F and RTXA02-R as primers. The amplified intact rtxA was ligated into SpeI-SphI-digested pDM4 to generate pKK1629 (Table 1) and pKK1629 was transferred into the rtxA mutant KK1604 to generate the rtxA revertant KK1606 by conjugation.
Preparation of Murine Bone-Marrow-Derived Dendritic Cells (BMDCs)
We generated BMDCs using the method originally described by Inaba et al. (28) with some modifications (8). In brief, the isolated bone-marrow cells were flushed out from the femurs and tibiae of mice, and red blood cells were depleted with RBC lysis buffer containing 0.15 M NH4Cl, 1 mM KHCO3, and 0.1 mM EDTA. For 7 days, cells (5 × 105 cells/mL) were cultured in petri dishes with 10 mL of Roswell Park Memorial Institute (RPMI)-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 2-mercaptoethanol (2-ME; 50 μM; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany), 10 mM of HEPES, 100 U/mL of penicillin, and 0.1 mg/mL of streptomycin (Corning and Invitrogen; Thermo Fisher Scientific, Inc.) in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF; 10 ng/mL; ProSpec, Rehovort, Israel). At 3 and 5 days of culture, 5 mL of fresh medium containing 10 ng/mL of GM-CSF were added. The loosely adherent DCs were harvested on day 7 for subsequent experiments.
In vitro Infection Protocol
Dendritic cells grown for 7 days were seeded onto a 24-well plate (1.5–2 × 106 cells/mL) in an antibiotic-free growth medium. Before infection, the bacteria were centrifuged at 2,420 × g for 3 min at room temperature, resuspended, and adjusted to 1.5–2 × 108 CFU/mL in antibiotic-free RPMI 1640 media. The DCs were infected with V. vulnificus WT and mutants at various multiplicities of infection (MOI; the ratio of bacteria number to BMDC number) for various durations. In particular, DCs were infected at an MOI of 1 for 30 or 60 min to evaluate the expression of surface markers and the secretion of cytokines, respectively or for 60 min at an MOI of 10 for co-culture with naïve CD4+ T cells. After infection, the cells were washed twice with PBS and incubated for 20 h in an antibiotic-containing growth medium at 37°C under 5% CO2.
In vitro Co-culture of CD4+ T Cells With DCs
Naïve CD4+ T cells isolated from the axillary, lateral axillary, inguinal, popliteal, and submandibular lymph nodes of mice were purified using magnetic beads (MACS; Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) and the purity of CD4+ T cells was confirmed to be >97% by flow cytometry. In a co-culture system, V. vulnificus-infected DCs (1 × 105 cells/mL) were mixed with naive CD4+ T cells (5 × 105 cells/mL) at a ratio of 1:5 in the presence of anti-CD3ε (1 μg/mL; cat. no. 553057) and CD28 (1 μg/mL; cat. no. 553294) monoclonal antibodies (mAbs) (BD Biosciences, San Diego, CA, USA). After 3 days, the cells were re-stimulated with 50 ng/mL of phorbol 12-myristate 13-acetate (PMA), 1 μg/mL of ionomycin, and 1 μL/mL of GolgiPlug for 6 h at 37°C and 5% CO2. After 6 h, the cells were harvested and stained with fluorescent antibodies for flow cytometric analysis.
In vivo Infection Protocol and Lamina Propria Cell Isolation
To ablate normal flora, mice were given drinking water containing rifampicin (50 μg/mL) for 24 h. Before infection with bacteria, food and water were eliminated from the cages of mice to empty their stomachs for 12 h. In the subsequent step, 100 μL of bacterial suspension containing 1 × 107 CFU of V. vulnificus was orally inoculated immediately following the administration of 50 μLof 8.5% (w/v) sodium bicarbonate (NaHCO3). Mice were sacrificed at day 2 post-infection for the isolation of cells from the lamina propria of small intestines, as previously described (29). In brief, the small intestines from the mice were washed in cold PBS to clear feces. Fat tissues and Peyer's patches were removed and the intestines were longitudinally cut, followed by washing with cold PBS. The intestines were then cut into pieces (2–3 cm) and incubated in RPMI medium containing 1 mM ethylenediaminetetraacetic acid (EDTA) with gentle stirring at 37°C for 15 min, followed by washing with warm PBS. The incubation in EDTA-containing medium was performed twice. The tissues were subsequently finely cut and incubated in RPMI containing 0.1 mg/mL of collagenase D (Roche Diagnostics, Basel, Switzerland) at 37°C for 30 min with gentle stirring. The incubated supernatants were collected using a 70-μm cell strainer, and the unfractionated cells were centrifuged at 400 × g for 3 min at 4°C. The lamina propria lymphocytes were isolated with 40 and 85% Percoll gradient media (GE Healthcare Life Sciences, Little Chalfont, UK) by gradient centrifugation.
Flow Cytometric Analysis
Antibodies used for both surface and intracellular staining were diluted at 1:250. The following mouse mAbs were used for flow cytometric analysis: CD4-FITC (cat. no. 553047), CD11c-FITC (cat. no. 553801), CD40-PE (cat. no. 553791), CD80-PE (cat. no. 553769), I-Ab-PE (cat. no. 553552) (BD Biosciences, San Diego, CA, USA), IL-17A-APC (cat. no. 17-7177; eBioscience, Inc., San Diego, CA, USA). To detect intracellular IL-17A, the cells were re-stimulated for 6 h with 50 ng/ml PMA, 1 μg/ml ionomycin, and 1 μl/ml Golgiplug at 37°C and 5% CO2. Subsequently, the cells were intracellularly stained after permeabilization of the cells using Cytofix/Cytoperm kits (BD Biosciences, San Diego, CA, USA). The stained cells were observed using a flow cytometer (FACSCalibur or BD Accuri C6 Plus; BD Biosciences, San Diego, CA, USA) gated on live CD11c+ or CD4+ cells. The data were analyzed using CellQuest software version 4.0.2 (BD Biosciences, San Diego, CA, USA).
Semi Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis
Total RNA obtained from the cells using TRIzol reagent was reverse transcribed into cDNA using a RocketScript™ Reverse Transcriptase kit (E-3162; Bioneer Corporation, Daejeon, Korea). PCR was conducted using 1 μl of each 5′ and 3′ primer, 1 μl of cDNA (50 ng). dH2O was then added to a final volume of 20 μl. PCR amplification of the cDNA was then performed using AccuPower® PCR PreMix (K-2016; Bioneer Corporation, Daejeon, Korea) with a thermal cycler (MJ Research, Inc., Watertown, MA, USA or Bioneer Corporation, Korea). The sequences of the PCR primers used in the present study were as follows: Murine IL-1β, forward 5′-CTAAAGTATGGGCTGGACTG-3′ and reverse 5′-AGCTTCAATGAAAGACCTCA-3′; murine IL-6, forward 5′-TGAACAACGATGATGCACTT-3′ and reverse 5′-CGTAGAGAACAACATAAGTC-3′; murine IL-23p19, forward 5′-AGCGGGACATATGAATCTAC-3′ and reverse 5′-TAAGCTGTTGGCACTAAGGG-3′; murine TGF-β, forward 5′-TATAGCAACAATTCCTGGCG-3′ and reverse 5′-TCCTAAAGTCAATGTACAGC-3′; murine β-actin, forward 5′-TGGAATCCTGTGGCATCCATGAAA-3′ and reverse 5′-TAAAACGCAGCTCAGTAACAGTCCG-3′. The temperature condition for PCR amplification was 95°C for 5 min; followed by 28–36 cycles consisting of 95°C for 30 s, 55–61°C for 30 s, and 72°C 30 s; plus a final cycle of 72°C for 5 min. The PCR reactions were performed for 28–36 cycles. After amplification, the products were separated on 1.5% (w/v) agarose gels stained with StainingSTAR (DyneBio, Gyeonggi-do, Korea).
Cytokine Assays
Dendritic cells were infected with V. vulnificus at the indicated MOIs and times, washed and seeded into a 96-well plate (2 × 104 cells/well), followed by further incubation for 20 h. The cell supernatant was obtained to measure the levels of secreted cytokines. The quantities of IL-1β, IL-6, and IL-23 in the culture supernatants were determined using Mouse ELISA Ready-Set-Go! kits (IL-1β; eBioscience, Inc., San Diego, CA, USA) and a Mouse IL-6 ELISA kit (BD Biosciences, San Diego, CA, USA) or sandwich ELISA with anti-mouse IL-23p19 monoclonal antibody (clone 5B2) for plate coating and biotinylated anti-mouse IL-12/23 p40 monoclonal antibody (clone C17.8). A standard curve was generated using recombinant IL-23 (eBioscience, Inc., San Diego, CA, USA). The levels of secreted IL-17A in the culture supernatants were determined using a Mouse ELISA Ready-Set-Go! kit (IL-17A; eBioscience, Inc).
Statistical Analysis
Statistical analysis was performed with unpaired Student's t-test for pairwise comparisons or one-way analysis of variance (ANOVA) with a Bonferroni t-test for multiple comparisons in Sigmaplot version 12.5 (Systat Software Inc., Washington, USA). P < 0.05 was considered statistically significant.
Results
Infection With rtxA Mutant Strain of V. vulnificus Affects DC Maturation and Activation
We previously demonstrated that infection with WT V. vulnificus results in the induction of DC maturation and activation, leading to Th17 cell stimulation (8). Although several studies have identified virulence factors of V. vulnificus, the specific virulence factors involved in Th17 responses are unknown. We evaluated the abilities of several virulence factors from V. vulnificus to induce Th17 cell responses by infecting DCs at a multiplicity of infection (MOI) of 1 for 30 min with the WT strain and mutant strains with defects in ahpCl, rtxA, and vvhA genes encoding peroxiredoxin, RtxA, and hemolysin, respectively. The expression of maturation/activation-related cell surface markers, including CD40, CD80, and major histocompatibility complex (MHC) II, was analyzed with flow cytometry. As shown in Figures 1A,B, expression levels of these markers were reduced in rtxA mutant-infected DCs compared to those in DCs infected with the WT strain. In particular, CD40 expression level was significantly reduced in rtxA mutant-infected DCs. On the contrary, no differences in CD40 expression level were observed between DCs infected with ahpCl and vvhA mutant strains and WT-infected DCs. These data suggest that RtxA may act as a virulence factor of V. vulnificus that induces maturation and activation of DCs.
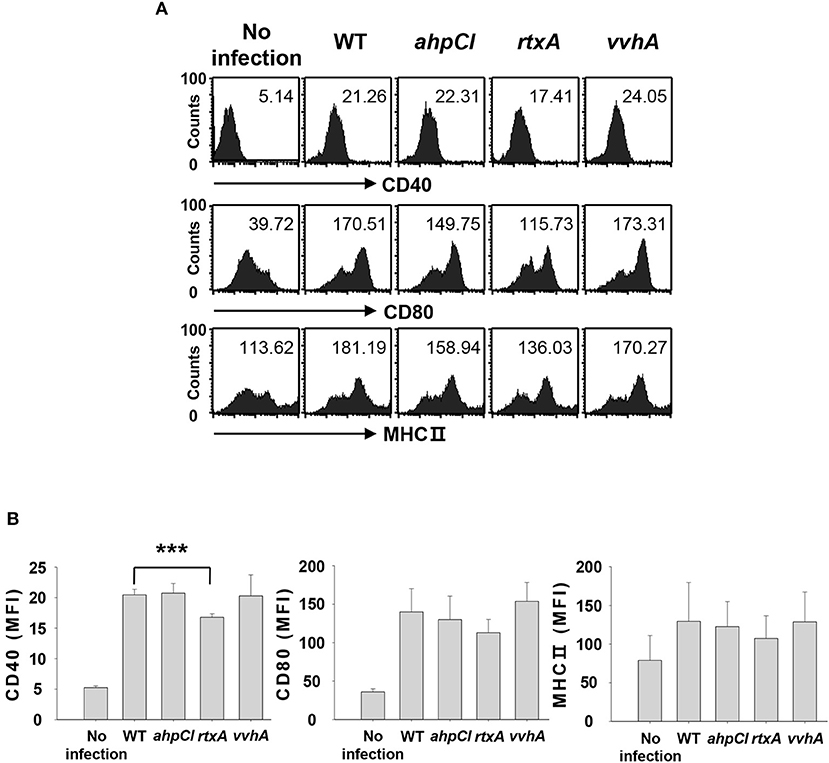
Figure 1. Vibrio vulnificus rtxA mutant was defective in inducing maturation and activation of DCs. (A) DCs were infected with WT and mutant ahpCl, rtxA, and vvhA mutant Vibrio vulnificus at an MOI of 1 for 30 min, followed by washing of cells with PBS and subsequent culturing for 20 h in the presence of antibiotics. After 20 h, the cells were stained with antibodies targeting CD40, CD80, and MHC II and their expressions were determined by flow cytometric analysis. The data shown in (A) are representative of three independent experiments, and bar graphs (B) represent the means ± SD of three independent experiments. ***p < 0.005 as determined by Student's t-test for pairwise comparisons. WT, wild type; ahpCl, ahpCl mutant; rtxA, rtxA mutant; vvhA, vvhA mutant.
DCs Infected With rtxA Mutant Strain Show Defective Secretion of Th17 Polarization-Related Cytokines
In our previous study, the expression and secretion of IL-1β and IL-6, the major Th17-polarizing cytokines, increased in DCs upon infection with the WT strain (8). To investigate the role of some virulence factors in the expression of cytokines related to Th17 cell induction, we infected DCs with WT and mutants of V. vulnificus at an MOI of 1 for 30 or 60 min and evaluated their expressions at the mRNA (Figure 2A) and protein (Figure 2B) levels. As shown in Figure 2A, the mRNA expressions of IL-6 and IL-23p19 were reduced in the rtxA mutant-infected group but not in cells infected with ahpCl and vvhA mutant strains, except for IL-1β expression in the vvhA mutant group. The secretion levels of IL-1β and IL-6 were significantly decreased in rtxA mutant-infected DCs compared to those in cells infected with other mutant strains (Figure 2B). In contrast, the expression level of transforming growth factor beta (TGF-β), a Th17 cell-related cytokine, was similar between mutant-infected and WT-infected DCs (Figure 2A). These results indicate that RtxA may be involved in Th17 cell induction caused by WT V. vulnificus.
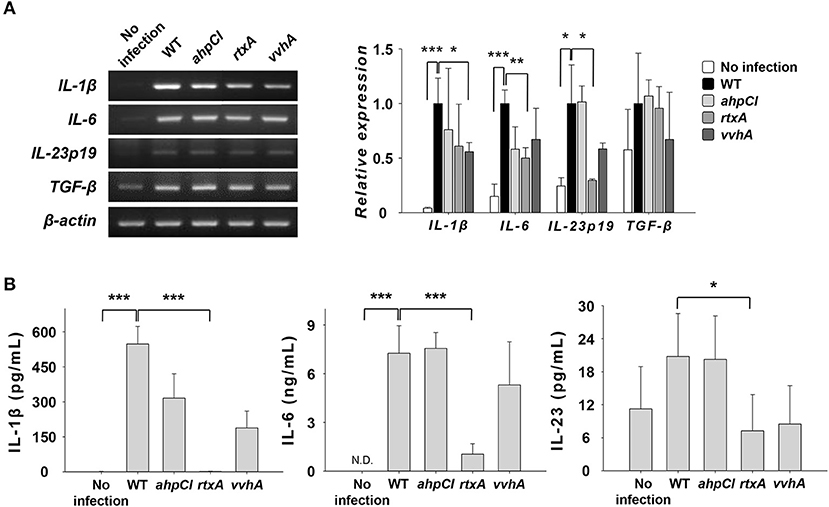
Figure 2. Vibrio vulnificus rtxA mutant was defective in inducing the secretion of Th17 polarization-inducing cytokines. (A) Total RNA was extracted from DCs infected with WT and mutant ahpCl, rtxA, and vvhA mutant Vibrio vulnificus at an MOI of 1 for 30 min. The expression levels of mRNAs for several Th17-polarizing cytokines were determined by RT-PCR. (B) DCs were infected for 60 min with WT and mutant ahpCl, rtxA, and vvhA mutant Vibrio vulnificus at an MOI of 1. The protein levels of IL-1β, IL-6, and IL-23 in the supernatants collected were determined by ELISA. The data shown in (A) are representative of three independent experiments, and bar graphs represent the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005 as determined by Student's t-test for pairwise comparisons. WT, wild type; ahpCl, ahpCl mutant; rtxA, rtxA mutant; vvhA, vvhA mutant.
Induction of Th17 Responses Is Reduced in vitro and in vivo After RtxA-Deficient V. vulnificus Infection
Dendritic cells are antigen-presenting cells involved in the polarization of naïve CD4+ T cells into each subset such as Th1, Th2, and Th17, depending on the cytokines they produce (30). To determine whether DCs infected with the rtxA mutant strain have a weaker ability to induce Th17 cell responses, we infected DCs with the WT or rtxA mutant strain at an MOI of 10 for 60 min, and subsequently co-cultured these cells with naïve CD4+ T cells at a ratio of 1:5 (DC: CD4+ T). We evaluated the population of IL-17-expressing CD4+ T (Th17) by flow cytometry (Figure 3A) and the levels of secreted IL-17A in the supernatant by ELISA (Figure 3B). As shown in Figures 3A,B, WT-infected DCs displayed an increased proportion of Th17 cells and secretion of IL-17A as compared to the uninfected DCs, whereas the increased in Th17 cell responses and secretion of IL-17A observed following by WT infection were significantly reduced in DCs infected with the rtxA mutant strain. These observations indicate that the ability of rtxA mutant-infected DCs to induce Th17 cell responses was lower than that of DCs infected with WT V. vulnificus. Consistent with the in vitro result, we observed a decrease in the population of Th17 cells in the small intestinal lamina propria of mice treated with the rtxA mutant as compared with those treated with the WT strain (Figure 3B). These results suggest that RtxA may play a critical role in the induction of Th17 responses by V. vulnificus.
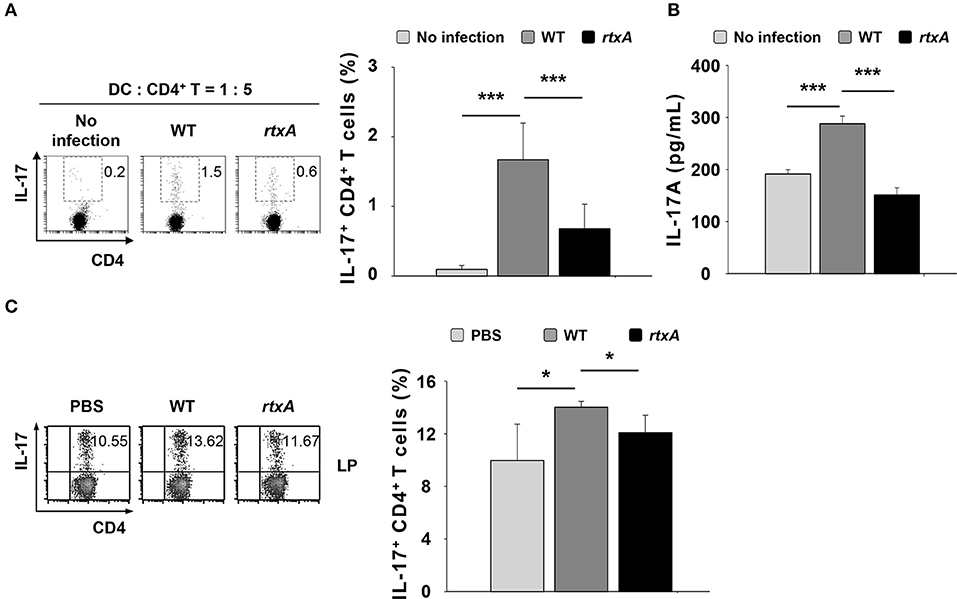
Figure 3. Vibrio vulnificus rtxA mutant was defective in inducing Th17 cell responses in vitro and in vivo. DCs were infected with WT and rtxA mutant V. vulnificus at an MOI of 10 for 60 min. After 20 h, DCs were co-cultured with naïve CD4+ T cells isolated from lymph nodes for 3 days in the presence of anti-CD3ε and anti-CD28 mAbs, followed by flow cytometric analysis of CD4 and IL-17 expression (A) or ELISA for IL-17A levels in the supernatants (B). (C) The expression of CD4 and IL-17 in the lamina propria samples of uninfected mice or mice infected with WT and rtxA mutant (1 × 107 CFU per mouse) were analyzed by flow cytometry. The data shown in (A,C) are representative of three independent experiments, and bar graphs represent the means ± SD of three independent experiments. Bars and error bars in (B) represent the mean ± SD of results performed in triplicate. *p < 0.05, ***p < 0.005 as determined by Student's t-test for pairwise comparisons. WT, wild type; rtxA, rtxA mutant.
V. vulnificus rtxA Revertant Infection Recovers the Reduced Induction of Th17 Responses After rtxA Mutant Infection
To confirm the involvement of RtxA in the induction of Th17 responses in vitro and in vivo, we used the revertant strain of rtxA mutant. DCs were infected with V. vulnificus WT, rtxA mutant, and rtxA revertant at an MOI of 1 for 30 or 60 min or at an MOI of 10 for 60 min (Figures 4A–D). As shown in Figure 4A, the reduced expressions of maturation/activation-related surface markers in rtxA mutant-infected DCs were increased in rtxA revertant-infected DCs. Likewise, the reduced mRNA (Figure 4B) and protein (Figure 4C) expressions of IL-1β and IL-6 in rtxA mutant-infected DCs were restored in rtxA revertant-infected DCs to the levels observed in WT-infected DCs. Both in vitro (Figure 4D) and in vivo (Figure 4E), the percentages of IL-17+ CD4+ cells in the rtxA revertant-infected group were restored to the levels comparable with those observed in the WT-infected group. These results confirmed the crucial role of RtxA in the induction of Th17 responses against V. vulnificus infection.
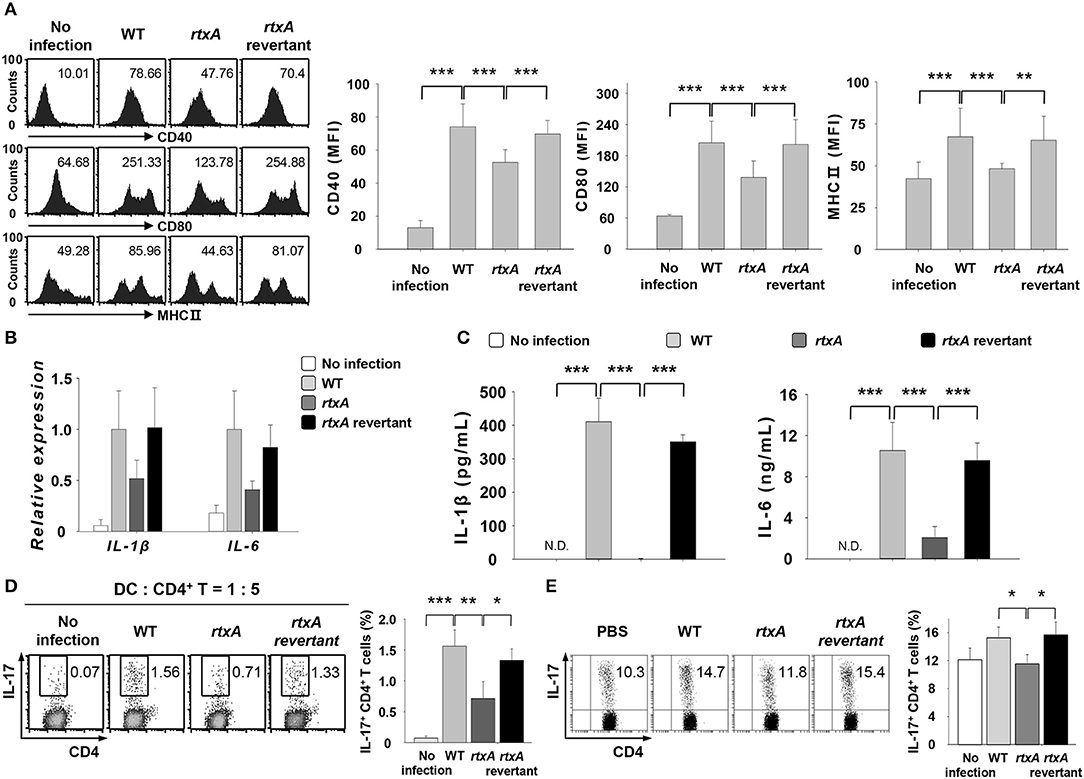
Figure 4. Vibrio vulnificus rtxA revertant restored the defect of the rtxA mutant by inducing the maturation and activation of DCs, secretion of Th17-polarizing cytokines, and Th17 cell responses. (A) DCs were infected with WT, rtxA mutant, and rtxA revertant V. vulnificus at an MOI of 1 for 30 min. (B) Total RNA was extracted from DCs infected with WT, rtxA mutant, and rtxA revertant strain. (C) DCs were infected for 60 min with V. vulnificus at an MOI of 1. (D) DCs were infected with WT, rtxA mutant, and rtxA revertant strain at an MOI of 10 for 60 min. After 20 h, DCs were co-cultured with naïve CD4+ T cells for 3 days in the presence of anti-CD3ε and anti-CD28 mAbs, followed by the flow cytometric analysis of CD4 and IL-17 expression. (E) The expression of CD4 and IL-17 in the lamina propria of uninfected mice or mice infected with WT, rtxA mutant, and rtxA revertant strains (1 × 107 CFU per mouse) were analyzed by flow cytometry. The data shown in (A,D,E) are representative of three independent experiments, and bar graphs represent the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005 as determined by one-way analysis of variance with a Bonferroni t-test for multiple comparisons. WT, wild type; rtxA, rtxA mutant.
Infection With V. vulnificus Mutant Carrying the Deletion of hlyU, an Activator of rtxA, Results in Consequences Similar to Those Observed With rtxA Mutant Infection
HlyU positively regulates the expression of rtxA by interfering with H-NS, while SmcR acts as a repressor of hlyU (31–33). To test if the role of rtxA is regulated by HlyU, we infected DCs with V. vulnificus WT or mutant strains lacking the genes associated with rtxA gene expression, hlyU and smcR at an MOI of 1 for 30 or 60 min or at an MOI of 10 for 60 min (Figures 5A–D). As shown in Figure 5, infection with the hlyU mutant also resulted in decreased secretion of IL-1β and IL-6, the two Th17-polarizing cytokines (Figure 5C), although the decreases in the expression of DC surface markers (Figure 5A) and cytokine mRNAs (Figure 5B) were not significant. In addition, the differentiation of naïve CD4+ T cells into IL-17-secreting cells was reduced following hlyU mutant infection (Figure 5D). No significant difference was observed between smcR mutant-infected DCs and WT-infected DCs. Taken together, these results imply that RtxA confers V. vulnificus with an ability to promote Th17 responses in vitro and in vivo.
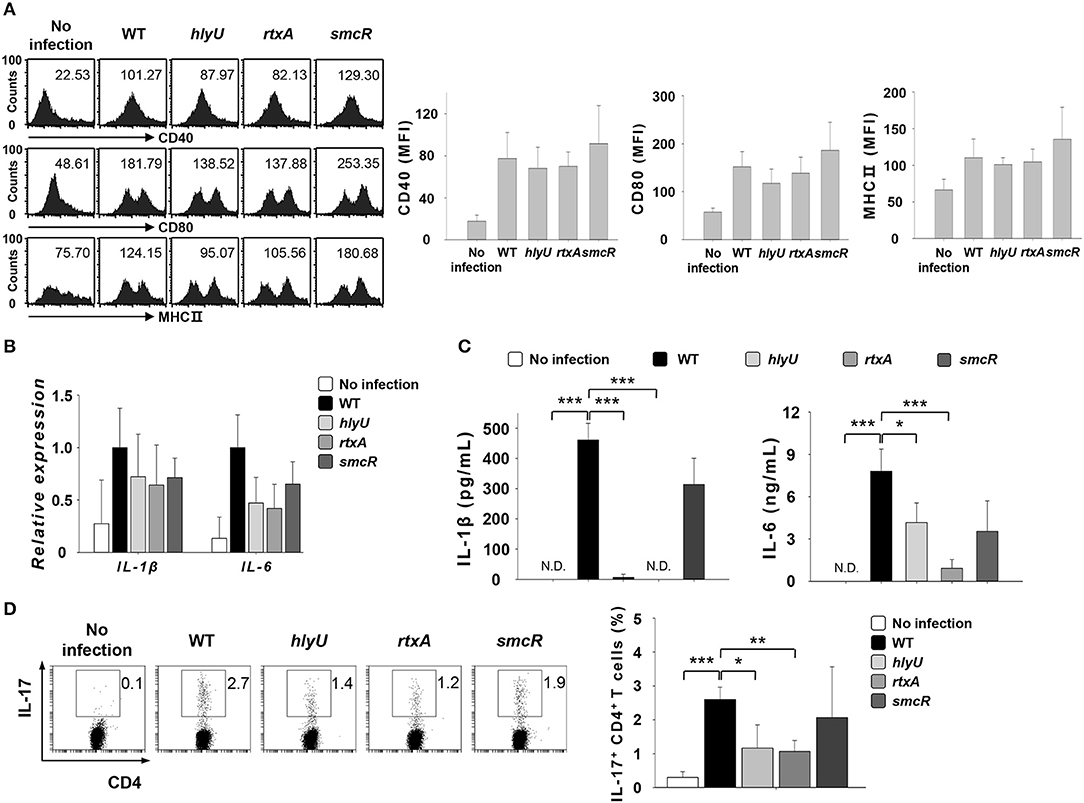
Figure 5. Vibrio vulnificus hlyU mutant reproduced the results observed for rtxA mutant. (A) DCs were infected with WT and hlyU, rtxA, and smcR mutant V. vulnificus at an MOI of 1 for 30 min. (B) Total RNA was extracted from DCs infected with WT and hlyU, rtxA, and smcR mutant strain. (C) DCs were infected for 60 min with WT and hlyU, rtxA, and smcR mutant strain at an MOI of 1. (D) DCs were infected with WT and hlyU, rtxA, and smcR mutant strain at an MOI of 10 for 60 min. After 20 h, DCs were co-cultured with naïve CD4+ T cells isolated from lymph nodes for 3 days in the presence of anti-CD3ε and anti-CD28 mAbs, followed by the flow cytometric analysis of CD4 and IL-17 expressions. The data shown in (A,D) are representative of three independent experiments, and bar graphs represent the means ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005 as determined by Student's t-test for pairwise comparisons. WT, wild-type; hlyU, hlyU mutant; rtxA, rtxA mutant; smcR, smcR mutant.
Discussion
V. vulnificus inhabits marine environments and can invade the host through the ingestion of contaminated seafood or via an open wound. Upon entry, it can cause a wide range of diseases, which include gastroenteritis and primary sepsis (1), and activates the innate and adaptive immune responses via diverse virulence factors (8, 9). Many studies have identified several virulence factors involved in the innate immune response activation (14, 34–37). However, few studies have focused on the identification of the virulence factors that induce the adaptive immune response. Therefore, in the present study, we investigated the involvement of RtxA of V. vulnificus in the induction of Th17 cell responses and demonstrated that the increase in the population of Th17 cells was related to RtxA from V. vulnificus. Adenylate cyclase toxin (ACT) of Bordetella pertussis, a member of RTX toxin family belonging to a different subgroup, induces suppressive and modulatory effects on the immune response through the inhibition of the production of pro-inflammatory cytokines or induction of Th17 cells (38, 39).
Our previous findings demonstrated that V. vulnificus induced Th17 cell responses by up-regulating the secretion of IL-1β and IL-6 from DCs, and that the induction of IL-6 was sufficient and necessary for the increased Th17 cell responses (8). Presently, RtxA was demonstrated to be responsible for the secretion of the pro-inflammatory cytokines, IL-1β and IL-6, and consequent Th17 responses using rtxA mutant and rtxA revertant strains of V. vulnificus. V. vulnificus RtxA reportedly protects bacteria from phagocytosis (16). Therefore, it is conceivable that the rtxA mutant is phagocytosed and destroyed by DCs, while WT V. vulnificus carrying the intact rtxA gene may evade immune responses and induce DC activation and the production of pro-inflammatory cytokines, such as IL-1β and IL-6. Thus, V. vulnificus RtxA may be involved in the induction of Th17 responses. How RtxA of V. vulnificus promotes IL-1β secretion may involve the activation of the NLRP3 inflammasome by RtxA (14). Additionally, in the previous study, B. pertussis ACT enhanced the production of IL-6 in DCs in the presence of low concentrations of LPS (40). The RtxA of V. vulnificus likely acts synergistically with other virulence factors on IL-6 production.
V. vulnificus HlyU is a homolog for the hemolysin gene regulator of V. cholerae and regulates the expression of the vvhA, vvpE, and rtxA genes (31–33). To investigate the molecular mechanisms by which rtxA is regulated in V. vulnificus, DCs were infected with hlyU mutant. The lack of HlyU failed to increase the production of IL-1β and IL-6 from DCs. Eventually, Th17 cell responses were also down-regulated. These results clearly demonstrate that the Th17-inducing capacity of RtxA is regulated by HlyU. However, the decrease in the expression of DC surface markers and the transcriptional expression of IL-1β and IL-6 were not statistically significant. These observed discrepancies are possibly due to the other pathways that regulate the expression of rtxA and the function of hlyU gene, which is related to other virulence factors like vvhA, vvpE, and smcR. The deletion of SmcR, a negative regulator of HlyU, could not increase the secretion of IL-1β and IL-6 or Th17 responses. The absence of effect by smcR mutation may be due to the saturated function of HlyU in WT V. vulnificus or to other rtxA gene-activating pathways.
The expression of some virulence factors, including RtxA, increases upon the contact of V. vulnificus with host cells (41, 42). This observation may be associated with the results shown in Figure 1, wherein no significant decrease in the expression of CD80 and MHC class II surface molecules was observed in rtxA mutant-infected DCs, even though the secretion of Th17-polarizing cytokines and Th17 responses were significantly reduced in the rtxA mutant-infected group (Figures 2, 3). Thus, further studies are needed to identify additional virulence factors other than RtxA that are likely to be involved in the upregulation of DC surface molecules and/or Th17 responses. Similar to a previous study that showed the effects of secreted outer membrane vesicles of V. cholerae on DC activation, the release of Th17 polarization-related cytokines, and the induction of inflammatory Th17 cells (43), other structural components and/or secreted factors of V. vulnificus may also be involved in Th17 responses induction.
Previous studies reported that the exposure of the immune cells to sub-lytic concentrations of Panton-Valentine leukocidin, a pore-forming toxin of Staphylococcus aureus, resulted in immune cell activation and the release of pro-inflammatory cytokines, rather than cell lysis (44, 45). Furthermore, the amount of secreted RtxA upon contact with host cells increased as the degree of contact increased (46). Therefore, V. vulnificus RtxA may exert different effects on the host based on the nature of contact between V. vulnificus and the host cell. The close contact between the host cells and a large number of V. vulnificus results in the production of high (over lytic) concentrations of the RTX toxin, leading to host cell lysis and spread of infection into the bloodstream of the infected host. However, weak contact between the host cells and fewer V. vulnificus cells results in the production of low (sublytic) concentrations of V. vulnificus RTX toxin, leading to the induction of Th17 cell-mediated responses that are likely to be involved in controlling V. vulnificus infection. The RTX toxin of V. vulnificus forms pores on the host cell membrane and causes cell lysis (13, 15), and the expression of rtxA gene is induced after the contact with the host both in vitro (15) and in vivo (17). Th17 cells play an important role in maintaining mucosal barriers and contribute to pathogen clearance at mucosal surfaces (47), although their roles in V. vulnificus infection are unknown.
In summary, our study demonstrates that RtxA, one of the key virulence factors of V. vulnificus, induces the secretion of IL-1β and IL-6 from V. vulnificus-infected DCs and contributes to the induction of Th17 cells in vitro. In addition, RtxA increases the population of Th17 cells in the small intestinal lamina propria. Overall, these data establish the importance of RtxA in adaptive immune responses against V. vulnificus.
Author Contributions
AL participated in the design of the study, performed all of the experiments, the data collection, and the analysis, and drafted the manuscript. MK conceived and designed the experiments. DC contributed reagents, and materials. KJ and SC constructed and provided all V. vulnificus strains. TK conceived the study and participated in its design and the coordination and also performed the data analysis and writing of the manuscript, and has full access to all the data in this study with the financial support.
Funding
This study was supported by the National Research Foundation of Korea (grant no. NRF-2017R1A2B2009442) and the Creative Materials Discovery Program through the National Research Foundation of Korea(NRF) funded by the Ministry of Science ICT and Future Planning (grant no. 2016M3D1A1021387), and also by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry(IPET) through Agriculture, Food and Rural Affairs Research Center Support Program, funded by Ministry of Agriculture, Food and Rural Affairs(MAFRA) (grant no. 710012-03-1-SB110).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Horseman MA, Surani S. A comprehensive review of Vibrio vulnificus: an important cause of severe sepsis and skin and soft-tissue infection. Int J Infect Dis. (2011) 15:e157–66. doi: 10.1016/j.ijid.2010.11.003
2. Amaro C, Fouz B, Biosca EG, Marco-Noales E, Collado R. The lipopolysaccharide O side chain of Vibrio vulnificus serogroup E is a virulence determinant for eels. Infect Immun. (1997) 65:2475–9.
3. Lee BC, Kim MS, Choi SH, Kim TS. Involvement of capsular polysaccharide via a TLR2/NF-kappaB pathway in Vibrio vulnificus-induced IL-8 secretion of human intestinal epithelial cells. Int J Mol Med. (2010) 25:581–91. doi: 10.3892/ijmm_00000380
4. Jeong KC, Jeong HS, Rhee JH, Lee SE, Chung SS, Starks AM, et al. Construction and phenotypic evaluation of a Vibrio vulnificus vvpE mutant for elastolytic protease. Infect Immun. (2000) 68:5096–106. doi: 10.1128/IAI.68.9.5096-5106.2000
5. Fan JJ, Shao CP, Ho YC, Yu CK, Hor LI. Isolation and characterization of a Vibrio vulnificus mutant deficient in both extracellular metalloprotease and cytolysin. Infect Immun. (2001) 69:5943–8. doi: 10.1128/IAI.69.9.5943-5948.2001
6. Baek WK, Lee HS, Oh MH, Koh MJ, Kim KS, Choi SH. Identification of the Vibrio vulnificus ahpCl gene and its influence on survival under oxidative stress and virulence. J Microbiol. (2009) 47:624–32. doi: 10.1007/s12275-009-0130-x
7. Lee JH, Kim MW, Kim BS, Kim SM, Lee BC, Kim TS, et al. Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J Microbiol. (2007) 45:146–52.
8. Lee A, Lim HX, Kim MS, Cho D, Jang KK, Choi SH, et al. Vibrio vulnificus infection induces the maturation and activation of dendritic cells with inflammatory Th17-polarizing ability. Int J Mol Med. (2018) 41:531–40. doi: 10.3892/ijmm.2017.3230
9. Wang MY, Liu XF, Xia J, Li Y, Geng JL, Hu CJ. Vibrio vulnificus VvhA induces Th1 and Tfh cells to proliferate against Vibrio vulnificus in a mouse model of infection. Future Microbiol. (2017) 12:953–65. doi: 10.2217/fmb-2017-0040(Wang,2017#1075)
10. Welch RA. RTX toxin structure and function: a story of numerous anomalies and few analogies in toxin biology. Curr Top Microbiol Immunol. (2001) 257:85–111. doi: 10.1007/978-3-642-56508-3_5
11. Linhartova I, Bumba L, Masin J, Basler M, Osicka R, Kamanova J, et al. RTX proteins: a highly diverse family secreted by a common mechanism. FEMS Microbiol Rev. (2010) 34:1076–112. doi: 10.1111/j.1574-6976.2010.00231.x
12. Jeong HG, Satchell KJ. Additive function of Vibrio vulnificus MARTX(Vv) and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. (2012) 8:e1002581. doi: 10.1371/journal.ppat.1002581
13. Lee BC, Choi SH, Kim TS. Vibrio vulnificus RTX toxin plays an important role in the apoptotic death of human intestinal epithelial cells exposed to Vibrio vulnificus. Microbes Infect. (2008) 10:1504–13. doi: 10.1016/j.micinf.2008.09.006
14. Toma C, Higa N, Koizumi Y, Nakasone N, Ogura Y, Mccoy AJ, et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-kappa B signaling. J Immunol. (2010) 184:5287–97. doi: 10.4049/jimmunol.0903536
15. Kim YR, Lee SE, Kook H, Yeom JA, Na HS, Kim SY, et al. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell Microbiol. (2008) 10:848–62. doi: 10.1111/j.1462-5822.2007.01088.x
16. Lo HR, Lin JH, Chen YH, Chen CL, Shao CP, Lai YC, et al. RTX toxin enhances the survival of Vibrio vulnificus during infection by protecting the organism from phagocytosis. J Infect Dis. (2011) 203:1866–74. doi: 10.1093/infdis/jir070
17. Chung KJ, Cho EJ, Kim MK, Kim YR, Kim SH, Yang HY, et al. RtxA1-induced expression of the small GTPase Rac2 plays a key role in the pathogenicity of Vibrio vulnificus. J Infect Dis. (2010) 201:97–105. doi: 10.1086/648612
18. Wright AC, Simpson LM, Oliver JD, Morris JG. Phenotypic evaluation of acapsular transposon mutants of Vibrio vulnificus. Infect Immun. (1990) 58:1769–73.
19. Kim SM, Park JH, Lee HS, Kim WB, Ryu JM, Han HJ, et al. LuxR homologue SmcR is essential for Vibrio vulnificus pathogenesis and biofilm detachment, and its expression is induced by host cells. Infect Immun. (2013) 81:3721–30. doi: 10.1128/IAI.00561-13
20. Jang KK, Lee ZW, Kim B, Jung YH, Han HJ, Kim MH, et al. Identification and characterization of Vibrio vulnificus plpA encoding a phospholipase A2 essential for pathogenesis. J Biol Chem. (2017) 292:17129–43. doi: 10.1074/jbc.M117.791657
21. Lee SJ, Jung YH, Kim JS, Lee HJ, Lee SH, Lee KH, et al. A Vibrio vulnificus VvpM induces IL-1β production coupled with necrotic macrophage death via distinct spatial targeting by ANXA2. Front Cell Infect. Microbiol. (2017) 7:352. doi: 10.3389/fcimb.2017.00352
22. Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic-engineering - transposon mutagenesis in gram-negative bacteria. Nat Biotechnol. (1983) 1:784–91. doi: 10.1038/nbt1183-784
23. Vieira J, Messing J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene (1982) 19:259–68. doi: 10.1016/0378-1119(82)90015-4
24. Milton DL, Otoole R, Horstedt P, Wolfwatz H. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol. (1996) 178:1310–9. doi: 10.1128/jb.178.5.1310-1319.1996
25. Sambrook J, Russell DW. Molecular Cloning: a Laboratory Manual, 3rd Ed., Cold Spring Harbor, NY: Cold Spring Harbor Laboratory (2001). 13.75–13.77.
26. Oka A, Sugisaki H, Takanami M. Nucleotide sequence of the kanamycin resistance transposon Tn903. J Mol Biol. (1981) 147:217–26. doi: 10.1016/0022-2836(81)90438-1
27. Jang KK, Gil SY, Lim JG, Choi SH. Regulatory characteristics of Vibrio vulnificus gbpa gene encoding a mucin-binding protein essential for pathogenesis. J Biol Chem. (2016) 291:5774–87. doi: 10.1074/jbc.M115.685321
28. Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. (1992) 176:1693–702. doi: 10.1084/jem.176.6.1693
29. Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc. (2007) 2:2307–11. doi: 10.1038/nprot.2007.315
30. Walsh KP, Mills KH. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. (2013) 34:521–30. doi: 10.1016/j.it.2013.07.006
31. Liu M, Alice AF, Naka H, Crosa JH. The HlyU protein is a positive regulator of rtxA1, a gene responsible for cytotoxicity and virulence in the human pathogen Vibrio vulnificus. Infect Immun. (2007) 75:3282–9. doi: 10.1128/IAI.00045-07
32. Liu MQ, Naka H, Crosa JH. HlyU acts as an H-NS antirepressor in the regulation of the RTX toxin gene essential for the virulence of the human pathogen Vibrio vulnificus CMCP6. Mol Microbiol. (2009) 72:491–505. doi: 10.1111/j.1365-2958.2009.06664.x
33. Shao CP, Lo HR, Lin JH, Hor LI. Regulation of cytotoxicity by quorum-sensing signaling in Vibrio vulnificus is mediated by SmcR, a repressor of hlyU. J Bacteriol. (2011) 193:2557–65. doi: 10.1128/JB.01259-10
34. Powell JL, Wright AC, Wasserman SS, Hone DM, Morris JG. Release of tumor necrosis factor alpha in response to Vibrio vulnificus capsular polysaccharide in in vivo and in vitro models. Infect Immun. (1997) 65:3713–8.
35. Shin NR, Lee DY, Shin SJ, Kim KS, Yoo HS. Regulation of proinflammatory mediator production in RAW264.7 macrophage by Vibrio vulnificus luxS and smcR. FEMS Immunol Med Microbiol. (2004) 41:169–76. doi: 10.1016/j.femsim.2004.03.001
36. Lee NY, Lee HY, Lee KH, Han SH, Park SJ. Vibrio vulnificus IlpA induces MAPK-mediated cytokine production via TLR1/2 activation in THP-1 cells, a human monocytic cell line. Mol Immunol. (2011) 49:143–54. doi: 10.1016/j.molimm.2011.08.001
37. Montero J, Gomez-Casado E, Garcia-Alcazar A, Meseguer J, Mulero V. Flagellin from Marinobacter algicola and Vibrio vulnificus activates the innate immune response of gilthead seabream. Dev Comp Immunol. (2014) 47:160–67. doi: 10.1016/j.dci.2014.07.003
38. Fedele G, Spensieri F, Palazzo R, Nasso M, Cheung GYC, Coote JG, et al. Bordetella pertussis commits human dendritic cells to promote a Th1/Th17 response through the activity of adenylate cyclase toxin and MAPK-pathways. PLoS ONE (2010) 5:e8734. doi: 10.1371/journal.pone.0008734
39. Dunne A, Ross PJ, Pospisilova E, Masin J, Meaney A, Sutton CE, et al. Inflammasome activation by adenylate cyclase toxin directs Th17 responses and protection against Bordetella pertussis. J Immunol. (2010) 185:1711–9. doi: 10.4049/jimmunol.1000105
40. Pádraig JR, Lavelle EC, Kingston HGM, Aoife PB. Adenylate cyclase toxin from bordetella pertussis synergizes with lipopolysaccharide to promote innate interleukin-10 production and enhances the induction of Th2 and regulatory T cells. Infect Immun. (2004) 72:1568–79. doi: 10.1128/IAI.72.3.1568-1579.2004
41. Lee BC, Lee JH, Kim MW, Kim BS, Oh MH, Kim KS, et al. Vibrio vulnificus rtxE is important for virulence, and its expression is induced by exposure to host cells. Infect Immun. (2008) 76:1509–17. doi: 10.1128/IAI.01503-07
42. Dey AK, Bhagat A, Chowdhury R. Host cell contact induces expression of virulence factors and VieA, a cyclic di-GMP phosphodiesterase, in Vibrio cholerae. J Bacteriol. (2013) 195:2004–10. doi: 10.1128/JB.02127-12
43. Chatterjee D, Chaudhuri K. Vibrio cholerae O395 outer membrane vesicles modulate intestinal epithelial cells in a NOD1 protein-dependent manner and induce dendritic cell-mediated Th2/Th17 cell responses. J Biol Chem. (2013) 288:4299–309. doi: 10.1074/jbc.M112.408302
44. Dragneva Y, Anuradha CD, Valeva A, Hoffmann A, Bhakdi S, Husmann M. Subcytocidal attack by staphylococcal alpha-toxin activates NFkB and induces interleukin-8 production. Infect Immun. (2001) 69:2630–5. doi: 10.1128/IAI.69.4.2630-2635.2001
45. Yoong P. Enhancement of bacterial virulence by antibody neutralization of immune-activating toxins. Virulence (2010) 1:409–13. doi: 10.4161/viru.1.5.12705
46. Guo RH, Lim JY, Tra My DN, Jo SJ, Park JU, Rhee JH, et al. Vibrio vulnificus RtxA1 toxin expression upon contact with host cells is RpoS-dependent. Front Cell Infect Microbiol. (2018) 8:70. doi: 10.3389/fcimb.2018.00070
Keywords: V. vulnificus, RTX toxin, dendritic cells, Th17, mouse
Citation: Lee A, Kim MS, Cho D, Jang KK, Choi SH and Kim TS (2018) Vibrio vulnificus RtxA Is a Major Factor Driving Inflammatory T Helper Type 17 Cell Responses in vitro and in vivo. Front. Immunol. 9:2095. doi: 10.3389/fimmu.2018.02095
Received: 21 February 2018; Accepted: 24 August 2018;
Published: 19 September 2018.
Edited by:
Amy Rasley, United States Department of Energy (DOE), United StatesReviewed by:
Dean T. Nardelli, University of Wisconsin–Milwaukee, United StatesVida A. Dennis, Alabama State University, United States
Copyright © 2018 Lee, Kim, Cho, Jang, Choi and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tae Sung Kim, dHNraW1Aa29yZWEuYWMua3I=