 Tue W. Kragstrup1,2*
Tue W. Kragstrup1,2* Thomas Andersen1Line D. Heftdal1
Thomas Andersen1Line D. Heftdal1 Malene Hvid1,3Jens Gerwien4Pallavur Sivakumar5Peter C. Taylor6
Malene Hvid1,3Jens Gerwien4Pallavur Sivakumar5Peter C. Taylor6 Ladislav Senolt7
Ladislav Senolt7 Bent Deleuran1,2,3
Bent Deleuran1,2,3- 1Department of Biomedicine, Aarhus University, Aarhus, Denmark
- 2Department of Rheumatology, Aarhus University Hospital, Aarhus, Denmark
- 3Department of Clinical Medicine, Aarhus University, Aarhus, Denmark
- 4Eli Lilly, Herlev, Denmark
- 5Immuno Oncology Translational Development, Celgene Corportation, Seattle, WA, United States
- 6Nuffield Department of Orthopedics, Rheumatology and Musculoskeletal Sciences, University of Oxford, Oxford, United Kingdom
- 7Institute of Rheumatology and Department of Rheumatology, First Faculty of Medicine, Charles University, Prague, Czechia
This review describes the IL-20 family of cytokines in rheumatoid arthritis (RA) and spondyloartrhitits (SpA) including psoriatic arthritis. The IL-20 receptor (R) cytokines IL-19, IL-20, and IL-24 are produced in both the peripheral blood and the synovial joint and are induced by Toll-like receptor ligands and autoantibody-associated immune complexes in monocytes. IL-19 seems to have anti-inflammatory functions in arthritis. In contrast, IL-20 and IL-24 increase the production of proinflammatory molecules such as monocyte chemoattractant protein 1 and are associated with bone degradation and radiographic progression. IL-22 is also associated with progression of bone erosions. This suggests that the IL-22RA1 subunit shared by IL-20, IL-22, and IL-24 is important for bone homeostasis. In line with this, the IL-22RA1 has been found on preosteoclasts in early RA. IL-26 is produced in high amounts by myofibroblasts and IL-26 stimulation of monocytes is an important inducer of Th17 cells in RA. This indicates a role for IL-26 as an important factor in the interactions between resident synovial cells and infiltrating leukocytes. Clinical trials that investigate inhibitors of IL-20 (fletikumab) and IL-22 (fezakinumab) in psoriasis and RA have been terminated. Instead, it seems that the strategy for modulating the IL-20 cytokine family should take the overlap in cellular sources and effector mechanisms into account. The redundancy encourages inhibition of more than one cytokine or one of the shared receptors. All IL-20 family members utilize the Janus kinase signaling pathway and are therefore potentially inhibited by drugs targeting these enzymes. Effects and adverse effects in ongoing clinical trials with inhibitors of IL-22 and the IL-22RA1 subunit and recombinant IL-22 fusion proteins will possibly provide important information about the IL-20 subfamily of cytokines in the future.
Rheumatoid Arthritis and Spondyloarthritis
Disease Characteristics
Rheumatoid arthritis (RA) and spondyloarthritis (SpA) are both immune-mediated rheumatic diseases characterized by chronic inflammation of the synovial joints. Both inflammatory joint diseases are included in this review as they present distinctive clinical features. RA is characterized by destructive polyarthritis and the involvement of multiple organs (1). SpA covers a group of diseases that affects the joints and entheses including ankylosing spondylitis, psoriatic arthritis, enteropathic arthritis, reactive arthritis, and undifferentiated spondyloarthritis (2). SpA can affect the joints of the axial skeleton and/or the peripheral joints. In both cases, extraarticular involvement is common, e.g., uveitis, inflammatory bowel disease (IBD), psoriasis, or enthesitis. The etiology of both RA and SpA is still largely unknown. In direct comparison, the RA pathogenesis involves more adaptive immune features such as autoreactive B cells and production of the autoantibodies rheumatoid factors (RFs) and anti-cyclic citrullinated peptide (CCP) antibodies, whereas SpA pathogenesis seems to be more driven by lymphocyte subsets producing IL-17A (3). In RA, the ultimate disease manifestation in the joint is bone erosions. These bone erosions are found close to the insertion site of the synovial membrane in connection with the formation of pannus tissue and the presence of osteoclasts (1, 4). In SpA, the typical structural change is new bone formation in the axial skeleton and entheses. However, peripheral joint disease in SpA can be destructive (2, 3, 5). Further, both diseases are associated with the development of osteoporosis because of the inflammatory activation of bone degradation (6).
Cytokines and Chemokines
In the immune system, cytokines are important signaling molecules that coordinate the immune response by mediating the communication between cells through specific receptors. These receptors can be found on cells that traditionally are considered as part of the immune system but also on what historically have been looked upon as non-immune cells such as epithelial cells and fibroblasts. In rheumatic disease, the regulation of cytokines is unbalanced. This involves both insufficient production of inhibitory cytokines and augmented production of proinflammatory cytokines that together contribute to the chronic inflammatory condition. Studying the pathogenesis of the rheumatic diseases has led to the development of biologic disease-modifying antirheumatic drugs (7, 8). TNFα inhibitors were the first validated biological therapy for RA. However, now several other anti-cytokine drugs, lymphocyte-targeting agents and small-molecule inhibitors of signal transduction pathways are available or in clinical trials (9). The purpose of the present review is to describe the role of the IL-20 cytokine family in RA and SpA.
The IL-20 Cytokine Family
The human IL-20 cytokine family consists of the cytokines IL-19, IL-20, IL-22, IL-24, and IL-26 in the IL-10 superfamily of cytokines (along with IL-10, IL-28, and IL-29) (10, 11). IL-19, IL-20, and IL-24 are also referred to as the IL-20R cytokines based on their shared binding to the receptor complexes containing the IL-20RB. Thus, all three cytokines bind the receptor complex IL-20RB/IL-20RA while only IL-20 and IL-24 also bind the receptor complex IL-20RB/IL-22RA1 (12–14). IL-22 uses the receptor complex IL-10RB/IL-22RA1 and IL-26 signals through IL-10RB/IL-20RA (Figure 1). Murine IL-26 is a pseudogene and the function of mouse IL-24 remains to be elucidated.
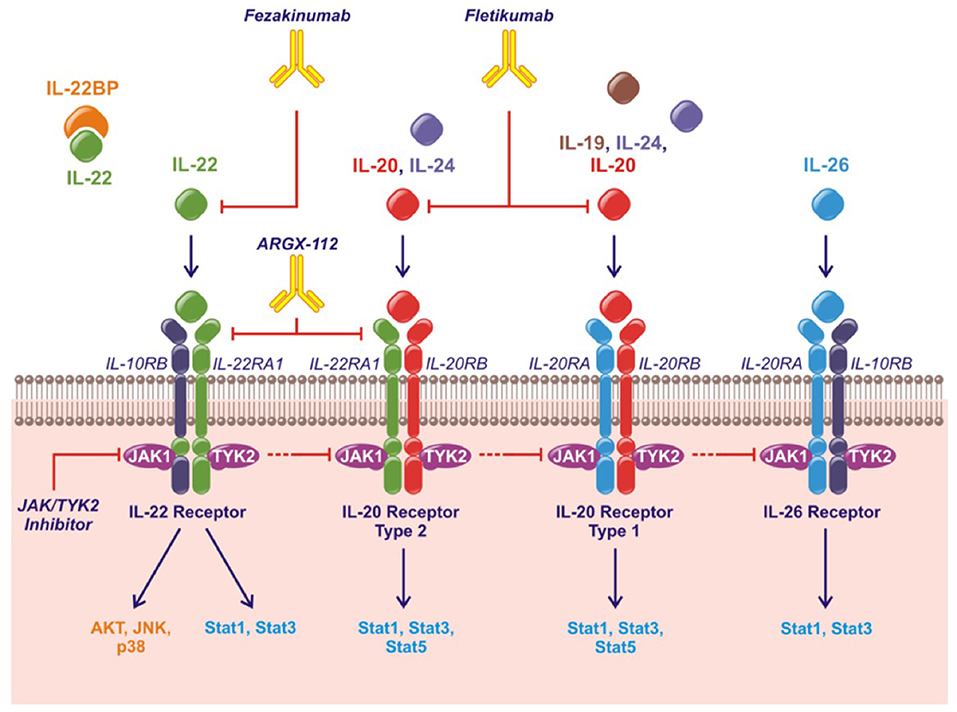
Figure 1. The IL-20 family of cytokines, their shared receptors and intracellular signaling pathways and therapeutic strategies approved or under investigation. Fezakinumab inhibits IL-22. Fletikumab inhibits IL-20. ARGX-112 inhibits the IL-22RA1 subunit. JAK/TYK2 inhibitors will prevent signaling from all the IL-20 family cytokines.
The IL-20 cytokine family signal through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway primarily activating STAT3. Further, IL-22 can activate Akt, extracellular signal-regulated kinases (ERKs), Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (Figure 1) (11).
IL-19, IL-20, and IL-24
Expression and Regulation
Cytokine Localization
IL-20 and IL-24 have been identified in mononuclear cells in the synovial membrane of RA patients by immunohistochemistry (15, 16). IL-19 has been found in vimentin+ and CD68+ synovial cells in the hyperplastic lining layers of RA synovial tissues (17). Further, the expression of IL-20 in RA synovial tissue was shown to be particularly associated with macrophages, neutrophil granulocytes and fibroblast-like synovial cells (18). IL-19, IL-20, and IL-24 have all been identified in both synovial fluid and plasma of patients with RA and SpA (15, 16, 18–20).
Cellular Sources
All three IL-20R cytokines are expressed by monocytes, fibroblasts and several epithelial cells. Furthermore, IL-19 and IL-24 are expressed by B cells, IL-20 production can be induced in dendritic cells and IL-24 is expressed by Th2 cells (11, 21, 22). Toll like receptor (TLR)2 and TLR4 agonists and IL-1β increase the production of all three cytokines (23–25) (Figure 2). The induction of the IL-20R cytokines through activation of TLR2 and TLR4 is interesting because several endogenous TLR2 and TLR4 agonists are produced in RA and SpA (e.g., HMGB1 and hyaluronic acid fragments) (26–29). In fact, this pathway may be the primary contributor to the increase in plasma levels of IL-20 and IL-24 in SpA. Myofibroblast pathways are thus activated in SpA, which lead to extracellular matrix turnover and new bone formation (30). In this process, factors from host cells such as fibrinogen, EDA fibronectin, hyaluronic acid, and tenascin C are released. These factors can all function as danger associated molecular patterns (DAMPs) (31). Further, all three cytokines are induced by immune complexes that activate FcγRs in PBMCs. More specifically, citrullinated fibrinogen-containing immune complexes were shown to activate FcγRIIa (Figure 2) (32). The association between the IL-20R cytokines and either IgM-RF and anti-CCP antibodies could be important because these autoantibodies and the generation of immune complexes is an ongoing process even in patients in clinical remission (1). In line with this, the concentration of IL-20 and IL-24 remained increased in some patients with early RA at follow-up after 6 months of treatment compared with HCs in this study (32). Thus, some patients have increased levels of IL-20 and IL-24 even when in clinical remission.
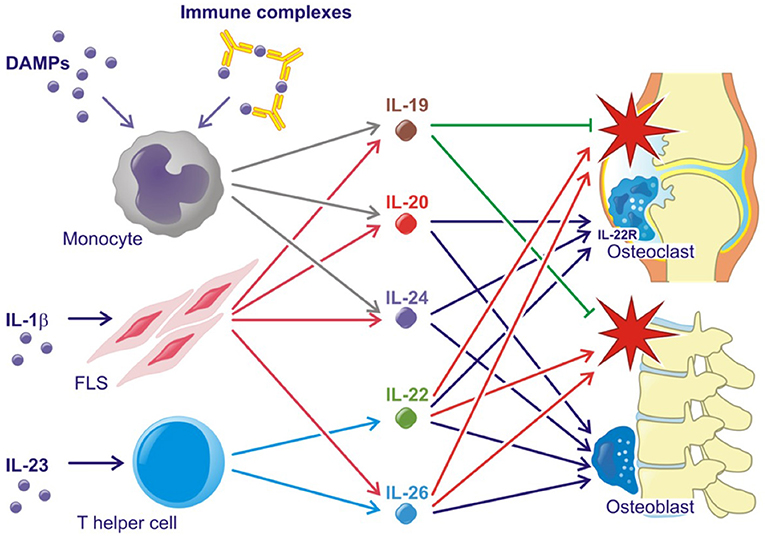
Figure 2. Simplified role of the IL-20 family cytokines in rheumatoid arthritis and spondyloarthritis. Red stars indicate inflammation. Lines with an arrow head indicate stimulation. Lines without arrow head indicate inhibition.
Receptor Expression
The three receptor subunits are primarily expressed by resident effector cells of target organs (e.g., keratinocytes, osteoclasts, and intestinal and airway epithelial cells) and not on cells traditionally associated with the immune system (21, 22). However, the IL-20RB has been identified on several peripheral blood leukocyte subsets in the expression pattern studies (21, 22), and the IL-20RA and IL-22RA1 can be induced in macrophages in the lung (13). In addition, some studies have reported effects of the IL-20R cytokines on cells of the immune system not known to express the receptor complexes (33–39). These findings have suggested (1) that IL-22RA1 and IL-20RA are only expressed by leukocytes under certain conditions, (2) that the these two receptor subunits are expressed by leukocytes below the detection limit in the expression pattern studies, or (3) that a not yet identified receptor subunit is expressed by leukocytes. In two studies, all three receptor subunits were found on FLSs in the synovial membrane (16, 17). However, in other studies FLSs did not express the receptors and were unresponsive to stimulation with IL-20 and IL-24 (24). Recently, expression of IL-20RA and IL-22RA1 have been shown on peripheral blood and synovial fluid monocyte subsets from RA patients (32). The percentage of monocytes expressing IL-20RA was not different when comparing RA synovial fluid, RA peripheral blood and HC peripheral blood. In contrast, the percentage of monocytes expressing IL-22RA1 was highest in RA synovial fluid and was also increased in RA peripheral blood compared with HC peripheral blood. The percentage of monocytes expressing both IL-20RA and IL-22RA1 was very low in both RA patients and HCs (32).
Function
The IL-20R axis was first associated with psoriasis (40–46), but later also with other immune-mediated inflammatory diseases such as inflammatory bowel disease (47–51) and arthritis. The IL-20R subunits have now been identified as risk genes for developing an immune-mediated inflammatory disease in three independent genetic studies (52–54).
Most studies have focused on pro-inflammatory roles of IL-20 and IL-24, whereas IL-19 seems to have anti-inflammatory functions. In evidence of this, transgenic mice overexpressing IL-20 (40) and IL-24 (42) develop psoriasis-like skin disease, while IL-19 overexpressing mice have a normal phenotype (13). In contrast, IL-19 knockout mice show increased susceptibility to experimentally induced colitis (47). In line with this, single-nucleotide polymorphisms of the IL-19 gene (but not the IL-20 or IL-24 genes) are protective in psoriasis (41). Likewise, single-nucleotide polymorphisms of the IL-20 and IL-24 genes (but not the IL-19 gene) are associated with juvenile idiopathic arthritis (55, 56). IL-19 has also been suggested to be protective in human colonic inflammation (57), while the role of IL-20 (48, 49) and IL-24 (50, 51) needs further study. Finally, IL-19 has several anti-inflammatory functions in atherosclerotic disease (58). Several studies have found induction of MCP-1 as a pathogenic mechanism of IL-20 and IL-24 function. MCP-1 expression is upregulated in transgenic mice overexpressing IL-24 and IL-20 increases the production of MCP-1 in kidney mesangial cells and intervertebral disc cells (42, 59, 60).
Supporting the tissue homeostasis function of the IL-20R cytokines, the receptor subunits IL-20RA and IL-22RA1 are found on several epithelial cells of target organs such as keratinocytes and lining cells of the gut and airways (13, 21, 22). The two cytokines are upregulated in wound healing and regulate tissue repair mechanisms (61–63). Supporting tissue homestatic properties in arthritis, IL-20 and IL-24 have been shown to increase osteoblast mineralization and are associated with chronic changes in the lumbar spine in SpA (Figure 2) (24). Further, the IL-22RA1 was recently found on osteoclast precursors and osteoclasts. In line with this, IL-20 and IL-24 plasma levels were associated with radiographic progression in early RA patients. Further, IL-20 and IL-24 stimulation led to an increased MCP-1 production in osteoclast cultures (32). MCP-1 is a chemoattractant protein that recruits CCR-2-expressing cells. This receptor is expressed by osteoclast precursors (64–66). Hereby, IL-20 and IL-24 could perpetuate bone destruction by stimulating the recruitment of more monocytes and osteoclast precursors to areas of ongoing bone resorption (Figure 2). These findings are in line with the identified role of IL-20 in osteoporosis (67) and osteoarthritis (68).
Potential Target for Disease Modification
IL-20 and IL-24 act upon a restricted number of epithelial cell populations and seem to have only moderate effects on leukocytes. Thus, inhibition of the two cytokines may not be associated with increased risk of infection (69). In this way, IL-20 and IL-24 could be potential treatment targets in RA and SpA. The anti-IL-20 drug candidate fletikumab (NN8226, NNC0109-0012) for the treatment of RA has been tested in clinical trials (Table 1). The drug was demonstrated to be safe and well tolerated in the phase 1 trial (NCT01038674) (70). In the Phase 2a trial, neutralization of IL-20 proved effective, especially in seropositive RA patients, which strengthens the link between IL-20 and RF (NCT01282255) (72). However, a Phase 2b trial with this drug candidate very recently failed to reach the primary outcome measure of ACR20 after 12 weeks in RA patients with inadequate responses to either anti-TNFα (NCT01636817) or methotrexate (NCT01636843) therapy. There are several potential reasons why this anti-IL-20 antibody failed in the clinical trial. First, inhibition of IL-20 alone might not be sufficient because of the redundancy in this cytokine family. In line with this, broad blockade of signaling from the IL-20 family of cytokines potently inhibit arthritis in a collagen-induced arthritis (CIA) model (75). Second, the NN8226 antibody (fletikumab) might not have been optimal. However, fletikumab was a human antibody (IgG4) with a high affinity (Kd: 37 pM) and a very high potency in blocking IL-20 mediated activity on IL-20RA/IL-20RB and IL-22RA1/IL20R2 expressing cells (IC50: 0.27 nM).It had a serum half-life of ~3 weeks in RA patients (70). Third, it is possible that the IL-20 axis plays a role only in a small subset of RA patients. Fourth, the primary end point of the clinical trials was disease activity after a short time period. Possibly, IL-20 inhibition could affect other outcomes such as progression of bone erosions after a longer time period. Finally, it could also mean that IL-20 does not play a central role in RA.
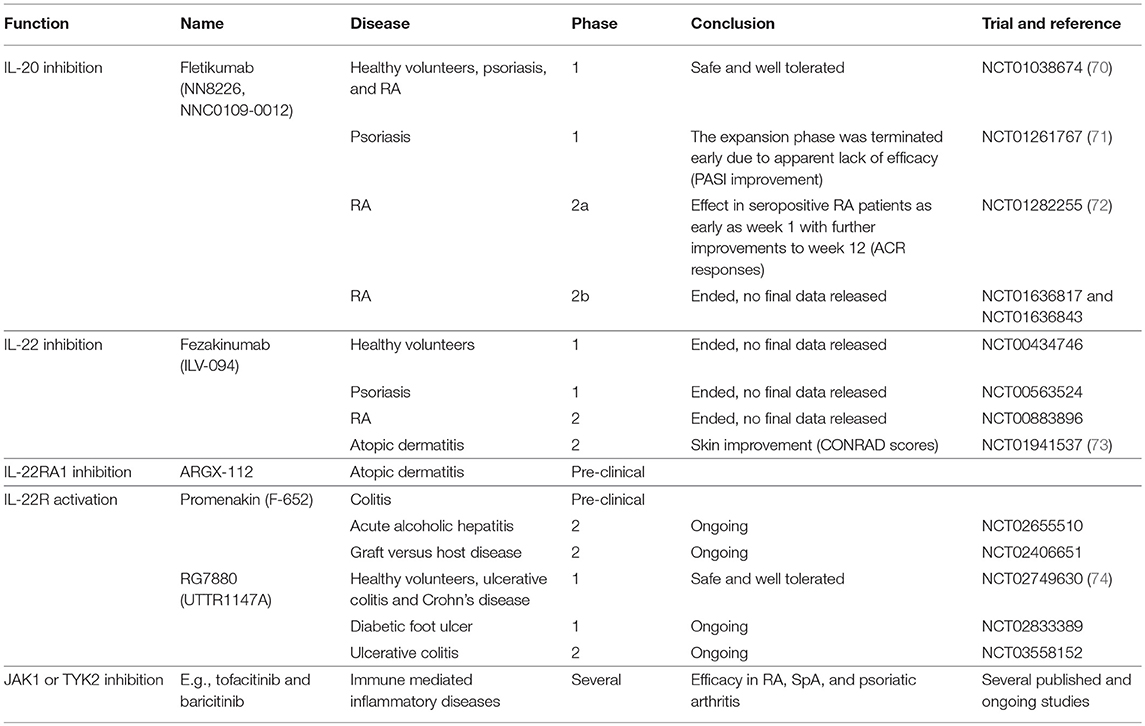
Table 1. Drugs targeting the IL-20 family of cytokines with relevance to immune mediated inflammatory arthritis.
Of note, the IL-20R axis signals via the JAK pathway and specifically utilizes the heterodimeric pairing of JAK1 and Tyrosine kinase 2, which in turn activate STAT3. Therefore, it is likely that JAK inhibitors such as tofacitinib (a selective inhibitor of JAK1 and 3) and baricitinib (a selective inhibitor of JAK1 and 2) interupt the intracellular signaling of IL-20R ligands as well as the signaling of other members of the type I/II cytokine family (Table 1) (76). Because the JAK inhibitors are multi-cytokine inhibitors, it is difficult to ascribe the magnitude of efficacy observed in any given immune-mediated inflammatory disorder to signaling via a particular cytokine receptor. However, it should be noted that both baricitinib and tofacitinib have well established efficacy in RA (76) and tofacitinib has demonstrated efficacy in a phase II study of ankylosing spondylitis and in psoriatic arthritis (77, 78).
IL-22
Expression and regulation
Another central effector cytokine in the IL-20 family of cytokines is IL-22. Apart from being produced by Th17 cells, IL-22 is also produced by the Th22 cell. Th17 and Th22 cells are closely related, both being CD4+ T helper cells developed under the influence of IL-1β, IL-6, and IL-23 (Figure 2). The development of Th17 and Th22 are dependent on different transcription factors, but they also show differences in their expression of surface receptors. While RAR-related orphan receptor gamma (RORγt) is the transcription factor responsible for Th17 induction, the aryl-hydro-carbon receptor (AHR) together with RORγt are the responsible transcription factors for the development of the Th22 phenotype. Th17 cells are identified through expression of CCR6, CCR4, but not CCR10. By contrast, Th22 cells express CCR4, CCR6 and also CCR10 (79, 80).
IL-22 signals through the heterodimeric receptor complex consisting of the ubiquitously expressed IL-10RB and the more restrictively expressed IL-22RA1 (81). IL-22RA1 is primarily present on epithelial cells and certain monocyte subsets, and very limited IL-22RA1 expression is found on other immune cells (21, 22). IL-22 functions are further regulated by the presence of IL-22 binding protein (BP), a soluble form of the IL-22RA1 (82). IL-22BP is able to bind IL-22, thereby antagonizing the effector functions of IL-22 (83, 84).
Function
The effect of IL-22 has been described as protective in some settings, where it promotes increased resistance to bacterial infection in the gut, maintains intestinal barrier functions and promotes the production of antimicrobial peptides (85). Further, T cell differentiation toward an IL-22-producing phenotype may also be favorable in alcoholic hepatitis (86). In contrast, in synovial tissue IL-22 is involved in pro-inflammatory reactions such as FLS proliferation and MCP-1 secretion (87).
In animal models, the IL-23-Th17 axis including IL-22 has been associated with several immune-mediated inflammatory conditions. IL-23 double-knockout mice were resistant to the development of CIA and to CNS inflammation in the experimental allergic encephalopathy (EAE) mouse model (88, 89). In line with this, IL-22 production was associated with increased joint destruction in the CIA model (Figure 2) (90). Recently, IL-23 was shown to act on RORγt+ CD3+CD4-CD8- T cells in order to develop SpA-like disease in mice. The mechanisms involved include IL-22 induced genes associated with new bone formation (Figure 2) (91). Animal data further suggest that the IL-22RA1 is the primary driver of disease in the autoinflammatory phenotype. Transgenic mice overexpressing IL-22 develop psoriasis-like disease, which is inhibited after breeding these mice with IL-22RA1 knockout mice (but not with IL-20RA knockout mice) (92). Furthermore, IL-22RA1 transgenic mice develop a systemic inflammatory condition. Eliminating the effect of all three cytokines in the IL-20RB knockout mice attenuates disease in a mouse psoriasis model (93), but upregulates antigen-specific T cell responses (94).
In humans, IL-22 has also been associated with diseases involving the IL-23-Th17 axis such as SpA including psoriatic arthritis (95). Adding to a potential role in these diseases characterized by new bone formation, IL-22 stimulates mineralization by ligament cells in odontologic disease (96). IL-22 has further been shown to be upregulated in RA patients and associated with radiographic progression and disease activity (97, 98).
Potential target for disease modification
Two studies of the IL-22 inhibitor fezakinumab (ILV-094) in psoriasis (NCT00563524) and RA (NCT00883896) were completed several years ago with no final data released (Table 1). This indicates that IL-22 might not have a central role in immune-mediated inflammatory arthritis. Very recently, fezakinumab showed clinical efficacy in atopic dermatitis (73). ZymoGenetics developed an IL-22RA1 antibody but the development was discontinued. More recently, ArGenX in collaboration with Leo Pharma have developed an IL-22RA1 antibody (ARGX-112) and plan clinical testing in atopic dermatitis (Table 1). Roche together with Genentech (RG7880, UTTR1147A) and Generon (Promenakin, F-652) develop IL-22 agonists for the treatment of ulcerative colitis with ongoing trials also in pancreatitis, acute alcoholic hepatitis, graft versus host disease and treatment of diabetic foot ulcers (Table 1). Treatment with these recombinant IL-22 fusion proteins seems to be safe (74, 99). No studies that interfere with the IL-22 receptor have been conducted in immune mediated inflammatory arthritis. However, modulation of IL-22 signaling by JAK inhibition could contribute to the clinical effect seen with these treatments (76, 100, 101).
IL-26
Expression and regulation
IL-26, also denominated AK155, is a highly cationic charged cytokine initially found in activated T-cells and NK cells as part of the IL-10 cytokine family, and later classified as a Th17 cytokine (102). Recent studies have revealed IL-26 expression in a number of other cell types, including alveolar macrophages (103), bronchial epithelial cells (104), synoviolin+ synovial cells from RA patients (36), and myofibroblasts of spondyloarthritis patients (105) suggesting that IL-26 production occurs in multiple cell types. There is no functional mouse IL-26 gene, which complicates the characterization of this cytokine in preclinical settings.
The IL-26 receptor is a heterodimer composed of IL-10RB and IL-20RA. The IL-10RB monomer is ubiquitously expressed, and shares with IL-10, IL-22, and interferons, while the more sparsely expressed IL-20RA monomer is shared with IL-19, IL-20, and IL-24 (106), and constitutes the limiting factor of the cells ability to respond to IL-26 (106, 107). Expression of IL-20RA is upregulated in psoriatic skin (40), but also reported present on osteoclasts and osteoblasts (108).
Function
The effector functions of IL-26 are still poorly defined. However, it seems that IL-26 primarily act as part of the anti-microbial response against bacteria by recruiting neutrophils to the site of infection (103). When IL-26 forms complexes with extracellular DNA, bacterial or self-DNA (109, 110), the IL-26-DNA complexes can be transported into the cytosol of myeloid cells or trigger type 1 interferon production by plasmacytoid dendritic cells and activate monocytes via STimulator of INterferon Genes (STING) and the inflammasome (109, 110). Elevated levels of IL-26 and IL-26-DNA complexes are seen in a variety of active inflammatory diseases (105).
In immune mediated inflammatory diseases, IL-26 is suggested to be both an important driver of Th17 differentiation and a key Th17 effector cytokine (Figure 2). Thus, IL-26 stimulated monocytes are reported to promote Th-17 cell generation from non-Th17-committed memory T-cells in RA (36), However, IL-26 is also produced by Th17 cells in colonic lesions of patients with active inflammatory bowel disease (IBD) (111).
In RA, IL-26 acts as a pro-inflammatory cytokine, which is constitutively expressed by synovial cells and induce cytokine secretion by myeloid cells leading to Th17 cell generation (Figure 2). Single-nucleotide polymorphisms (SNPs) associated with RA, as well as other inflammatory disorders, like IBD, have been identified in the IL26 gene (112–114).
In SpA, levels of IL-26 are elevated in synovial fluid compared with plasma which suggests that IL-26 is produced locally in the inflamed joint. IL-26 was primarily produced by α-smooth-muscle-actin expressing fibroblasts, called myofibroblasts. This subtype of fibroblast is specifically upregulated in SpA (115) and is a contributor to fibrosis in several immune mediated diseases (116). Osteoblasts treated with IL-26 show increased mineralization and IL-26 could therefore play a pro-fibrotic role in SpA as well as induce inflammation (Figure 2). Supporting this association, a fibrotic effect of IL-26 was also reported in graft versus host disease (117).
Potential target for disease modification
The role of IL-26 in the innate immune defense against bacterial infections complicates the use of the cytokine as a therapeutic target of chronic immune mediated inflammatory diseases. However, the fibrotic properties of the cytokine makes the cytokine interesting as a therapeutic target. Thus, TNFα and IL-17A inhibitors mostly target inflammation while only slowing down tendon calcification at a moderate level.
No medical therapeutics have currently been developed against IL-26. This might partly be due to the lack of IL-26 in mice and rats which complicates the initial pharmaceutical experiments (118). IL-26 signals through JAK1 and Tyrosine kinase 2 and STAT1 and 3 (106, 119). IL-26 is therefore also inhibited by drugs targeting the JAK molecules, e.g., tofacitinib and baricitinib (Table 1) (76).
Conclusion
IL-19 seems to have an anti-inflammatory role in arthritis. IL-20 and IL-24 have been associated with bone degradation and radiographic progression. Potentiation of MCP-1 from osteoclasts could be a mechanism contributing to this, but other effects of IL-20 and IL-24 on osteoclasts cannot be excluded. IL-22 has been associated with progression of bone erosion and IL-26 was shown to induce several proinflammatory cytokines in RA. The strategy for modulation should take into account the effects of all IL-20 family members. This redundancy encourages inhibition of more than one cytokine or one of the shared receptors in this cytokine family. However, the roles of this cytokine family could be limited in human immune-mediated arthritis or only relevant in a subset of patients or for certain aspects of the diseases. All of the IL-20 family members signal through the JAK-STAT pathway, and therefore are potential targets for drugs that inhibit this pathway. Effects and adverse effects in ongoing clinical trials with inhibitors of IL-22 and the IL-22RA1 subunit and recombinant IL-22 fusion proteins will possibly give important information about the IL-20 subfamily of cytokines in the future.
Author Contributions
TK, TA, and LH drafted the manuscript. MH, JG, PS, PT, LS, and BD made substantial contributions to the first draft. TK finalized the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
JG and PS are former employees in Novo Nordisk working on the anti-IL-20 drug candidate fletikumab. LS was principal investigator on the phase 2a study of the anti-IL-20 drug candidate fletikumab for the treatment of RA. JG is now employed by Eli Lilly. PS is now employed by Celgene.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Graphic design by Teis Jensen (dGotaWxsdXN0cmF0aW9uQG91dGxvb2suY29t). This work was supported by the Danish Rheumatism Association, the Aage Bang foundation, and the Faculty of Health at Aarhus University. LS was supported by a project of the Ministry of Health of the Czech Republic for conceptual research development No. 023728.
References
1. Scott DL, Wolfe F, Huizinga TWJ. Rheumatoid arthritis. Lancet (2010) 376:1094–108. doi: 10.1016/S0140-6736(10)60826-4
2. Dougados M, Baeten D. Spondyloarthritis. Lancet (2011) 377:2127–37. doi: 10.1016/S0140-6736(11)60071-8
3. Lories RJ, Baeten DLP. Differences in pathophysiology between rheumatoid arthritis and ankylosing spondylitis. Clin Exp Rheumatol. (2009) 27:S10–4.
4. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nat Rev Rheumatol. (2012) 8:656–64. doi: 10.1038/nrrheum.2012.153
5. Goldring SR. Osteoimmunology and bone homeostasis: relevance to spondyloarthritis. Curr Rheumatol Rep. (2013) 15:342–6. doi: 10.1007/s11926-013-0342-2
6. Will R, Palmer R, Bhalla AK, Ring F, Calin A. Osteoporosis in early ankylosing spondylitis: a primary pathological event? Lancet (1989) 2:1483–5.
7. Burmester GR, Feist E, Dörner T. Emerging cell and cytokine targets in rheumatoid arthritis. Nat Rev Rheumatol. (2014) 10:77–88. doi: 10.1038/nrrheum.2013.168
8. Taylor PC. Developing anti-TNF and biologic agents. Rheumatology (2011) 50:1351–3. doi: 10.1093/rheumatology/ker235
9. Choy EH, Kavanaugh AF, Jones SA. The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol. (2013) 9:154–63. doi: 10.1038/nrrheum.2013.8
10. Hofmann SR, Rösen-Wolff A, Tsokos GC, Hedrich CM. Biological properties and regulation of IL-10 related cytokines and their contribution to autoimmune disease and tissue injury. Clin Immunol. (2012) 143:116–27. doi: 10.1016/j.clim.2012.02.005
11. Rutz S, Wang X, Ouyang W. The IL-20 subfamily of cytokines - from host defence to tissue homeostasis. Nat Rev Immunol. (2014) 14:783–95. doi: 10.1038/nri3766
12. Dumoutier L, Leemans C, Lejeune D, Kotenko SV, Renauld JC. Cutting edge: STAT activation by IL-19, IL-20 and mda-7 through IL-20 receptor complexes of two types. J Immunol. (2001) 167:3545–9. doi: 10.4049/jimmunol.167.7.3545
13. Parrish-Novak J, Xu W, Brender T, Yao L, Jones C, West J, et al. Interleukins 19, 20, and 24 signal through two distinct receptor complexes. Differences in receptor-ligand interactions mediate unique biological functions. J Biol Chem. (2002) 277:47517–23. doi: 10.1074/jbc.M205114200
14. Logsdon NJ, Deshpande A, Harris BD, Rajashankar KR, Walter MR. Structural basis for receptor sharing and activation by interleukin-20 receptor-2 (IL-20R2) binding cytokines. Proc Natl Acad Sci USA. (2012) 109:12704–9. doi: 10.1073/pnas.1117551109
15. Kragstrup TW, Otkjaer K, Holm C, Jørgensen A, Hokland M, Iversen L, et al. The expression of IL-20 and IL-24 and their shared receptors are increased in rheumatoid arthritis and spondyloarthropathy. Cytokine (2008) 41:16–23. doi: 10.1016/j.cyto.2007.10.004
16. Hsu Y-H, Li H-H, Hsieh M-Y, Liu M-F, Huang K-Y, Chin L-S, et al. Function of interleukin-20 as a proinflammatory molecule in rheumatoid and experimental arthritis. Arthritis Rheum. (2006) 54:2722–33. doi: 10.1002/art.22039
17. Sakurai N, Kuroiwa T, Ikeuchi H, Hiramatsu N, Maeshima A, Kaneko Y, et al. Expression of IL-19 and its receptors in RA: potential role for synovial hyperplasia formation. Rheumatology (2008) 47:815–20. doi: 10.1093/rheumatology/ken061
18. Šenolt L, Prajzlerová K, Hulejová H, Šumová B, Filková M, Veigl D, et al. Interleukin-20 is triggered by TLR ligands and associates with disease activity in patients with rheumatoid arthritis. Cytokine (2017) 97:187–92. doi: 10.1016/j.cyto.2017.06.009
19. Scrivo R, Conigliaro P, Riccieri V, Di Franco M, Alessandri C, Spadaro A, et al. Distribution of interleukin-10 family cytokines in serum and synovial fluid of patients with inflammatory arthritis reveals different contribution to systemic and joint inflammation. Clin Exp Immunol. (2015) 179:300–8. doi: 10.1111/cei.12449
20. Valentina M, Jan F, Peder NL, Bo Z, Hongjie D, Pernille K. Cytokine detection and simultaneous assessment of rheumatoid factor interference in human serum and synovial fluid using high-sensitivity protein arrays on plasmonic gold chips. BMC Biotechnol. (2015) 15:73. doi: 10.1186/s12896-015-0186-0
21. Wolk K, Kunz S, Asadullah K, Sabat R. Cutting edge: immune cells as sources and targets of the IL-10 family members? J Immunol. (2002) 168:5397–402. doi: 10.4049/jimmunol.168.11.5397
22. Nagalakshmi ML, Murphy E, McClanahan T, de Waal Malefyt R. Expression patterns of IL-10 ligand and receptor gene families provide leads for biological characterization. Int Immunopharmacol. (2004) 4:577–92. doi: 10.1016/j.intimp.2004.01.007
23. Kragstrup TW, Andersen T, Holm C, Schiøttz-Christensen B, Jurik AG, Hvid M, et al. Toll-like receptor 2 and 4 induced interleukin-19 dampens immune reactions and associates inversely with spondyloarthritis disease activity. Clin Exp Immunol. (2015) 180:233–42. doi: 10.1111/cei.12577
24. Kragstrup TW, Andersen MN, Schiøttz-Christensen B, Jurik AG, Hvid M, Deleuran B. Increased interleukin (IL)-20 and IL-24 target osteoblasts and synovial monocytes in spondyloarthritis. Clin Exp Immunol. (2017) 189:342–51. doi: 10.1111/cei.12973
25. Alanärä T, Karstila K, Moilanen T, Silvennoinen O, Isomäki P. Expression of IL-10 family cytokines in rheumatoid arthritis: elevated levels of IL-19 in the joints. Scand J Rheumatol. (2010) 39:118–26. doi: 10.3109/03009740903170823
26. Oktayoglu P, Em S, Tahtasiz M, Bozkurt M, Ucar D, Yazmalar L, et al. Elevated serum levels of high mobility group box protein 1 (HMGB1) in patients with ankylosing spondylitis and its association with disease activity and quality of life. Rheumatol Int. (2013) 33:1327–31. doi: 10.1007/s00296-012-2578-y
27. Duruöz MT, Turan Y, Cerrahoglu L, Isbilen B. Serum hyaluronic acid levels in patients with ankylosing spondylitis. Clin Rheumatol. (2008) 27:621–6. doi: 10.1007/s10067-007-0757-0
28. Goh FG, Midwood KS. Intrinsic danger: activation of Toll-like receptors in rheumatoid arthritis. Rheumatology (2012) 51:7–23. doi: 10.1093/rheumatology/ker257
29. Huang Q-Q, Pope RM. The role of Toll-like receptors in rheumatoid arthritis. Curr Rheumatol Rep. (2009) 11:357–64. doi: 10.1007/s11926-009-0051-z
30. Beyer C, Distler JHW. Changing paradigms in spondylarthritis: the myofibroblast signature. Arthritis Rheum. (2012) 65:24–7. doi: 10.1002/art.37703
31. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
32. Kragstrup TW, Greisen SR, Nielsen MA, Rhodes C, Stengaard-Pedersen K, Hetland ML, et al. The interleukin-20 receptor axis in early rheumatoid arthritis: novel links between disease-associated autoantibodies and radiographic progression. Arthritis Res Ther. (2016) 18:61. doi: 10.1186/s13075-016-0964-7
33. Liao S-C, Cheng Y-C, Wang Y-C, Wang C-W, Yang S-M, Yu C-K, et al. IL-19 induced Th2 cytokines and was up-regulated in asthma patients. J Immunol. (2004) 173:6712–8. doi: 10.4049/jimmunol.173.11.6712
34. Jordan WJ, Eskdale J, Boniotto M, Lennon GP, Peat J, Campbell JDM, et al. Human IL-19 regulates immunity through auto-induction of IL-19 and production of IL-10. Eur J Immunol. (2005) 35:1576–82. doi: 10.1002/eji.200425317
35. Oral HB, Kotenko SV, Yilmaz M, Mani O, Zumkehr J, Blaser K, et al. Regulation of T cells and cytokines by the interleukin-10 (IL-10)-family cytokines IL-19, IL-20, IL-22, IL-24 and IL-26. Eur J Immunol. (2006) 36:380–8. doi: 10.1002/eji.200425523
36. Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. (2012) 10:e1001395. doi: 10.1371/journal.pbio.1001395
37. Hoffman C, Park S-H, Daley E, Emson C, Louten J, Sisco M, et al. Interleukin-19: a constituent of the regulome that controls antigen presenting cells in the lungs and airway responses to microbial products. PLoS ONE (2011) 6:e27629. doi: 10.1371/journal.pone.0027629
38. Gallagher G, Eskdale J, Jordan W, Peat J, Campbell J, Boniotto M, et al. Human interleukin-19 and its receptor: a potential role in the induction of Th2 responses. Int Immunopharmacol. (2004) 4:615–26. doi: 10.1016/j.intimp.2004.01.005
39. Buzas K, Oppenheim JJ, Zack Howard OM. Myeloid cells migrate in response to IL-24. Cytokine (2011) 55:429–34. doi: 10.1016/j.cyto.2011.05.018
40. Blumberg H, Conklin D, Xu WF, Grossmann A, Brender T, Carollo S, et al. Interleukin 20: discovery, receptor identification, and role in epidermal function. Cell (2001) 104:9–19. doi: 10.1016/S0092-8674(01)00187-8
41. Kõks S, Kingo K, Rätsep R, Karelson M, Silm H, Vasar E. Combined haplotype analysis of the interleukin-19 and−20 genes: relationship to plaque-type psoriasis. Genes Immun. (2004) 5:662–7. doi: 10.1038/sj.gene.6364141
42. He M, Liang P. IL-24 transgenic mice: in vivo evidence of overlapping functions for IL-20, IL-22, and IL-24 in the epidermis. J Immunol. (2010) 184:1793–8. doi: 10.4049/jimmunol.0901829
43. Wei C-C, Chen W-Y, Wang Y-C, Chen P-J, Lee JY-Y, Wong T-W, et al. Detection of IL-20 and its receptors on psoriatic skin. Clin Immunol. (2005) 117:65–72. doi: 10.1016/j.clim.2005.06.012
44. Otkjaer K, Kragballe K, Johansen C, Funding AT, Just H, Jensen UB, et al. IL-20 gene expression is induced by IL-1beta through mitogen-activated protein kinase and NF-kappaB-dependent mechanisms. J Invest Dermatol. (2007) 127:1326–36. doi: 10.1038/sj.jid.5700713
45. Otkjaer K, Kragballe K, Funding AT, Clausen JT, Noerby PL, Steiniche T, et al. The dynamics of gene expression of interleukin-19 and interleukin-20 and their receptors in psoriasis. Br J Dermatol. (2005) 153:911–8. doi: 10.1111/j.1365-2133.2005.06800.x
46. Kunz S, Wolk K, Witte E, Witte K, Doecke W-D, Volk H-D, et al. Interleukin (IL)-19, IL-20 and IL-24 are produced by and act on keratinocytes and are distinct from classical ILs. Exp Dermatol. (2006) 15:991–1004. doi: 10.1111/j.1600-0625.2006.00516.x
47. Azuma Y-T, Matsuo Y, Kuwamura M, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. Interleukin-19 protects mice from innate-mediated colonic inflammation. Inflamm Bowel Dis. (2010) 16:1017–28. doi: 10.1002/ibd.21151
48. Fonseca-Camarillo G, Furuzawa-Carballeda J, Llorente L, Yamamoto-Furusho JK. IL-10– and IL-20–expressing epithelial and inflammatory cells are increased in patients with ulcerative colitis. J Clin Immunol. (2013) 33:640–8. doi: 10.1007/s10875-012-9843-4
49. Yamamoto-Furusho JK, De-León-Rendón JL, la Torre de MG, Alvarez-León E, Vargas-Alarcón G. Genetic polymorphisms of interleukin 20 (IL-20) in patients with ulcerative colitis. Immunol Lett. (2012) 149:50–3. doi: 10.1016/j.imlet.2012.11.008
50. Camarillo GF, Furuzawa-Carballeda J, Martínez-Benítez B, Barreto-Zúñiga R, Yamamoto-Furusho JK. Role of the interleukin 24 in patients with ulcerative colitis. Inflamm Bowel Dis. (2011) 17:2209–10. doi: 10.1002/ibd.21635
51. Andoh A, Shioya M, Nishida A, Bamba S, Tsujikawa T, Kim-Mitsuyama S, et al. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J Immunol. (2009) 183:687–95. doi: 10.4049/jimmunol.0804169
52. McGovern A, Schoenfelder S, Martin P, Massey J, Duffus K, Plant D, et al. Capture Hi-C identifies a novel causal gene, IL20RA, in the pan-autoimmune genetic susceptibility region 6q23. Genome Biol. (2016) 17:212. doi: 10.1186/s13059-016-1078-x
53. Wu J, Yang S, Yu D, Gao W, Liu X, Zhang K, et al. CRISPR/cas9 mediated knockout of an intergenic variant rs6927172 identified IL-20RA as a new risk gene for multiple autoimmune diseases. Genes Immun. (2018) 17:160. doi: 10.1038/s41435-018-0011-6
54. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature (2014) 506:376–81. doi: 10.1038/nature12873
55. Omoyinmi E, Forabosco P, Hamaoui R, Bryant A, Hinks A, Ursu S, et al. Association of the IL-10 gene family locus on chromosome 1 with Juvenile Idiopathic Arthritis (JIA). PLoS ONE (2012) 7:e47673. doi: 10.1371/journal.pone.0047673
56. Fife MS, Gutierrez A, Ogilvie EM, Stock CJW, Samuel JM, Thomson W, et al. Novel IL10 gene family associations with systemic juvenile idiopathic arthritis. Arthritis Res Ther. (2006) 8:R148. doi: 10.1186/ar2041
57. Yamamoto-Furusho JK, Alvarez-León E, Fragoso JM, Gozalishvilli A, Vallejo M, Vargas-Alarcón G. Protective role of interleukin-19 gene polymorphisms in patients with ulcerative colitis. Hum Immunol. (2011) 72:1029–32. doi: 10.1016/j.humimm.2011.08.013
58. England RN, Autieri MV. Anti-inflammatory effects of interleukin-19 in vascular disease. Int J Inflam. (2012) 2012:253583. doi: 10.1155/2012/253583
59. Li H-H, Cheng H-H, Sun K-H, Wei C-C, Li C-F, Chen W-C, et al. Interleukin-20 targets renal mesangial cells and is associated with lupus nephritis. Clin Immunol. (2008) 129:277–85. doi: 10.1016/j.clim.2008.07.006
60. Huang K-Y, Lin R-M, Chen W-Y, Lee C-L, Yan J-J, Chang M-S. IL-20 may contribute to the pathogenesis of human intervertebral disc herniation. Spine (2008) 33:2034–40. doi: 10.1097/BRS.0b013e31817eb872
61. Poindexter NJ, Williams RR, Powis G, Jen E, Caudle AS, Chada S, et al. IL-24 is expressed during wound repair and inhibits TGFalpha-induced migration and proliferation of keratinocytes. Exp Dermatol. (2010) 19:714–22. doi: 10.1111/j.1600-0625.2010.01077.x
62. Bosanquet DC, Harding KG, Ruge F, Sanders AJ, Jiang WG. Expression of IL-24 and IL-24 receptors in human wound tissues and the biological implications of IL-24 on keratinocytes. Wound Repair Regen. (2012) 20:896–903. doi: 10.1111/j.1524-475X.2012.00840.x
63. Sa SM, Valdez PA, Wu J, Jung K, Zhong F, Hall L, et al. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J Immunol. (2007) 178:2229–40. doi: 10.4049/jimmunol.178.4.2229
64. Komano Y, Nanki T, Hayashida K, Taniguchi K, Miyasaka N. Identification of a human peripheral blood monocyte subset that differentiates into osteoclasts. Arthritis Res Ther. (2006) 8:R152. doi: 10.1186/ar2046
65. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity (2003) 19:71–82. doi: 10.1016/S1074-7613(03)00174-2
66. Matsubara R, Kukita T, Ichigi Y, Takigawa I, Qu P-F, Funakubo N, Miyamoto H, Nonaka K, Kukita A. Characterization and identification of subpopulations of mononuclear preosteoclasts induced by TNF-α in combination with TGF-β in rats. PLoS ONE (2012) 7:e47930. doi: 10.1371/journal.pone.0047930
67. Hsu Y-H, Chen W-Y, Chan C-H, Wu C-H, Sun Z-J, Chang M-S. Anti-IL-20 monoclonal antibody inhibits the differentiation of osteoclasts and protects against osteoporotic bone loss. J Exp Med. (2011) 208:1849–61. doi: 10.1084/jem.20102234
68. Hsu Y-H, Yang Y-Y, Huwang M-H, Weng Y-H, Jou I-M, Wu P-T, et al. Anti-IL-20 monoclonal antibody inhibited inflammation and protected against cartilage destruction in murine models of osteoarthritis. PLoS ONE (2017) 12:e0175802. doi: 10.1371/journal.pone.0175802
69. Leng R-X, Pan H-F, Tao J-H, Ye D-Q. IL-19, IL-20 and IL-24: potential therapeutic targets for autoimmune diseases. Expert Opin Ther Targets (2011) 15:119–26. doi: 10.1517/14728222.2011.534461
70. Lundblad MS, Overgaard RV, Göthberg M, Fjording MS, Watson E. Clinical pharmacokinetics of the anti-interleukin-20 monoclonal antibody NNC0109-0012 in healthy volunteers and patients with psoriasis or rheumatoid arthritis. Adv Ther. (2015) 1–11. doi: 10.1007/s12325-015-0191-7
71. Gottlieb AB, Krueger JG, Sandberg Lundblad M, Göthberg M, Skolnick BE. First-in-human, phase 1, randomized, dose-escalation trial with recombinant anti-IL-20 monoclonal antibody in patients with psoriasis. PLoS ONE (2015) 10:e0134703. doi: 10.1371/journal.pone.0134703
72. Šenolt L, Leszczynski P, Dokoupilová E, Göthberg M, Valencia X, Hansen BB, et al. Efficacy and Safety of Anti-Interleukin-20 Monoclonal Antibody in Patients With Rheumatoid Arthritis: A Randomized Phase IIa Trial. Arthrit Rheumatol. (2015) 67:1438–48. doi: 10.1002/art.39083
73. Guttman-Yassky E, Brunner PM, Neumann AU, Khattri S, Pavel AB, Malik K, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. (2018) 78:872–881.e6. doi: 10.1016/j.jaad.2018.01.016
74. Rothenberg ME, Wang Y, Lekkerkerker A, Danilenko DM, Maciuca R, Erickson R, et al. Randomized phase I healthy volunteer study of UTTR1147A (IL-22Fc): a potential therapy for epithelial injury. Clin Pharmacol Ther. (2018) 33:747. doi: 10.1002/cpt.1164
75. Liu X, Zhou H, Huang X, Cui J, Long T, Xu Y, et al. A broad blockade of signaling from the IL-20 family of cytokines potently attenuates collagen-induced arthritis. J Immunol. (2016) 197:3029–37. doi: 10.4049/jimmunol.1600399
76. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
77. van der Heijde D, Deodhar A, Wei JC, Drescher E, Fleishaker D, Hendrikx T, et al. Tofacitinib in patients with ankylosing spondylitis: a phase II, 16-week, randomised, placebo-controlled, dose-ranging study. Ann Rheum Dis. (2017) 76:1340–7. doi: 10.1136/annrheumdis-2016-210322
78. Mease P, Hall S, FitzGerald O, van der Heijde D, Merola JF, Avila-Zapata F, et al. Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med. (2017) 377:1537–50. doi: 10.1056/NEJMoa1615975
79. Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. (2009) 10:857–63. doi: 10.1038/ni.1767
80. Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nat Immunol. (2009) 10:864–71. doi: 10.1038/ni.1770
81. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rbeta) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J Biol Chem. (2001) 276:2725–32. doi: 10.1074/jbc.M007837200
82. Kotenko SV, Izotova LS, Mirochnitchenko OV, Esterova E, Dickensheets H, Donnelly RP, et al. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J Immunol. (2001) 166:7096–103. doi: 10.4049/jimmunol.166.12.7096
83. Jones BC, Logsdon NJ, Walter MR. Structure of IL-22 bound to its high-affinity IL-22R1 chain. Structure (2008) 16:1333–44. doi: 10.1016/j.str.2008.06.005
84. Dumoutier L, Lejeune D, Colau D, Renauld JC. Cloning and characterization of IL-22 binding protein, a natural antagonist of IL-10-related T cell-derived inducible factor/IL-22. J Immunol. (2001) 166:7090–5. doi: 10.4049/jimmunol.166.12.7090
85. Liang SC, Tan X-Y, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. (2006) 203:2271–9. doi: 10.1084/jem.20061308
86. Støy S, Sandahl TD, Dige AK, Agnholt J, Rasmussen TK, Grønbæk H, et al. Highest frequencies of interleukin-22-producing T helper cells in alcoholic hepatitis patients with a favourable short-term course. PLoS ONE (2013) 8:e55101. doi: 10.1371/journal.pone.0055101
87. Ikeuchi H, Kuroiwa T, Hiramatsu N, Kaneko Y, Hiromura K, Ueki K, et al. Expression of interleukin-22 in rheumatoid arthritis: potential role as a proinflammatory cytokine. Arthritis Rheum. (2005) 52:1037–46. doi: 10.1002/art.20965
88. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
89. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
90. Marijnissen RJ, Koenders MI, Smeets RL, Stappers MHT, Nickerson Nutter C, Joosten LAB, et al. Increased expression of interleukin-22 by synovial Th17 cells during late stages of murine experimental arthritis is controlled by interleukin-1 and enhances bone degradation. Arthritis Rheum. (2011) 63:2939–48. doi: 10.1002/art.30469
91. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4–CD8– entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
92. Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med. (2009) 87:523–36. doi: 10.1007/s00109-009-0457-0
93. Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, Abbondanzo S, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. (2006) 203:2577–87. doi: 10.1084/jem.20060244
94. Wahl C, Müller W, Leithäuser F, Adler G, Oswald F, Reimann J, et al. IL-20 receptor 2 signaling down-regulates antigen-specific T cell responses. J Immunol. (2009) 182:802–10. doi: 10.4049/jimmunol.182.2.802
95. Mitra A, Raychaudhuri SK, Raychaudhuri SP. Functional role of IL-22 in psoriatic arthritis. Arthritis Res Ther. (2012) 14:R65. doi: 10.1186/ar3781
96. Kato-Kogoe N, Nishioka T, Kawabe M, Kataoka F, Yamanegi K, Yamada N, et al. The promotional effect of IL-22 on mineralization activity of periodontal ligament cells. Cytokine (2012) 59:41–8. doi: 10.1016/j.cyto.2012.03.024
97. da Rocha LF, Duarte ÂLBP, Dantas AT, Mariz HA, Pitta IDR, Galdino SL, et al. Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol. (2012) 39:1320–5. doi: 10.3899/jrheum.111027
98. Leipe J, Schramm MA, Grunke M, Baeuerle M, Dechant C, Nigg AP, et al. Interleukin 22 serum levels are associated with radiographic progression in rheumatoid arthritis. Ann Rheum Dis. (2011) 70:1453–7. doi: 10.1136/ard.2011.152074
99. Tang K-Y, Lickliter J, Huang Z-H, Xian Z-S, Chen H-Y, Huang C, et al. Safety, pharmacokinetics, and biomarkers of F-652, a recombinant human interleukin-22 dimer, in healthy subjects. Cell Mol Immunol. (2018) 13:21. doi: 10.1038/s41423-018-0029-8
100. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov. (2014) 13:21–38. doi: 10.1038/nrd4176
101. Cotter DG, Schairer D, Eichenfield L. Emerging therapies for atopic dermatitis: JAK inhibitors. J Am Acad Dermatol. (2018) 78:S53–62. doi: 10.1016/j.jaad.2017.12.019
102. Donnelly RP, Sheikh F, Dickensheets H, Savan R, Young HA, Walter MR. Interleukin-26: An IL-10-related cytokine produced by Th17 cells. Cytokine Growth Factor Rev. (2010) 21:393–401. doi: 10.1016/j.cytogfr.2010.09.001
103. Che KF, Tengvall S, Levänen B, Silverpil E, Smith ME, Awad M, et al. Interleukin-26 in antibacterial host defense of human lungs. Effects on neutrophil mobilization. Am J Respir Crit Care Med. (2014) 190:1022–31. doi: 10.1164/rccm.201404-0689OC
104. Che KF, Kaarteenaho R, Lappi-Blanco E, Levänen B, Sun J, Wheelock Å, et al. Interleukin-26 production in human primary bronchial epithelial cells in response to viral stimulation: modulation by Th17 cytokines. Mol Med. (2017) 23:1. doi: 10.2119/molmed.2016.00064
105. Heftdal LD, Andersen T, Jæhger D, Woetmann A, Østgård R, Kenngott EE, et al. Synovial cell production of IL-26 induces bone mineralization in spondyloarthritis. J Mol Med. (2017) 95:779–87. doi: 10.1007/s00109-017-1528-2
106. Sheikh F, Baurin VV, Lewis-Antes A, Shah NK, Smirnov SV, Anantha S, et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J Immunol. (2004) 172:2006–10. doi: 10.4049/jimmunol.172.4.2006
107. Hör S, Pirzer H, Dumoutier L, Bauer F, Wittmann S, Sticht H, et al. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. (2004) 279:33343–51. doi: 10.1074/jbc.M405000200
108. Hsu Y-H, Chiu Y-S, Chen W-Y, Huang K-Y, Jou I-M, Wu P-T, et al. Anti-IL-20 monoclonal antibody promotes bone fracture healing through regulating IL-20-mediated osteoblastogenesis. Sci Rep. (2016) 6:24339. doi: 10.1038/srep24339
109. Poli C, Augusto JF, Dauvé J, Adam C, Preisser L, Larochette V, et al. IL-26 Confers proinflammatory properties to extracellular DNA. J Immunol. (2017) 198:3650–61. doi: 10.4049/jimmunol.1600594
110. Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol. (2015) 16:970–9. doi: 10.1038/ni.3211
111. Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut (2009) 58:1207–17. doi: 10.1136/gut.2007.130112
112. Silverberg MS, Cho JH, Rioux JD, McGovern DPB, Wu J, Annese V, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. (2009) 41:216–20. doi: 10.1038/ng.275
113. Padua D, Mahurkar-Joshi S, Law IKM, Polytarchou C, Vu JP, Pisegna JR, et al. A long noncoding RNA signature for ulcerative colitis identifies IFNG-AS1 as an enhancer of inflammation. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G446–57. doi: 10.1152/ajpgi.00212.2016
114. Vandenbroeck K, Cunningham S, Goris A, Alloza I, Heggarty S, Graham C, et al. Polymorphisms in the interferon-gamma/interleukin-26 gene region contribute to sex bias in susceptibility to rheumatoid arthritis. Arthritis Rheum. (2003) 48:2773–8. doi: 10.1002/art.11236
115. Yeremenko N, Noordenbos T, Cantaert T, van Tok M, van de Sande M, Cañete JD, et al. Disease-specific and inflammation-independent stromal alterations in spondyloarthritis synovitis. Arthritis Rheum. (2012) 65:174–85. doi: 10.1002/art.37704
116. LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. (2013) 19:1047–53. doi: 10.1038/nm.3218
117. Ohnuma K, Hatano R, Aune TM, Otsuka H, Iwata S, Dang NH, et al. Regulation of pulmonary graft-versus-host disease by IL-26+CD26+CD4 T lymphocytes. J Immunol. (2015) 194:3697–712. doi: 10.4049/jimmunol.1402785
118. Fickenscher H, Pirzer H. Interleukin-26. Int Immunopharmacol. (2004) 4:609–13. doi: 10.1016/j.intimp.2004.01.004
Keywords: cytokine, rheumatoid arthritis, spondyloarthritis, interleukin, IL-10 family, fibroblast, osteoclast, autoantibody
Citation: Kragstrup TW, Andersen T, Heftdal LD, Hvid M, Gerwien J, Sivakumar P, Taylor PC, Senolt L and Deleuran B (2018) The IL-20 Cytokine Family in Rheumatoid Arthritis and Spondyloarthritis. Front. Immunol. 9:2226. doi: 10.3389/fimmu.2018.02226
Received: 21 June 2018; Accepted: 07 September 2018;
Published: 25 September 2018.
Edited by:
Jan Niess, Universitätsspital Basel, SwitzerlandReviewed by:
Raymond P. Donnelly, United States Food and Drug Administration, United StatesPavel Strnad, Universität Ulm, Germany
Copyright © 2018 Kragstrup, Andersen, Heftdal, Hvid, Gerwien, Sivakumar, Taylor, Senolt and Deleuran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tue W. Kragstrup, a3JhZ3N0cnVwQGJpb21lZC5hdS5kaw==