 Mehmet Akce
Mehmet Akce Mohammad Y. Zaidi2,3
Mohammad Y. Zaidi2,3 Gregory B. Lesinski
Gregory B. Lesinski- 1Department of Hematology and Oncology, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA, United States
- 2Department of General Surgery, Indiana University, Bloomington, IN, United States
- 3Division of Surgical Oncology, Department of Surgery, Emory University School of Medicine, Atlanta, GA, United States
Pancreatic cancer has a dismal prognosis and effective treatment options are limited. It is projected to be the second most common cause of cancer related mortality in the United States by 2030 and there is urgent unmet need for novel systemic treatment options. Immunotherapy with antibodies targeting PD-1, PD-L1, CTLA-4 has not shown clinical activity in unselected pancreatic cancer, emphasizing the need for combination immunotherapy approaches or other therapeutic strategies. As such, chimeric antigen receptor (CAR) T cell therapy represents an emerging therapeutic option for pancreatic cancer. This modality utilizes genetically engineered T cells that are redirected to specific cancer-associated antigens to elicit potent cytotoxic activity. This review summarizes the available preclinical data and highlights early phase clinical trials using CAR T cell approaches in pancreatic cancer, a disease state that is gaining attention as a conduit for cell therapy. Future directions in application of CAR T cell therapy are also considered including its ability to be directed against novel epitopes and combined with other therapeutic regimens.
Introduction
Pancreatic cancer is the fourth most common cause of cancer related mortality in the United States, and estimates project it to become the second most common cause of cancer related mortality by 2030 (1, 2). Approximately 60% of patients present with advanced disease and median overall survival ranges between 8.5 and 11.1 months with the current therapeutic approaches (3, 4). The majority of patients who present with localized disease eventually develop metastasis. Therefore, development of effective systemic therapies is key in improving the outcomes of patients with pancreatic cancer. The therapeutic options for systemic therapy are limited and clinical activity is at best modest. Immunotherapy with checkpoint inhibitors has emerged as a novel therapeutic option in many solid and hematologic malignancies. However, trials in pancreatic cancer with checkpoint inhibitors have only shown activity in a small subset of patients with micro-satellite instability of their cancer, which increases mutation burden and the likelihood of tumors harboring neoantigens. The limited success of immunotherapy in pancreatic adenocarcinoma is multi-factorial, but certainly influenced by a notoriously immunosuppressive tumor microenvironment (TME) and lack of tumor-associated neo-antigens to stimulate an immune response in most patients. Therefore, systemic therapies for pancreatic cancer remain a significant unmet need.
Adoptive cellular therapy using tumor antigen-specific T cells has endured a remarkable evolution from its roots in the setting of malignant melanoma. The initial development of this approach was led by Dr. Steven Rosenberg and his colleagues in the 1980s using TILs isolated from melanoma patients (5–7). Decades of subsequent translational and clinical research by many talented investigators led to continued refinement of adoptive T cell therapy approaches and the critical factors that can be manipulated to maximize their efficacy. These advances include a multitude of factors including pre-conditioning regimens, optimal ex vivo expansion protocols, and co-administration of cytokines, among many others [Reviewed in (7, 8)]. Further refinement of antigen specificity was achieved in the 1990s following the development of gene transfer techniques that enabled introduction of chimeric antigen receptors (CARs) into T cells (7, 9, 10).
This review addresses the potential for CAR T cell therapy in the setting of pancreatic cancer. Herein we summarize both preclinical and early-phase clinical experience in CAR-mediated redirection of T cells. Key antigens of relevance to pancreatic ductal adenocarcinoma (PDAC) are discussed, along with innovative future directions of research occurring in this rapidly moving field.
CAR-T Cells Represent a Promising Therapeutic Modality
Adoptive transfer of lymphocytes continues to evolve as a treatment modality for advanced cancer. This general approach can leverage the versatility of T cells and their ability to be redirected toward relevant tumor antigens via engineered T cell receptors (TCRs) or CARs. Redirecting cell specificity via CARs represents one sophisticated approach that has gained traction in clinical care of hematologic malignancy. To generate the appropriate cell therapy product, T cells are collected from patient peripheral blood by leukapheresis and redirected to a specific antigen via viral expression of a Chimeric Antigen Receptor (CAR; Figure 1). To date, this approach has been widely utilized as an individualized therapy with genetic modification of autologous T cells from patients, although “off-the-shelf” CAR T cell approaches are beginning to emerge using T cells from allogeneic donors. The CAR constructs, when incorporated into T cells, mimic TCR activation, and redirect specificity and effector function toward an intended antigen, with the important advantage of eliciting recognition in a non-MHC-restricted manner (11).
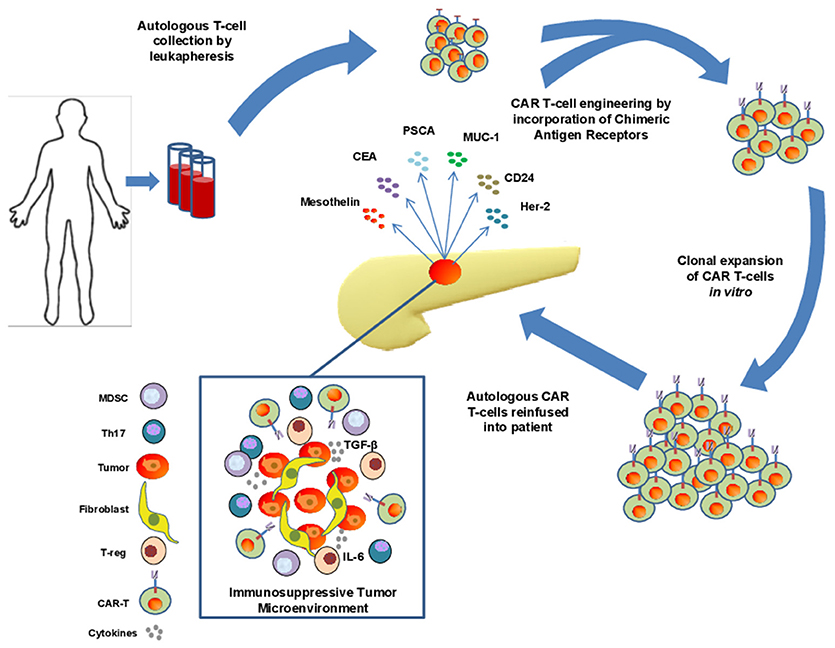
Figure 1. Isolation, engineering, and challenges of CAR T cell therapy in pancreatic adenocarcinoma (PDAC). T cells are collected from peripheral blood of patients with PDAC via leukapheresis and engineered to express chimeric antigen receptors directed toward a specific tumor antigen. These cells are subsequently expanded before reinfusion into patients. Significant challenges exist for these cells to infiltrate the immunosuppressive tumor microenvironement of PDAC including the presence of dense stroma and myofibroblast cells, immunosuppressive cytokines such as IL-6 and TGF-β, and the presence of immunosuppressive immune cell types such as Th17 cells, MDSCs, and suppressive T-regs.
The design of CARs continues to evolve, whereby the first-generation constructs contained an extracellular ligand-binding domain, such as a single chain variable fragment (scFv) that is directed toward a specific antigen, along with the CD3ζ or Fc receptor γ signaling domain (10, 11). Subsequent second generation or third generation CARs contain one or more costimulatory domains, respectively such as CD28, 4-1BB, ICOS or OX40 to selectively modify the function and/or persistence of the resultant CAR T cells (12–17). For example, inclusion of the CD28 domain can afford a more rapid expansion of the CAR T cells, while the 41BB domain is typically used to enhance in vivo persistence of the cells, albeit at a slower rate of expansion. These particular domains may also lead to a differential effect on redirected CD4+ or CD8+ T cell subsets. In a general sense, the CD28 molecule is recognized to more selectively promote expansion of naïve and CD4+ T cells, while the 41BB domain is more relevant to facilitating expansion of memory and CD8+ T cell subsets (17). Finally, development of antigen-redirected T cells has already entered the realm of an imaginative transition to utilizing fourth generation CAR constructs. These include highly sophisticated engineering that incorporates the ability of redirected T cells to produce immunomodulatory cytokines (e.g., IL-12, IL-18, IL-21) or antibodies (e.g., anti-PD-1), optimize trafficking via expression of chemokine receptors, and converting immune suppressive signals rich within the tumor microenvironment into activating signals that further enhance T cell activity. These and other constructs incorporating safety-intended suicide switches and receptors conferring bi-specificity and other signaling domains are in the early phases of both pre-clinical and clinical testing. These fourth generation CAR constructs and their application in solid tumors is the topic of a detailed, informative review by Knochelmann et al. (15).
Once CAR T cells are generated and undergo clonal expansion in culture, cells can be infused into patients to achieve antigen-directed, T cell mediated antitumor immune responses. In order to maximize in vivo expansion and decrease inhibitory lymphocytes that may persist systemically, patients are given a chemotherapy pre-conditioning regimen for lymphodepletion prior to infusion of CAR T cells (13, 14). Bulk T cell populations are typically reinfused as a cellular product for yield and logistical reasons, however the relative ratio of CD4+ vs. CD8+ T cells that are redirected toward antigen via CAR constructs may impact both the effector function and persistence of the cells. This emphasizes the complexity of implementing CAR T cell therapeutic approaches, and from a biological perspective, the importance of T cell help in generating effective antitumor immune responses.
Although dramatic efficacy can be achieved in patients following CAR T cell therapy, there is a risk for serious adverse events. In particular, cytokine release syndrome and neurological toxicity have been observed in patients, and illustrate the consequences of potent immune recognition of antigenic targets and the resulting robust immune response. In the context of pediatric ALL and other disease settings, administration of antibodies that block the interleukin-6 (IL-6) receptor are effective for reversal of the cytokine release syndrome (18), and allow patients to tolerate CAR T cell therapy. A provocative report by Fraietta et al. further showed that serum IL-6 correlated with CAR T cell expansion and that IL-6/STAT3 signaling may be important for proliferation of the redirected T cells in the setting of chronic lymphocytic leukemia (19). These data highlight the complexity of cytokine signaling in mediating toxicity and potential response related to CAR T cell therapy. More recent pre-clinical studies by Giavridis et al. report that IL-1 blockade may also be an effective means to control this cytokine release syndrome secondary to CAR T cell therapy (20). However, the concerns with neurotoxicity highlight the need for careful antigen selection, and patient monitoring during the course of these therapies.
Clinical Efficacy of Car T Cell Therapy in Hematologic Malignancy has Provided a Firm Foundation for Growth
A wealth of clinical experience with CAR T cell therapy has been derived from hematologic malignancy. Clinical efficacy of CAR T cell therapy was shown when Kochenderfer et al. reported a partial response with anti-CD19 CAR T cell therapy in a patient with treatment-refractory stage IVB follicular lymphoma (21). This was the first reported patient who achieved a clinical response with anti-CD19 CAR T cell therapy and it lasted 8 months before the patient developed progressive disease. Porter et al. utilized anti-CD19 CAR T cell therapy in a patient with refractory CLL and achieved a complete remission 3 weeks after treatment, which persisted for at least 10 months (22). CAR T cells persisted for 6 months at high levels in the peripheral blood and bone marrow. Two other patients treated with the same regimen achieved clinical response. These early findings prompted further development of a prospective studies in hematologic malignancies and produced overall response rates ranging from 52 to 92%. Complete response rates ranging from 43 to 90% were reported in subsequent clinical trials with anti-CD19 CAR T cell therapy in treatment refractory advanced lymphomas (18, 23–29). The ZUMA-1 and 2 trials, and ongoing ZUMA-3 and 4 trials reported high and durable response rates in patients with relapsed refractory large B cell lymphomas and with relapsed refractory pre-B cell ALL. Additionally, the phase II ELIANA trial of CTL019 CAR T cell therapy reported 75% relapse free probability at 6 months after remission, 89% probability of survival at 6 months, and 79% probability of survival at 12 months in pediatric and young adult patients with relapsed and refractory ALL (26, 30–33). This clinical trial provided the foundation for development of Tisagenlecleucel, CTL019, (Kymriah) which as a result was approved by the FDA for treatment of relapsed refractory B-cell precursor ALL in pediatric and young adult patients. This was followed by FDA-approval of axicabtagene ciloleucel (axi-cel; Yescarta), which targets CD19 in adult patients with relapsed refractory large B cell lymphoma after 2 lines of prior therapy (34).
Paving the Road for Car T Cells in PDAC With Preclinical Studies
A number of published studies demonstrate efficacy of CAR T cell therapy platforms in preclinical models of pancreatic cancer. The results to date are derived from multiple in vivo models, including transplantable human tumors, patient-derived xenografts, and in a more limited number of cases, immune competent mice with spontaneously-arising, or transplanted syngeneic tumors. One priority for the field remains the need to discover and validate new, bona-fide pancreatic cancer-specific antigens to minimize potential on-target, off-tumor adverse events. However, the existing data with several candidate antigens has infused some momentum into the field (Figure 2). For example, pre-clinical work on CAR T cells in pancreatic cancer targeting the tumor antigen mesothelin subsequently led to human studies. Mesothelin is a glycoprotein over-expressed on a variety of tumor cells including pancreatic cancer. This antigen is present on the cell surface, first as a 69-kDa protein, that later becomes cleaved via the furin protease into a soluble 32 kDa N-terminal fragment and a 40-kDa C-terminal fragment that remains membrane bound (44, 45). From a functional standpoint, data indicate mesothelin positively regulates tumor growth and invasion. The protein has relatively limited expression in normal tissue, where it is localized to mesothelial cells, including cells of the pleura, pericardium, peritoneum, fallopian tubes, trachea, tonsils, and cornea where its function is unclear (44, 46–48). One intriguing observation qualifying its role as an antigen of interest is that mesothelin-specific CD4+ and CD8+ T cell reactivity is evident in patients with pancreatic cancer (48–50). Mesothelin-directed CAR T cell therapy has been used widely across pre-clinical models in a variety of malignancies (51–54), including elegant studies in pancreatic cancer by Beatty et al. whereby T cells were redirected via mRNA electroporation of mesothelin-targeted CAR with CD3z and 4-1BB signaling domains to elicit a transient CAR expression for safety (55).
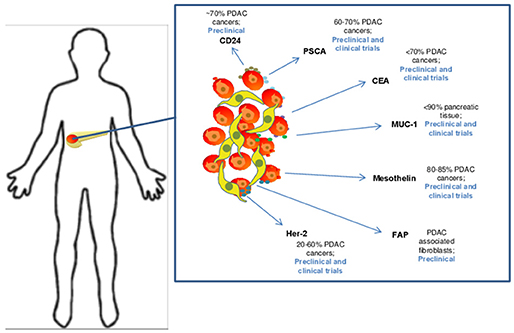
Figure 2. Candidate target antigens for CAR T cell therapy in pancreatic ductal adenocarcinoma. Several target antigens for CAR T cell therapy have been identified and are being studied in both preclinical and clinical trails. These antigens are expressed at varying levels on PDAC cells and include CD24 (35), Prostate stem cell antigen (PSCA) (36), CEA (37), MUC-1 (38), Mesothelin (39), FAP (40), and Her-2 (41).
An assortment of preclinical studies targeting antigens in preclinical models of PDAC continue to emerge (Figure 2). Among these are interesting data in which CAR T cells were redirected against CEA (CEACAM5), a long-standing antigen of interest for immune response in advanced gastrointestinal malignancy. The expression profile of CEA has made it a target for vaccine trials (56, 57). It is expressed in up to 70% of pancreatic cancers, leading to its potential as a tumor specific antigenic target. In a murine model of PDAC, CEA-redirected CAR T cells produced long-term anti-tumor responses with no evidence of damage to normal tissues with lower levels of CEA expression (58). Targeting of other tumor antigens including PSCA, HER2 neu CD24 and MUC1 has also demonstrated impressive activity in preclinical PDAC models (36, 59–62). Other innovative approaches for redirecting T cells are in development as our understanding into the complexity of tumor antigens in PDAC improves. For example, a recent study by Posey et al. directly addressed the poor expression of cancer-specific antigens on the cell surface by developing a CAR that recognized a Th glycoform of the MUC1 antigen. This creative work showed CAR T cells redirected against this aberrantly glycosylated antigen were efficacious in xenograft models of PDAC and leukemia (62). These data highlight the potential for re-invigorated antigen discovery when considering finer structure of proteins and their modifications that can be preferentially expressed on PDAC cells.
Overcoming Suppressive “Roadblocks” for Effective Car T Cell Therapy in Pancreatic Cancer
One notable challenge for CAR T cell therapy in preclinical models of PDAC is the inherent histologic properties and profound immunosuppressive capacity of this tumor type (Figure 1). For example, the desmoplastic reaction present in a majority of pancreatic tumors leads to a dense stroma that in theory, can serve as a physical barrier for drug delivery and penetration of endogenous or engineered T effector cells. Various components of the stroma can also serve as a source of abundant inhibitory cytokines, including IL-6 (63). In the context of other cytokines in the tumor microenvironment, such as TGF-β or IL-10 (42, 64) a cytokine milieu emerges which promotes expansion and enhanced suppressive function of cells such as T-regs, Th17 and myeloid derived suppressor cells (MDSC). Together these soluble and cellular factors can limit the function of any cytotoxic T cells that may be directed toward tumor antigens.
Overcoming this immunosuppressive microenvironment is a potential strategy to improve CAR T cell survival, trafficking and persistence in PDAC, and numerous pre-clinical studies addressing this concept are underway. For example, heparanase is under investigation as a means to overcome the physical barriers of the desmoplastic reaction, and in preclinical studies, was shown to increase antitumor activity and tumor infiltration of CAR redirected T lymphocytes (65). To address inefficient trafficking of CAR T cells into the tumor microenvironment, several studies have employed targeted approaches redirecting these cells to prominent cell surface molecules that are of relevance for pancreatic cancer. For example, the chemokine receptor CCR2 was successfully transduced into mesoCAR T cells, which increased T cell infiltration and antitumor activity compared to conventional mesoCAR T cell administration in immunodeficient mice with large mesothelin expressing tumors (66). Likewise, fibroblast activation protein (FAP) has been leveraged as a potential target antigen in the context of CAR T cells. This antigen is particularly unique given its expression on myofibroblast cells present within the pancreatic stroma. In an eloquent proof of principle study, Tran et al. developed a novel CAR T cell approach whereby cells were redirected to interact with FAP, and adoptively transferred in to mouse models bearing a variety of subcutaneous tumors including pancreatic adenocarcinoma. This approach was associated with cachexia and lethal bone toxicity, which most likely would limit its utilization as a universal target with CAR T cell therapy (40).
Another principal mediator of immune suppression is upregulation of immune checkpoint receptors including PD-1 on the surface of antigen-experienced T cells. Increased expression of PD-1 is associated with T cell exhaustion, and blocking the interaction of PD-1 with its ligands PD-L1 and PD-L2 has shown impressive clinical activity in oncology, leading to FDA-approval for several antibodies targeting this immunosuppressive axis (67). Similarly, CAR T cells upon infusion can upregulate the inhibitory, PD-1 receptor on their surface (68). Similar to unmanipulated T lymphocytes, PD-1 upregulation on CAR T cells can signify exhaustion of effector function, limiting the efficacy of CAR T cell based therapy. These observations provide rationale to combine CAR T cell therapy with antibodies targeting the PD-1/PD-L1 axis (69). This concept is being explored clinically in combination immunotherapy trials, and in sophisticated, fourth-generation CAR design strategies that allow for genetic ablation of PD-1 in antigen redirected cells (70–72) or even production of PD-1/PD-L1-targeted agents by the modified T cells themselves (73, 74). These advanced design approaches highlight the adaptability of T cells as a therapeutic platform and how they can be manipulated to overcome redundant mechanisms of immune suppression in cancer.
Clincial Experience With Car T Cells in Pancreatic Cancer
Preliminary results of case reports using CAR T cell therapy in pancreas and other non-hematologic malignancies encouraged development of multiple clinical trials in pancreatic cancer with CAR T cell therapy. Multiple different antigens specific to the tumor are being targeted in current ongoing clinical trials in non-hematologic malignancies including pancreas cancer (Table 1). Several antigens including CEA, HER2, MUC1, CD133, prostate stem cell antigen (PSCA) and mesothelin are prominent targets using this approach in pancreatic cancer (Figure 1). The challenges in developing CAR T cells in non-hematologic malignancies include lack of cell surface tumor-specific molecules that could be targeted by genetically engineered CAR T cells (75). The expression of the targeted antigen in normal and tumor tissues raises concern regarding potential off target toxicity (76, 77). Another challenge present clinically is the profound degree of immunosuppression that limits both the persistence of CAR T cells and their ability to effectively traffic to the tumor sites. Lymphodepletion has been shown in prior trials to enhance expansion, cytokine release, and persistence of the CAR T cells. Cyclophosphamide alone or in combination with fludarabine has been utilized in various CAR T cell therapy trials in hematologic malignancies as preconditioning regimens in order to achieve lymphodepletion prior to CAR T cell therapy. The success of CAR T cells depends on their ability to expand ex vivo while loss of central memory and naïve T cell subsets can be seen in heavily treated lymphoma patients due to multiple rounds of cytotoxic chemotherapy. This may also be a factor of relevance in patients with PDAC who may be candidates for experimental trials with CAR T cells, as most patients will have received systemic chemotherapy as standard of care. A recent report has shown that the quality of patient T cells could be a variable in the success of CAR T cell manufacturing, particularly loss of CD27/CD28 expression in heavily pre-treated lymphoma patients (78). Considering many lines of prior systemic therapy in non-hematologic malignancies prior to CAR T cell therapies similar challenges could be encountered in this area as well.
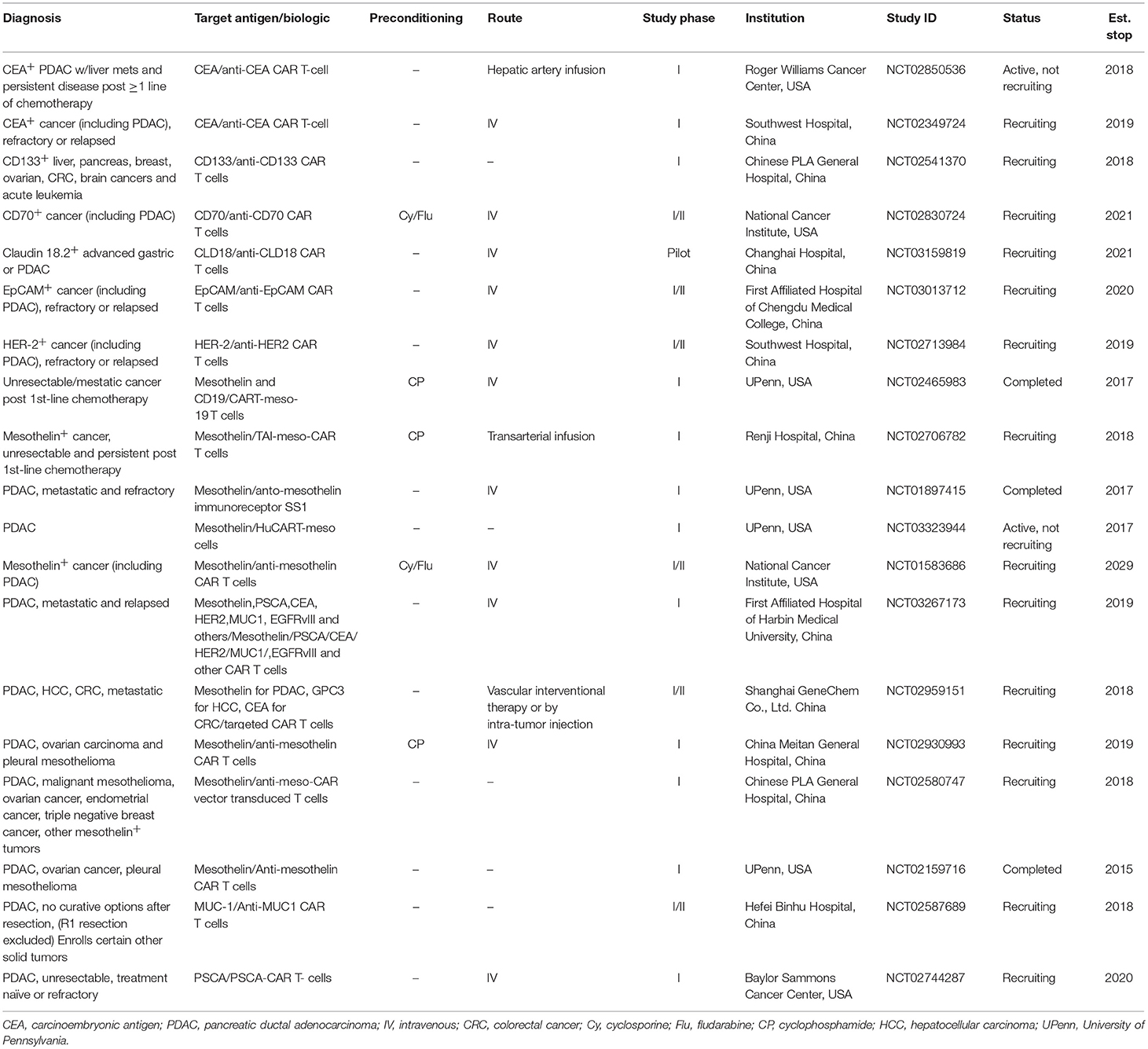
Table 1. Recently completed and ongoing clinical trials with CAR T cell therapy in pancreatic cancer.
Data from pre-clinical studies have permitted translation of CAR T cell therapy into several clinical trials for patients with pancreatic cancer (Table 1). Largely, target antigens used in patient trials have overlapped with the pre-clinical studies leading up to them. To date, the most experience with CAR T cells in PDAC patients has been gained through targeting mesothelin. Approximately 80–85% of pancreatic carcinomas express mesothelin and as a result, it has become a prominent target for CAR T cell therapy trials (39, 44). At the time of this review article, 10 separate clinical trials using CAR T cells directed against mesothelin have been completed or are in progress. The first results using mesothelin-directed CAR T cells (CARTmeso cells) were reported by Beatty et al. in a case report from two patients, one with malignant pleural mesothelioma (MPM) and a second with pancreatic cancer (55). In this trial, a major goal was to ensure safety given potential for expression of target antigens on normal tissues. To accomplish this, the authors developed a strategy for transient CAR expression via mRNA electroporation encoding an anti-mesothelin ss1 scFv CAR. Interestingly, in the patient with pancreatic cancer, the T cells were obtained from his monozygotic twin brother, activated, expanded with mRNA electroporation encoding the CAR. The CARTmeso cells were administered as eight doses of intravenous infusion followed by two intratumoral injections for the pancreatic cancer patient. No lymphodepleting therapy was given to either patient. Results indicated the CARTmeso transgene was detectable in peripheral blood in both patients after intravenous CARTmeso administration at pre-specified time points per the study; in the ascitic fluid 3 days after the initial intravenous infusion and 13 days after first intratumoral injection in the patient with pancreatic cancer. CARTmeso transcripts were also detected in the pre and post-pancreatic tumor biopsy tissue in the patient with the first intratumoral injection of CARTmeso cells. This finding indicated CARTmeso cell trafficking into the tumor tissue indeed occurred following intravenous administration. The patient with MPM achieved partial response, which lasted 6 months, and the patient with pancreatic cancer achieved stable disease.
In a more recent phase I study of CARTmeso cells by the same group, 6 patients with treatment refractory metastatic pancreatic cancer were administered CARTmeso cells intravenously 3 times per week for 3 weeks. In this trial, no dose limiting toxicity, cytokine release syndrome or neurological complications were encountered, while stable disease was reported in two of the patients (79). These findings were complemented by decreased FDG uptake on PET computed tomography, suggestive of activity against the tumor in these patients. This initial encouraging safety data with a more conservative, transient expression approach of CAR targeting mesothelin has prompted further attempts at targeting this antigen using more stable introduction of lentiviral constructs into T cells (NCT02159716). Several other trials are currently open to patients with mesothelin-expressing PDAC to further evaluate CARs directed at this antigen (NCT01583686, NCT02465983) (48).
Clinical experience with targeting other antigens via CAR T cells continues to emerge (Table 1). For example, multiple clinical trials are underway with CEA-directed CAR T cell therapy that include patients with pancreatic cancer (NCT02850536, NCT02349724). However, existing data with targeting CEA emphasize cautionary discretion is needed in subsequent clinical studies. In a feasibility study, anti-CEA CAR T cell therapy resulted in significant respiratory toxicities in patients with advanced CEACAM5+ gastrointestinal malignancies, including pancreatic cancer. These results necessitated premature closure of the study (80). Clinical trials with CAR T cells redirected against HER2 neu have also gained traction in the setting of PDAC, yet will proceed with caution given potential for toxicity. Namely, one clinical study was terminated early, after the first patient (with colorectal cancer) died as a result of pulmonary toxicity, cytokine storm, and multiorgan failure arising following infusion of the HER2 neu targeted CAR T cells (81). (NCT00924287) Other trials are ongoing in HER2 neu positive malignancies with CAR T cells redirected against this antigen (NCT02713984, NCT00889954) that will be informative once completed for assessment of both safety and potential for toxicity.
Equipping CAR T cells to engage tumors via specificity for other antigens in the setting of PDAC will continue to progress into clinical trials. This is a rapidly moving field that has extended beyond mesothelin, CEA and HER2 neu and into additional antigens including PSCA (36, 59, 60), and in MUC-1 (Table 1), among others. Of paramount importance will be careful consideration of on-target toxicities and antitumor efficacy, given the limited experience of adapting CAR T cells into solid tumor oncology.
While our understanding of CAR T cell-mediated adverse events remains in its early stages, combination immunotherapy approaches continue to enter clinical trials at a remarkably rapid pace (82). This momentum has been influenced by the success of immune checkpoint blockade, particularly with antibodies targeting PD-1 and CTLA-4 (67). There is a high level of interest in the field to develop treatment strategies that administer these antibodies, which can reverse T cell exhaustion with the antigen-specificity of CAR T cell therapy. Although a majority of this work is occurring in hematologic malignancy, its application to PDAC or other solid tumors is becoming accelerated. Novel studies are ongoing utilizing autologous CAR T cells redirected to recognize mesothelin and simultaneously engineered to express antibodies targeting PD-1 and CTLA-4 for recurrent and refractory non-hematologic malignancies (NCT03182803, NCT03030001). Another clinical trial is recruiting patients with non-hematologic tumors to evaluate the novel combination of autologous MUC1-redirected CAR T cells engineered to express antibodies targeting CTLA-4 and PD-1 (NCT03179007). Finally, sophisticated gene editing technologies such as CRISPR/Cas9 are being utilized to further engineer CAR T cells that lack genes encoding factors such as PD-1 that mediate an exhausted phenotype and function (71, 72) or alternatively aid in the design of universal, “off-the-shelf” CAR T cell approaches that lack self-recognition factors (83, 84). Other gene engineering strategies are also in development which incorporate suicide genes to improve the safety of these cell therapy strategies so that the therapeutic effect can be turned on or off, on demand (85).
Time for Custom Design: Future Advances for Car T Cell Therapy in Pancreatic Cancer
Further advances in customizing the design of CAR T cells continues to emerge and may overcome challenges related to persistence, features of the tumor microenvironment and optimizing phenotype (Figure 3). Guedan et al. addressed some of these challenges by studying how specific intracellular signaling domains (ICDs) such as ICOS or 41BB can be leveraged to enhance the persistence and effector phenotype, respectively (14). These data suggest CAR T cells can be customized to overcome limitations simply by adjusting the ICD of choice. Likewise, our understanding of the optimal cellular compartment to use as a “starting point” for generating CAR T cells continues to mature. While many approaches involve redirecting CD8+ T cells to take advantage of their effector function by redirecting mixed CD4+ and CD8+ populations or even more precise phenotypes such as T cells expressing high levels of the CD26 surface marker (43). These CD26high cells are apparently licensed with a natural capacity to traffic to and elicit antitumor activity in solid tumors including PDAC as shown in preclinical models. Other phenotypically distinct T cell subsets may also be well-suited for use in the setting of CAR T cell therapy. Other subsets including tissue-resident, stem-like T cells could also be considered as candidate cellular templates for CAR-mediated modification. In particular, a CD8+ T cell subpopulation expressing CXCR5 and the transcription factor, TCF-1 has been identified in mice that gives rise to new CD8+ effector cells from models of chronic LCMV infection and in tumors (86). In theory, these cells could be adaptable as a platform upon which to facilitate redirection of antigen specificity and in vivo expansion. Finally, CAR T cells are being designed that capitalize upon these abundant immunosuppressive features within the PDAC microenvironment that in theory can be leveraged as a switch to engage the full cytotoxic potential of CAR T cells. In a clever report, Sukumaran et al. rendered T cells responsive to immunologic features present exclusively at the tumor site by using a trio of CARs that recognize PSCA, TGF-β, and IL-4. These cells were capable of transmitting signals needed for activation, co-stimulation and cytokine support. T cells redirected via these novel constructs elicited antitumor activity against PDAC in vivo that was accompanied by T cell expansion and persistence (42). Other preclinical studies continue to provide a foundation for combinatorial therapy involving CAR T cells in PDAC, including combinatorial approaches with innate immune agonists such as STING (87), oncolytic viruses (88), or using FITC-labeled, tumor-directed antibodies to increase CAR T cell recognition (89).
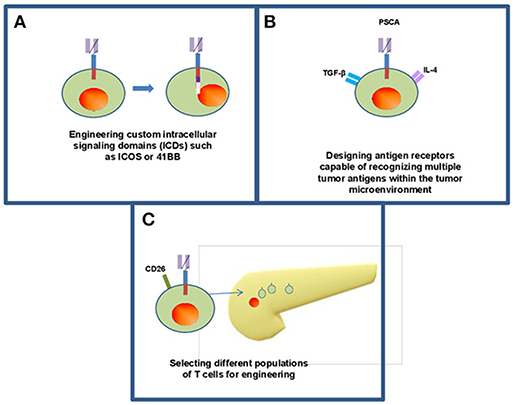
Figure 3. Frontiers in CAR T cell therapy for pancreatic ductal adenocarcinoma. Continuing research within CAR T cell therapy for solid organ malignancy has opened new methods for engineering T cells including enhancing intracellular signaling domains (A) to enhance persistence of CAR T cells within the body as well as efficacy against tumors. (B) Some groups have designed CARs that recognize multiple antigens within the tumor microenvironment while maintaining their antitumor activity within the body (42). (C) There is opportunity to utilize individual phenotypically defined populations of T cells including CD26 high cells (43).
Conclusions
The impressive results of CAR T cell therapy in hematologic malignancies and preliminary positive outcomes in non-hematologic tumors including pancreatic cancer prompted multiple clinical trials with CAR T cell therapy in this disease setting. Further understanding of the tumor microenvironment, improving off target effects of CAR T cell therapy, reasons of failures with single agent immunotherapy agents, and incorporation of novel agents in combination with CAR T cell therapy may help accomplish effective treatment outcomes in pancreatic cancer. While the extension of CAR T cell therapy into PDAC is just beginning, the field is moving rapidly, and embracing innovative technologies to mitigate potential toxicity and elicit antigen-direct tumor killing.
Author Contributions
MA, GL, and BE-R study concepts. MA, GL, and BE-R study design. MA, GL, BE-R, and EW quality control of data and algorithms. MA, GL, MZ, and BE-R manuscript preparation. MA, GL, BE-R, MZ, and EW manuscript editing. MA, MZ, GL, BE-R, and EW manuscript review.
Funding
NIH/NCI under award number P30CA138292 and 1R01CA208253-01 (GL).
Conflict of Interest Statement
GL, Consultant, ProDa Biotech, LLC, and research support from Merck, Inc.; Vaccinex, Inc. BE-R, Research support from Novartis, BMS, and Merck.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. (2017) 67:7–30. doi: 10.3322/caac.21387
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. (2013) 369:1691–703. doi: 10.1056/NEJMoa1304369
4. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. (2011) 364:1817–25. doi: 10.1056/NEJMoa1011923
5. Boon T, Cerottini JC, Van den Eynde B, van der Bruggen p, Van Pel A. Tumor antigens recognized by T lymphocytes. Annu Rev Immunol. (1994) 12:337–65. doi: 10.1146/annurev.iy.12.040194.002005
6. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science (1991) 254:1643–7.
7. Yang JC, Rosenberg SA. Adoptive T-Cell therapy for cancer. Adv. Immunol. (2016) 130:279–94. doi: 10.1016/bs.ai.2015.12.006
8. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature (2017) 545:423–31. doi: 10.1038/nature22395
9. Eshhar Z, Waks T, Gross G. The emergence of T-bodies/CAR T cells. Cancer J. (2014) 20:123–6. doi: 10.1097/PPO.0000000000000027
10. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. (1993) 90:720–4. doi: 10.1073/pnas.90.2.720
11. Srivastava S, Riddell SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol. (2018) 200:459–68. doi: 10.4049/jimmunol.1701155
12. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
13. Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood (2014) 124:1070–80. doi: 10.1182/blood-2013-10-535245
14. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel R, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight (2018) 3:96976. doi: 10.1172/jci.insight.96976
15. Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. (2018) 9:1740. doi: 10.3389/fimmu.2018.01740
16. Shen CJ, Yang YX, Han EQ, Cao N, Wang YF, Wang Y, et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J Hematol Oncol. (2013) 6:33. doi: 10.1186/1756-8722-6-33
17. Zhang H, Snyder KM, Suhoski MM, Maus MV, Kapoor V, June CH, et al. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J Immunol. (2007) 179:4910–8. doi: 10.4049/jimmunol.179.7.4910
18. Maude SL, Frey N, Shaw A, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
19. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
20. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M, CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
21. Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood (2010) 116:4099–102. doi: 10.1182/blood-2010-04-281931
22. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
23. Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. J Clin Oncol. (2017) 35:1803–13. doi: 10.1200/JCO.2016.71.3024
24. Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood (2012) 119:2709–20. doi: 10.1182/blood-2011-10-384388
25. Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. (2015) 33:540–9. doi: 10.1200/JCO.2014.56.2025
26. Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. (2017) 25:285–95. doi: 10.1016/j.ymthe.2016.10.020
27. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. (2017) 377:2531–44. doi: 10.1056/NEJMoa1707447
28. Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood (2017) 130:1800–8. doi: 10.1182/blood-2017-03-769620
29. Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak O, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. (2017) 377:2545–54 doi: 10.1056/NEJMoa1708566
30. Buechner J, Grupp SA, Maude SL, Boyer M, Bittencourt H, Laetsch TW, et al. Global registration trial of efficacy and safety of CTL019 in pediatric and young adult patients with relapsed/refractory (r/r) acute lymphoblastic leukemia (ALL): update to the interim analysis. Eur Hematol Assoc Ann Congr. (2017) 102:s476.
31. Wang M, Lundry Locke F, Munoz J, Goy A, Holmes HE, Siddiqi T, et al. ZUMA-2: A phase 2 multi-center study evaluating the efficacy of KTE-C19 (Anti-CD19 CAR T cells) in patients with relapsed/refractory Mantle cell lymphoma (R/R MCL). Ann Oncol. (2016) 27(Suppl. 6):945TiP. doi: 10.1093/annonc/mdw375.40
32. Shah B, Castro J, Gökbuget N, José Kersten M, Hagenbeek T, Wierda W, et al. ZUMA-3: a phase 1/2 multi-center study evaluation the safety and efficacy of KTE-C19 anti-CD19 CAR T cells in adult patients with relapsed/refractory B precursor acute lymphoblastic leukemia (R/R ALL). Ann Oncol. (2016) 27(Suppl. 6):415TiP. doi: 10.1093/annonc/mdw368.58
33. Wayne AS, Sender LS, Lee DW, Handgretinger R, Brown PA, Jiang Y, et al. ZUMA-4: a phase 1/2 multicenter study evaluating the safety and efficacy of KTE-C19 (anti-CD19 CAR T cells) in pediatric and adolescent subjects with relapsed/refractory B-precursor acute lymphoblastic leukemia (r/r ALL). J Clin Oncol. (2016) 34(15 Suppl.):TPS7075. doi: 10.1200/JCO.2016.34.15_suppl.TPS7075
34. FDA approves second CAR T-cell therapy. Cancer Discov. (2018) 8:5–6. doi: 10.1158/2159-8290.CD-NB2017-155
35. Jacob J, Bellach J, Grutzmann R, Alldinger I, Pilarsky C, Dietel M, et al. Expression of CD24 in adenocarcinomas of the pancreas correlates with higher tumor grades. Pancreatology (2004) 4:454–60. doi: 10.1159/000079824
36. Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. (2001) 61:4320–4.
37. Gansauge S, Gansauge F, Beger HG Molecular oncology in pancreatic cancer. J Mol Med. (1996) 74:313–20.
38. Qu CF, Li Y, Song YJ, Rizvi SM, Raja C, Zhang D, et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by (213)Bi-C595 radioimmunoconjugate. Br J Cancer (2004) 91:2086–93. doi: 10.1038/sj.bjc.6602232
39. Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. (2001) 7:3862–8.
40. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. (2013) 210:1125–35. doi: 10.1084/jem.20130110
41. Komoto M, Nakata B, Amano R, Yamada N, Yashiro M, Ohira M, et al. HER2 overexpression correlates with survival after curative resection of pancreatic cancer. Cancer Sci. (2009) 100:1243–7. doi: 10.1111/j.1349-7006.2009.01176.x
42. Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. (2018) 8:972–87. doi: 10.1158/2159-8290.CD-17-1298
43. Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun. (2017) 8:1961. doi: 10.1038/s41467-017-01867-9
44. Morello A, Sadelain M, Adusumilli S. mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. (2016) 6:133–46. doi: 10.1158/2159-8290.CD-15-0583
45. Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res. (2014) 74:2907–12. doi: 10.1158/0008-5472.CAN-14-0337
46. Chang K, Pastan I, Willingham MC. Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int J Cancer (1992) 50:373–81. doi: 10.1002/ijc.2910500308
47. Jirsova K, Neuwirth A, Kalasova S, Vesela V, Merjava S. Mesothelial proteins are expressed in the human cornea. Exp Eye Res. (2010) 91:623–9. doi: 10.1016/j.exer.2010.08.002
48. O'Hara M, Stashwick C, Haas AR, Tanyi JL. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy (2016) 8:449–60. doi: 10.2217/imt.16.4
49. Chen Y, Ayaru L, Mathew S, Morris E, Pereira SP, Behboudi S. Expansion of anti-mesothelin specific CD4+ and CD8+ T cell responses in patients with pancreatic carcinoma. PLoS ONE (2014) 9:e88133. doi: 10.1371/journal.pone.0088133
50. Thomas AM, Santarsiero LM, Lutz ER, Armstrong TD, Chen YC, Huang LQ, et al. Mesothelin-specific CD8(+) T cell responses provide evidence of in vivo cross-priming by antigen-presenting cells in vaccinated pancreatic cancer patients. J Exp Med. (2004) 200:297–306. doi: 10.1084/jem.20031435
51. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. (2014) 6:261ra151. doi: 10.1126/scitranslmed.3010162
52. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. (2009) 106:3360–5. doi: 10.1073/pnas.0813101106
53. Jiang H, Song B, Wang P, Shi B, Li Q, Fan M, et al. Efficient growth suppression in pancreatic cancer PDX model by fully human anti-mesothelin CAR-T cells. Protein Cell (2017) 8:926–31. doi: 10.1007/s13238-017-0472-9
54. Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther. (2012) 20:633–43. doi: 10.1038/mt.2011.256
55. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. (2014) 2:112–20. doi: 10.1158/2326-6066.CIR-13-0170
56. Marshall J. Carcinoembryonic antigen-based vaccines. Semin Oncol. (2003) 30(3 Suppl. 8):30–6. doi: 10.1016/S0093-7754(03)00233-1
57. Berinstein NL. Carcinoembryonic antigen as a target for therapeutic anticancer vaccines: a review. J Clin Oncol. (2002) 20:2197–207. doi: 10.1200/JCO.2002.08.017
58. Chmielewski M, Hahn O, Rappl G, Nowak M, Schmidt-Wolf IH, Hombach AA, et al. T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology (2012) 143:1095–107.e2. doi: 10.1053/j.gastro.2012.06.037
59. Abate-Daga D, Lagisetty KH, Tran E, Zheng Z, Gattinoni L, Yu Z, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. (2014) 25:1003–12. doi: 10.1089/hum.2013.209
60. Katari UL, Keirnan JM, Worth AC, Hodges SE, Leen AM, Fisher WE, et al. Engineered T cells for pancreatic cancer treatment. HPB (2011) 13:643–50. doi: 10.1111/j.1477-2574.2011.00344.x
61. Maliar A, Servais C, Waks T, Chmielewski M, Lavy R, Altevogt P, et al. Redirected T cells that target pancreatic adenocarcinoma antigens eliminate tumors and metastases in mice. Gastroenterology (2012) 143:1375–84.e5. doi: 10.1053/j.gastro.2012.07.017
62. Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
63. Varghese AM. Chimeric antigen receptor (CAR) T and other T cell strategies for pancreas adenocarcinoma. Chin Clin Oncol. (2017) 6:66. doi: 10.21037/cco.2017.09.04
64. Batchu RB, Gruzdyn OV, Mahmud EM, Chukr F, Dachepalli R, Manmari SK, et al. Inhibition of Interleukin-10 in the tumor microenvironment can restore mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery (2018) 163:627–32. doi: 10.1016/j.surg.2017.10.056
65. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
66. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
67. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
68. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
69. Abate-Daga D, Hanada K, Davis JL, Yang JC, Rosenberg SA, Morgan RA. Expression profiling of TCR-engineered T cells demonstrates overexpression of multiple inhibitory receptors in persisting lymphocytes. Blood (2013) 122:1399–410. doi: 10.1182/blood-2013-04-495531
70. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
71. Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-Specific CAR T Cells. Hum Gene Ther. (2018). doi: 10.1089/hum.2017.234. [Epub ahead of print].
72. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. (2017) 23:2255–66. doi: 10.1158/1078-0432.CCR-16-1300
73. Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. (2017) 23:6982–92. doi: 10.1158/1078-0432.CCR-17-0867
74. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
75. Tran E, Longo DL, Urba WJ. A milestone for CAR T cells. N Engl J Med. (2017) 377:2593–6. doi: 10.1056/NEJMe1714680
76. Zhang BL, Qin DY, Mo ZM, Li Y, Wei W, Wang YS, et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci China Life Sci. (2016) 59:340–8. doi: 10.1007/s11427-016-5027-4
77. Beatty GL. Engineered chimeric antigen receptor-expressing T cells for the treatment of pancreatic ductal adenocarcinoma. Oncoimmunology (2014) 3:e28327. doi: 10.4161/onci.28327
78. Petersen CT, Hassan M, Morris AB, Jeffery J, Lee K, Jagirdar N, et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv. (2018) 2:210–23. doi: 10.1182/bloodadvances.2017011254
79. Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
80. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
81. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
82. Popovic A, Jaffee EM, Zaidi N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J Clin Invest. (2018) 128:3209–18. doi: 10.1172/JCI120775
83. Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia (2018) 32:1970–83. doi: 10.1038/s41375-018-0065-5
84. Torikai H, Cooper LJ. Translational Implications for off-the-shelf immune cells expressing chimeric antigen receptors. Mol Ther. (2016) 24:1178–86. doi: 10.1038/mt.2016.106
85. Singh N, Shi J, June CH, Ruella M. Genome-editing technologies in adoptive T cell immunotherapy for cancer. Curr Hematol Malig Rep. (2017) 12:522–9. doi: 10.1007/s11899-017-0417-7
86. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
87. Smith TT, Moffett HF, Stephan SB, Opel CF, Dumigan AG, Jiang X, et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J Clin Invest. (2017) 127:2176–91. doi: 10.1172/JCI87624
88. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight (2018) 3:99573. doi: 10.1172/jci.insight.99573
Keywords: pancreas cancer, mesothelin, adoptive T cell therapy, CAR T cells, genetically engineered T cells
Citation: Akce M, Zaidi MY, Waller EK, El-Rayes BF and Lesinski GB (2018) The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 9:2166. doi: 10.3389/fimmu.2018.02166
Received: 08 May 2018; Accepted: 03 September 2018;
Published: 25 September 2018.
Edited by:
Masoud H. Manjili, Virginia Commonwealth University, United StatesReviewed by:
Chrystal M. Paulos, Hollings Cancer Center, United StatesUlrike Koehl, Hannover Medical School, Germany
Copyright © 2018 Akce, Zaidi, Waller, El-Rayes and Lesinski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gregory B. Lesinski, Z3JlZ29yeS5iLmxlc2luc2tpQGVtb3J5LmVkdQ==