 Kim Blom1*
Kim Blom1* Angelica Cuapio1
Angelica Cuapio1 J. Tyler Sandberg1
J. Tyler Sandberg1 Renata Varnaite1
Renata Varnaite1 Jakob Michaëlsson1
Jakob Michaëlsson1 Niklas K. Björkström1
Niklas K. Björkström1 Johan K. Sandberg1
Johan K. Sandberg1 Jonas Klingström1Lars Lindquist2,3Sara Gredmark Russ1,2
Jonas Klingström1Lars Lindquist2,3Sara Gredmark Russ1,2 Hans-Gustaf Ljunggren1*
Hans-Gustaf Ljunggren1*- 1Department of Medicine Huddinge, Center for Infectious Medicine, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
- 2Department of Infectious Diseases, Karolinska University Hospital, Stockholm, Sweden
- 3Unit of Infectious Diseases, Department of Medicine Huddinge, Karolinska Institutet, Karolinska University Hospital, Stockholm, Sweden
Tick-borne encephalitis virus (TBEV) is a flavivirus that belongs to the Flaviviridae family. TBEV is transmitted to humans primarily from infected ticks. The virus causes tick-borne encephalitis (TBE), an acute viral disease that affects the central nervous system (CNS). Infection can lead to acute neurological symptoms of significant severity due to meningitis or meningo(myelo)encephalitis. TBE can cause long-term suffering and has been recognized as an increasing public health problem. TBEV-affected areas currently include large parts of central and northern Europe as well as northern Asia. Infection with TBEV triggers a humoral as well as a cell-mediated immune response. In contrast to the well-characterized humoral antibody-mediated response, the cell-mediated immune responses elicited to natural TBEV-infection have been poorly characterized until recently. Here, we review recent progress in our understanding of the cell-mediated immune response to human TBEV-infection. A particular emphasis is devoted to studies of the response mediated by natural killer (NK) cells and CD8 T cells. The studies described include results revealing the temporal dynamics of the T cell- as well as NK cell-responses in relation to disease state and functional characterization of these cells. Additionally, we discuss specific immunopathological aspects of TBEV-infection in the CNS.
Introduction
Tick-borne encephalitis virus (TBEV) is a flavivirus that belongs to the Flaviviridae family. Flaviviruses comprise many human pathogens including the commonly known Dengue virus (DENV), Japanese encephalitis virus (JEV), West Nile virus (WNV), Yellow fever virus (YFV), and Zika virus (ZIKV) (1). With respect to TBEV, three subtypes of the virus exist: European (TBEV-Eu), Siberian (TBEV-Sib), and Far Eastern (TBEV-FE) (2).
TBEV is transmitted to humans primarily from infected ticks, mainly from the Ixodes family. The virus can also be transmitted from unpasteurized dairy products from infected livestock (3–5). Infection with TBEV causes tick-borne encephalitis (TBE), an acute viral infection that affects the central nervous system (CNS) with often severe long-term neurological consequences (3, 4, 6, 7). The first TBE-like disease was described as early as in the eighteenth century in Scandinavian church records (8). Traditionally, the disease is described as a syndrome with a biphasic course beginning with an influenza-like illness followed by a second neuroinvasive phase with neurological symptoms of variable severity, ranging from meningitis to severe meningoencephalitis with or without myelitis (3, 4, 6) (Figure 1). It shall be noted, however, that also monophasic patterns of disease development have been described (9). Upon infection, virus is detected in serum in the first phase of the disease but rarely in the second phase (10).
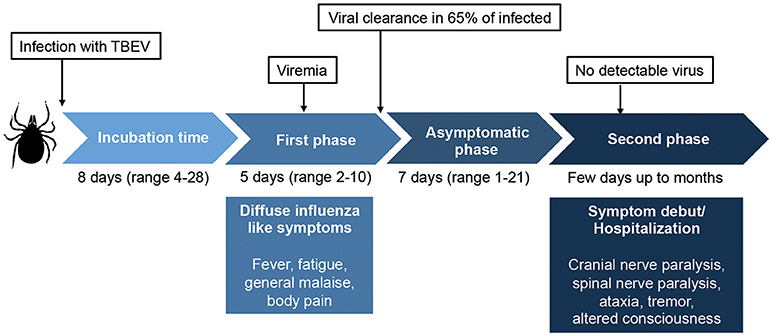
Figure 1. Overview of the classic biphasic disease-pattern of human TBEV infection. The viremic first phase includes influenza-like symptoms and occurs around 1 week after virus transmission. It is estimated that 65–70% of infected individuals clear the virus after this phase, but for one third of the patients, an asymptomatic disease phase follows before the second phase of disease begins. In this phase, symptoms of meningitis or encephalitis occur, including fever, headache, tremor, nystagmus, altered state of consciousness, cranial nerve paralysis, and spinal nerve paralysis. Classically, no virus is detected in sera or plasma in the second phase of disease. Around 30% of patients that enter the second phase of disease will suffer from long lasting sequeale, with a decreased quality of life. Figure compiled from Lindquist and Vapalahti (3), Taba et al. (4), and Haglund and Gunther (6).
Due to increased geographic distribution of TBEV as well as a marked increase in morbidity in many areas, TBEV-infection has more recently caught attention as a public health problem. TBE is now observed in large parts of Europe as well as in northern Asia (3, 4). The main risk areas for TBE in Europe are primarily parts of central and eastern Europe as well as the Baltic and Nordic countries. With respect to central Europe, risk areas extend from Switzerland in the west into northern Italy and the Balkan countries (11). The incidence of TBEV-infection in endemic countries varies from year to year (12–14), however, an overall upsurge has been reported in certain parts of Europe, including the borders between Austria, Slovenia, and Italy (15, 16). These changes have been related to climatic, ecological, environmental, and socioeconomic factors that all can lead to an increased risk of human exposure to infected ticks (17–20).
The total number of annual cases has been estimated to be up to 13,000, and as such the infection constitutes the most important tick-borne viral disease (4). More than 30% of patients with clinical symptoms from TBEV-infection develop prolonged sequelae, some of which may become life-long including neuropsychiatric symptoms, severe headaches, and a general decrease in quality of life (3, 4, 6, 7). The mortality rates differ between the strains. Infection with the Far Eastern strain (TBEV-FE) has a mortality rate of 5–35%, whereas the other two strains (TBEV-Eu and TBEV-Sib) have mortality rates of 1–3% (3, 4). There is no specific treatment (e.g., antivirals) for TBE; rather, symptomatic treatment is the only available option (3, 4, 9).
Of importance, TBE may be prevented by vaccination. There are in total four licensed vaccines to TBE. Two vaccines based on TBE-Eu subtype are licensed in Europe and two are licensed in Russia. Additionally, a TBEV-vaccine based on the Far Eastern subtype is produced and marketed in China. All vaccines are based on formalin-inactivated strains of TBEV (3, 4, 21, 22). In areas where the disease is highly endemic, WHO recommends that vaccination should be offered to all groups above 1 year of age (4, 23). Primary vaccination against TBE includes three doses of the vaccine within the first year, followed by revaccinations every third to fifth year to maintain immunity. Vaccination is generally considered effective and TBE incidence has decreased substantially in TBEV-endemic regions with successful vaccination-programs (24). Randomized controlled trials in large populations have shown high immunogenicity with often-strong antibody production and acceptable rates of adverse events following vaccination (25–28). Breakthrough TBE after vaccination is generally considered rare (4). However, over the last years, vaccine failures have been reported, in particular in middle-aged and elderly individuals, who have completed the primary vaccination (29–31).
Infection with TBEV triggers humoral and cell-mediated immune responses. A confirmed diagnosis of TBE is established by the detection of specific IgM and IgG in serum. IgM antibodies have been observed in sera very early in symptomatic TBE disease, whereas IgG antibodies peak in the convalescent phase of disease (32). IgG antibodies can persist over lifetime and prevent TBE (4, 33). Early after clinical disease onset, TBEV-specific antibodies can also be found in the cerebrospinal fluid (CSF) (32, 33). In contrast to the humoral immune response, the cell-mediated immune responses elicited to natural infection have been rather poorly studied until recently. The latter responses may contribute both to host resistance against infection as well as to pathological reactions affecting the target organ of the virus, i.e., primarily the CNS.
Here, we review recent progress in studies of the cell-mediated immune response to human TBEV infection. A particular emphasis is devoted to natural killer (NK) cell- and T cell-mediated responses. Responses to TBEV are discussed in context of cell-mediated immune responses toward other flavivirus infections. We also discuss some immunopathological aspects of TBE with a particular emphasis on cell-mediated immune reactions in the CNS. Cell-mediated immune reactions in the CNS may contribute to neural damage with severe consequences of brain function, and could in the worst cases lead to fatal outcome. First, however, some aspects of the TBEV itself are covered.
TBEV and Other Flaviviruses
All flaviviruses are enveloped and have a positive-sense single stranded RNA genome, which per se acts as messenger RNA upon entrance in the host cell. The RNA encodes for a polyprotein, which is co- and post-translationally cleaved by viral and cellular proteases into three structural proteins; capsid (C), precursor membrane (prM) and envelope glycoproteins (E), and seven non-structural proteins including NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 (34). Flaviviruses enter the cell through clathrin-dependent endocytosis upon attachment of the E protein to a receptor. Heparan sulfate has been identified as such receptor for TBEV (35); however, there are most likely also other yet not identified receptors for the virus. Following cell entry, the flavivirus is delivered to endosomes (36), in which the low pH triggers the E protein to fuse with the endosomal membrane and the nucleocapsid is released into the cytosol. The assembly of immature flavivirus virions (36–38), including TBEV (39, 40), occurs in the ER, and the viral particles are transported to the Golgi apparatus. The virion particles are immature until the envelope protein is rearranged and prM is cleaved by the host enzyme furin in the acidic environment in the Golgi apparatus. Immature particles are non-infectious and proteolytic cleavage of prM is a prerequisite for viral infectivity. However, studies have shown that complete cleavage of prM is not necessary for viral infectivity (41–43).
In general, species of flaviviruses have many similarities, but their preferred host cells differ. TBEV is shown to replicate 10,000-fold higher in human neuronal cells as compared with epithelial cells (44). A similar infection pattern has recently been shown for ZIKV (45).
NK Cells
NK cells are innate lymphocytes, though recent studies have revealed “adaptive” features of these cells (46, 47). They are perhaps best known for their ability to kill virus-infected and tumor cells. NK cell cytotoxicity is regulated by the expression of numerous activating and inhibitory receptors that sense ligands on neighboring healthy and altered cells. Several activating receptors recognize molecules that are up-regulated on cells during conditions of cellular stress, such as viral infection and transformation [reviewed in (48, 49)] whereas many inhibitory receptors, e.g., human killer cell Ig-like receptors (KIR) bind to HLA class I molecules. Additionally, NK cells have an important role in producing cytokines and chemokines, as well as by other means interacting with other immune and non-immune cells.
Human NK cells are classically defined as CD3− (T cell receptor negative), CD56+ cells and represent about 15% of peripheral blood lymphocytes. These cells have for long been divided into two main subsets; CD56bright and CD56dim cells (50). The CD56bright NK cells are thought to be less mature and are commonly known as primarily cytokine-producing cells with low cytotoxic ability, whereas CD56dim NK cells are best known for their potent cytotoxic activity upon target cell recognition (51). However, the latter are also ample cytokine producers upon interaction with target cells (51). Both “natural” and antibody-mediated NK cell cytotoxicity is mediated by exocytosis of cytoplasmic granules containing perforin and granzymes (52). Cytotoxic responses may also to various degrees involve TRAIL- and Fas-ligand-mediated induction of apoptosis (53, 54). CD56dim NK cells frequently express CD16 (FcγRIII), KIRs, and CD57, which regulate their function and define distinct stages of NK cell maturation (55), whereas CD56bright NK cells largely lack expression of these molecules.
The Role of NK Cells in Human TBEV Infection
Direct evidence for a protective role of NK cells has been found in experimental models of several viral infections, including cytomegalovirus and influenza, and a number of studies have indicated that they play a role also in protection against viral infections in humans. For example, NK cell-deficiencies in humans result in severe herpes virus infections in childhood and adolescence (56). NK cells may also have a protective role in human TBEV-infection. At the same time, responses mediated by these cells may be associated with development of symptoms in the course of TBEV-infection. Although there is only little known about NK cells in TBE, NK cells have been detected in CSF of patients with TBE (57), an observation that indicates transmigration through the blood brain barrier (BBB).
To gain a better understanding of the NK cell response to human TBEV-infection, we recently performed a longitudinal study providing an in-depth analysis of the human NK cell response to acute TBEV-infection in a well-defined cohort of TBE patients. The study had an emphasis on NK cell responses during the second stage of disease from which clinical samples were available. NK cell activation, as measured by expression of the proliferation marker Ki67, was apparent at the time of hospitalization (58) (illustrated in Figure 2). Concomitant with the increase in NK cell activation in the acute stage of disease, augmented levels of IL-12, IL-15, IL-18, IFN-γ, and TNF were detected in patient plasma. In parallel with high levels of activation, the activated NK cells expressed less perforin, granzyme B, and Bcl-2. By 3 weeks after hospitalization, the NK cell activation decreased to levels seen in healthy controls. This TBEV-induced NK cell activation was restricted predominantly to more differentiated CD57+CD56dim NK cells. Functionally, CD56dim NK cells responded poorly to target cells at the time of hospitalization, but they recovered functional capacity to healthy control levels during the convalescent phase. The poor functionality of NK cell responses was exclusive for target cell recognition, since NK cell responses induced by IL-18 and IL-12 remained unchanged throughout the disease (58).
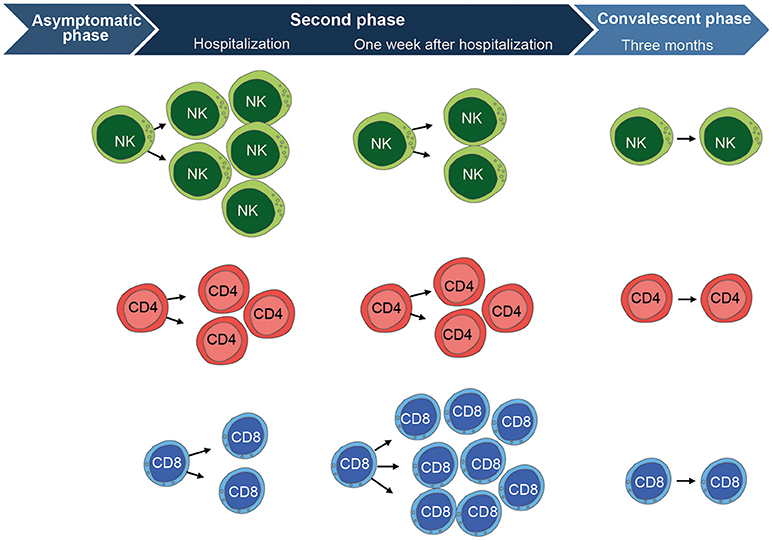
Figure 2. Overview of the cell-mediated immune response to TBEV-infection. CD56dim NK cells (mainly highly differentiated CD57+ cells) are highly activated at the time of hospitalization during the second phase of disease. They express significantly higher levels of Ki67, CD38, and produce less cytokines in response to target cells as compared to the convalescent phase. NK cells then become fully normalized, comparable to healthy control levels, at the convalescent phase as assessed 3 months after hospitalization. CD4 T cells show a similar pattern as the CD56dim NK cells. They are activated at hospitalization and express significantly higher levels of Ki67, CD38, HLA-DR, and Granzyme B as compared to the convalescent phase. CD4 T cells have retracted to normal healthy control levels at the convalescent phase. CD8 T cells show a different pattern and peak in activation at 1 week after hospitalization. They express significantly higher levels of Ki67, CD38, HLA-DR, Granzyme B, Perforin, PD-1, T-bet, and Eomesodermin as compared CD8 T cells at the convalescent phase. The CD8 T cell activation subsequently return to normal healthy control levels at the convalescent phase, just as is the case for CD4 T cells and NK cells. Figure compiled from Blom et al. (58) and Blom et al. (59).
NK Cell Responses Toward Other Acute Flavivirus Infections in Humans
To be able to interpret the above-mentioned NK cell responses to acute TBEV infection, it is important to understand NK cell responses to other acute virus infections, including acute flavivirus infections. In this respect, NK cells have to various extent been studied ex vivo in other acute flavivirus infections, including DENV (60, 61) and WNV (62, 63) as well as hepatitis C virus (HCV), a distant relative within the Flaviviridae family (64, 65). NK cells have also been studied after vaccination with the live attenuated YFV 17D vaccine (66–68). They have been suggested to influence disease severity and outcome, and to contribute to viral control in these infections, even though underlying mechanisms are not well studied.
In this context, it was observed that the absolute number of NK cells in patients with a mild form of infection with DENV was higher as compared to patients with the more severe form of the infection, dengue hemorrhagic fever (DHF) (69). Reduced numbers of NK cells in the circulation may be indicative of migration toward peripheral target organs. Furthermore, a higher frequency of NK cells expressing CD69 early on during the infection in children developing severe DHF has been reported (70). In recent studies of DENV-infection, we found NK cells to be robustly activated during the first week after symptom debut. Here, the response seemed to be confined largely to the CD56bright subset of NK cells and less mature CD56dim NK cells (our own unpublished studies). Noteworthy in the context of acute TBEV infection, activation of NK cells may also occur very early, even before the onset of symptoms. This possibility is supported by the observation that the highest levels of NK cell activation in most TBEV infected patients were observed already at the time of hospitalization (58). This notion is corroborated in studies of YFV vaccinated individuals. An early response by NK cells was observed in study subjects vaccinated with YFV-17D, where expression of both Ki67 and CD69 was increased on NK cells as early as 1 week after vaccination (66). Accumulation of adaptive-like NK cells expressing the activating receptor CD94/NKG2C has been reported in some human viral infections (71–73); however, no expansion of NKG2C+ NK cells in blood has been observed in TBEV-infection or any other flavivirus infection (58). It can, however, not be excluded that this type of expansion could occur locally, e.g., at the site of infection.
In addition to the observed activation of NK cells in vivo in different flavivirus infections, a protective role of NK cells is also supported by in vitro data. For example, primary activated human NK cells have been shown to inhibit WNV-infection of Vero cells (63) and IFNα-activated NK cells can kill HCV-infected hepatoma cells in vitro (65). In addition, flavivirus-infected target cells have been reported to display virus-mediated up-regulation of MHC class I (74), and could thereby theoretically evade lysis from NK cells by engaging inhibitory receptors. The dampened NK cell responses to target cells in acute TBEV-infection further support this notion (58). On the other hand, increased MHC class I expression could result in enhanced T cell responses. In such a scenario, one may speculate that flaviviruses may have been driven more toward escape from innate immunity rather than from adaptive T cell immunity (75).
T Cells
In contrast to NK cells, CD8 and CD4 T cells recognize specific foreign peptide sequences presented by HLA class I and II molecules, respectively (76). Like NK cells, major functions of CD8 T cells are to kill infected cells through the release of perforin and granzymes, and to secrete cytokines such as IFN-γ, TNF, and IL-2. The cytotoxic T cell response to acute infection can typically be divided into three phases; priming and expansion, resolution and contraction, and memory formation. During the first phase, naïve CD8 T cells divide and differentiate into effector cells acquiring high cytotoxic ability (77). Following viral clearance, the effector T cell population contracts and the majority of the pathogen-specific T cells enter apoptosis. A small pool of pathogen specific T cells (5–10%) survives as memory cells in the third stage (78). Memory T cells are a principal component of immunity against intracellular pathogens such as viruses. They are distinguished by their capacity to survive long-term, and undergo rapid and robust proliferation and acquisition of effector function upon antigen re-exposure (78). Memory T cells can vary in their phenotype, localization, and function allowing them to protect the host against a broad array of potential insults.
Distinct stages of CD8 T cell differentiation are defined by the expression of specific surface markers such as the isoforms of CD45 and expression of the homing receptor CCR7. These stages of differentiation are useful in the characterization of responses to, e.g., anti-viral responses. The set stages define CD45RA+CCR7+ as naive (TN), CD45RA−CCR7+ as central memory (TCM), CD45RA−CCR7− as effector memory (TEM), and CD45RA+CCR7− as effector memory RA (TEMRA) CD8 T cells (79, 80). TCM cells primarily reside in secondary lymphoid organs, possess the greatest proliferative potential among the memory T cell subsets and can rapidly expand and differentiate following re-challenge. TCM cells have higher sensitivity to antigenic stimulation, are less dependent on co-stimulation and provide better feedback to DCs and B cells compared to TN cells. TEM cells can migrate between tissues and secondary lymphoid organs and provide immune surveillance.
The Role of T Cells in Human TBEV Infection
Due to difficulties in identifying the acute phase of viral infection in humans, T cell responses to viral infections have to a large extent been addressed in studies of pathogens causing chronic infections such as HIV-1, EBV, HCV, and CMV (81–85). Such responses can be very robust, as exemplified by the massive clonal expansion of antigen-specific CD8 T cells seen in many infections (83, 85). Based on these studies, it has also become clear that the resulting populations of human CD8 T cells display striking phenotypic differences, as determined by the expression profiles of surface markers (79–81). In contrast to many other infections, including some flavivirus infections (86–89), there are only few studies of T cell responses to TBEV-infection in humans. This hold true for acute as well as cases with prolonged TBE disease. Noteworthy, however, one report has shown that TBEV-specific CD4 T cells from naturally infected patients show a higher level of polyfunctionality in response to antigen in the convalescent phase of disease, as compared to TBE-vaccine specific T cells (90).
The general lack of studies more systematically characterizing the human T cell response to TBEV-infection prompted us to study the primary T cell-mediated immune response in patients diagnosed with TBE with a particular emphasis of CD8 T cells (59). Similar to our study on NK cells (58), the T cell study focused on responses during the second stage of disease from which clinical samples were available. During this phase, CD8 T cells were strongly activated, as detected by increased expression of Ki67, within 1 week of hospitalization (illustrated in Figure 2). A large part of these CD8 T cells expressed high levels of perforin and granzyme B, and low levels of the anti-apoptotic protein Bcl-2. In contrast to CD8 T cells, CD4 T cells showed only low or at most moderate levels of activation. The TBEV-antigen specific CD8 T cells had a TEM PD-1+ phenotype throughout the course of disease. TBEV-specific CD8 T cells were predominantly Eomes+Ki67+T-bet+ in the acute stage of disease. This pattern was replaced by an Eomes−Ki67−T-bet+ profile in the convalescent phase of disease. TBEV-specific CD8 T cells were mainly monofunctional in the acute stage of disease, and tended to become more polyfunctional in the convalescent phase when clinical symptoms retracted (59).
T Cell Responses Toward Other Acute Flavivirus Infections in Humans
To be able to better interpret the above-mentioned T cell responses to acute TBEV infection, we compared the present results with T cell responses to other acute virus infections, including infections by other flaviviruses. The live attenuated YFV-vaccine strain can replicate after vaccination leading to a detectable viral load similar to a mild infection. Thus, this vaccine can be utilized as a controlled model to study mild acute viral infection in humans. CD8 T cells become activated within 1–2 weeks after vaccination with the YFV vaccine (86, 91, 92). YFV antigen-specific CD8 T cells predominantly display a TEM PD-1+ phenotype, which transition into a TEMRA PD-1− memory phenotype (86). With respect to DENV-infection, a high level of functionality of DENV-specific T cells is associated with a better disease outcome (93). Similarly, patients hospitalized with (severe) TBE show a low level of T cell functionality in the acute stage of disease (59), indicating the importance of high function among virus-specific T cells for beneficial disease outcome. CD8 T cells have been shown primarily to respond with IFN-γ to JEV in asymptomatic JEV-exposed donors (87).
Activation of CD4 T cells with an optimal magnitude, specificity and kinetics may be a requirement for viral clearance and protective immunity. In the immune response induced by the YFV vaccine, activation of CD4 T cells (peak at 10 days after vaccination) precedes that of CD8 T cells, and this may be of importance to elicit strong immunological memory (86). Furthermore, CD4 T cell release of IFN-γ may have an impact on disease outcome since CD4 T cells, and not CD8 T cells, were shown to dominate the IFN-γ response in recovered Japanese encephalitis (JE) patients. In addition, a high quality polyfunctional CD4 T cell response can be associated with better disease outcome in JE patients (87). In murine models, a perforin-dependent mechanism by the CD8 T cells has been shown to clear WNV from infected neurons, thereby suggesting an immunopathological role of T cells in mice (88). In this context, it is of interest to note that TBEV-specific T cells have a high content of both perforin and granzyme B (59), but whether the same perforin-dependent mechanism is causing immunopathogenesis in acute infection with TBEV remains to be investigated.
Cross-Reactivity Within the Family of Flaviviruses
Immunological cross-reactivity between TBEV and other species within the flavivirus family may also contribute to disease (94, 95). Antibody-dependent enhancement (ADE) is a described phenomenon that can occur when non-neutralizing antibodies facilitate virus entry into host cells, leading to increased infectivity in the cells. ADE is commonly observed in vitro in cell culture-based models (96), but it is questioned as to which degree this phenomenon occurs in vivo. A recent study from non-human primates in vivo, did not observe increased ZIKV titers after prior infection with heterologous flaviviruses (97). Protective cross-reactivity of flaviviruses has been reported as well, opposing increased pathogenesis upon pre-exposure to other species of flaviviruses (98). TBEV has been suggested to cause both pathogenic and protective cross-reactivity. Polyclonal sera against members of the TBE serocomplex (including TBEV, Kyasanur Forest disease virus, Omsk hemorrhagic fever virus, and Langat virus) enhance viral replication of TBEV in vitro (96). However, it has recently been shown that antibodies generated from TBEV infection or from the TBEV vaccine can mediate cross neutralization against other, if not most, of the members of the TBE virus complex (99). Furthermore, sera from some individuals vaccinated against TBEV and JEV neutralized WNV, and the neutralization was enhanced by YFV vaccination in some recipients (95), altogether indicating that previous flavivirus exposure may sometimes provide a degree of protection to new flaviviruses.
Cell-mediated immunity and cross-reactivity caused by TBEV and other flaviviruses has been less well studied. Recently, a study demonstrated that JEV- and JE vaccine-specific T cells cross-react with DENV (87, 100). In line with this, it was also recently shown that vaccination with YFV vaccine could induce ZIKA-specific T cells, thereby suggesting cross-protection of flavivirus-specific T cells (101). The latter phenomena opened up a discussion as to the possibility of utilizing the YFV vaccine to protect against Zika virus infection (101). In the present context, it remains to be investigated whether YFV vaccination elicits protective cross-reactive immunity also toward TBEV.
Immunopathological Aspects of TBEV Infection in the CNS
In contrast to the significant interest in emerging infections such as the recent Zika pandemic [reviewed in (45, 102, 103)], studies of the immune response toward TBEV-infection as such, and TBEV-induced immunopathology in particular, have been rather limited.
Immune and none-immune mechanisms have been proposed contribute to the crossing of TBEV over the BBB and invasion of the CNS [reviewed in (104)]. Cytokines may facilitate this process. Cytokines such as TNF-a and IL-6 have an impact on endothelial cell permeability that may induce a BBB disruption (105, 106), leading to crossover of the virus into the CNS. A distinct mechanism by which the TBEV could possibly cross the BBB is the Trojan Horse mechanism (107), by which TBEV-infected immune cells such as dendritic cells, neutrophils, monocytes, macrophages, and T cells would migrate into the parenchymal compartment causing infection of neurons or other cells in the brain and the spinal cord. Yet, an alternative route is invasion via the olfactory epithelium (108, 109).
After the landmark discovery of the lymphatic system present in the meninges that connects the CNS to the peripheral blood (110, 111), the classical concept of the CNS as an immune privileged site has been replaced by a view of an immune regulated site. Hence, under normal conditions a continuous transmigration of lymphocytes, monocytes, DCs and macrophages occurs. They may serve to detect any kind of infection or injury in the brain [reviewed in (112)]. Similar to other CNS infections, increased frequencies of T cells have been reported in CSF of TBE patients (57). Hence, activated T cells are crossing the BBB in TBE; however, the role of T cells at this site not well understood (113, 114). On one hand they could contribute toward clearing viral infection but on the other hand they may mediate immunopathology within the CNS. Corroborating the latter speculations are findings in which granzyme B+ CD8 T cell infiltrates have been linked to cell-death in infected human neuronal tissue (113) and, in parallel, mice with CD8 T cell deficiency have been shown to have prolonged survival upon infection with TBEV compared to mice with adoptively transferred CD8 T cells to immuno-competent mice (114). Furthermore, studies of post-mortem tissue of TBE patients have shown a predominance of macrophages/microglia and CD3+ T cells (both CD4+ and CD8+) in brain parenchyma (113). As seen in other flavivirus infections, macrophages and microglia also play a role in tissue destruction in human TBE.
In relation to NK cells and their possible role in causing immunopathogenesis, it is of interest to note that also these cells have been detected in the cerebrospinal fluid (CSF), albeit in low numbers, in patients with severe TBE meningitis or encephalitis (57). Activated NK cells may be protective, but they may also, like T cells, take part in immunopathological reactions as they are known to participate in direct killing of infected cells, indirect killing through cytokines or chemokines, or by the recruitment of inflammatory cells into the tissues (115, 116). Although recent results support a role for NK cells in clinical TBEV-infection, more studies are needed to provide a better understanding of the role NK cells play in pathogenic processes of human TBE infection.
Knowledge and experience gained in the field of the immunopathogenesis of other diseases affecting the CNS and its immunological compartments could be helpful in understanding TBE-specific diseases patterns. For example, in multiple sclerosis (MS), an inflammatory disease with pathology affecting the CNS (117), the concentration of the Sphingosine-1-Phosphate (S1P) in CSF is elevated and S1P-signaling is altered. In MS, binding of S1P to S1P1-receptors expressed on lymphocytes leads to invasion of autoreactive T cells into the CNS, the latter contributing to the hallmarks of the disease including demyelination and neurodegeneration (118). Interestingly, during the phase of acute infection in TBEV-infected patients, the levels of S1P in blood and CSF are highly elevated (119). This increase might promote a proinflammatory response. An increased production of extracellular S1P can be regulated by modulators of the S1P pathway, such as fingolimod, which is an immunomodulatory drug used in the treatment of MS (118). Therefore, therapeutic options used in other CNS diseases that share common immunopathogenic mechanisms with TBE could be used as models to aid in the development of new strategies for TBE treatment.
Concluding Remarks
In the present review, we have focused our attention to recent insights into the cell-mediated immune response to human TBEV infection, with an emphasis on studies of NK cell and CD8 T cell mediated responses. Until recently, the latter have been poorly studied. As yet, however, much more needs to be learnt with respect to these responses and research in this area should be encouraged. We have also addressed some aspects of TBEV CNS pathogenesis, a process still far from understood in detail. Clearly, however, cell-mediated immune responses likely play an important role in this process. As TBE continues to be an increasing global health problem and challenge, much more research is needed into this emerging disease. Several areas of research of the TBEV itself, and the clinical disease TBE merit further studies. Not the least, the specific organ pathogenesis caused by TBEV and the immune response, including infiltrating immune cells, needs more investigation. Furthermore, the possibility of antiviral treatment and other possible treatment modalities needs much more thorough investigation to prevent disease development and the often severe sequeale following infection of humans with TBEV.
Author Contributions
KB, AC, and H-GL wrote the manuscript. All other authors provided valuable contributions and insights into the manuscript.
Funding
The authors were founded by the Swedish Research Council, the Swedish Foundation for Strategic Research, Karolinska Institutet and the Stockholm County Council.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Calisher CH. Antigenic classification and taxonomy of flaviviruses (family Flaviviridae) emphasizing a universal system for the taxonomy of viruses causing tick-borne encephalitis. Acta Virol. (1988) 32:469–78.
2. Heinz FX, Collett MS, Purcell RH, Gould EA, Howard CR, Houghton M, et al. Family Flaviviridae. San Diego, CA: Academic Press (2000).
3. Lindquist L, Vapalahti O. Tick-borne encephalitis. Lancet (2008) 371:1861–71. doi: 10.1016/S0140-6736(08)60800-4
4. Taba P, Schmutzhard E, Forsberg P, Lutsar I, Ljostad U, Mygland A, et al. EAN consensus review on prevention, diagnosis and management of tick-borne encephalitis. Eur J Neurol. (2017) 24:1214–e61. doi: 10.1111/ene.13356
5. Kohl I, Kozuch O, Eleckova E, Labuda M, Zaludko J. Family outbreak of alimentary tick-borne encephalitis in Slovakia associated with a natural focus of infection. Eur J Epidemiol. (1996) 12:373–5. doi: 10.1007/BF00145300
6. Haglund M, Gunther G. Tick-borne encephalitis–pathogenesis, clinical course and long-term follow-up. Vaccine (2003) 21(Suppl. 1):S11–8. doi: 10.1016/S0264-410X(02)00811-3
7. Mansfield KL, Johnson N, Phipps LP, Stephenson JR, Fooks AR, Solomon T. Tick-borne encephalitis virus - a review of an emerging zoonosis. J Gen Virol. (2009) 90:1781–94. doi: 10.1099/vir.0.011437-0
9. Mickiene A, Laiskonis A, Gunther G, Vene S, Lundkvist A, Lindquist L. Tickborne encephalitis in an area of high endemicity in lithuania: disease severity and long-term prognosis. Clin Infect Dis. (2002) 35:650–8. doi: 10.1086/342059
10. Puchhammer-Stockl E, Kunz C, Mandl CW, Heinz FX. Identification of tick-borne encephalitis virus ribonucleic acid in tick suspensions and in clinical specimens by a reverse transcription-nested polymerase chain reaction assay. Clin Diagn Virol. (1995) 4:321–6. doi: 10.1016/0928-0197(95)00022-4
11. ECDC. Epidemiological situation of tick-borne encephalitis in the European Union and European Free Trade Association countries (2012).
12. Donoso Mantke O, Escadafal C, Niedrig M, Pfeffer M. Working Group For Tick-Borne Encephalitis Virus C. Tick-borne encephalitis in Europe, 2007 to 2009. Euro Surveill. (2011) 16.
13. Schuler M, Zimmermann H, Altpeter E, Heininger U. Epidemiology of tick-borne encephalitis in Switzerland, 2005 to 2011. Euro Surveill. (2014) 19. doi: 10.2807/1560-7917.ES2014.19.13.20756
14. Heinz FX, Stiasny K, Holzmann H, Grgic-Vitek M, Kriz B, Essl A, et al. Vaccination and tick-borne encephalitis, central Europe. Emerg Infect Dis. (2013) 19:69–76. doi: 10.3201/eid1901.120458
15. Amicizia D, Domnich A, Panatto D, Lai PL, Cristina ML, Avio U, et al. Epidemiology of tick-borne encephalitis (TBE) in Europe and its prevention by available vaccines. Hum Vaccin Immunother. (2013) 9:1163–71. doi: 10.4161/hv.23802
16. Heinz FX, Stiasny K, Holzmann H, Kundi M, Sixl W, Wenk M, et al. Emergence of tick-borne encephalitis in new endemic areas in Austria: 42 years of surveillance. Euro Surveill. (2015) 20:9–16. doi: 10.2807/1560-7917.ES2015.20.13.21077
17. Randolph SE, EDEN-TBD sub-project team. Human activities predominate in determining changing incidence of tick-borne encephalitis in Europe. Euro Surveill. (2010) 15:24–31. doi: 10.2807/ese.15.27.19606-en
18. Zeman P, Pazdiora P, Benes C. Spatio-temporal variation of tick-borne encephalitis (TBE) incidence in the Czech Republic: is the current explanation of the disease's rise satisfactory? Ticks Tick Borne Dis. (2010) 1:129–40. doi: 10.1016/j.ttbdis.2010.05.003
19. Gray JS, Dautel H, Estrada-Pena A, Kahl O, Lindgren E. Effects of climate change on ticks and tick-borne diseases in europe. Interdiscip Perspect Infect Dis. (2009) 2009:593232. doi: 10.1155/2009/593232
20. Sumilo D, Asokliene L, Bormane A, Vasilenko V, Golovljova I, Randolph SE. Climate change cannot explain the upsurge of tick-borne encephalitis in the Baltics. PLoS ONE (2007) 2:e500. doi: 10.1371/journal.pone.0000500
21. Lehrer AT, Holbrook MR. Tick-borne encephalitis vaccines. J Bioterror Biodef. (2011) 2011:3. doi: 10.4172/2157-2526.S1-003
24. Schondorf I, Beran J, Cizkova D, Lesna V, Banzhoff A, Zent O. Tick-borne encephalitis (TBE) vaccination: applying the most suitable vaccination schedule. Vaccine (2007) 25:1470–5. doi: 10.1016/j.vaccine.2006.10.028
25. Loew-Baselli A, Poellabauer EM, Pavlova BG, Fritsch S, Firth C, Petermann R, et al. Prevention of tick-borne encephalitis by FSME-IMMUN vaccines: review of a clinical development programme. Vaccine (2011) 29:7307–19. doi: 10.1016/j.vaccine.2011.07.089
26. Beran J, Xie F, Zent O. Five year follow-up after a first booster vaccination against tick-borne encephalitis following different primary vaccination schedules demonstrates long-term antibody persistence and safety. Vaccine (2014) 32:4275–80. doi: 10.1016/j.vaccine.2014.06.028
27. Loew-Baselli A, Konior R, Pavlova BG, Fritsch S, Poellabauer E, Maritsch F, et al. Safety and immunogenicity of the modified adult tick-borne encephalitis vaccine FSME-IMMUN: results of two large phase 3 clinical studies. Vaccine (2006) 24:5256–3. doi: 10.1016/j.vaccine.2006.03.061
28. Ehrlich HJ, Pavlova BG, Fritsch S, Poellabauer EM, Loew-Baselli A, Obermann-Slupetzky O, et al. Randomized, phase II dose-finding studies of a modified tick-borne encephalitis vaccine: evaluation of safety and immunogenicity. Vaccine (2003) 22:217–23. doi: 10.1016/S0264-410X(03)00563-2
29. Andersson CR, Vene S, Insulander M, Lindquist L, Lundkvist A, Gunther G. Vaccine failures after active immunisation against tick-borne encephalitis. Vaccine (2010) 28:2827–31. doi: 10.1016/j.vaccine.2010.02.001
30. Lotric-Furlan S, Bogovic P, Avsic-Zupanc T, Jelovsek M, Lusa L, Strle F. Tick-borne encephalitis in patients vaccinated against this disease. J Intern Med. (2017) 282:142–55. doi: 10.1111/joim.12625
31. Stiasny K, Holzmann H, Heinz FX. Characteristics of antibody responses in tick-borne encephalitis vaccination breakthroughs. Vaccine (2009) 27:7021–6. doi: 10.1016/j.vaccine.2009.09.069
32. Gunther G, Haglund M, Lindquist L, Skoldenberg B, Forsgren M. Intrathecal IgM, IgA and IgG antibody response in tick-borne encephalitis. Long-term follow-up related to clinical course and outcome. Clin Diagn Virol. (1997) 8:17–29. doi: 10.1016/S0928-0197(97)00273-0
33. Holzmann H. Diagnosis of tick-borne encephalitis. Vaccine (2003) 21(Suppl 1):S36–40. doi: 10.1016/S0264-410X(02)00819-8
34. Murray CL, Jones CT, Rice CM. Architects of assembly: roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat Rev Microbiol. (2008) 6:699–708. doi: 10.1038/nrmicro1928
35. Kroschewski H, Allison SL, Heinz FX, Mandl CW. Role of heparan sulfate for attachment and entry of tick-borne encephalitis virus. Virology (2003) 308:92–100. doi: 10.1016/S0042-6822(02)00097-1
36. Smit JM, Moesker B, Rodenhuis-Zybert I, Wilschut J. Flavivirus cell entry and membrane fusion. Viruses (2011) 3:160–71. doi: 10.3390/v3020160
37. Mackenzie JM, Westaway EG. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J Virol. (2001) 75:10787–99. doi: 10.1128/JVI.75.22.10787-10799.2001
38. Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, et al. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe (2009) 5:365–75. doi: 10.1016/j.chom.2009.03.007
39. Miorin L, Romero-Brey I, Maiuri P, Hoppe S, Krijnse-Locker J, Bartenschlager R, et al. Three-dimensional architecture of tick-borne encephalitis virus replication sites and trafficking of the replicated RNA. J Virol. (2013) 87:6469–81. doi: 10.1128/JVI.03456-12
40. Bily T, Palus M, Eyer L, Elsterova J, Vancova M, Ruzek D. Electron tomography analysis of tick-borne encephalitis virus infection in human neurons. Sci Rep. (2015) 5:10745. doi: 10.1038/srep10745
41. Junjhon J, Lausumpao M, Supasa S, Noisakran S, Songjaeng A, Saraithong P, et al. Differential modulation of prM cleavage, extracellular particle distribution, and virus infectivity by conserved residues at nonfurin consensus positions of the dengue virus pr-M junction. J Virol. (2008) 82:10776–91. doi: 10.1128/JVI.01180-08
42. Stadler K, Allison SL, Schalich J, Heinz FX. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol. (1997) 71:8475–81.
43. Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, et al. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science (2008) 319:1834–37. doi: 10.1126/science.1153264
44. Ruzek D, Vancova M, Tesarova M, Ahantarig A, Kopecky J, Grubhoffer L. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J Gen Virol. (2009) 90:1649–58. doi: 10.1099/vir.0.010058-0
45. Miner JJ, Diamond MS. Zika virus pathogenesis and tissue tropism. Cell Host Microbe (2017) 21:134–42. doi: 10.1016/j.chom.2017.01.004
46. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. (2010) 11:1127–35. doi: 10.1038/ni.1953
47. O'Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity (2015) 43:634–45. doi: 10.1016/j.immuni.2015.09.013
48. Bryceson YT, Chiang SC, Darmanin S, Fauriat C, Schlums H, Theorell J, et al. Molecular mechanisms of natural killer cell activation. J Innate Immun. (2011) 3:216–26. doi: 10.1159/000325265
49. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
50. Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. (2001) 22:633–40. doi: 10.1016/S1471-4906(01)02060-9
51. Fauriat C, Long EO, Ljunggren HG, Bryceson YT. Regulation of human NK-cell cytokine and chemokine production by target cell recognition. Blood (2010) 115:2167–76. doi: 10.1182/blood-2009-08-238469
52. Cichicki F, Schlums H, Theorell J, Tesi B, Miller JS, Ljunggren HG, et al. Diversification and functional specialization of human NK cell subsets. Curr Top Microbiol Immunol. (2016) 395:63–94. doi: 10.1007/82_2015_487
53. Screpanti V, Wallin RP, Grandien A, Ljunggren HG. Impact of FASL-induced apoptosis in the elimination of tumor cells by NK cells. Mol Immunol. (2005) 42:495–9. doi: 10.1016/j.molimm.2004.07.033
54. Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab Invest. (2012) 92:967–77. doi: 10.1038/labinvest.2012.54
55. Bjorkstrom NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood (2010) 116:3853–64. doi: 10.1182/blood-2010-04-281675
56. Biron CA, Byron KS, Sullivan JL. Severe herpesvirus infections in an adolescent without natural killer cells. N Engl J Med. (1989) 320:1731–5. doi: 10.1056/NEJM198906293202605
57. Tomazic J, Ihan A. Flow cytometric analysis of lymphocytes in cerebrospinal fluid in patients with tick-borne encephalitis. Acta Neurol Scand. (1997) 95:29–33. doi: 10.1111/j.1600-0404.1997.tb00064.x
58. Blom K, Braun M, Pakalniene J, Lunemann S, Enqvist M, Dailidyte L, et al. NK cell responses to human tick-borne encephalitis virus infection. J Immunol. (2016) 197:2762–71. doi: 10.4049/jimmunol.1600950
59. Blom K, Braun M, Pakalniene J, Dailidyte L, Beziat V, Lampen MH, et al. Specificity and dynamics of effector and memory CD8 T cell responses in human tick-borne encephalitis virus infection. PLoS Pathog. (2015) 11:e1004622. doi: 10.1371/journal.ppat.1004622
60. Azeredo EL, De Oliveira-Pinto LM, Zagne SM, Cerqueira DI, Nogueira RM, Kubelka CF. NK cells, displaying early activation, cytotoxicity and adhesion molecules, are associated with mild dengue disease. Clin Exp Immunol. (2006) 143:345–56. doi: 10.1111/j.1365-2249.2006.02996.x
61. Lim DS, Yawata N, Selva KJ, Li N, Tsai CY, Yeong LH, et al. The combination of type I IFN, TNF-alpha, and cell surface receptor engagement with dendritic cells enables NK cells to overcome immune evasion by dengue virus. J Immunol. (2014) 193:5065–75. doi: 10.4049/jimmunol.1302240
62. Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med. (2015) 7:297ra115. doi: 10.1126/scitranslmed.aac5722
63. Zhang M, Daniel S, Huang Y, Chancey C, Huang Q, Lei YF, et al. Anti-West Nile virus activity of in vitro expanded human primary natural killer cells. BMC Immunol. (2010) 11:3. doi: 10.1186/1471-2172-11-3
64. Alter G, Jost S, Rihn S, Reyor LL, Nolan BE, Ghebremichael M, et al. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+ NK cells in acute HCV infection may predict viral clearance. J Hepatol. (2011) 55:278–88. doi: 10.1016/j.jhep.2010.11.030
65. Stegmann KA, Bjorkstrom NK, Ciesek S, Lunemann S, Jaroszewicz J, Wiegand J, et al. Interferon alpha-stimulated natural killer cells from patients with acute hepatitis C virus (HCV) infection recognize HCV-infected and uninfected hepatoma cells via DNAX accessory molecule-1. J Infect Dis. (2012) 205:1351–62. doi: 10.1093/infdis/jis210
66. Marquardt N, Ivarsson MA, Blom K, Gonzalez VD, Braun M, Falconer K, et al. The human NK cell response to yellow fever virus 17D Is primarily governed by NK cell differentiation independently of NK cell education. J Immunol. (2015) 195:3262–72. doi: 10.4049/jimmunol.1401811
67. Nascimento Silva JR, Camacho LA, Siqueira MM, Freire Mde S, Castro YP, Maia Mde L, et al. Mutual interference on the immune response to yellow fever vaccine and a combined vaccine against measles, mumps and rubella. Vaccine (2011) 29:6327–34. doi: 10.1016/j.vaccine.2011.05.019
68. Neves PC, Matos DC, Marcovistz R, Galler R. TLR expression and NK cell activation after human yellow fever vaccination. Vaccine (2009) 27:5543–49. doi: 10.1016/j.vaccine.2009.07.028
69. Wahid SF, Sanusi S, Zawawi MM, Ali RA. A comparison of the pattern of liver involvement in dengue hemorrhagic fever with classic dengue fever. Southeast Asian J Trop Med Public Health (2000) 31:259–63.
70. Green S, Pichyangkul S, Vaughn DW, Kalayanarooj S, Nimmannitya S, Nisalak A, et al. Early CD69 expression on peripheral blood lymphocytes from children with dengue hemorrhagic fever. J Infect Dis. (1999) 180:1429–35. doi: 10.1086/315072
71. Bjorkstrom NK, Lindgren T, Stoltz M, Fauriat C, Braun M, Evander M, et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J Exp Med. (2011) 208:13–21. doi: 10.1084/jem.20100762
72. Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood (2004) 104:3664–71. doi: 10.1182/blood-2004-05-2058
73. Petitdemange C, Becquart P, Wauquier N, Beziat V, Debre P, Leroy EM, et al. Unconventional repertoire profile is imprinted during acute chikungunya infection for natural killer cells polarization toward cytotoxicity. PLoS Pathog. (2011) 7:e1002268. doi: 10.1371/journal.ppat.1002268
74. Lobigs M, Mullbacher A, Regner M. MHC class I up-regulation by flaviviruses: immune interaction with unknown advantage to host or pathogen. Immunol Cell Biol. (2003) 81:217–23. doi: 10.1046/j.1440-1711.2003.01161.x
75. Fernandez-Garcia MD, Mazzon M, Jacobs M, Amara A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe (2009) 5:318–28. doi: 10.1016/j.chom.2009.04.001
76. Townsend A, Ohlen C, Bastin J, Ljunggren HG, Foster L, Karre K. Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature (1989) 340:443–8. doi: 10.1038/340443a0
77. Kaech SM, Ahmed R. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat Immunol. (2001) 2:415–22. doi: 10.1038/87720
78. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science (1996) 272:54–60. doi: 10.1126/science.272.5258.54
79. Champagne P, Ogg GS, King AS, Knabenhans C, Ellefsen K, Nobile M, et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature (2001) 410:106–11. doi: 10.1038/35065118
80. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
81. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, et al. Rowland-Jones. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. (2002) 8:379–85. doi: 10.1038/nm0402-379
82. Appay V, Rowland-Jones SL. Lessons from the study of T-cell differentiation in persistent human virus infection. Semin Immunol. (2004) 16:205–12. doi: 10.1016/j.smim.2004.02.007
83. Callan MF, Tan L, Annels N, Ogg GS, Wilson JD, O'Callaghan CA, et al. Direct visualization of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J Exp Med. (1998) 187:1395–402. doi: 10.1084/jem.187.9.1395
84. Lauer GM, Ouchi K, Chung RT, Nguyen TN, Day CL, Purkis DR, et al. Comprehensive analysis of CD8(+)-T-cell responses against hepatitis C virus reveals multiple unpredicted specificities. J Virol. (2002) 76:6104–13. doi: 10.1128/JVI.76.12.6104-6113.2002
85. Roos MT, van Lier RA, Hamann D, Knol GJ, Verhoofstad I, van Baarle D, et al. Changes in the composition of circulating CD8+ T cell subsets during acute epstein-barr and human immunodeficiency virus infections in humans. J Infect Dis. (2000) 182:451–8. doi: 10.1086/315737
86. Blom K, Braun M, Ivarsson MA, Gonzalez VD, Falconer K, Moll M, et al. Temporal dynamics of the primary human T cell response to yellow fever virus 17D as it matures from an effector- to a memory-type response. J Immunol. (2013) 190:2150–8. doi: 10.4049/jimmunol.1202234
87. Turtle L, Bali T, Buxton G, Chib S, Chan S, Soni M, et al. Human T cell responses to Japanese encephalitis virus in health and disease. J Exp Med. (2016) 213:1331–52. doi: 10.1084/jem.20151517
88. Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. (2006) 80:119–29. doi: 10.1128/JVI.80.1.119-129.2006
89. Hatch S, Endy TP, Thomas S, Mathew A, Potts J, Pazoles P, et al. Intracellular cytokine production by dengue virus-specific T cells correlates with subclinical secondary infection. J Infect Dis. (2011) 203:1282–91. doi: 10.1093/infdis/jir012
90. Aberle JH, Schwaiger J, Aberle SW, Stiasny K, Scheinost O, Kundi M, et al. Human CD4+ T helper cell responses after tick-borne encephalitis vaccination and infection. PLoS ONE (2015) 10:e0140545. doi: 10.1371/journal.pone.0140545
91. Miller JD, van der Most RG, Akondy RS, Glidewell JT, Albott S, Masopust D, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity (2008) 28:710–22. doi: 10.1016/j.immuni.2008.02.020
92. Akondy RS, Monson ND, Miller JD, Edupuganti S, Teuwen D, Wu H, et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J Immunol. (2009) 183:7919–30. doi: 10.4049/jimmunol.0803903
93. Jeewandara C, Adikari TN, Gomes L, Fernando S, Fernando RH, Perera MK, et al. Functionality of dengue virus specific memory T cell responses in individuals who were hospitalized or who had mild or subclinical dengue infection. PLoS Negl Trop Dis. (2015) 9:e0003673. doi: 10.1371/journal.pntd.0003673
94. Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol. (2016) 17:1102–8. doi: 10.1038/ni.3515
95. Mansfield KL, Horton DL, Johnson N, Li L, Barrett AD, Smith DJ, et al. Flavivirus-induced antibody cross-reactivity. J Gen Virol. (2011) 92:2821–9. doi: 10.1099/vir.0.031641-0
96. Phillpotts RJ, Stephenson JR, Porterfield JS. Antibody-dependent enhancement of tick-borne encephalitis virus infectivity. J Gen Virol. (1985) 66 (Pt 8):1831–7.
97. McCracken MK, Gromowski GD, Friberg HL, Lin X, Abbink P, De La Barrera R, et al.Impact of prior flavivirus immunity on Zika virus infection in rhesus macaques. PLoS Pathog. (2017) 13:e1006487. doi: 10.1371/journal.ppat.1006487
98. Hoke CH, Nisalak A, Sangawhipa N, Jatanasen S, Laorakapongse T, Innis BL, et al. Protection against Japanese encephalitis by inactivated vaccines. N Engl J Med. (1988) 319:608–14. doi: 10.1056/NEJM198809083191004
99. McAuley AJ, Sawatsky B, Ksiazek T, Torres M, Korva M, Lotric-Furlan S, et al. Cross-neutralisation of viruses of the tick-borne encephalitis complex following tick-borne encephalitis vaccination and/or infection. NPJ Vaccines (2017) 2:5. doi: 10.1038/s41541-017-0009-5
100. Turtle L, Tatullo F, Bali T, Ravi V, Soni M, Chan S, et al. Cellular immune responses to live attenuated Japanese Encephalitis (JE) vaccine SA14-14-2 in adults in a JE/dengue co-endemic area. PLoS Negl Trop Dis. (2017) 11:e0005263. doi: 10.1371/journal.pntd.0005263
101. Blom K, Sandberg JT, Lore K, Ljunggren HG. Prospects for induction of CD8 T cell-mediated immunity to Zika virus infection by yellow fever virus vaccination. J Intern Med. (2017) 282:206–8. doi: 10.1111/joim.12638
102. Fauci AS, Morens DM. Zika virus in the Americas–yet another arbovirus threat. N Engl J Med. (2016) 374:601–4. doi: 10.1056/NEJMp1600297
103. Ferguson NM, Cucunuba ZM, Dorigatti I, Nedjati-Gilani GL, Donnelly CA, Basanez MG, et al. EPIDEMIOLOGY. Countering the Zika epidemic in Latin America. Science (2016) 353:353–4. doi: 10.1126/science.aag0219
104. Spindler KR, Hsu TH. Viral disruption of the blood-brain barrier. Trends Microbiol. (2012) 20:282–90. doi: 10.1016/j.tim.2012.03.009
105. de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol. (1996) 64:37–43. doi: 10.1016/0165-5728(95)00148-4
106. Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. (2010) 37:26–32. doi: 10.1016/j.nbd.2009.07.031
107. Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology (2009) 385:425–33. doi: 10.1016/j.virol.2008.11.047
108. McMinn PC. The molecular basis of virulence of the encephalitogenic flaviviruses. J Gen Virol. (1997) 78 (Pt 11):2711–22.
109. Avsic-Zupanc T, Poljak M, Maticic M, Radsel-Medvescek A, LeDuc JW, Stiasny K, et al. Laboratory acquired tick-borne meningoencephalitis: characterisation of virus strains. Clin Diagn Virol. (1995) 4:51–9. doi: 10.1016/0928-0197(94)00062-Y
110. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature (2015) 523:337–41. doi: 10.1038/nature14432
111. Aspelund A, Antila S, Proulx ST, Karlsen TV, Karaman S, Detmar M, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. (2015) 212:991–9. doi: 10.1084/jem.20142290
112. Negi N, Das BK. CNS: not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. (2018) 37:57–68. doi: 10.1080/08830185.2017.1357719
113. Gelpi E, Preusser M, Laggner U, Garzuly F, Holzmann H, Heinz FX, et al. Inflammatory response in human tick-borne encephalitis: analysis of postmortem brain tissue. J Neurovirol. (2006) 12:322–7. doi: 10.1080/13550280600848746
114. Ruzek D, Salat J, Palus M, Gritsun TS, Gould EA, Dykova I, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology (2009) 384:1–6. doi: 10.1016/j.virol.2008.11.023
115. Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. (1999) 17:189–220. doi: 10.1146/annurev.immunol.17.1.189
116. Guidotti LG, Chisari FV. Immunobiology and pathogenesis of viral hepatitis. Annu Rev Pathol. (2006) 1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230
117. Sospedra M, Martin R. Immunology of multiple sclerosis. Annu Rev Immunol. (2005) 23:683–747. doi: 10.1146/annurev.immunol.23.021704.115707
118. Martin R, Sospedra M. Sphingosine-1 phosphate and central nervous system. Curr Top Microbiol Immunol. (2014) 378:149–70. doi: 10.1007/978-3-319-05879-5_7
Keywords: cell-mediated immunity, flavivirus, NK cells, T cells, tick-borne encephalitis, tick-borne encephalitis virus, viral immunopathogenesis
Citation: Blom K, Cuapio A, Sandberg JT, Varnaite R, Michaëlsson J, Björkström NK, Sandberg JK, Klingström J, Lindquist L, Gredmark Russ S and Ljunggren H-G (2018) Cell-Mediated Immune Responses and Immunopathogenesis of Human Tick-Borne Encephalitis Virus-Infection. Front. Immunol. 9:2174. doi: 10.3389/fimmu.2018.02174
Received: 27 March 2018; Accepted: 03 September 2018;
Published: 26 September 2018.
Edited by:
Alan Chen-Yu Hsu, University of Newcastle, AustraliaReviewed by:
Alessandro Marcello, International Centre for Genetic Engineering and Biotechnology, ItalyManuela Zlamy, Innsbruck Medical University, Austria
Copyright © 2018 Blom, Cuapio, Sandberg, Varnaite, Michaëlsson, Björkström, Sandberg, Klingström, Lindquist, Gredmark Russ and Ljunggren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kim Blom, a2ltLmJsb21Aa2kuc2U=
Hans-Gustaf Ljunggren, aGFucy1ndXN0YWYubGp1bmdncmVuQGtpLnNl