 Huensuk Kim1,2,3
Huensuk Kim1,2,3 Christoph Schaniel1,2,3,4,5*
Christoph Schaniel1,2,3,4,5*- 1Black Family Stem Cell Institute, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 2Department of Cell, Developmental and Regenerative Biology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 3Graduate School of Biomedical Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 4Department of Pharmacological Sciences, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Mount Sinai Institute for Systems Biomedicine, Icahn School of Medicine at Mount Sinai, New York, NY, United States
The advent of induced pluripotent stem cells (iPSCs) together with recent advances in genome editing, microphysiological systems, tissue engineering and xenograft models present new opportunities for the investigation of hematological diseases and cancer in a patient-specific context. Here we review the progress in the field and discuss the advantages, limitations, and challenges of iPSC-based malignancy modeling. We will also discuss the use of iPSCs and its derivatives as cellular sources for drug target identification, drug development and evaluation of pharmacological responses.
Introduction
Hematological diseases and cancers are devastating diseases with a high economic and social burden. Generally basic and preclinical cancer research relies on model systems in order to understand the cellular and molecular mechanisms of the malignant state at the cellular, organ and organism level. The hope is that the information gained from such model systems will be helpful in devising precise, effective, and personalized therapeutic strategies. Prototypically, these model systems include immortalized cell lines and genetically engineered, mutant mice. More recently, advanced patient-derived models such as conditionally reprogrammed cells (CRs) (1–3), patient-derived tumor xenografts (PDXs) (4), CRs combined with PDXs (5), and three-dimensional patient derived organoid cell cultures (6–9), engineered tissues (10–12), and microphysiological systems (MPSs) (13–20) have attracted the interest of the biomedical research community. One particular (r)evolution in modern era biomedical research arose with the breakthrough, Noble-prize awarded discovery of induced pluripotent stem cell (iPSC) generation from somatic cells (21–24). These iPSCs are akin embryonic stem cells, and can be maintained indefinitely in a self-renewing, undifferentiated pluripotent state in culture and be directed to differentiate to any cell type in the body, provided the right cues. Thus, the derivation of iPSCs from patient cells provides a new tool in the arsenal for investigation of disease and cancer pathogenesis, drug development and precision medicine (Figure 1).
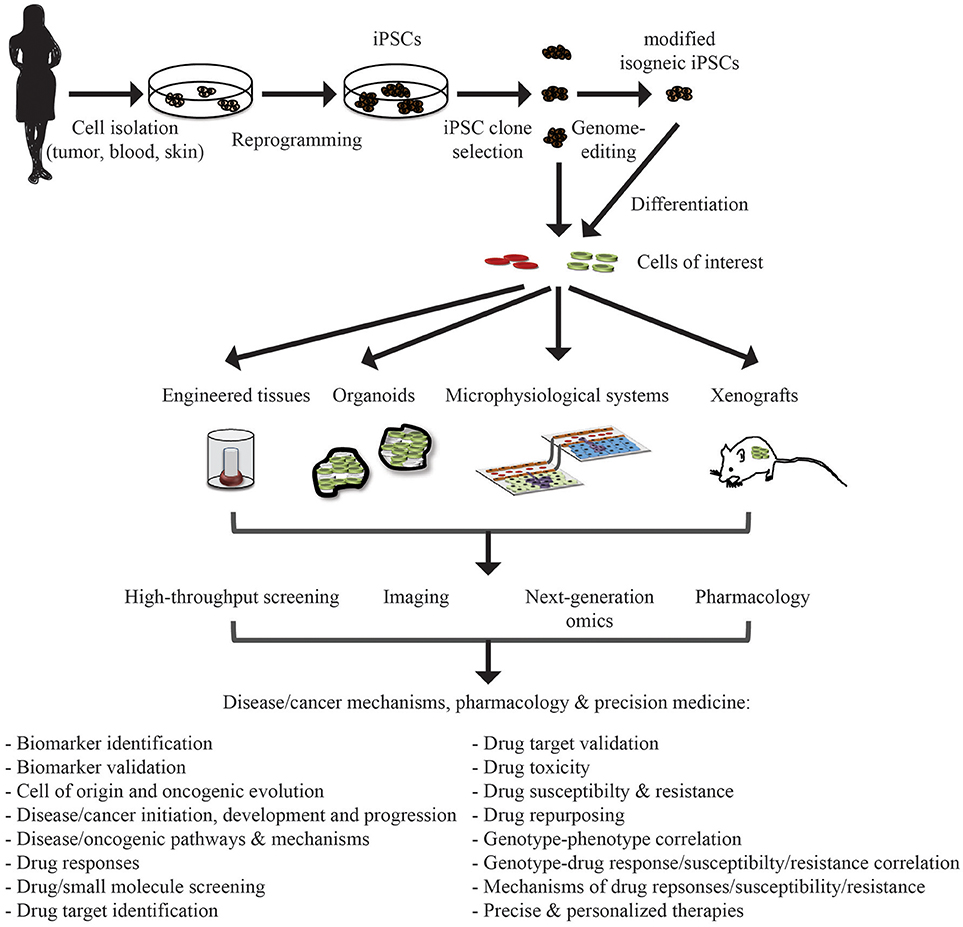
Figure 1. Application of iPSC in disease/cancer modeling, pharmacology, and precision medicine. Patient samples can be collected from a variety of tissue source depending on the need and reprogrammed to iPSCs. These iPSCs can be genome-modified to introduce or correct specific mutations or lesions to generate isogenic iPSC lines for comparative analysis. These iPSC lines can be differentiated into cells of interest (cell type of origin of the malignancy or unrelated cells that may be affected by adverse drug events/toxicity). These differentiated cells can then be integrated, also with other cell types, into engineered tissues, organoids, and microphysiological systems, or xenografted into appropriate in vivo model systems. These systems can then be interrogated for understanding disease/cancer mechanisms and signaling pathways, drug discovery and evaluation, and deriving precise personalized therapies.
Induced Pluripotent Stem Cell Models of Hematological Diseases, Blood Cell Cancers and Non-Hematopoietic Cancers
The use of iPSCs in the study of hematological diseases, cancer, and tumorigenicity is gaining momentum. It started with the generation of iPSCs from a human melanoma and a human prostate cancer cell line in 2008 (25). Since then numerous malignant cell lines have been reprogrammed that represent among other organs the brain, intestine, liver, lung, pancreas, prostate, and skin, as well as the blood (26–37) (Table 1).
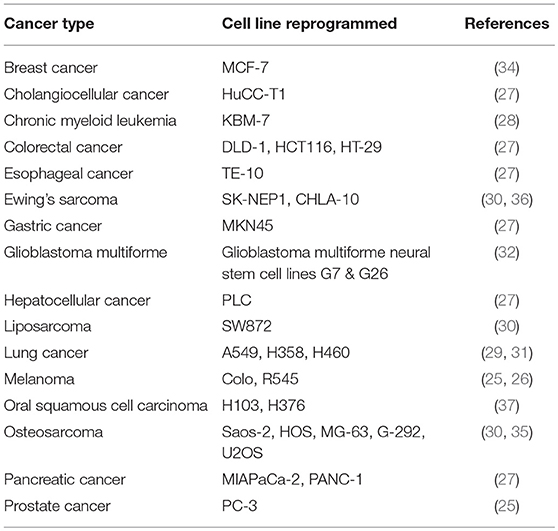
Table 1. Human cancer cell line-derived iPSCs.
The reprogramming of cancer cell lines was soon followed by the generation of iPSCs representing various hematological diseases, blood cell cancers, and non-hematopoietic cancers (38–77) (Table 2). These iPSCs were derived from primary patient cells, cancerous tissues or patient cells harboring known oncogenic lesions. In Table 2 we summarize whether functional assays were performed in attempt to phenocopy the disease/malignancy, describe the phenotypes observed and whether the studies used genome editing to either create or correct disease/cancer-associated mutations.
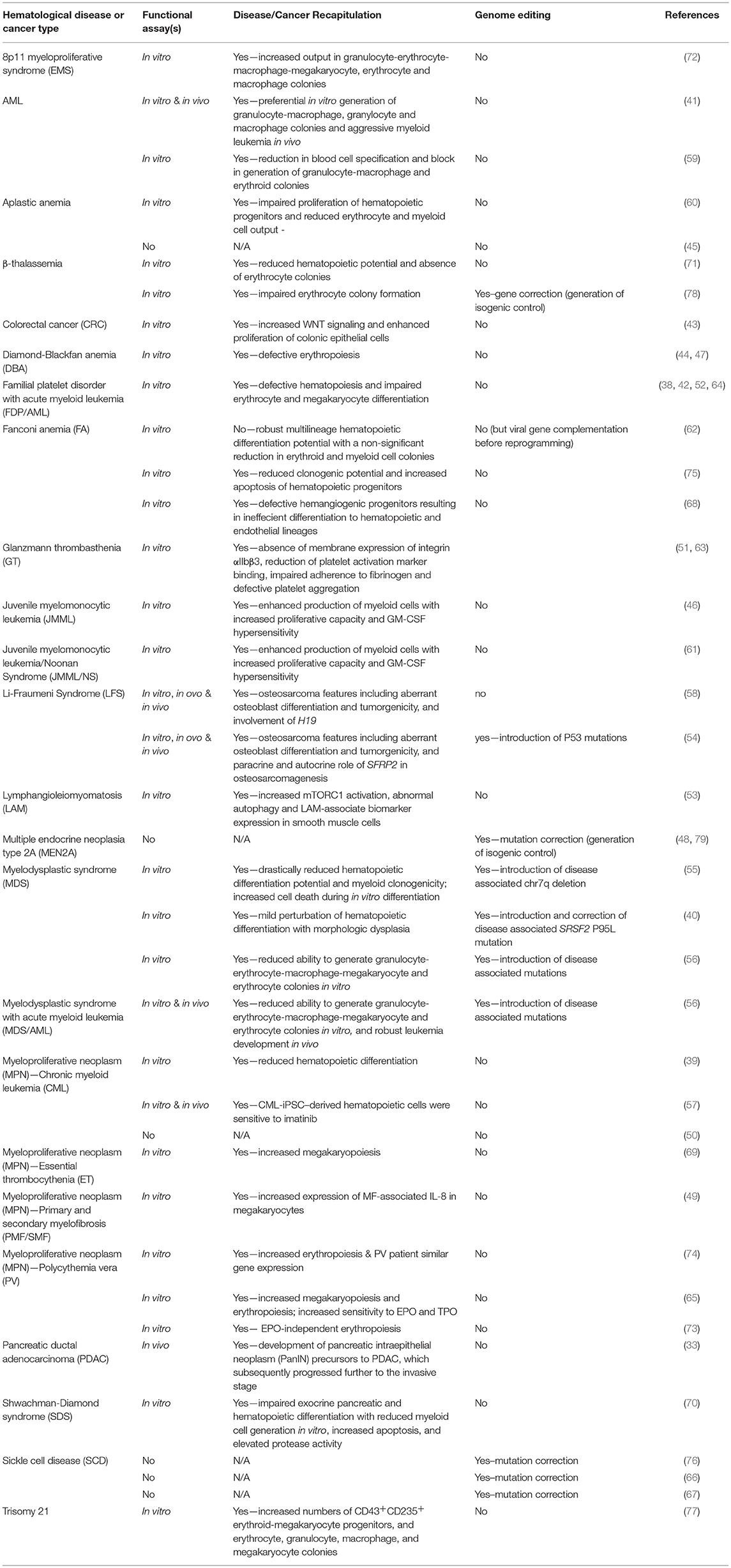
Table 2. Current patient-specific iPSC models of hematological diseases and cancer.
Advantages of iPSCs
One of the main advantages of the iPSC technology is that hematological disease-associated and malignant lesions can be studied with human cells and in the genomic context of the patient. This is of considerable importance given that certain non-human models are not reflective of the human condition. An example is familial platelet disorder with a tendency to develop acute myeloid leukemia (FPD/AML) that is caused by inherited monoallelic mutations in RUNX1 (80). FPD/AML presents with mild to moderate thrombocytopenia and bleeding due to impaired proplatelet formation, platelet activation defects, abnormal megakaryocyte differentiation and polyploidization, and a predisposition to develop AML (81). Neither, mouse nor zebrafish models of RUNX1 mutations do develop a bleeding disorder or leukemia. In contrast, FDP/AML-iPSC derived “early wave” and “second wave” hematopoietic stem/progenitor cells showed aberrant hematopoiesis as occurs in FDP/AML patients (38, 42, 52, 64). Additionally, a person's genomic background greatly influences disease/cancer severity and progression as well as therapeutic response. Second, iPSCs provide a self-renewable, cryopreservable source of cells that are scalable to fulfill any need in cell numbers for cellular, biochemical, molecular, and other downstream applications. Third, with the appropriate cues and protocols iPSCs can be differentiated in vitro to many, in the future hopefully all cell types present in the body, enabling the study of multi-cell type affected diseases/cancers with one patient iPSC source. As an example, Tulpule et al. were able to show that Shwachman-Diamond syndrome (SDS)-iPSCs were impaired in both exocrine pancreatic and hematopoietic differentiation with reduced myeloid cell generation in vitro, increased apoptosis, and elevated protease activity recapitulating SDS patient phenotypes (70). Forth when the somatic cells used to generate iPSCs are isolated from primary hematological diseases/cancers or metastatic tumor specimens of non-germ line malignancies through biopsy, a bone marrow aspirate or blood sampling, normal cells will be inadvertently co-isolated along the malignant cells. Thus, the same reprogramming event can simultaneously generate paired malignant and normal iPSCs that share the same genetic background with exception of the disease-associated/cancerous lesion(s) in the malignant iPSCs. Distinguishing the normal iPSCs from the disease/cancer iPSCs has to be done retrospectively through genetic analysis (33, 55, 73). Alternatively, isogenic normal iPSCs can be established independently through a separate reprogramming experiment with somatic cells obtained from a non-malignant area adjacent to the tumor, a biopsy from an unaffected tissue such as the skin or from blood in the case of non-hematological disorders or cancers (33, 82). Another advantage of the iPSC technology is that reprogramming of malignant cells might establish iPSCs that represent various stages of disease progression, as cancers are often associated with serial accumulation of specific malignant mutations/lesions. Papapetrou et al. elegantly demonstrated this by using bone marrow or peripheral blood from four patients in different risk categories of myelodysplastic syndrome (MDS) or MDS/AML (56). They were successful in generating a library of iPSC lines that represents various disease stages including normal/healthy, preleukemia, low-risk MDS, high-risk MDS, and MDS/AML. The derived iPSC lines carried the respective gene mutations and chromosomal abnormalities found in the patients' bone marrow or peripheral blood cells used for reprogramming. Moreover, hematopoietic differentiation of these iPSC lines representing the various disease stages captured corresponding cellular phenotypes of graded severity and disease specificity.
Limitations and Challenges of iPSC Modeling of Human Malignancies
Modeling hematological diseases and cancers with patient-specific iPSCs could face various hurdles due to technical, genomic stability and epigenome resetting challenges. It has been reported that some cancer cells are refractory to reprogramming (83, 84). This can have several reasons. For one, certain cancer cells and cells representing diverse stages may be difficult or even impossible to obtain and maintain for reprogramming purposes. Second, hematological diseases and cancers are often heterogeneous in nature, and reprogramming may preferentially select for cells with certain mutations and chromosomal aberrations and not others. Thus, the possibility exists that the panel of iPSC lines generated might not represent the entire heterogeneous composition of the patients' malignancy. Third, some cancer-associated mutations or genetic lesions might interfere with the reprogramming process itself or prevent maintenance of the pluripotent state. Fourth, even if iPSCs from patients with certain genetic lesions could be established, the specific lesions may render the cell genomically unstable. This will lead to acquisition of additional mutations and genomic abnormalities, which no longer reflect the cancer's genomic footprint and make the cells useless for proper disease modeling. Examples of unsuccessful reprogramming include the inability to establish iPSC lines from highly purified leukemic blast cells from patients with cytogenetically different subtypes of B cell-ALL (B-ALL) (84), and form Fanconi anemia (FA)-fibroblasts in one case (83). It is noteworthy to mention that FA-iPSC lines have been successfully generated (62, 68, 75). However, the FA pathway facilitates efficient reprogramming (62) and FA cells are genomically instable and predisposed to apoptosis (85). The latter is reflected by the observation of Yung et al. who showed that their FA-iPSC lines acquired significant additional abnormalities (hyperploidy) (75). The success in generating FA-iPSC likely might be dependent on the reprogramming condition—hypoxia appears better than normoxia (62) -, which FA-associated gene (fifteen genes constitute the FA complementation group) is mutated or even the kind of mutation. The derivation of AML-iPSCs, although successful for three AML patients with rearrangements in KMT2A/MLL (41, 59), has failed for AMLs with different mutations or lesions as well as KMT2A/MLL leukemic aberrations (41, 59). Stanford et al. also reported that TSC2-deficiency represents a barrier to reprogramming (53), while TSC2-happloinsufficient allowed iPSC generation with TSC2+/−-iPSC-derived smooth muscle cells recapitulating Lymphangioleiomyomatosis (LAM) features including increased mTORC1 activation, abnormal autophagy and LAM-associate biomarker expression (53).
Another possible limitation is the inability to derive cells of a defined cell type and developmental stage characteristic of the malignancy from iPSCs. Although protocols for generation of many general cell types have been established, the signaling cues and in vitro differentiation protocols for certain specialized cells, and developmental and maturation staged are still not fully understood. This is further complicated by the fact that differentiation and maturation efficiency is never 100% and, in most cases, the differentiation and maturation stage of a given cell within a population cannot easily be discriminated, thus, potentially hampering the correlation of disease phenotypes with the cellular phenotypes present in the culture. This issue could be resolved by introduction of stage-specific reporter genes via genome editing or by detailed stepwise characterization of the stages of differentiation and maturation in order to identify the exact stage at which the disease phenotype manifests. Additionally, the constant technological advances in single cell analyses at the cellular and molecular level will greatly improve disease modeling and mechanistic studies.
Cell reprogramming is associated with resetting of the starting cell's epigenetic landscape to that of a pluripotent stem cell. This resetting might eliminate characteristic features of the disease/cancer cell phenotype that might not be recreated upon differentiation, thus producing a significant difference between the disease/cancer iPSC model and the original disease/cancer cell. Here, it is worth bringing forth the theory that the initial oncogenic insult to the cancer-initiating cell might (re)program the epigenome toward a specific cancer cell fate (86). This potentially important aspect of malignancy could well be lost in iPSCs as reprogramming to iPSCs is accompanied by genome-wide epigenetic resetting (see Epigenome, Cancer, and iPSCs). Additionally, if one agrees with Sánchez-García's tumor stem cell reprogramming viewpoint that cancer cell properties can reemerge upon differentiation and that this property is to a fixed, uni-differentiated cell fate then this may not reemerge in an iPSC model due to the fact that iPSCs by definition possess pluripotent differentiation ability. On the other hand, such a resetting might be looked at favorably in certain diseases/cancers of “pure” epigenetic origin for which one could envision of using cells differentiated from these epigenetically reset iPSCs as a regenerative therapy.
Last but not least, modeling systemic processes in vitro is a challenge, as generally iPSC are maintained isolated as functionally autonomous entities in two-dimensional culture systems and not physiological integrated within the disease/tumor microenvironment. Recent progress and use of tissue engineering, three-dimensional organoids, MPS and in vivo xenografts offers a window to more sophisticated modeling that enables incorporation of malignant cells with cellular and extracellular components of the disease/tumor microenvironment, nutrient supply, and mimicking of blood/lymph flow thus attempting to recapitulate the in vivo architecture and physiological condition in which the malignant cells reside and grow.
Epigenome, Cancer, and iPSCs
Hematological diseases and cancers are profoundly influenced by changes in the epigenome and associated with a specific epigenetic profile. Since reprogramming to pluripotency is achieved through a stepwise resetting of the epigenetic landscape of the starting cell to that of a self-renewing, pluripotent iPSC (87), it is foreseeable that under certain circumstances this could have a negative impact on specific disease/cancer iPSC-based models. For example, iPSCs derived from non-small cell lung cancer (NSCLC) cell lines reset the NSCLC-associated transcriptional and methylation pattern of associated oncogenes and tumor suppressors (31). Similarly, Zhang et al. showed that reprogramming of sarcoma cell lines with complex, abnormal karyotypes to iPSCs resets the sarcoma transcriptional and epigenetic pattern and that the derived iPSCs gained self-renewal and multi-lineage differentiation potential (30). Neither of these studies examined whether the cancer-associated epigenetic profile reminiscent of the original cancer cell could be reestablished upon differentiation. Comparably, iPSCs generated from patients with AML carrying MLL rearrangements retained the leukemic mutations but also reset leukemic DNA methylation and gene expression patterns (41). However, leukemic DNA methylation and gene expression profiles reemerged in AML-iPSC-derived hematopoietic cells. Similarly, human glioblastoma-derived iPSCs remain highly malignant after differentiation into neural progenitors and pancreatic ductal adenocarcinoma (PDAC)-iPSCs establish secondary pancreatic-cancer in patient-derived xenografts (see also below) (32, 33). These examples suggest that cancer cell properties, albeit reset in iPSCs, can reemerge upon differentiation to the appropriate cancer cell type.
Remarkably, a recent report showed that the cellular context could significantly impact on the genetic information and behavior of malignant cells (88). Hashimoto et al. reprogrammed mouse colon tumor cells with loss of Apc. The reprogrammed tumor cells, Apc-iPSCs, displayed iPSC-like morphology and gene expression but lacked pluripotency and showed a trophectoderm-differentiation bias. Surprisingly, the majority of genes affected by the Apc mutation in Apc-iPSCs were different than those affected in the colon. Genetic Apc-rescue coupled with a subsequent deletion strategy revealed neoplastic growth specific to intestinal cells but not other cell types in vivo. It is noteworthy though that the majority of Apc-iPSC-derived colonic legions remained in a pretumoral microadenoma stage and did not develop into full blown macroscopic colon tumors. These findings imply that disease cell properties and biological consequences of tumor-causing mutations are strongly depending on the cellular context and underscore that epigenetic regulation, which is critical for cell fate determination and fixing the malignant cell state in cancer (see also our discussion of this issue in Limitations and challenges of iPSC modeling of human malignancies), exerts great influence on disease development and progression.
Genome Editing
Genetic modification of human pluripotent stem cells through conventional homologous recombination is hampered by extremely rare recombination events (89). Recent advances in genome editing technologies (zinc finger nucleases (ZFN), transcription activator-like effector nucleases (TALENS) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) with the Cas9 nuclease) that enable precise genetic modifications at the single nucleotide level efficiently are gaining wide use in iPSC disease modeling, including the investigation of hematological diseases and cancers (90–92). Genome editing can be used to correct or introduce disease-associated mutations, individually or in combinations, into patient-specific iPSCs or normal iPSCs, respectively, thus enabling systemic interrogation of gene function and disease development (89, 93). In both correction or introduction of mutation cases, iPSCs will be generated that bear the same genomic background and only differ in the specific genetic alteration, thus, providing ideal, isogenic iPSC pairs for comparative analysis. Genome editing through non-homologous end-joining will generate frame-shift mutations through introduction of small, random nucleotide insertions or deletions (indels) and, hence, is well suited for monoallelic or biallelic inactivation of haploinsufficient or classical tumor-suppressor genes. On the other hand, homology-directed repair (HDR) utilizing co-delivery of homologous donor DNA template to guide the homologous recombination-mediated repair process, will generate precise modifications and, thus, can be used to study point mutations in disease/cancer-associate genes or associated regulatory regions. For example, using ZFN or TALEN-based HDR, several groups succeeded in correcting of the causative, single-nucleotide mutations in HBB in sickle cell disease (SCD) and β-thalassemia iPSC lines (66, 67, 76, 78). Ma et al. showed that two distinct β-thalassemia major patient-corrected iPSC lines showed increased erythrocyte colony formation of hematopoietic progenitors compared to their isogenic, mutant iPSCs (78). Papapetrou et al. have conducted some of the most elegant gene editing for hematological malignancy modeling (56). Using correction or introduction of mutations via CRISPR/Cas9 in combination with patient-specific diseased or normal iPSCs, they modeled various disease progression stages ranging from normal/healthy, preleukemic, low-risk MDS, high-risk MDS to MDS/AML (56) as well as the contribution of the splicing factor SRSF2p.P95L mutant to MDS alone or in the context of MDS with del(7q) (40).
Genome editing systems can also be used to introduce or revert large-scale genetic lesions often associated with specific malignancies, including chromosomal deletions, inversions and translocations (55, 94–97). Brunet et al. used ZFNs and TALENs in human cells, including embryonic stem cell-derived mesenchymal precursors to generate t(11;22)(q24;q12) EWSR1-FLI1 fusion and t(2;5)(p23;q35) NMP1-ALK fusion genomic translocations associated with Ewing sarcoma and anaplastic large cell lymphoma, respectively, or to revert the t(2;5)(p23;q35) NMP1-ALK translocation (95). Torres-Ruiz et al. using CRISPR/Cas9 successfully recreated the t(11;22)(q24;q12) EWSR1-FLI1 fusion translocation in iPSCs (97). Using the adeno-associated vector-mediated gene targeting of an HSV-tk transgene approach, Papapetrou et al. generated various deletions of chromosome 7q that let them to identify an approximately 20 Mb region spanning 7q32.3-7q36.1 as the critical region in del(7q)-associated MDS (55). We together with our colleagues and the late Ihor R. Lemischka previously generated iPSCs from a Li-Fraumeni syndrome (LFS) family to investigate the oncogenic role of mutant TP53 in the development of LFS-osteosarcoma (58). In a follow up-study we identified SFRP2 as an autocrine and paracrine factor involved in P53 mutation-mediated osteosarcomagenesis. Using genome-editing we confirmed a correlation between various P53 mutations and increased SFRP2 expression in iPSC and embryonic stem cell derived osteoblasts (54) and Kim et al. (under review).
Integration of iPSCs With Tissue Engineering, Three-Dimensional Organoids and Microphysiological Systems
Diseases and cancers do not occur in a two-dimensional vacuum of malignant cells in culture but rather involve complex interactions and communication with neighboring cells and the microenvironment. Cells in the niche and the extracellular matrix provide anchor, biomechanical support and spatiotemporally regulated biochemical signals and nutrients needed for disease initiation, progression and survival. The use of tissue engineering, three-dimensional organoids and MPS attempts to more faithfully mimic the in vivo cellular milieu, architectural structure, spatial organization and physiological parameters than two-dimensional culture systems ever could. Integration of directed differentiation of iPSCs with tissue engineering, organoid cultures MPS are being developed for many complex tissues such as the heart, liver, kidney, intestine, eye, and brain (98, 99).
Organoids derived from primary resected tumors or biopsies are hailed to create opportunities to build large biobanks with relevant patient material for cancer research, drug evaluation and therapy development (100–109). With the goal of modeling human diseases of the large intestine, Chen et al. developed an efficient colonic organoid (CO) strategy using embryonic stem cells and iPSCs (43). Through a stepwise differentiation protocol following progressive normal development of definitive endoderm to hindgut endoderm to subsequently COs, using patient-specific colorectal cancer familial adenomatous polyposis (FAP)-iPSCs that carry a germline nonsense mutation in APC causing early termination of translation, they were able to demonstrate enhanced WNT signaling and increased epithelial cell proliferation. Additionally, they used these FAP-iPSC COs as a platform for testing drugs (see iPSCs in drug development & pharmacology).
As discussed in iPSCs in xenograft models, Zaret et al. modeled PDAC development using PDAC-iPSCs in combination with in vivo transplantation (33). In order to establish an in vitro model of early stage human pancreatic cancer, they harvest the PanIN structures from the developing PDAC-iPSC-derived teratomas and set up organoid cultures. The formed organoids retained PDAC-associated marker expression and served as a platform for biomarker identification.
MPS, also known as microfluidic organ-on-a-chip, offer a precise means to integrate cells, including iPSC-derived cell types and 3-dimensional constructs or organoids, into an in vitro dynamic system that further incorporates vascular flow and micro-biofabrication that mimics the systematic architectural and spatial compositions and interactions among different cell-types, tissues and organs in the body. Use of MPS in cancer research is gaining traction to investigate complex cancer, growth, tumor-niche interactions, metastatic invasion, and drug delivery, efficacy and toxicity (13–20). However, the incorporation of iPSCs or derived progenies into MPS is just beginning (110–113). Advances in generating higher-order MPS that are able to link individual systems into a physiome- or body-on-a-chip (114, 115) coupled with inline detectors and fluorescent reporters (116–119) will enable dynamic, real-time interrogation of cellular, molecular, and biomechanical parameters of disease pathogenesis (initiation and progression) and drug responses.
iPSCs in Xenograft Models
Patient-derived xenografts (PDXs) have become a prominent model system as they are presumed to more faithfully capture the cellular, molecular and physiological characteristics of primary and metastatic malignancies (120, 121). Additionally, PDX-models are gaining attraction in such field as biomarker identification, drug development and assessment of drug responses (122).
Transplantation of iPSCs or derived cells into appropriate animal models can provide a more physiological, three-dimensional in vivo environment and, hence, expand their experimental utility. PDAC has a very poor prognosis and until the elegant study by Zaret et al. lacked a human cell model of early disease progression (33). Subcutaneous, injection of iPSCs into immunocompromised mice is a process used to assess the pluripotency of iPSCs through the formation of teratomas. When Zaret et al. injected PDAC-iPSCs, ductal structures formed within the developing teratomas that had a more prominent architectural organization compared to controls. Detailed cellular and molecular characterization of these structures let to the conclusion that they resembled PanIN-stage like structures that eventually further progressed to an invasive PDAC stage.
Majeti et al. established an AML model based on iPSCs generated form patients with rearrangements of the KMT2A/MLL locus (41). Using intravenous or orthotopic transplantation into immunocompromised mice to evaluate leukemia formation in vivo they found that the ability to give rise to leukemia in vivo is dependent on transplantation of AML-iPSC-derived hematopoietic cells as AML-iPSCs lacked leukemic potential. Additionally, despite retaining the leukemic-driver mutations, AML-iPSCs reset the leukemic DNA methylation and gene expression patterns. Surprisingly, hematopoietic differentiation of these AML-iPSCs and leukemia formation was sufficient to reestablish the leukemic DNA methylation and gene expression profile strongly suggesting that the genetic mutations/rearrangements of the KMT2A/MLL locus in AML-iPSCs reactivate a leukemic program in the context of hematopoietic cells (41).
It was recently reported that copy number alterations recurrently observed in primary human tumors gradually disappeared in PDXs, suggesting that events undergoing positive selection in humans can become dispensable during propagation in mice (123). In light of this observation and its critical implications for PDX-based disease/cancer modeling, cytogenetic analyses of PDX-donor cells after in vivo transplantation and propagation appears important in order to know whether the attempted PDX-model accurately retains the genetic lesions present in the original malignant cells or if they evolve, and if they evolve whether the evolution is specific to the patient or the host.
iPSCs in Drug Development and Pharmacology
The cost of drug development from discovery, through clinical trials to approval and marketing is in excess of $2.6 billion (124). As costly as clinical trials are, drug failures are key contributors to development costs. Induced PSCs and derived cells are gaining attraction and are being more widely used in translational-research settings, including discovery and validation of biomarkers and therapeutic targets, compound screening for drug discovery and drug repurposing, and preclinical drug susceptibility, efficacy and toxicity studies (33, 39, 41, 43, 57, 65, 72, 73, 110, 125–131). Of particular usefulness is that many different cell type, including cardiomyocytes, hepatocytes, neurons, and hematopoietic cells, can readily be generated from a diverse set (age, gender, race/ethnicity) of iPSCs from healthy individuals or patients with a given disease/cancer. This has been exemplified in the use of iPSCs in drug toxicity screening. Therapeutically effective drugs can cause serious unintended adverse events that limit or even prohibit their use. Several groups have used iPSC-derived cardiomyocytes to model and investigate anticancer drug-induced cardiotoxicity (132–137). In one case, cardiomyocytes generated from iPSCs from breast cancer patients were able to recapitulate patient-specific doxorubicin-induced cardiotoxicity at the cellular level (134). Another application is the evaluation of drug susceptibility and variable responses of phenotypic distinct cell populations, cancer subclones or patients (39, 41, 57, 65, 72, 73). Primary or acquired-drug resistance is a serious clinical problem. Induced PSCs derived either from drug-sensitive and drug-resistance patients or from cells of the same patients at the drug-sensitive and drug-resistant stage and iPSC derived cells might help decipher the mechanisms underlying drug-resistance. Examples along this line are from Bedel et al. (39) and Kumano et al. (57). They derived iPSC lines from CML patients that carry the abnormal Philadelphia chromosome that resulted from a translocation between chromosome 22 and 9 leading to the fused, oncogenic BCR-ABL tyrosine kinase. While both groups reported that the generated CML-iPSC lines were resistant to the tyrosine kinase inhibitor imatinib, which is used to treat CML patients, Bedel et al. (39) found that CD34+ hematopoietic progenitors obtained from their patient's CML-iPSCs were partially sensitive to imatinib and Kumano et al. (57) found imatinib-sensitivity in CML-iPSC derived CD34− hematopoietic cells but not CD34+ hematopoietic progenitors, which recapitulated the pathophysiological feature of initial CML of that patient. In depth molecular characterization at the epigenome, transcriptome and proteome level will be necessary to discover the signaling networks responsible for the observed behavior. Induced PSCs and derived cells also present an opportunity for phenotypic drug testing and screening. This can be especially attractive for diseases with no previously characterized targets or drug treatment strategies. However, such phenotypic drug testing and screening requires the ability to identify cellular phenotypes or functional properties, such as proliferation, apoptosis, activation of a specific signaling pathway, a distinct metabolic profile that correlate with patient phenotypes and responses and thus can serve as surrogate readouts of therapeutic effectiveness (43, 110, 117, 130, 138). Undoubtedly, the next stage in drug discovery and pharmacological testing will expand on the integration of iPSC-based model systems with three-dimensional organoids and MPS (43, 110).
Concluding Remarks
iPSC technology started a new, exciting era in biomedicine. The ease by which patient-specific iPSCs from various primary or metastatic somatic tissues and blood of patients with hematological diseases and cancers can be derived provides a self-renewable, scalable and cryopreservable source of cells with the patient's genetic background. iPSCs are readily enable to genome-editing in order to either correct or introduce known or suspected disease-associated mutations. This novel tool enables attempts to successfully recapitulate various pathological disease states and features associated with malignancies in a patient-specific context. Integration of iPSC-based disease and cancer models with advanced, bioengineered physiological systems, in vivo PDX models, automated high-throughput-screening tools and next-generation omics approaches will lead to a greater mechanistic understanding of disease/cancer, the relationship between malignant cells and their microenvironment, and drug responses. Undoubtedly, iPSC technology is revolutionizing the way we approach disease modeling, preclinical cancer research, drug development and precision medicine.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to acknowledge the late Ihor R. Lemischka for his support and mentorship over the years. HK is the recipient of an NCI Pre- to Post-doctoral Transition Award (1F99CA212489).
References
1. Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. (2012) 180:599–607. doi: 10.1016/j.ajpath.2011.10.036
2. Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci USA. (2012) 109:20035–40. doi: 10.1073/pnas.1213241109
3. Liu X, Krawczyk E, Suprynowicz FA, Palechor-Ceron N, Yuan H, Dakic A, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. (2017) 12:439–51. doi: 10.1038/nprot.2016.174
4. Lai Y, Wei X, Lin S, Qin L, Cheng LLi P. Current status and perspectives of patient-derived xenograft models in cancer research. J Hematol Oncol. (2017) 10:106. doi: 10.1186/s13045-017-0470-7
5. Borodovsky A, McQuiston TJ, Stetson D, Ahmed A, Whitston D, Zhang J, et al. Generation of stable PDX derived cell lines using conditional reprogramming. Mol Cancer (2017) 16:177. doi: 10.1186/s12943-017-0745-1
6. Sachs NClevers H. Organoid cultures for the analysis of cancer phenotypes. Curr Opin Genet Dev. (2014) 24:68–73. doi: 10.1016/j.gde.2013.11.012
7. Fatehullah A, Tan SHBarker N. Organoids as an in vitro model of human development and disease. Nat Cell Biol. (2016) 18:246–54. doi: 10.1038/ncb3312
8. Kretzschmar KClevers H. Organoids: modeling development and the stem cell niche in a dish. Dev Cell (2016) 38:590–600. doi: 10.1016/j.devcel.2016.08.014
9. Weeber F, Ooft SN, Dijkstra KKVoest EE. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem Biol. (2017) 24:1092–100. doi: 10.1016/j.chembiol.2017.06.012
10. Hutmacher DW, Horch RE, Loessner D, Rizzi S, Sieh S, Reichert JC, et al. Translating tissue engineering technology platforms into cancer research. J Cell Mol Med. (2009) 13:1417–27. doi: 10.1111/j.1582-4934.2009.00853.x
11. Hutmacher DW, Loessner D, Rizzi S, Kaplan DL, Mooney DJClements JA. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. (2010) 28:125–33. doi: 10.1016/j.tibtech.2009.12.001
12. Holzapfel BM, Wagner F, Thibaudeau L, Levesque JP, Hutmacher DW. Concise review: humanized models of tumor immunology in the 21st century: convergence of cancer research and tissue engineering. Stem Cells (2015) 33:1696–704. doi: 10.1002/stem.1978
13. Hu S, Liu G, Chen W, Li X, Lu W, Lam RH, et al. Multiparametric biomechanical and biochemical phenotypic profiling of single cancer cells using an elasticity microcytometer. Small (2016) 12:2300–11. doi: 10.1002/smll.201503620
14. Portillo-Lara R, Annabi N. Microengineered cancer-on-a-chip platforms to study the metastatic microenvironment. Lab Chip. (2016) 16:4063–81. doi: 10.1039/c6lc00718j
15. Fan Q, Liu R, Jiao Y, Tian C, Farrell JD, Diao W, et al. A novel 3-D bio-microfluidic system mimicking in vivo heterogeneous tumour microstructures reveals complex tumour-stroma interactions. Lab Chip (2017) 17:2852–60. doi: 10.1039/c7lc00191f
16. Armbrecht L, Gabernet G, Kurth F, Hiss JA, Schneider G, Dittrich PS. Characterisation of anticancer peptides at the single-cell level. Lab Chip (2017) 17:2933–40. doi: 10.1039/c7lc00505a
17. Low LA, Tagle DA. Tissue chips - innovative tools for drug development and disease modeling. Lab Chip (2017) 17:3026–36. doi: 10.1039/c7lc00462a
18. Huang YL, Segall JE, Wu M. Microfluidic modeling of the biophysical microenvironment in tumor cell invasion. Lab Chip (2017) 17:3221–33. doi: 10.1039/c7lc00623c
19. Caballero D, Blackburn SM, de Pablo M, Samitier J, Albertazzi L. Tumour-vessel-on-a-chip models for drug delivery. Lab Chip (2017) 17:3760–71. doi: 10.1039/c7lc00574a
20. Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS, et al. Human organ chip models recapitulate orthotopic lung cancer growth, therapeutic responses, and tumor dormancy in vitro. Cell Rep. (2017) 21:508–16. doi: 10.1016/j.celrep.2017.09.043
21. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
22. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science (2007) 318:1917–20. doi: 10.1126/science.1151526
23. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) 131:861–72. doi: 10.1016/j.cell.2007.11.019
24. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature (2008) 451:141–6. doi: 10.1038/nature06534
25. Lin SL, Chang DC, Chang-Lin S, Lin CH, Wu DT, Chen DT, et al. Mir-302 reprograms human skin cancer cells into a pluripotent ES-cell-like state. RNA (2008) 14:2115–24. doi: 10.1261/rna.1162708
26. Utikal J, Maherali N, Kulalert W, Hochedlinger K. Sox2 is dispensable for the reprogramming of melanocytes and melanoma cells into induced pluripotent stem cells. J Cell Sci. (2009) 122(Pt 19):3502–10. doi: 10.1242/jcs.054783
27. Miyoshi N, Ishii H, Nagai K, Hoshino H, Mimori K, Tanaka F, et al. Defined factors induce reprogramming of gastrointestinal cancer cells. Proc Natl Acad Sci USA. (2010) 107:40–5. doi: 10.1073/pnas.0912407107
28. Carette JE, Pruszak J, Varadarajan M, Blomen VA, Gokhale S, Camargo FD, et al. Generation of iPSCs from cultured human malignant cells. Blood (2010) 115:4039–42. doi: 10.1182/blood-2009-07-231845
29. Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. (2011) 71:4640–52. doi: 10.1158/0008-5472.CAN-10-3320
30. Zhang X, Cruz FD, Terry M, Remotti F, Matushansky I. Terminal differentiation and loss of tumorigenicity of human cancers via pluripotency-based reprogramming. Oncogene (2013) 32:2249–60, 60 e1–21. doi: 10.1038/onc.2012.237
31. Mahalingam D, Kong CM, Lai J, Tay LL, Yang H, Wang X. Reversal of aberrant cancer methylome and transcriptome upon direct reprogramming of lung cancer cells. Sci Rep. (2012) 2:592. doi: 10.1038/srep00592
32. Stricker SH, Feber A, Engstrom PG, Caren H, Kurian KM, Takashima Y, et al. Widespread resetting of DNA methylation in glioblastoma-initiating cells suppresses malignant cellular behavior in a lineage-dependent manner. Genes Dev. (2013) 27:654–69. doi: 10.1101/gad.212662.112
33. Kim J, Hoffman JP, Alpaugh RK, Rhim AD, Reichert M, Stanger BZ, et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. (2013) 3:2088–99. doi: 10.1016/j.celrep.2013.05.036
34. Corominas-Faja B, Cufi S, Oliveras-Ferraros C, Cuyas E, Lopez-Bonet E, Lupu R, et al. Nuclear reprogramming of luminal-like breast cancer cells generates Sox2-overexpressing cancer stem-like cellular states harboring transcriptional activation of the mTOR pathway. Cell Cycle (2013) 12:3109–24. doi: 10.4161/cc.26173
35. Choong PF, Teh HX, Teoh HK, Ong HK, Choo KB, Sugii S, et al. Heterogeneity of osteosarcoma cell lines led to variable responses in reprogramming. Int J Med Sci. (2014) 11:1154–60. doi: 10.7150/ijms.8281
36. Moore JBt, Loeb DM, Hong KU, Sorensen PH, Triche TJ, Lee DW, et al. Epigenetic reprogramming and re-differentiation of a Ewing sarcoma cell line. Front Cell Dev Biol. (2015) 3:15. doi: 10.3389/fcell.2015.00015
37. Verusingam ND, Yeap SK, Ky H, Paterson IC, Khoo SP, Cheong SK, et al. Susceptibility of Human Oral Squamous Cell Carcinoma (OSCC) H103 and H376 cell lines to Retroviral OSKM mediated reprogramming. PeerJ. (2017) 5:e3174. doi: 10.7717/peerj.3174
38. Antony-Debre I, Manchev VT, Balayn N, Bluteau D, Tomowiak C, Legrand C, et al. Level of RUNX1 activity is critical for leukemic predisposition but not for thrombocytopenia. Blood (2015) 125:930–40. doi: 10.1182/blood-2014-06-585513
39. Bedel A, Pasquet JM, Lippert E, Taillepierre M, Lagarde V, Dabernat S, et al. Variable behavior of iPSCs derived from CML patients for response to TKI and hematopoietic differentiation. PLoS ONE (2013) 8:e71596. doi: 10.1371/journal.pone.0071596
40. Chang CJ, Kotini AG, Olszewska M, Georgomanoli M, Teruya-Feldstein J, Sperber H, et al. Dissecting the contributions of cooperating gene mutations to cancer phenotypes and drug responses with patient-derived iPSCs. Stem Cell Reports (2018) 10:1610–24. doi: 10.1016/j.stemcr.2018.03.020
41. Chao MP, Gentles AJ, Chatterjee S, Lan F, Reinisch A, Corces MR, et al. Human AML-iPSCs reacquire leukemic properties after differentiation and model clonal variation of disease. Cell Stem Cell (2017) 20:329–44 e7. doi: 10.1016/j.stem.2016.11.018
42. Connelly JP, Kwon EM, Gao Y, Trivedi NS, Elkahloun AG, Horwitz MS, et al. Targeted correction of RUNX1 mutation in FPD patient-specific induced pluripotent stem cells rescues megakaryopoietic defects. Blood (2014) 124:1926–30. doi: 10.1182/blood-2014-01-550525
43. Crespo M, Vilar E, Tsai SY, Chang K, Amin S, Srinivasan T, et al. Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing. Nat Med. (2017) 23:878–84. doi: 10.1038/nm.4355
44. Doulatov S, Vo LT, Macari ER, Wahlster L, Kinney MA, Taylor AM, et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci Transl Med. (2017) 9:eaah5645. doi: 10.1126/scitranslmed.aah5645
45. Espinoza JL, Elbadry MI, Chonabayashi K, Yoshida Y, Katagiri T, Harada K, et al. Hematopoiesis by iPSC-derived hematopoietic stem cells of aplastic anemia that escape cytotoxic T-cell attack. Blood Adv. (2018) 2:390–400. doi: 10.1182/bloodadvances.2017013342
46. Gandre-Babbe S, Paluru P, Aribeana C, Chou ST, Bresolin S, Lu L, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood (2013) 121:4925–9. doi: 10.1182/blood-2013-01-478412
47. Garcon L, Ge J, Manjunath SH, Mills JA, Apicella M, Parikh S, et al. Ribosomal and hematopoietic defects in induced pluripotent stem cells derived from Diamond Blackfan anemia patients. Blood (2013) 122:912–21. doi: 10.1182/blood-2013-01-478321
48. Hadoux J, Feraud O, Griscelli F, Opolon P, Divers D, Gobbo E, et al. Generation of an induced pluripotent stem cell line from a patient with hereditary multiple endocrine neoplasia 2A (MEN2A) syndrome with RET mutation. Stem Cell Res. (2016) 17:154–57. doi: 10.1016/j.scr.2016.06.008
49. Hosoi M, Kumano K, Taoka K, Arai S, Kataoka K, Ueda K, et al. Generation of induced pluripotent stem cells derived from primary and secondary myelofibrosis patient samples. Exp Hematol (2014) 42:816–25. doi: 10.1016/j.exphem.2014.03.010
50. Hu K, Yu J, Suknuntha K, Tian S, Montgomery K, Choi KD, et al. Efficient generation of transgene-free induced pluripotent stem cells from normal and neoplastic bone marrow and cord blood mononuclear cells. Blood (2011) 117:e109–19. doi: 10.1182/blood-2010-07-298331
51. Hu L, Du L, Zhao Y, Li W, Ouyang Q, Zhou D, et al. Modeling Glanzmann thrombasthenia using patient specific iPSCs and restoring platelet aggregation function by CD41 overexpression. Stem Cell Res. (2017) 20:14–20. doi: 10.1016/j.scr.2017.02.003
52. Iizuka H, Kagoya Y, Kataoka K, Yoshimi A, Miyauchi M, Taoka K, et al. Targeted gene correction of RUNX1 in induced pluripotent stem cells derived from familial platelet disorder with propensity to myeloid malignancy restores normal megakaryopoiesis. Exp Hematol. (2015) 43:849–57. doi: 10.1016/j.exphem.2015.05.004
53. Julian LM, Delaney SP, Wang Y, Goldberg AA, Dore C, Yockell-Lelievre J, et al. Human pluripotent stem cell-derived TSC2-haploinsufficient smooth muscle cells recapitulate features of lymphangioleiomyomatosis. Cancer Res. (2017) 77:5491–502. doi: 10.1158/0008-5472.CAN-17-0925
54. Kim HS, Yoo S, Bernitz JM, Yuan Y, Gomes AM, Daniel MG, et al. Oncogenic role of sFRP2 in P53-mutant osteosarcoma development via autocrine and paracrine mechanism. bioRxiv (2018). doi: 10.1101/246454. [Epub ahead of print].
55. Kotini AG, Chang CJ, Boussaad I, Delrow JJ, Dolezal EK, Nagulapally AB, et al. Functional analysis of a chromosomal deletion associated with myelodysplastic syndromes using isogenic human induced pluripotent stem cells. Nat Biotechnol. (2015) 33:646–55. doi: 10.1038/nbt.3178
56. Kotini AG, Chang CJ, Chow A, Yuan H, Ho TC, Wang T, et al. Stage-specific human induced pluripotent stem cells map the progression of myeloid transformation to transplantable leukemia. Cell Stem Cell (2017) 20:315–28 e7. doi: 10.1016/j.stem.2017.01.009
57. Kumano K, Arai S, Hosoi M, Taoka K, Takayama N, Otsu M, et al. Generation of induced pluripotent stem cells from primary chronic myelogenous leukemia patient samples. Blood (2012) 119:6234–42. doi: 10.1182/blood-2011-07-367441
58. Lee DF, Su J, Kim HS, Chang B, Papatsenko D, Zhao R, et al. Modeling familial cancer with induced pluripotent stem cells. Cell (2015) 161:240–54. doi: 10.1016/j.cell.2015.02.045
59. Lee JH, Salci KR, Reid JC, Orlando L, Tanasijevic B, Shapovalova Z, et al. Brief report: human acute myeloid leukemia reprogramming to pluripotency is a rare event and selects for patient hematopoietic cells devoid of leukemic mutations. Stem Cells (2017) 35:2095–102. doi: 10.1002/stem.2655
60. Melguizo-Sanchis D, Xu Y, Taheem D, Yu M, Tilgner K, Barta T, et al. iPSC modeling of severe aplastic anemia reveals impaired differentiation and telomere shortening in blood progenitors. Cell Death Dis. (2018) 9:128. doi: 10.1038/s41419-017-0141-1
61. Mulero-Navarro S, Sevilla A, Roman AC, Lee DF, D'Souza SL, Pardo S, et al. Myeloid dysregulation in a human induced pluripotent stem cell model of PTPN11-associated juvenile myelomonocytic leukemia. Cell Rep. (2015) 13:504–15. doi: 10.1016/j.celrep.2015.09.019
62. Muller LU, Milsom MD, Harris CE, Vyas R, Brumme KM, Parmar K, et al. Overcoming reprogramming resistance of Fanconi anemia cells. Blood (2012) 119:5449–57. doi: 10.1182/blood-2012-02-408674
63. Orban M, Goedel A, Haas J, Sandrock-Lang K, Gartner F, Jung CB, et al. Functional comparison of induced pluripotent stem cell- and blood-derived GPIIbIIIa deficient platelets. PLoS ONE (2015) 10:e0115978. doi: 10.1371/journal.pone.0115978
64. Sakurai M, Kunimoto H, Watanabe N, Fukuchi Y, Yuasa S, Yamazaki S, et al. Impaired hematopoietic differentiation of RUNX1-mutated induced pluripotent stem cells derived from FPD/AML patients. Leukemia (2014) 28:2344–54. doi: 10.1038/leu.2014.136
65. Saliba J, Hamidi S, Lenglet G, Langlois T, Yin J, Cabagnols X, et al. Heterozygous and homozygous JAK2(V617F) states modeled by induced pluripotent stem cells from myeloproliferative neoplasm patients. PLoS ONE (2013) 8:e74257. doi: 10.1371/journal.pone.0074257
66. Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells (2011) 29:1717–26. doi: 10.1002/stem.718
67. Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng (2014) 111:1048–53. doi: 10.1002/bit.25018
68. Suzuki NM, Niwa A, Yabe M, Hira A, Okada C, Amano N, et al. Pluripotent cell models of fanconi anemia identify the early pathological defect in human hemoangiogenic progenitors. Stem Cells Transl Med. (2015) 4:333–8. doi: 10.5966/sctm.2013-0172
69. Takei H, Edahiro Y, Mano S, Masubuchi N, Mizukami Y, Imai M, et al. Skewed megakaryopoiesis in human induced pluripotent stem cell-derived haematopoietic progenitor cells harbouring calreticulin mutations. Br J Haematol. (2018) 181:791–802. doi: 10.1111/bjh.15266
70. Tulpule A, Kelley JM, Lensch MW, McPherson J, Park IH, Hartung O, et al. Pluripotent stem cell models of Shwachman-Diamond syndrome reveal a common mechanism for pancreatic and hematopoietic dysfunction. Cell Stem Cell (2013) 12:727–36. doi: 10.1016/j.stem.2013.04.002
71. Varela I, Karagiannidou A, Oikonomakis V, Tzetis M, Tzanoudaki M, Siapati EK, et al. Generation of human beta-thalassemia induced pluripotent cell lines by reprogramming of bone marrow-derived mesenchymal stromal cells using modified mRNA. Cell Reprogram (2014) 16:447–55. doi: 10.1089/cell.2014.0050
72. Yamamoto S, Otsu M, Matsuzaka E, Konishi C, Takagi H, Hanada S, et al. Screening of drugs to treat 8p11 myeloproliferative syndrome using patient-derived induced pluripotent stem cells with fusion gene CEP110-FGFR1. PLoS ONE (2015) 10:e0120841. doi: 10.1371/journal.pone.0120841
73. Ye Z, Liu CF, Lanikova L, Dowey SN, He C, Huang X, et al. Differential sensitivity to JAK inhibitory drugs by isogenic human erythroblasts and hematopoietic progenitors generated from patient-specific induced pluripotent stem cells. Stem Cells (2014) 32:269–78. doi: 10.1002/stem.1545
74. Ye Z, Zhan H, Mali P, Dowey S, Williams DM, Jang YY, et al. Human-induced pluripotent stem cells from blood cells of healthy donors and patients with acquired blood disorders. Blood (2009) 114:5473–80. doi: 10.1182/blood-2009-04-217406
75. Yung SK, Tilgner K, Ledran MH, Habibollah S, Neganova I, Singhapol C, et al. Brief report: human pluripotent stem cell models of fanconi anemia deficiency reveal an important role for fanconi anemia proteins in cellular reprogramming and survival of hematopoietic progenitors. Stem Cells (2013) 31:1022–9. doi: 10.1002/stem.1308
76. Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood (2011) 118:4599–608. doi: 10.1182/blood-2011-02-335554
77. Maclean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, et al. Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci USA. (2012) 109:17567–72. doi: 10.1073/pnas.1215468109
78. Ma N, Liao B, Zhang H, Wang L, Shan Y, Xue Y, et al. Transcription activator-like effector nuclease (TALEN)-mediated gene correction in integration-free beta-thalassemia induced pluripotent stem cells. J Biol Chem. (2013) 288:34671–9. doi: 10.1074/jbc.M113.496174
79. Hadoux J, Desterke C, Feraud O, Guibert M, De Rose RF, Opolon P, et al. Transcriptional landscape of a RET(C634Y)-mutated iPSC and its CRISPR-corrected isogenic control reveals the putative role of EGR1 transcriptional program in the development of multiple endocrine neoplasia type 2A-associated cancers. Stem Cell Res. (2018) 26:8–16. doi: 10.1016/j.scr.2017.11.015
80. Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. (1999) 23:166–75. doi: 10.1038/13793
81. Bellissimo DC, Speck NA. RUNX1 Mutations in Inherited and Sporadic Leukemia. Front Cell Dev Biol. (2017) 5:111. doi: 10.3389/fcell.2017.00111
82. Salci KR, Lee JH, Laronde S, Dingwall S, Kushwah R, Fiebig-Comyn A, et al. Cellular reprogramming allows generation of autologous hematopoietic progenitors from AML patients that are devoid of patient-specific genomic aberrations. Stem Cells (2015) 33:1839–49. doi: 10.1002/stem.1994
83. Raya A, Rodriguez-Piza I, Guenechea G, Vassena R, Navarro S, Barrero MJ, et al. Disease-corrected haematopoietic progenitors from Fanconi anaemia induced pluripotent stem cells. Nature (2009) 460:53–9. doi: 10.1038/nature08129
84. Munoz-Lopez A, Romero-Moya D, Prieto C, Ramos-Mejia V, Agraz-Doblas A, Varela I, et al. Development refractoriness of MLL-rearranged human B cell acute leukemias to reprogramming into pluripotency. Stem Cell Rep. (2016) 7:602–18. doi: 10.1016/j.stemcr.2016.08.013
85. Taniguchi TD'Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood (2006) 107:4223–33. doi: 10.1182/blood-2005-10-4240
86. Vicente-Duenas C, Hauer J, Ruiz-Roca L, Ingenhag D, Rodriguez-Meira A, Auer F, et al. Tumoral stem cell reprogramming as a driver of cancer: Theory, biological models, implications in cancer therapy. Semin Cancer Biol. (2015) 32:3–9. doi: 10.1016/j.semcancer.2014.02.001
87. Papp B, Plath K. Reprogramming to pluripotency: stepwise resetting of the epigenetic landscape. Cell Res. (2011) 21:486–501. doi: 10.1038/cr.2011.28
88. Hashimoto K, Yamada Y, Semi K, Yagi M, Tanaka A, Itakura F, et al. Cellular context-dependent consequences of Apc mutations on gene regulation and cellular behavior. Proc Natl Acad Sci USA. (2017) 114:758–63. doi: 10.1073/pnas.1614197114
89. Hockemeyer D, Jaenisch R. Induced pluripotent stem cells meet genome editing. Cell Stem Cell (2016) 18:573–86. doi: 10.1016/j.stem.2016.04.013
90. Chen S, Sun H, Miao K, Deng CX. CRISPR-Cas9: from genome editing to cancer research. Int J Biol Sci. (2016) 12:1427–36. doi: 10.7150/ijbs.17421
91. Moses C, Garcia-Bloj B, Harvey AR, Blancafort P. Hallmarks of cancer: The CRISPR generation. Eur J Cancer (2018) 93:10–8. doi: 10.1016/j.ejca.2018.01.002
92. Ratan ZA, Son YJ, Haidere MF, Uddin BMM, Yusuf MA, Zaman SB, et al. CRISPR-Cas9: a promising genetic engineering approach in cancer research. Ther Adv Med Oncol. (2018) 10:1758834018755089. doi: 10.1177/1758834018755089
93. Kim HS, Bernitz JM, Lee DF, Lemischka IR. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. (2014) 23:2673–86. doi: 10.1089/scd.2014.0167
94. Brunet E, Simsek D, Tomishima M, DeKelver R, Choi VM, Gregory P, et al. Chromosomal translocations induced at specified loci in human stem cells. Proc Natl Acad Sci USA. (2009) 106:10620–5. doi: 10.1073/pnas.0902076106
95. Piganeau M, Ghezraoui H, De Cian A, Guittat L, Tomishima M, Perrouault L, et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. (2013) 23:1182–93. doi: 10.1101/gr.147314.112
96. Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, et al. In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature (2014) 516:423–7. doi: 10.1038/nature13902
97. Torres-Ruiz R, Martinez-Lage M, Martin MC, Garcia A, Bueno C, Castano J, et al. Efficient recreation of t(11;22) EWSR1-FLI1(+) in human stem cells using CRISPR/Cas9. Stem Cell Rep. (2017) 8:1408–20. doi: 10.1016/j.stemcr.2017.04.014
98. Lancaster MA, Knoblich JA. Organogenesis in a dish: modeling development and disease using organoid technologies. Science (2014) 345:1247125. doi: 10.1126/science.1247125
99. Liu C, Oikonomopoulos A, Sayed N, Wu JC. Modeling human diseases with induced pluripotent stem cells: from 2D to 3D and beyond. Development (2018) 145:dev156166. doi: 10.1242/dev.156166
100. Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology (2011) 141:1762–72. doi: 10.1053/j.gastro.2011.07.050
101. Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell (2014) 159:176–87. doi: 10.1016/j.cell.2014.08.016
102. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell (2015) 160:324–38. doi: 10.1016/j.cell.2014.12.021
103. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell (2015) 161:933–45. doi: 10.1016/j.cell.2015.03.053
104. Huang L, Holtzinger A, Jagan I, BeGora M, Lohse I, Ngai N, et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat Med. (2015) 21:1364–71. doi: 10.1038/nm.3973
105. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell (2018) 172:373–86 e10. doi: 10.1016/j.cell.2017.11.010
106. Mazzocchi AR, Rajan SAP, Votanopoulos KI, Hall AR, Skardal A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci Rep. (2018) 8:2886. doi: 10.1038/s41598-018-21200-8
107. Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science (2018) 359:920–26. doi: 10.1126/science.aao2774
108. Drost J, Clevers H. Organoids in cancer research. Nat Rev Cancer (2018) 18:407–18.doi: 10.1038/s41568-018-0007-6
109. Seidlitz T, Merker SR, Rothe A, Zakrzewski F, von Neubeck C, Grutzmann K, et al. Human gastric cancer modelling using organoids. Gut (2018). doi: 10.1136/gutjnl-2017-314549. [Epub ahead of print]
110. Mathur A, Loskill P, Shao K, Huebsch N, Hong S, Marcus SG, et al. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci Rep. (2015) 5:8883. doi: 10.1038/srep08883
111. Giobbe GG, Michielin F, Luni C, Giulitti S, Martewicz S, Dupont S, et al. Functional differentiation of human pluripotent stem cells on a chip. Nat Methods (2015) 12:637–40. doi: 10.1038/nmeth.3411
112. Marsano A, Conficconi C, Lemme M, Occhetta P, Gaudiello E, Votta E, et al. Beating heart on a chip: a novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip (2016) 16:599–610. doi: 10.1039/c5lc01356a
113. Musah S, Mammoto A, Ferrante TC, Jeanty SSF, Hirano-Kobayashi M, Mammoto T, et al. Mature induced-pluripotent-stem-cell-derived human podocytes reconstitute kidney glomerular-capillary-wall function on a chip. Nat Biomed Eng. (2017) 1:0069. doi: 10.1038/s41551-017-0069
114. Maschmeyer I, Lorenz AK, Schimek K, Hasenberg T, Ramme AP, Hubner J, et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip (2015) 15:2688–99. doi: 10.1039/c5lc00392j
115. Edington CD, Chen WLK, Geishecker E, Kassis T, Soenksen LR, Bhushan BM, et al. Interconnected microphysiological systems for quantitative biology and pharmacology studies. Sci Rep. (2018) 8:4530. doi: 10.1038/s41598-018-22749-0
116. Zhao WN, Cheng C, Theriault KM, Sheridan SD, Tsai LH, Haggarty SJ. A high-throughput screen for Wnt/beta-catenin signaling pathway modulators in human iPSC-derived neural progenitors. J Biomol Screen (2012) 17:1252–63. doi: 10.1177/1087057112456876
117. Lapp H, Bruegmann T, Malan D, Friedrichs S, Kilgus C, Heidsieck A, et al. Frequency-dependent drug screening using optogenetic stimulation of human iPSC-derived cardiomyocytes. Sci Rep (2017) 7:9629. doi: 10.1038/s41598-017-09760-7
118. Jung KB, Lee H, Son YS, Lee JH, Cho HS, Lee MO, et al. In vitro and in vivo imaging and tracking of intestinal organoids from human induced pluripotent stem cells. FASEB J. (2018) 32:111–22. doi: 10.1096/fj.201700504R
119. Bjork S, Ojala EA, Nordstrom T, Ahola A, Liljestrom M, Hyttinen J, et al. Evaluation of optogenetic electrophysiology tools in human stem cell-derived cardiomyocytes. Front Physiol. (2017) 8:884. doi: 10.3389/fphys.2017.00884
120. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. (2012) 9:338–50. doi: 10.1038/nrclinonc.2012.61
121. Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res. (2013) 73:5315–9. doi: 10.1158/0008-5472.CAN-13-1069
122. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. (2015) 21:1318–25. doi: 10.1038/nm.3954
123. Ben-David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. (2017) 49:1567–75. doi: 10.1038/ng.3967
124. DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: New estimates of R&D costs. J Health Econ. (2016) 47:20–33. doi: 10.1016/j.jhealeco.2016.01.012
125. Lee G, Ramirez CN, Kim H, Zeltner N, Liu B, Radu C, et al. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. (2012) 30:1244–8. doi: 10.1038/nbt.2435
126. Engle SJ, Puppala D. Integrating human pluripotent stem cells into drug development. Cell Stem Cell (2013) 12:669–77. doi: 10.1016/j.stem.2013.05.011
127. Engle SJ, Vincent F. Small molecule screening in human induced pluripotent stem cell-derived terminal cell types. J Biol Chem. (2014) 289:4562–70. doi: 10.1074/jbc.R113.529156
128. Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. (2014) 7:1–11. doi: 10.1016/j.celrep.2014.03.019
129. Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science (2014) 345:688–93. doi: 10.1126/science.1250127
130. Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell Rep (2014) 9:810–21. doi: 10.1016/j.celrep.2014.09.055
131. Miyauchi M, Koya J, Arai S, Yamazaki S, Honda A, Kataoka K, et al. ADAM8 is an antigen of tyrosine kinase inhibitor-resistant chronic myeloid leukemia cells identified by patient-derived induced pluripotent stem cells. Stem Cell Rep. (2018) 10:1115–30. doi: 10.1016/j.stemcr.2018.01.015
132. Liang P, Lan F, Lee AS, Gong T, Sanchez-Freire V, Wang Y, et al. Drug screening using a library of human induced pluripotent stem cell-derived cardiomyocytes reveals disease-specific patterns of cardiotoxicity. Circulation (2013) 127:1677–91. doi: 10.1161/CIRCULATIONAHA.113.001883
133. Chaudhari U, Nemade H, Wagh V, Gaspar JA, Ellis JK, Srinivasan SP, et al. Identification of genomic biomarkers for anthracycline-induced cardiotoxicity in human iPSC-derived cardiomyocytes: an in vitro repeated exposure toxicity approach for safety assessment. Arch Toxicol. (2016) 90:2763–77. doi: 10.1007/s00204-015-1623-5
134. Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. (2016) 22:547–56. doi: 10.1038/nm.4087
135. Maillet A, Tan K, Chai X, Sadananda SN, Mehta A, Ooi J, et al. Modeling doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci Rep. (2016) 6:25333. doi: 10.1038/srep25333
136. Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. (2017) 9:eaaf2584. doi: 10.1126/scitranslmed.aaf2584
137. Louisse J, Wust RCI, Pistollato F, Palosaari T, Barilari M, Macko P, et al. Assessment of acute and chronic toxicity of doxorubicin in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol In Vitro (2017) 42:182–90. doi: 10.1016/j.tiv.2017.04.023
Keywords: cancer, blood disorders, hematopoietic malignancies, induced pluripotent stem cells, model systems
Citation: Kim H and Schaniel C (2018) Modeling Hematological Diseases and Cancer With Patient-Specific Induced Pluripotent Stem Cells. Front. Immunol. 9:2243. doi: 10.3389/fimmu.2018.02243
Received: 29 June 2018; Accepted: 10 September 2018;
Published: 28 September 2018.
Edited by:
Rhodri Ceredig, National University of Ireland Galway, IrelandReviewed by:
Kay L. Medina, Mayo Clinic, United StatesEncarnacion Montecino-Rodriguez, University of California, Los Angeles, United States
Geoffrey Brown, University of Birmingham, United Kingdom
Copyright © 2018 Kim and Schaniel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Schaniel, Y2hyaXN0b3BoLnNjaGFuaWVsQG1zc20uZWR1