 Magdalena Radwanska1
Magdalena Radwanska1 Nick Vereecke
Nick Vereecke Violette Deleeuw
Violette Deleeuw Joar Pinto
Joar Pinto Stefan Magez
Stefan Magez- 1Laboratory for Biomedical Research, Ghent University Global Campus, Incheon, South Korea
- 2Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium
Salivarian trypanosomes are single cell extracellular parasites that cause infections in a wide range of hosts. Most pathogenic infections worldwide are caused by one of four major species of trypanosomes including (i) Trypanosoma brucei and the human infective subspecies T. b. gambiense and T. b. rhodesiense, (ii) Trypanosoma evansi and T. equiperdum, (iii) Trypanosoma congolense and (iv) Trypanosoma vivax. Infections with these parasites are marked by excessive immune dysfunction and immunopathology, both related to prolonged inflammatory host immune responses. Here we review the classification and global distribution of these parasites, highlight the adaptation of human infective trypanosomes that allow them to survive innate defense molecules unique to man, gorilla, and baboon serum and refer to the discovery of sexual reproduction of trypanosomes in the tsetse vector. With respect to the immunology of mammalian host-parasite interactions, the review highlights recent findings with respect to the B cell destruction capacity of trypanosomes and the role of T cells in the governance of infection control. Understanding infection-associated dysfunction and regulation of both these immune compartments is crucial to explain the continued failures of anti-trypanosome vaccine developments as well as the lack of any field-applicable vaccine based anti-trypanosomosis intervention strategy. Finally, the link between infection-associated inflammation and trypanosomosis induced anemia is covered in the context of both livestock and human infections.
Introduction
Human African Trypanosomosis and Animal African Trypanosomosis are two well-known diseases that affect sub-Saharan Africa and have historically prevented the development of vast lands of the African continent into highly productive agricultural areas. However, the first salivarian pathogenic trypanosome to be discovered was T. evansi, a parasite identified by Dr. Griffith Evans in 1880, in horses and camels suffering from a disease called Surra on the Indian subcontinent (1). Almost 140 years after this initial discovery, a wealth of world-wide epidemiological data on pathogenic trypanosomes shows they are present on four different continents. Molecular parasite mechanisms, that allow the escape from the hosts' immune and non-immune defense systems, have been discovered and various interactions in the context of vector biology have been described. However, in the end the data available today has still not given us a way to intervene in trypanosomosis transmission by means of an effective anti-parasite vaccination strategy. Hence, control still relies on a combination of active case diagnosis and treatment, as well as vector control (2, 3). In this review we cover the classification of trypanosomes, which has recently become under scrutiny (4), as well as new discoveries with respect to genetic exchange between trypanosomes that takes place in the insect vector (5, 6). In addition, the paper provides an update on recent discoveries with respect to the B cell destructive potential of trypanosomes (7, 8), T cell biology (9), and the impact of trypanosomosis on red blood cell (RBC) homeostasis and infection-associated anemia (10). Throughout the data review, both animal trypanosomosis (AT) and human trypanosomosis (HT) have been considered. However, as most recent data shows, this “artificial” distinction might be less useful than previously thought, as atypical human trypanosomosis (a-HT), which can be caused by various animal trypanosomes, is now gaining more and more attention in the field (11).
Setting the Scene for Salivarian Trypanosomosis
Trypanosomes are unicellular protozoan organisms of the class Kinetoplastida that cause a wide range of infections in a broad range of hosts. The latter includes not just mammals but also fish (12), birds (13), and reptiles (14), while insect vectors actually should be considered not just as transmission “tools” but also as definite hosts. Indeed, it is only here that sexual reproduction stages have been reported, as comprehensively outlined in a recent review by Gibson W. (5). In mammals, both salivarian and stercorarian trypanosomes cause diseases that affect the health status of the infected host in multiple ways. While the stercorarian trypanosomes are an important group of parasites, the main focus of this review is directed toward the pathogenic salivarian trypanosomes that cause infections in human, livestock, and game animals. These infections are marked by the extracellular nature of the infecting agent, causing pathologies and health complications that are very different from the features that characterize intracellular pathogenic infections such as those caused by the stercorarian T. cruzi parasite. An additional complication that arises when describing trypanosomosis, is the use of the term African Trypanosomosis. This denomination is very often used in an incorrect way. Indeed, as will be described in this review, all major pathogenic salivarian trypanosome infections do occur on the African continent. However several of the pathogens responsible for these diseases have moved “out of Africa” and infections are progressing throughout the world. A last introductory remark for this paper is the fact that Human African Trypanosomosis or HAT has recently been brought under control in a very significant manner by huge consorted international efforts of the last decennium (15). Hence, this might give the impression that trypanosomosis has become a disease of the past. This however could very well be a wrong assumption for three main reasons. First, there are no reports that suggest that AT is near to being controlled on a world-wide scale. Second, the most aggressive form of HAT caused by T. b. rhodesiense has a zoonotic origin, so as long as human infective trypanosomes are present in a wildlife reservoir, re-emergence of the disease remains a risk (16, 17). This holds true even if the majority of infections caused by T. b gambiense are being brought under control. Third, reports of a-HT in- and outside Africa show that “African” trypanosomosis is only part of the problem (11). Hence, for now trypanosome diseases still remain a threat to human health and to agriculture systems of emerging economies. In the absence of any vaccine strategy preventing the spread of these infections, continued research into host-parasite interactions is needed. This will provide a better understanding of trypanosome diseases itself, the mechanisms of disease resistance, modes of immune evasion, and ultimately the reasons for continued failure of vaccination attempts.
Classification of the Main Pathogenic Salivarian Trypanosomes
Trypanosomes belong to the sub-kingdom Protozoa, the order Kinetoplastida, the family Trypanosomatidae, and genus Trypanosoma. The large numbers of different species belonging to this genus have been classified in several subgenera according to their morphology. For the salivarian pathogenic trypanosomes the subgenera include Trypanozoon, Duttonella, Nannomonas, and Pycnomonas, of which the first three account for the vast majority of human and animal infections and are the subject of this review. Their combined geographic spread covers most of the developing world (Figure 1).
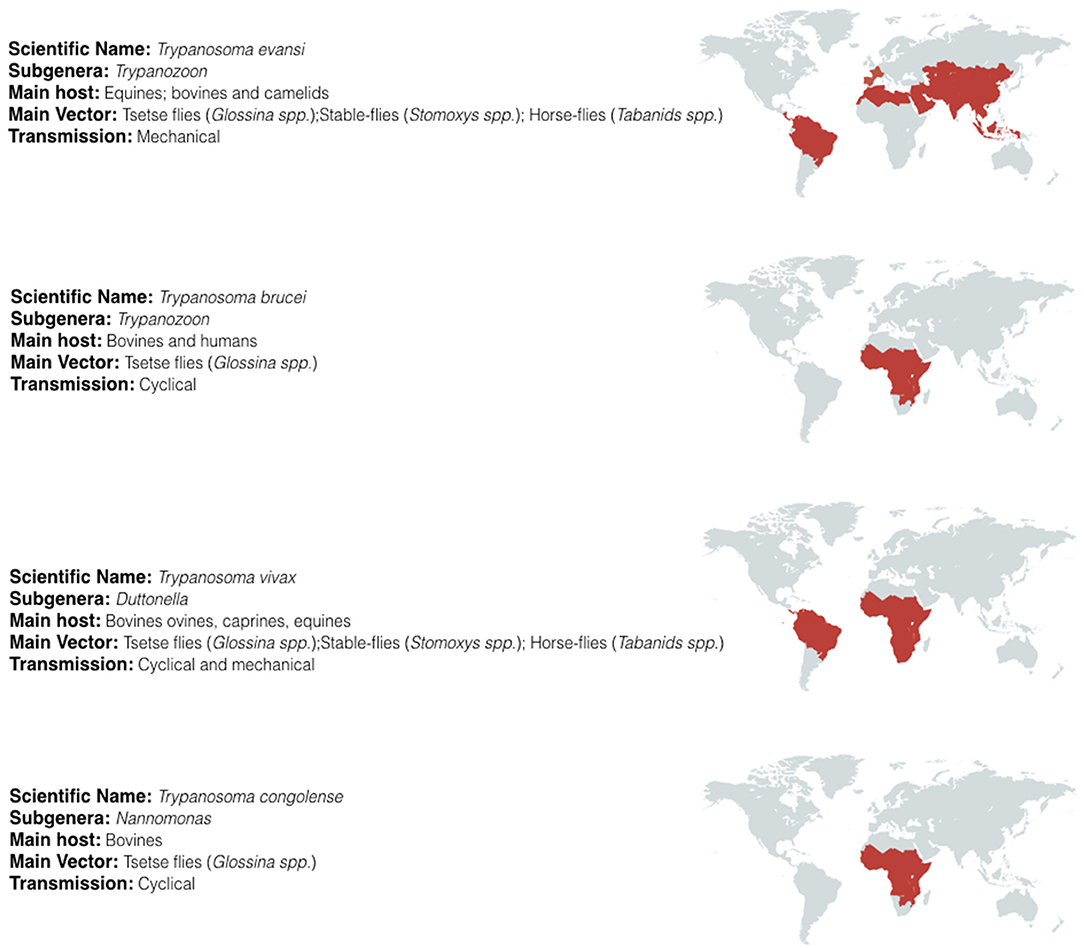
Figure 1. Geographic distribution of salivarian trypanosomosis. Salivarian trypanosomosis is a worldwide problem caused in large by Trypanosoma evansi, Trypanosoma brucei (including the human infective subspecies T. b. gambiense and T. b. rhodesiense), Trypanosoma vivax and Trypanosoma congolense. T. brucei, and T. congolense infections are limited to the sub-Saharan tsetse belt. In contrast, as T. vivax and T. evansi can be mechanically transmitted, these parasites have migrate beyond the tsetse belt, out of Africa and into South America and Asia [adapted from (18–22)].
The first trypanosome subgenus, Trypanozoon, is composed of several Trypanosoma species, which are human and animal infective and includes the first pathogenic trypanosome ever to be discovered i.e. Trypanosoma evansi. Today, T. evansi is a parasite that is considered to have mainly a veterinary importance (1), causing the disease Surra in a wide range of economically important mammals such as horses, cattle, goats, buffalos, dogs, and camels. In addition, the parasite can be found in game animals such as deer, wild pigs, and capybaras, representing a reservoir that often might escape attention. Today, T. evansi is found across Central and South America, North Africa, the Russian territories, the Indian subcontinent, China, and Southeast Asia (23). Transmission mainly occurs mechanically through the bite of bloodsucking insects from the family Tabanidae (genus Tabanus) (24), Chrysops (25), Atylotus (26), and Muscidae (genus Stomoxys and Haematobia) (18). It is this mechanical transmission that has allowed the parasite to move beyond the tsetse fly region and out of Africa. Morphologically, T. evansi has long been considered as a monomorphic parasite with the main bloodstream form appearing as so called “long slender” forms. However, the appearance of short intermediate forms has also been reported, but only in blood smears of infected cat and monkey (27). Important is that the parasite has a peculiar kinetoplast, characterized by either a reduced (lack of maxi-circles and homogenous mini-circles) or a total absence of kinetoplast DNA (kDNA) (28). This deficiency is thought to lock T. evansi in the bloodstream form, as they are unable to transcribe the kDNA genes required to perform the oxidative phosphorylation required for the developmental processes in the midgut of the tsetse (29). For long, this altered kDNA characteristic has been used to differentiate T. evansi from African T. brucei subspecies. Recently however, genetic analysis of a large battery of both T. evansi and T. brucei parasites has shown that the situation is more complex, and that many T. evansi parasites are closely related to T. brucei, even more closely than the relation between T. evansi parasites from different geographic locations (30–32). In addition, these data suggest that T. evansi arose multiple times from a different T. brucei ancestor. Hence, this has sparked a debate about the nomenclature of the Trypanozoon parasites. While it has been suggested by some to consider T. evansi as a T. brucei variant, a most recent revision has been proposed based on the proper application of the principles of biological nomenclature. This proposal suggests to rename all T. brucei subspecies as T. evansi subspecies, and even adopt the use of T. evansi gambiense and T. evansi rhodesiense for human infective African Trypanosomes (4). Important in the context of the parasite-host interplay of T. evansi is the notion that several human T. evansi infections have been reported in- and outside Africa (11). However, despite the wide geographic distribution of T. evansi, the reports on human non-African trypanosomosis are overall extremely rare. However, it cannot be excluded that one of the reasons for the scarce amount of data on a-HT is simply due to the lack of proper diagnostic practices that are able to correctly identify human trypanosomosis in T. evansi endemic areas.
The second Trypanozoon subspecies to be discovered was T. brucei, endemic to sub-Saharan Africa and transmitted by biting flies of the genus Glossina, commonly known as tsetse, with tsetse meaning “fly” in the Tswana language of Southern Africa. T. brucei parasites present a major health problem for humans, as the causative agent of the disease called HAT, or sleeping sickness. Domestic animals such as cattle, pigs, small ruminants, and game animals are also common hosts for T. brucei, in which the latter serve as a natural reservoir of the parasite. Three morphological indistinguishable subspecies of T. brucei are known (namely T. b. gambiense, T. b. rhodesiense, and T. b. brucei.) with T. b. gambiense and T. b. rhodesiense responsible for human trypanosome diseases in West/Central and East Africa, respectively. Uganda is one of the only countries where the two forms of HAT appear in adjacent regions. T. b. brucei, on the other hand, is unable to infect humans and is responsible for animal trypanosomosis only. The correct identification of the T. brucei subspecies is nearly impossible when solely based on their morphology or geographical origin. Indeed, all three subspecies appear as pleomorphic bloodstream parasites, having both long slender and short stumpy forms. However, molecular approaches have shown that this group is highly heterogeneous (33). The exclusive presence of the serum resistance associated (SRA) gene in T. b. rhodesiense has been used as a marker for the identification of this subspecies (34, 35). T. b. gambiense specific identification can be done through PCR amplification of the T. b. gambiense-specific glycoprotein (TgsGP) gene that encodes for a receptor-like glycoprotein, which is also involved in normal human serum resistance (36).
The third species within the subgenera of Trypanozoon, responsible for livestock infections, is Trypanosoma equiperdum. This parasite can be sexually transmitted amongst species from the family Equidae (horses and donkey), causing a venereal disease known as Dourine. Due to this transmission mode, the parasite has also acquired a wide geographic distribution. As for T. evansi, to which it is very closely related, also the classification of T. equiperdum as a separate species has been under scrutiny for many years (37), and several authors have suggested that there is no scientific argument to make a species level distinction between the two.
The second trypanosome subgenus responsible for salivarian trypanosomosis is Dutonella, which is mainly composed of two species, i.e., T. vivax and T. uniforme. Due to the global socio-economic impact caused by T. vivax infections in domestic animals, most of the studies of this subgenus have been carried out on this particular parasite species. This trypanosome is a pathogenic parasite species mainly found in Africa and South America (38). So far, only a single human infection has ever been reported (11). In Africa, transmission occurs in large through the bite of tsetse flies, having the highest infection rate of any tsetse-transmitted trypanosome species (39, 40). This high infectivity could be attributed to the relatively simple cycle of development in the vector's mouthparts. The transmission beyond Africa is mainly carried out mechanically by hematophagous flies from the genus Stomoxys and Tabanus, which transmit the disease to domestic animals such as cattle and goats, as well as to endemic wild animals such as capybaras, deer, and bubaline antelopes. Despite its large economic impact, especially in South America, T. vivax remains one of the less studied animal-infective Trypanosoma species. The main issue with T. vivax in the context of experimental parasitology and immunology research is the fact that virtually no parasites of this species are capable to be grown in mice. Hence, virtually all laboratory infections in mouse models (incl. Tv700, STIB 719, STIB 731-A, ILRAD 560, IL 1392) are being executed with the derivatives of Y468 T. vivax clone that was originally isolated in Nigeria that happened to “grow well” in mice after extensive adaptation (41). Hence, it remains to be seen if the TvY486 T. vivax reference strain is a good representative parasite for the general population of parasites found both in Africa and South America regarding host-parasite interaction mechanisms.
The third subgenus of pathogenic salivarian trypanosomes is Nannomonas, encompassing three species of animal-infective trypanosomes, i.e., Trypanosoma simiae, Trypanosoma godfreyi, and Trypanosoma congolense. The two first species are mainly infective to mammals belonging to the Suidae family (domestic pigs, warthogs etc.) while T. congolense has a broader range of hosts including livestock and game animals, but is generally accepted to be non-infective to humans. It should however be mentioned that a mixed T. b. gambiense/T. congolense infection has been reported in a human (42) and that in vitro testing of human serum-induced trypanolysis has shown a resistance phenotype in several stocks (43). T. congolense is the major tsetse transmitted pathogenic salivarian livestock trypanosome present in sub-Saharan Africa and causes large economical losses in the countries where it is endemic. The disease caused by this parasite is referred to as Nagana, meaning depressed spirit in the Zulu language of Southern Africa. During transmission, T. congolense develops in the midgut and proboscis of the tsetse vector. Mechanical transmission can occur, involving mainly Tabanus and Stomoxys species (44). In general, T. congolense is considered to be a monomorphic parasite. The host-parasite interaction as well as immunopathology associated with T. congolense infections has been better studied than in the case of T. evansi and T. vivax. However, it is important to point out the fact that while the molecular parasite surface structure has been well-described and compared to T. brucei (45), the regulation and kinetics of surface coat variation, as well as the infection of the coat with the immune system have never been analyzed in detail. Hence, most statements about these interactions and regulations are based on the assumption that T. congolense should behave the same way as T. brucei. A second scientific issue that plagues the T. congolense research literature is the fact that it is often used as a “chronic” model in comparison to “acute” T. brucei infections. This artifact originates from the fact that a specific chronic T. congolense clone, i.e., Tc13, has been used in major immunological investigations for almost three decades (46). In contrast, the vast majority of experimental host-parasite research in T. brucei brucei and T. brucei rhodesiense models has been done with much more virulent T. b. AnTat 1 or LouTat 1 clones (47–49). While these studies by themselves all resulted in valid experimental data, it should be said that there are virulent T. congolense strains, resulting in infections in mice which display very similar survival times as the T. brucei clones mentioned above (50). Unfortunately, these more virulent T. congolense isolates have not been systematically used in comparative studies with T. brucei. Hence, reports that compare highly virulent T. brucei infections with low virulent T. congolense infections, and subsequently provide conclusion in which infection outcome is linked to the species-specific background of the parasite, should be taken with utmost caution. Of note is that the high genetic heterogeneity of T. congolense has led to the division of this parasite in three different subgroups i.e., Savannah, Kilifi, and Forest within the same species (51).
General Life Cycle of Salivarian Trypanosomes and Interaction With Both Insect and Mammalian Hosts
The life cycle of cyclically transmitted salivarian trypanosomes inside their vector shows the plasticity of those parasites to adapt to new environments (Figure 2). Today, most of the knowledge on parasite transmission comes from the T. brucei model in which infection of tsetse flies occurs when the non-dividing short stumpy bloodstream form parasites are taken up during a fly's blood meal, reaching the fly's midgut and transforming into procyclic trypomastigotes (52). Once the parasite infection is established in the fly's midgut, the parasites migrate anteriorly to the proventriculus of the fly. Here, elongated trypomastigotes start to divide asymmetrically into both long and short epimastigotes of which the latter migrate toward the salivary glands and attach to the epithelial cells of the gland. Next, a final division occurs giving rise to the host-infective metacyclic forms. Once in the host's bloodstream, metacyclic parasites transform into long slender shaped bloodstream form parasites, which further divide by binary fission and represent the active dividing parasite form during the mammalian infection stage. It is at this stage that trypanosomes express the Variant Surface Glycoprotein (VSG) (55). This coat protein is encoded by a battery of over 1,000 different genes, mosaics and pseudogenes (56) and serves as an antibody decoy defense system (57). Indeed, VSGs are highly immunogenic and induce VSG-specific antibody responses. Hence, by regularly altering VSG variant expression, the parasite avoids efficient immune recognition and destruction (58). Genetic regulation of antigenic variation has been studied in detail and has been shown to involve various mechanisms of DNA recombination and transcription regulation of VSG genes (59, 60). With respect to mechanisms of genetic variation in trypanosomes, there is now ample evidence supporting the fact that T. brucei parasite mating or sexual reproduction does take place in the tsetse (5, 6). Hence, while the tsetse is conventionally referred as the insect vector for trypanosomosis, it should actually be considered as the definite host for the parasite. To put it in other words: mammals are merely the vessel that is used to ensure that trypanosomes are able to migrate from one tsetse to the next, and in addition provide long-term reservoirs that allow trypanosomes to survive seasonal periods in which fly populations are diminished. Sexual reproduction inside the insect vector offers the parasite in theory the chance of generating new hybrids, combining different parental characteristics. Important to note however is the fact that the effectiveness of trypanosome infection in the fly rapidly decreases with the age of the fly, hence also affecting the chance to generate hybrid descendants. Using both green and red fluorescent trypanosomes to study hybrid formation, it was shown that midgut and salivary gland infection rates were highest when flies were exposed to parasites in their first feed (53). Waiting 21/2 weeks for a first parasite exposure reduced the infection success by half. Interestingly, exposing tsetse flies to two different trypanosome lines in a consecutive feeding experiment resulted most often in the establishment of the first infection only, as if the primary infection was able to push the vector to mount a protective immune response preventing secondary infection. Under natural circumstances, this would greatly reduce the chance of hybrids being formed, although the experimental conditions used above showed that in all combinations tested, hybrid formation did take place (53). As will be outlined later in this review, even the rarest hybrid formation events can have a significant impact on the transition from AAT to HAT, as it allows generation of a continuous pool of new human infective T. rhodesiense parasite strains (54). With respect to the vector immunity mentioned above, several studies published in the recent past have made contributions to the understanding of the mechanism underlying tsetse anti-trypanosome immunity. It is interesting to note that tsetse immunity development per se requires the presence of the obligate symbiont Wigglesworthia in the larval stage of the fly, transmitted through maternal milk gland secretion (61, 62). This finding complements the notion that the development of a fully functional innate immune system of the mature adult tsetse fly depends on the establishment of a bacterial microbiome population, and that the immaturity of the immune system is responsible for the high susceptibility to trypanosome infections during a first blood feeding (63). The fly immunity itself relies on multiple mechanisms. Indeed, the action of scavenger receptor peptidoglycan-recognition protein LB (PGRP-LP) is crucial for the colonization of the fly by its Wigglesworthia symbiont, and in addition has a direct trypanocidal activity on both procyclic and bloodstream form trypanosomes (64, 65). In addition, anti-trypanosome immunity relies on activation of the immune deficiency regulated pathway and antimicrobial peptides (66, 67), as well as reactive oxygen species (ROS) mediated defenses (68), which provides combined protective immunity at the level of the midgut and hemocoel. Interesting here is that for some time the peritrophic matrix, which is a chitinous protective layer lining the insect gut, has been considered as a physical barrier that could provide protection against invading infections. However, RNA interference-based reversed genetic approaches have shown that the matrix is a true immunological regulator. Its integrity is necessary to build a proper immune context in the defense against different microbes, including trypanosomes, through its role in the expression of the antimicrobial peptide attacin as well as dual oxidase and iNOS, both involved in the production of reactive oxygen intermediates (ROIs) (65). Finally, the tsetse fly's specific TsetseEP protein was shown to provide anti-trypanosome protection at the level of the midgut (69). Interestingly, starvation of flies reduces immune responsiveness and increases susceptibility toward trypanosome infections both in young and older flies (70–72).
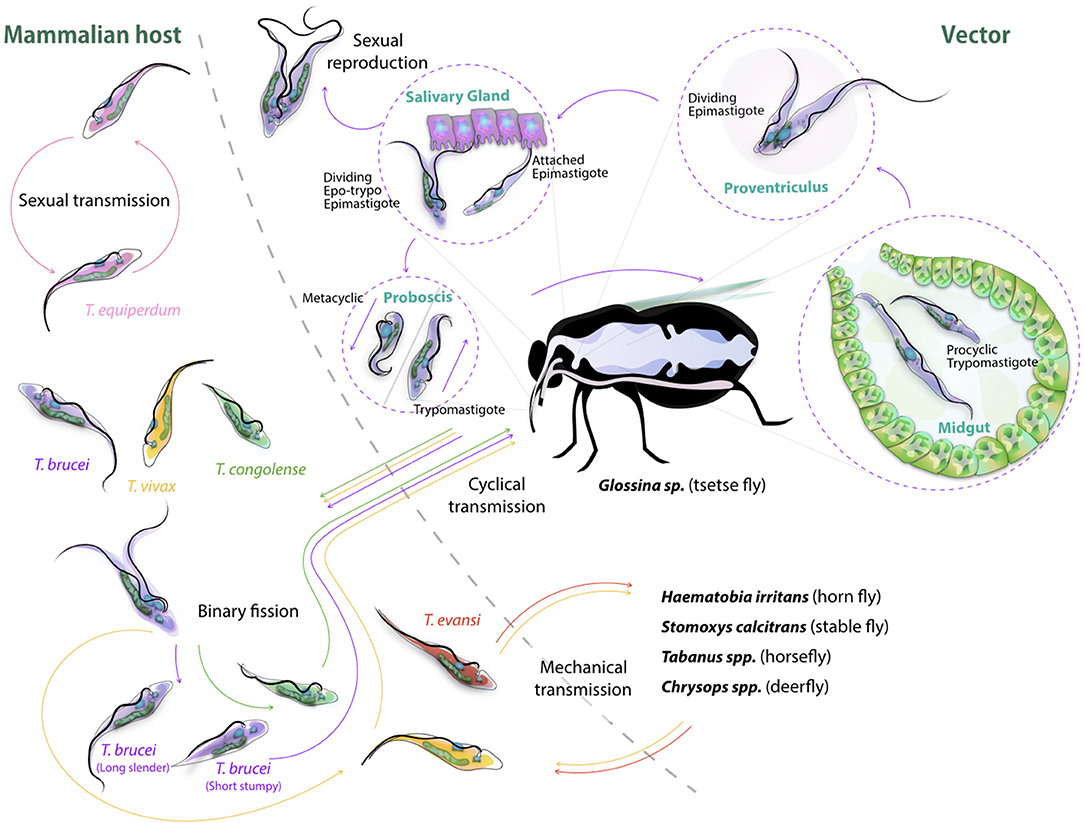
Figure 2. Cyclic transmission of salivarian trypanosomes mediated by tsetse and other biting flies. Most lifecycle data of African Trypanosomes is based on the T. brucei cycle involving the tsetse (52). Here, short stumpy form parasite are taken up by the fly, progress through the body of the insect vector passing via the midgut, proventiculus and salivary gland, to be re-injected though the proboscis as infective metacyclic trypomastigotes into a new target. In the bloodstream, differentiation to long slender forms occurs followed by binary fission proliferation. Differentiation back to short stumpy forms will complete the cycle. While T. congolense and T. vivax can follow a similar cyclic transmission mode, the latter also is known to be spread through mechanical transmission (53, 54), as is the case for T. evansi (1). T. equiperdum spreads through sexual transmission only (37). Sexual reproduction of trypanosomes itself has been reported to take part in the teste, and has been reported for both T. brucei and T. congolense (5).
Naturally, from a human and economic point of view, it is the mammalian infection stage that has attracted most attention in the past. It is only more recently that the parasite-vector interaction and biology has received more detailed attention and that also non-T. brucei infections have been studied in tsetse (73, 74). These reports show that in fact both T. congolense and T. vivax are much more effective in establishing tsetse infections than T. brucei. In particular, for T. congolense it has been shown that this parasite is particularly effective in reaching the proboscis of the fly, where the trypomastigote-epimastigote transformation takes place. In this case, migration from the foregut to the mouthparts appeared to occur with high efficiency. In contrast, T. brucei is much less efficient in colonizing the tsetse, as most parasites do not survive the migration from the foregut to the salivary glands. Investigating both T. brucei and T. congolense infections in parallel have suggested that T. brucei adopted to final survival in the salivary gland, as this niche would not be preoccupied by the much more efficiently growing T. congolense parasites. Hence, despite the fact that both parasites use the same transmission vector, and that also for T. congolense meiotic reproduction has been reported in the teste vector (75), there are remarkable differences in the way the two trypanosomes infect and occupy the body of the insect host. In recent years, specific attention has also been given to the immunological events that take place at the bite site of the tsetse, in order to explain how successful mammalian infections are initiated. Here, it has become clear that there is a crucial role for tsetse saliva components in preventing local blood clothing, vasodilation, and neutrophil influx, all leading to the successful establishment of a primary infection and allowing metacyclic saliva parasites to be transformed successfully into long slender bloodstream form parasites (76, 77).
Besides the biological vector transmission described above, mechanical transmission is a second way of ensuring parasites can move from one mammalian host to the next. This mode has been described for T. congolense, T. evansi, and T. vivax, but only the latter two have successfully used this way of transmission to migrate out of Africa, into other continents. The main vectors that have been reported today for both trypanosome species are the horsefly, stable fly, horn fly, and deerfly. In all cases, mechanical transmission occurs when a fly with blood-contaminated mouthparts, containing living bloodstream form parasites, rapidly changes feeding hosts allowing the parasites to be transmitted without any intermediate insect-specific forms. To date, virtually no information is available on possible immune interactions that could make this way of parasite transmission more or less successful. Interestingly, also nothing is known about the immune events that aid in parasite transfer back to the fly. Indeed, it is remarkable that livestock parasitaemia levels are often extremely low, presenting blood parasite load levels that are hardly detectable by microscopy. Yet, even when circulating parasite numbers are extremely low, flies still manages to successfully pick up parasites while taking very small blood meal volumes. Whether this is due to the fact that parasitaemia in the skin microvasculature is uniquely high as compared to the general blood circulation, or whether fly saliva has unique and potent trypanosome chemoattractant, remains to be elucidated.
Human Trypanosomosis in Africa is Only Part of the Problem
In 2009, the number of reported HAT cases dropped below 10.000 for the first time in 50 years, and the most recent figures available for 2015 indicate that the global incidence of HAT has dropped below 3.000. It is now estimated that by 2020 HAT will no longer be considered as a major human health problem in Africa and hence will also no longer be listed as a neglected disease (78). With HAT being caused by either Trypanosoma brucei gambiense (>97%) or Trypanosoma brucei rhodesiense (<3%) (79), these numbers and assumptions are mainly based on the current West/Central African situation. Considering however that T. b. rhodesiense is a zoonotic parasite with a cattle and wildlife reservoir, re-emergence of HAT is going to remain a crucial concern in Africa. Indeed, whether or not the reservoir of T. b rhodesiense has been brought under control is hard to verify due to the insufficient systemic reporting on T. b. rhodesiense game and cattle infections. In addition, beyond the borders of the African continent the existence of non-T. brucei human trypanosomosis could become a future problem as T. evansi infections spread around the globe. Despite the reports of T. evansi infections in humans (80–83), this parasite is still not widely considered as a human pathogen. The lack of interest in these infections, combined with the continued spread of these trypanosomes mainly in South America, the Indian subcontinent, and Asia, risks of exposing humans to a new type of “unconventional” disease that will require a whole new approach to trypanosomosis world-wide. In addition, the lack of experimental studies on T. evansi infections as compared to T. brucei infections makes it harder to link the discussion about the genetic classification of T. evansi as a T. brucei subspecies to data dealing with the cellular and molecular mechanisms that govern host-pathogen interactions, pathology development, and zoonotic behavior.
Taken the importance of the zoonotic aspect of most remaining human trypanosome infections, it is clear that (African) Animal Trypanosomosis (AAT) per se deserves particular attention. In fact, there is a wealth of information available with respect to geographic distribution of AAT, immune pathologies including anemia and vaccine efforts as well as failures. These reports outnumber the scientific data published on host-pathogen aspect of human trypanosomosis. However, this contrasts very much the situation with respect to the actual understanding of trypanosome diseases. Indeed, while many have focused on experimental models to understand human trypanosomosis in the past, much less effort has been made to understand the fundamentals of animal trypanosomosis. A fine example to illustrate this is the issue of antigenic variation. For more than four decades, scientists have been studying antigenic variation of the VSG coat used by trypanosomes to deliver a protective response against the host antibody defense system. All work that included genetic approaches as well as molecular biology approaches combined with computer modeling, has been done for T. brucei, largely ignoring the fact that the main cattle parasites i.e., T. congolense and T. vivax are very different trypanosomes that might use other systems of regulation and defense. For example, comparative studies of the VSG repertoire conducted in T. congolense and T. vivax indicated that the scale of the VSG recombination differs between two species, being more frequent in T. congolense than in T. vivax. Moreover, T. vivax populations were shown to be consistent with clonal reproduction (84), while the T. congolense ability to undergo sexual reproduction generates opportunities for allelic recombination among VSG genes (85). Hence, to date we are left with molecular tools that are very suitable to study a wide range of host-parasite interactions in mice which could model human infections, but at the same time we have very little experimental models and tools to study AT. One striking example is T. equiperdum, which is hardly studied in comparison to other infections due to the fact that it causes a disease characterized by sexual transmission in horses, a feature so far not reported in mice. In fact, only a single report is available on experimental sexual transmission of trypanosomosis in mice, and this report is dealing with T. b. gambiense (86). However, whether sexual transmission of T. brucei gambiense is a real issue in human trypanosomosis, remains a matter of debate (87) and more species-specific research is needed to resolve these issues.
Adaptations of Animal Infective Trypanosomes to Human Host
Humans are considered naturally resistant to pathogenic animal trypanosomes such as T. b. brucei, T. evansi, T. equiperdum, T. congolense, and T. vivax. The mechanism of human resistance to those trypanosomes is based on the presence of cytolytic factors in the high density lipoprotein (HDL) fraction of normal human serum (NHS) (88) and is attributed to two fractions called trypanosome lytic factor 1 and 2 (TLF1 and TLF2), with the latter being the major active compound (89). The human infective T. brucei subspecies T. b. gambiense and T. b. rhodesiense are known to be resistant to TLF lysis through various mechanisms. A number of reports demonstrating T. evansi infections in humans has triggered a new wave of interest in molecular mechanisms underlying human infectivity in the context of the transition from an animal infective trypanosome to a zoonotic pathogen causing disease in humans. While TLF1 and TLF2 have slightly different compositions, they both contain the haptoglobin related protein (HPR) and Apolipoprotein L1 (ApoL1), of which the latter is considered the main active lytic compound. Furthermore, TLF2 is complexed with IgM molecules and has a much higher molecular mass than TLF1, but is lipid poor (90). With respect to the mechanisms of lysis and resistance to NHS, initial data was obtained in an experimental model for T. b. rhodesiense. Here, it was shown that resistant and susceptible sub-clones can be derived from a common ancestor by passaging trypanosomes in either the presence or absence of human serum. This work gave rise to the discovery of the SRA gene, encoding a truncated VSG and located within the VSG expression site (91). Transfer of the gene to T. b. brucei showed that the presence of this single gene indeed was enough to confer resistance to NHS (92). Subsequently, the identity of SRA allowed to characterize ApoL1 as the active serum compound (93) that acts through both lysosomal and mitochondrial membrane permeabilization (94). The inhibitory mechanism of SRA was shown to rely on its capacity to block the membrane pore-forming capacity of ApoL1 upon entering the acid compartments of the lysosomal system (95–98). Finally, the haptoglobin-hemoglobin receptor (HpHbR), localized in the flagellar pocket, was identified as the trypanosome receptor involved in TLF1 uptake (99), while the mechanism for TLF2 uptake has still not been elucidated (100). Indeed, HpHbR knock down T. b. brucei parasites remain sensitive to both NHS and TLF2 lysis (101). However, as both TLFs contain the same active ApoL1 compound, the SRA is capable of blocking all NHS activity. Interestingly, the SRA gene is only present in T. b. rhodesiense and hence has been used in a string of different diagnostic approaches for HAT, including PCR and LAMP, targeting not just human samples but also livestock and game reservoirs as well as the tsetse vector, in various regions in Eastern Africa. Of note is a report in which a Ugandan HAT survey indicated that 20% of parasitologicaly confirmed T. b. rhodesiense cases resulted from infections that could not be detected by any of the SRA PCR methods described so far. Whether this indicates that SRA-negative human infective T. b. rhodesiense parasites exist, implying these parasites have alternative NHS resistance mechanisms, or whether the negative results were due to SRA gene polymorphism that prevented PCR primer annealing, remains a matter of debate (102). The existence of such SRA gene polymorphisms has been studied in the context of human disease severity (54). Most recently, using SRA as a genetic tracer, it has been shown that genetic recombination between T. b rhodesiense and the much larger pool of T. b. brucei animal trypanosomes allows for the continuous generation of new SRA-positive human infective parasites (103, 104). This once again indicates that in order to really bring HAT under control, AAT and particularly the animal T. b. rhodesiense reservoir has to be eliminated as well.
Interestingly, more than 97% of all human trypanosomosis infections are caused by SRA-negative T. b. gambiense (79), a parasite that is subdivided in Group 1 and Group 2 T. b. gambiense (105). While Group 1 parasites have an invariant phenotype toward NHS resistance, Group 2 parasites show a variable degree of resistance, modulated by the actual exposure to NHS. Neither Group 1, nor Group 2 parasites have the SRA gene (8), indicating that there are multiple ways to mount resistance to NHS. Literature of the last 5 years has mainly focused on the Group 1 T. b. gambiense resistance, showing that these parasites have acquired a combination of defense systems which allows them to resist the lytic action of NHS. This includes a specific point mutation in the HpHbR in combination with reduced receptor expression, reducing TLF1 uptake (106, 107). However, as this receptor is not a main player in TLF2 uptake, T. b. gambiense needs additional mechanisms to survive in human blood. These involve the alteration of lipid membrane fluidity by the T. b. gambiense-specific glycoprotein (TgsGP) (108, 109), as well as increased cysteine protease activity in the digestive vacuoles of the parasite. The latter is believed to directly affect ApoL1 activity (110). Today, it remains unclear how T. b. gambiense Group 2 parasites deal with the toxicity of NHS.
T. evansi parasites express neither SRA, nor TgsGP, but now have been reported in several instances as human pathogens causing a-HT (11, 18, 80, 82, 111). Similarly to T. b. rhodesiense and T. b. gambiense Group 2 parasites, a remarkable phenotypic plasticity of T. evansi has been described upon exposure to NHS, with resistance occurring after prolonged NHS exposure and being absent when NHS selective pressure is removed. Screenings of various T. evansi isolates indicated that some even display natural resistance to NHS, while others were found to be fully sensitive (112). In line with these observations in T. evansi, even T. b. brucei NHS resistance has now been reported. In the latter, a switch to a resistant phenotype was recorded to occur upon repetitive exposure to NHS or TLF1 in the absence of the SRA gene (113). However, a fragment homolog to SRA, named SRA basic copy (SRAbc), was found in the T. brucei brucei TREU927/4 strain, which exhibits low resistance to NHS (114). Similarly, one of the human infective T. evansi isolates was shown to contain the SRAbc homolog (115). Interestingly, the T. b. brucei parasites exhibiting increased NHS resistance had a significant reduction in TLF1 uptake, which coincided with downregulation of T. b. brucei HpHbR mRNA levels (100). Hence, it seems that the resistance mechanism in these parasites shows a mixed but attenuated phenotype of those found in either T. b. gambiense or T. b. rhodesiense, as if the latter could have been selected as “optimized” derivatives of T. b. brucei semi-resistant predecessors.
With respect to human T. evansi, it is important to highlight that at least one human infective case has been attributed to the lack of functional ApoL1 (81). Indeed, a frameshift mutation, found in both ApoL1 alleles of the patient, resulted in the ability of trypanosomes to establish infection and to survive in the human bloodstream. This strongly suggests that human trypanosome resistance in large relies on a non-classical immune mechanism, i.e., lipid membrane disruption by TLF. However, a Vietnamese a-HT T. evansi victim was shown to have fully functional ApoL1 alleles and a normal concentration of serum ApoL1 (116). Hence, this shows that there is an additional role for the immune system in the overall defense against trypanosomes, most likely involving a combination of the action of antibodies, cytokines, and complement factors. In at least two confirmed human T. evansi infections, the infected individuals were shown to have a compromised immune system. One case relates to a pregnant woman from Mumbai, India suffering from HIV/AIDS, anemia, and upper respiratory tract infection (117). The second case relates to the above mentioned Vietnamese woman who had just given birth (116), with pregnancy itself being known as a unique immune condition that is modulated by fetus development resulting in immune alterations that in some cases in facilitation of parasite growth. Important is that also in experimental T. evansi infections in mice, in particular IgM antibodies are crucial for parasitaemia control (118). This indicates that when “natural resistance” such as the resistance conferred by ApoL1 fails, the antibody-mediated immune response does provide a second defense barrier against the progressing of infection.
Finally, it should also be mentioned that several reports in the past have indicated the existence of a-HT caused by the stercorarian T. lewisi parasite (119). Here, resistance to NHS lysis was correlated with resistance to human ApoL1 as well. Hence, it seems that while multiple mechanisms have been acquired by various trypanosomes to block the lysosomal pore-forming catalytic activity of NHS, this activity itself is executed in large by a single factor, i.e., ApoL1. This finding itself has attracted scientific attention over the last years with respect to primate evolution (120), and has resulted in the findings that (i) ApoL1 is the common lytic factor in human, gorilla, and baboon primate sera (7, 8, 121), (ii) the chimpanzee, orangutan, and macaque, which are susceptible to all T. brucei subspecies, lack functional ApoL1 (121), and (iii) the baboon ApoL1 variant is capable of killing even T. b. gambiense and T. b. rhodesiense, as opposed to the human ApoL1 (122, 123). The latter finding has prompted an attempt to generate genetically modified TLF transgenic livestock that would be able to resist all known pathogenic trypanosome species (122).
The Role of B Cells and Antibodies in Salivarian Trypanosomosis
As outlined above, VSG switching and antigenic variation, including the generation of VSG mosaic genes, have generally be considered as the major defense systems that parasites have developed against the host's adaptive immune system (56, 124, 125). However, in recent years it has become clear that trypanosomes have developed several precautionary adaptations that provide a rescue in case they do get recognized by antibodies. The reason for this is obvious: even if different VSG variants exhibit different hypervariable loops, and even if mosaic VSG present new epitopes to the immune system, there is no reason to assume that the overall pool of infection-induced antibodies is not at all capable of detecting new parasite variants. In fact, the existence of the cross-reactive nature of anti-VSG antibodies has been used since the beginning of trypanosome immunology research. Here, the VSG of the first arising variant or living cloned parasites, expressing a single VSG, have been used to monitor fluctuating anti-VSG titers throughout infection for weeks or months (57, 126). More interestingly maybe is the fact that reinfection models have shown that weeks into a primary infection, mice can be killed by a secondary infection with virulent trypanosomes expressing exactly the same VSG as the primary infection (127). In order to understand these results, three major host-parasite interactions mechanisms have to be considered. First, trypanosomes have developed a very efficient way of driving endocytosis. This system allows to continuously clear surface bound antibodies from the VSG coat, preventing antibody-mediated lysis (128). Secondly, while antibody/complement-mediated lysis has long been proposed to result in antibody-mediated trypanosome killing, it is important to note that AKR mice, which are natural C5 complement knockout mice, are able to clear peaks of parasitaemia in a similar way as other mice. This shows that the lack of complement-mediated lysis does not prevent the immune system of controlling peak stage parasite levels (129). Third, there is now ample evidence that trypanosomes cause a B cell depletion pathology, which is initiated by the very rapid disappearance of immature B cells in the bone marrow, as well as transitional and IgM+ marginal zone B cells from the spleen, followed by a gradual depletion of Follicular B cells (FoB) (127, 130). This has now been reported for T. b. gambiense and T. congolense infections (50, 131). Mechanisms involved in the depletion process have been linked to IFNγ-mediated inflammation (132), NK-mediated B cell destruction (133), and direct cell-cell contact-mediated B cell apoptosis (111, 127). Once FoB cell depletion is accomplished by the parasite, it becomes impossible for the host to generate new efficient antibody responses against newly arising VSG variants (Figure 3). In addition, it hampers anti-VSG memory recall responses against previously encountered variants, hence making the host susceptible to secondary infections with old variants. This might also explain the accumulation of mosaic VSG variants during later stages of infection (56). Indeed, the question whether later variants are immunologically distinct (or not) from their ancestral variants, which has so far not been answered, might not be important at all. Possibly, these variants arise simply from the continuous gene rearrangements that are ongoing at the telemoric ends of the VSG expression sites and are tolerated, due to the lack of antibody-mediated elimination by the host, rather than being produced in order to evade the already existing antibodies.
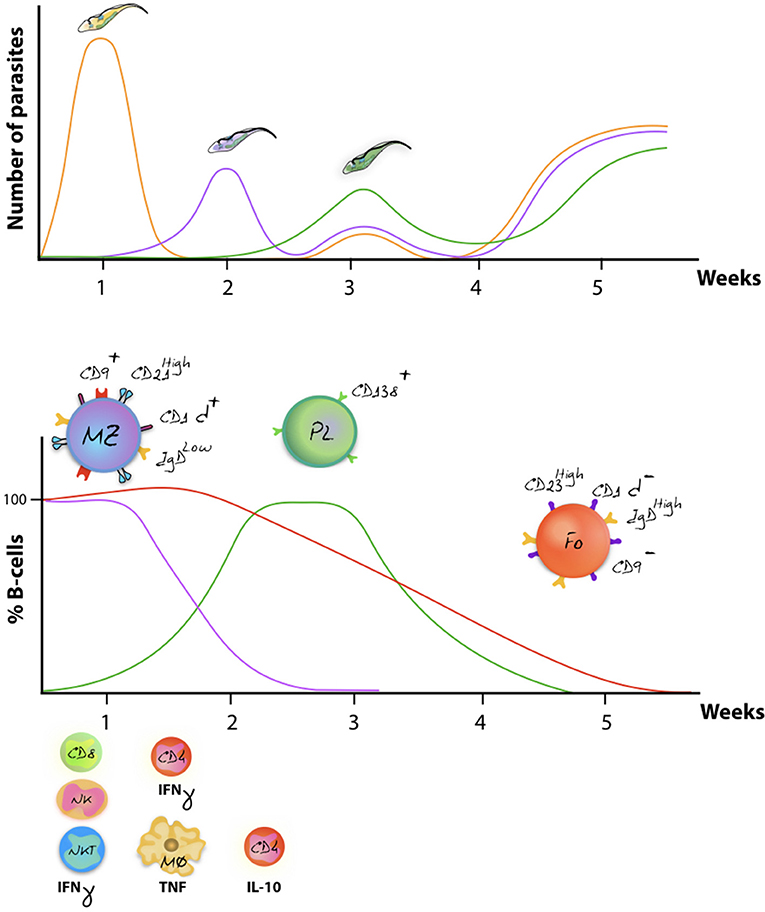
Figure 3. Antigenic variation and host immune destruction are closely linked. Antigenic variation of the trypanosome VSG surface coat enables trypanosomes to escape specific antibody-mediated destruction, resulting in immunologicaly distinct parasites occurring at regular intervals (upper panel). To prevent total eradication, trypanosomes undermine the immune system by ablation of the B cell compartment. In mice, this results in abrogation of an efficient antibody mediated immune defense system, allowing different parasite variants to occur simultaneously (schematically represented as the week 3 situation). Despite to co-occurrence of several variants, later-stage parasitaemia peaks usually have a reduced magnitude in terms of actual parasite numbers as various non-B cell defense systems aid in parasiteamia control [upper panel, adapted from (134)]. The lower panel schematically represent the finding that onset of infection is followed by a rapid depletion of the MZ B cell compartment (purple), followed by a gradual destruction of the FoB cell compartment (red) (116, 119). While the initially host immune response generates effector Plasma B cells, later waves of newly arising parasite variants fail to be efficiently depleted due to the impaired capacity of the host to deliver a renewed Plasma B cell response (green). Overall immunopathology is initiated by excessive production of IFNγ during the first week of infection, involving mainly CD8+ T cells, NK cells, and NKT cells. By 7 days post infection, IFNγ production is taken over by CD4+ T cells, while activated macrophages now produce excessive amounts of TNF that contribute to pathology (135, 136). Later-on in infection, production of IL-10 has been documented to counteract the initial inflammation (137).
B cell dysfunction, associated to trypanosomosis, also has a secondary detrimental effect on the mammalian host, i.e., the elimination of vaccine-induced memory recall responses. Indeed, in an experimental model for DTPa vaccination, it was shown that T. b. brucei is capable of destroying immunological memory rendering vaccinated mice susceptible to infections with Bordetella pertussis (130). This was not a result of an infection-associated immunosuppression, as it persisted after anti-trypanosome drug treatment and the elimination of active trypanosome infections. This detrimental effect of trypanosomes on non-related vaccine efficacy has also been reported in other models and natural infections. Although not thoroughly studied in human infections, one study has shown that antibody titers induced by the anti-measles vaccine are significantly downregulated in HAT patients, and that curative HAT treatment did not result in a restauration of antibody titers (Figure 4). For obvious ethical reasons, this study stopped short of assessing whether or not the remaining titers would still confer protection. In addition, the study did not address the question whether vaccine-induced memory recall responses were affected (138). With respect to AT, more data is available in particular with respect to T. evansi and T. congolense infections. Indeed, such infections in pigs were shown to abrogate protective immune responses generated against the classical swine fever vaccine (139). They were characterized by significantly reduced antibody responses, leukopenia, and high fever. Similarly, T. evansi infections in water buffalos, vaccinated against Pasteurella multocida (hemorrhagic septicemia), showed impaired capacity to mount a humoral and cell-mediated immune response upon challenge (140). When cattle, harboring T. congolense and T. vivax, were given a Brucella abortus vaccine or were vaccinated against contagious bovine pleuropneumonia, specific antibody responses to the vaccine were shown to be severely depressed (135, 141–143). Similarly, T. congolense infected goats vaccinated with Bacillus anthracis showed a profoundly diminished anti-anthrax antibody responses (144), while T. congolense infected cattle were shown to suffer from immunosuppression and failed recall responses in a foot-and-mouth vaccination setting (145). Taken together, these studies, conducted in natural hosts for animal trypanosomes such as cattle, water buffalos, goats, and pigs, confirmed studies in mice showing that trypanosome infection induces severe impairment of B cell responses and antibody production to a number of non-trypanosome related commercial veterinary vaccines.
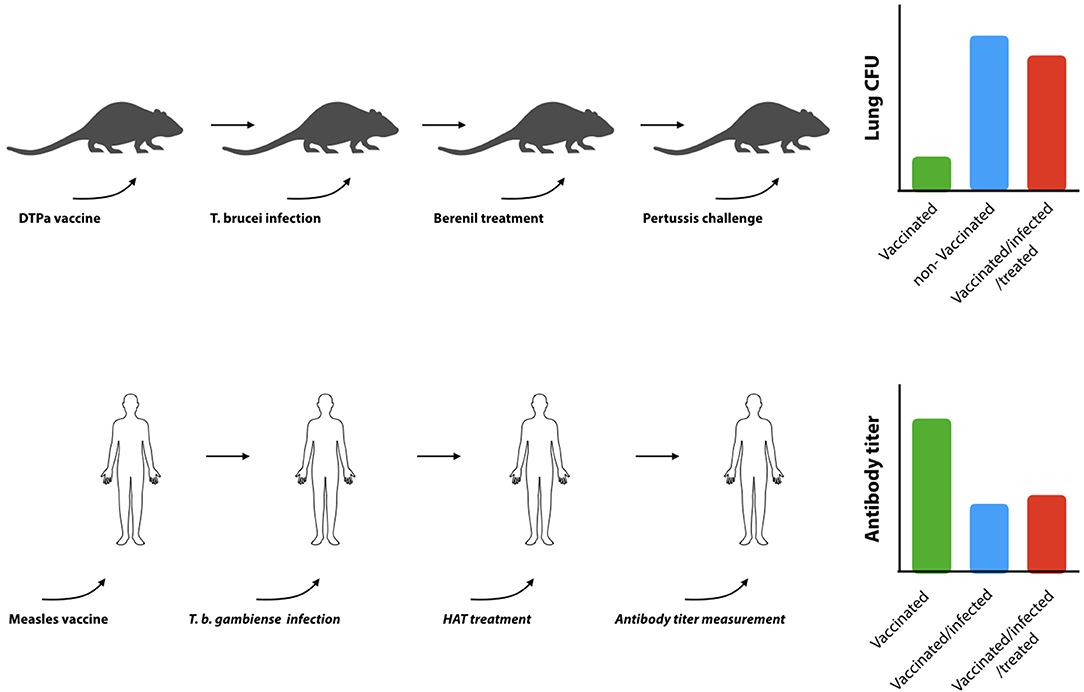
Figure 4. Trypanosomosis-induced B cell destruction results in prolonged B cell dysfunction. Experimental infections in mice have shown that trypanosomes can destroy non-related DTPa induced vaccine responses (upper panel). Vaccinated mice that have been confronted with trypanosomes fail to clear diphtheria bacteria from their lungs, even when the bacterial challenge is performed after the trypanosome infection has been cleared by drug treatment [adapted from (119)]. Indeed is was shown that while the commercial vaccine Boostrix® provides significant protection against a B. pertussis challenge (green bar: vaccinated, blue bar: non-vaccinated), exposure to trypanosomes abrogated vaccine-induced protection (red bar). In humans (lower panel), trypanosome infections were shown to suppress vaccine induced anti-measles antibodies. Serum antibody titers in vaccinated T. gambiense HAT patients (blue bar) were shown to be significantly lower as compared to vaccinated control individuals (green bar), and specific antibody titers did not recover after curative anti-HAT treatment (red bar) (121).
The Role of T Cells and T Cell-Derived Cytokines in Salivarian Trypanosomosis
Taken the extracellular nature of salivarian trypanosome infections, initial thoughts on the control of infection were naturally focused on the role of antibodies and B cells. However, already early in the 1980's it became obvious that while the virulence of experimental trypanosomosis was not linked to the expression of a specific VSG variant, or the use of a specific MHC-II type, CD4+ T cells played an absolutely crucial role in infection control (146). Twenty-five years after this initial discovery, it was shown that major T cell responses against cryptic T cell epitopes play a major role in trypanosomosis control (147, 148). This observation caused a major paradigm shift in the way trypanosomosis control is thought to occur, as it shows that T cell help is not just needed to support effective B cell functioning and antibody production, but that it plays a crucial second B cell-independent role during the progression of infection. This role of T cell biology might initially have been underestimated, as a multitude of studies had shown that trypanosomosis, both in mice and cattle, results in the occurrence of T cell immunosuppression (49, 149). However, these reports mostly referred to suppression of T cell proliferation and not to cytokine secretion. To date, the active disease controlling role of T cells is mainly attributed to the cytokine production in which polarization toward a Th1-type response is crucial for initial parasitaemia control (137). Detailed analysis of both T. brucei and T. congolense infections has indeed shown that early IFNγ production is crucial for the control of the onset of infection (Figure 3). This hypothesis was initially driven by the description of the cytokine production profile of CD4+T cells, and was later corroborated by the use of neutralizing anti-IFNγ antibodies as well as the use of IFNγ knockout mice (150, 151). The latter were shown to have an impaired control of the first peak of parasitaemia, followed by their inability to clear increasing parasite numbers, leading to the early death of the mice using the C57Bl/6 model (152). In experimental T. congolense infections, it was demonstrated that while hyper-susceptible BALB/c mice preferentially mount an infection associated Th2-type response against the Tc13 T. congolense parasite, Th1-biased C57Bl/6 mice were able to survive for up to 6 months when infected with the same clone (136). Also here it was shown that altering immune balances, by treatment with neutralizing anti-cytokine (or cytokine receptor) monoclonal antibodies, drastically alters the outcome of infection (153). Most recently, it was reported for T. brucei that the CD4+ IFNγ response is preceded by the production of this cytokine by NK and NKT cells, followed by a marked upregulation of IFNγ production by CD8+ T cells (154). This was observed in both the spleen and the liver of infected mice. However, by 10 days post infection it was clear that these cell populations were either drastically reduced in numbers or became totally depleted, leaving the CD4+ T cells to execute the major task of cytokine production. A comprehensive report on the overall role of IFNγ in various trypanosome infection models was recently published by Wu et al. (155). Interestingly, to date only one single trypanosome factor has been identified as being able to induce IFNγ production by T cells, CD8+ T cells in particular. This molecule, named TLTF (trypanosome lymphocyte triggering factor) (10) has been characterized in T. brucei and T. evansi parasites (156), but a homolog has not been described for other trypanosomes. TLTF was shown to be capable of inducing IFNγ production by astrocytes, suggesting a direct role of the molecule in the pathology development of sleeping sickness (157). This hypothesis has been further supported by the finding that IFNγ deficient mice show reduced CD4+ and CD8+ T cell influx into the brain parenchyma of T. brucei infected mice (158). Important is that when the role of T cells and IFNγ are considered within a trypanosomosis context, IL-10 was shown to be the main counter regulator of infection-associated inflammation in both T. brucei and T. congolense models. The source of the latter was proposed to be the regulatory T cells (9). However, a most recent effort to understand the full kinetics of IL-10 production during trypanosomosis, using the IL-10 reporter mouse system, has indicated that a whole range of cells is capable of producing IL-10 during infection and that most notably CD4+ T cells, that do not have a defined regulatory phenotype, are the main producers of this cytokine (159).
As for the role of IFNγ in parasitaemia control, early reports characterized this cytokine as a trypanosome growth factor (160). In contrast, a number of follow-up reports have indicated that IFNγ is crucial for inhibition of trypanosome growth, as well as for trypanosomosis-associated NO production and TNF production that both limit trypanosome growth (152, 161, 162). Both these factors have subsequently been identified as direct players in the control of peak parasitaemia levels in T. brucei as well as T. congolense infections. Using TNF knockout mice, it was shown that parasitaemia control in relatively resistant C57Bl/6 mice depends on TNF production during the first peak of parasitaemia (163, 164). This confirmed initial reports in which neutralizing anti-TNF antibodies had a negative effect on T. brucei parasitaemia control (165). Subsequently, a comparative infection model using BALB/c, C57Bl/6, C3H/HeN, and CBA/Ca mice showed that while TNF production per se was required for proper parasitaemia control, infected mice responded to TNF-associated inflammation with the shedding of TNF-R2 receptors. This, in turn resulted in limiting the TNF-mediated infection-associated immunopathology (166). Based on these results, and placing these observations in the wider context of trypanosome infections using various gene-deficient mouse models, it has been proposed that while soluble TNF could plays a pivotal role in parasitaemia control, it is the membrane-bound form of the cytokine that has a major impact on progressing inflammation and inducing pathology (167). When analyzing the possible direct effect of TNF on trypanosomes, results showed a differential outcome depending upon the model studied. First, it was reported for the T. brucei brucei AnTat 1.1 clone that TNF has a direct trypanolytic effect. The latter is mediated by a lectin binding domain of the TNF molecule that is located at the opposite site of the molecule as compared to the mammalian receptor binding site (168). Interestingly, the lectin specificity of TNF exhibits a high affinity for complex branched mannose sugars, such as those found on the trypanosome VSG molecule (169). Using a model for T. brucei gambiense, these findings were independently confirmed in an in vitro co-culture system (170). In contrast, experimental mouse studies with the human infective T. brucei rhodesiense LouTat 1 clone did not show a direct effect of TNF (171), suggesting that various trypanosome stocks can exhibit different levels of susceptibility to host cytokine-mediated growth regulation, as might be the case for IFNγ. Studies in trypanotolerant vs. trypanosusceptible cattle, using a T. congolense model, confirmed the possible role of TNF in parasitaemia control in natural infections (172), but here no link was made to a possible direct trypanolytic effect of the cytokine. Also, for the T. congolense infection mouse model, the direct action of TNF has not been reported but here TNF-R1 signaling has been associated to the combined release of soluble TNF and NO by activated macrophages. Those two combined were shown to play a crucial role in parasitaemia control, in conjunction with infection-induced anti-trypanosome antibodies (118). Interesting to note here is that in contrast to the limited knowledge on the molecular mechanisms of trypanosomosis-driven T cell IFNγ production, detailed research into the mechanisms of macrophage-derived TNF induction have been able to provide a more in-depth understanding of the host-parasite interplay. Indeed, first the VSG-GPI anchor was identified as the major trypanosome molecule responsible for TNF induction, with the trypanosome-specific galactose side-branch of the anchor playing a major role in macrophage activation (173, 174). Subsequently, it was shown that IFNγ could prime macrophages to become responsive to GPI-VSG and trypanosome stimulation (175). These results are in line with in vivo data showing that a rise in serum IFNγ values precedes the induction of infection-associated TNF (154). However, when macrophages are primed with VSG, prior to IFNγ, the cells are de-sensitize with respect to cytokine secretion (175). This may represent a way to prevent hyper-inflammation during the phase that corresponds to peak parasitaemia clearance, in which massive amounts of VSG are being released into the hosts' circulation. This information has been used to try and design an anti-disease vaccination approach for trypanosomosis (176). Although it was shown that repeated exposure to VSG-GPI prior to infection could indeed reduce the infection-associated pathology related to excessive macrophage activation, the results indicated that the vaccine approach did not result in the buildup of immunological memory involving antibodies or B cells. Instead, the reduction of inflammation, following the repeated GPI exposure related to an alteration of macrophage phenotypes, shifted the balance from infection-induced inflammatory type 1 cells to more anti-inflammatory type 2 cells or macrophages. This response was short-lived. Moreover, as the beneficial effect of the VSG-GPI treatment was observed to the same extent in both B cell deficient mice and in wild-type littermates, it became clear the approach could no longer be considered as a possible vaccine approach (177). In addition to the VSG-GPI, trypanosome DNA that contains high levels of unmethylated CpG sequences has been reported to be responsible for driving NF-κB an MAPK signaling pathways, resulting in the transcription of pro-inflammatory cytokine genes including tnf, in case of T. brucei infection(178). As for infection with T. evansi, the information on the role of IFNγ is limited, but data has shown that in addition to anti-trypanosome antibodies, this cytokine is required for parasitaemia control (179). Most recently, a comparative cytokine analysis using different T. evansi infections confirmed the induction of IFNγ in all infections (180). For T. vivax infections, there is a general lack of published information on the role of T cells and their cytokines in the control of onset in infection, but it appears reasonable to assume that a similar cytokine environment is needed to obtain the best parasitaemia control possible. As for human (T. b. rhodesiense) and cattle (T. congolense) infection, the role of IFNγ in inducing inflammatory pathology has been confirmed in both (181, 182), and as already outlined above, IFNγ has been shown to play a major role in the actual cerebral complications that characterize human sleeping sickness (183).
Anemia is a Major Pathological Feature of Animal Trypanosomosis
From the data reviewed above, it has become clear that the IFNγ/TNF driven immunopathology is a hallmark of trypanosomosis in both human and livestock. When focusing on the latter it has been amply documented that TNF-linked infection-associated anemia is the major pathological feature that marks both T. congolense and T. vivax trypanosomosis. In fact, the classification of so-called trypanotolerant and trypanosusceptible animals is based on the relative capacity to control anemia during infection which consequently is directly linked to whether or not animals remain productive while infected with trypanosomes (184). Mechanisms that underlie this tolerance are complex and to date still not clearly understood, but most likely include differences in erythropoeitic potential and hemodilution (185), factors involved in erythrolysis (186), eryhtrophagocytosis (187), the regulation of the erythropoietin homeostasis (188), the host's potential to raise neutralizing antibodies against secreted trypanosome virulence factors (189), and the general mechanism involved in inflammation control such as the regulation of the IFNγ/IL-10 balance as well as other cytokines (167). With respect to cytokine regulation during infection, it is worth mentioning that MIF (Macrophage migration Inhibitory Factor) was recently shown to be one of the key trypanosomosis-associated inflammation inducing cytokines involved in the promotion of so-called anemia of inflammation, in which classically activated macrophages also play a major role (190). Indeed, it was shown that MIF promotes the storage of iron in liver myeloid cells, subsequently depriving iron from the eythropoeitic system and preventing the maturation of RBCs. Important here is that the T. congolense C57Bl/6 mouse model has been proven useful over the years to unravel the molecular mechanisms that govern anemia pathology. Gene expression profiling studies in these mice have pinpointed infection-induced deregulation of erythropoiesis as well as the involvement of stress-induced acute phase responses linked to T. congolense-associated anemia (191). More recently, the C57Bl/6 model was also shown to be suitable for unraveling the pathology of T. vivax infections (129, 192, 193). In addition, the use of various knockout mouse strains generated on the C56Bl/6 background, such as TNF−/− and Lymphytoxin−/− (194), TNFp75−/− (166), IFNγR−/− (152), IL-10−/− (195), and Gal3−/− (196) have all been used to study specific aspects of the inflammatory cascade that drive trypanosomosis-associated inflammation. One of the most recent finding here has shown that the reduced PCV, associated with experimental mouse trypanosomosis, is not just the result of reduced circulating RBC numbers, but also of increased plasma volume, leading to hemodilution. The reduction of platelet concentrations in T. congolense-infected mice has also been reported in T. congolense-infected cattle and sheep (197, 198) suggesting that hemodilution could be a major pathogenic characteristic of livestock trypanosomosis, as well.
To date, the characteristics of anemia are probably the most and best described features of AT. Whenever new surveillance efforts result in the discovery of new infection foci, anemia is usually the parameter that is used to describe disease severity. This is the case for T. evansi, T. congolense, and T. vivax independently of the geographic location of infection or host species. As described for T. congolense, T. evansi-induced anemia was linked to infection-associated deregulation of the iron metabolism as well (199), although the number of detailed mechanistic studies available for this infection are rare. Interestingly, trypanosomosis-associated anemia has even been observed in more rare infection cases of wildlife in the Australian little red flying fox infected with T. minasense and T. rangeli (200). Together, all these reports indicate that the general occurrence of trypanosomosis-associated anemia is most likely due to the multitude of inflammatory pathways, and linked to various aspects of impaired RBC development and clearance. Taken this into account, it is very intriguing that reports of severe infection-associated anemia are missing in the case of HAT. Indeed, most reports documenting HAT-associated anemia describe the induction of RBC lysis and acute anemia as the result of treatment rather than the infection itself. This is particularly the case for T. b. gambiense infections (201–204). For T. b. rhodesiense rare reports do exist on the occurrence of infection-associated anemia and hemolysis (205), and models have been developed using vervet monkeys to investigate this aspect of acute HAT pathology in details (206). However, at the same time other reports have shown the lack of any correlation between T. b. rhodesiense severity in humans and the presence or absence of anemia (207–209). Hence, it seems that the human immune system might have found unique ways of dealing with trypanosomosis both in terms of dealing with parasite killing through the TLF1/2 ApoL1 mechanisms, as well as the control of infection-associated anemia.
Conclusion
Over the past years, international joint efforts to control Human African Trypanosomosis have resulted in a drastic reduction in the number of confirmed disease cases. In addition, it might be feasible that by 2020 the incidence of this neglected tropic disease will be reduced to levels where it is no longer considered as a public health threat. However, at the same time the incidence of animal trypanosomosis is on the rise, affecting animal productivity through the detrimental health effects caused by excessive host-parasite immune interactions. This has now become a world-wide issue that affects agricultural infrastructures in emerging economy countries, not just in Africa but also in Asia and South America. In addition, AT even threatens the livestock industry of developed countries when sudden outbreaks occur due to importation of the disease either through infected vectors or host animals. To date, not a single vaccine strategy is available to control AT. Detrimental infection-associated mechanisms undermine both T cell and B cell compartments, making it virtually impossible to develop methods that allow the generation of long-term sustainable immunological memory. It appears that in the evolutionary battle between the trypanosome and the host immune system, the parasite has not only been able to find ways to evade innate trypanosome killing mechanisms, but has also acquired tools to undermine the crucial immunological defense and memory systems of the host. Only recently, it has become obvious that one of the major strengths of the African Trypanosomes' defense system is the capacity of genetic exchanges between individual parasites, while residing in the insect vector. Through this mechanism, new human-infective parasites can be generated and this by combining resistant genes from existing T. b. rhodesiense parasites with the vast repertoire of the T. b. brucei animal parasite reservoir. In addition, it has become clear that mechanisms of genetic plasticity also have allowed T. b. brucei to transform into T. equiperdum and T. evansi, parasites that have now successfully conquered large areas of the developing world, as they no longer need the tsetse vector for transmission. In particular, the latter risks to become a future health problem, as human infective cases of T. evansi have already been reported in India, Vietnam, Egypt, and Algeria. Together, these results show that while classic T. gambiense trypanosomosis is becoming rare, AT and new atypical human trypanosomosis remain a serious risk for the human population. Future research into the host-parasite interactions and trypanosomosis- associated inflammation of these new atypical infections should allow to obtain a better understanding of problems, including vaccine failure, and hopefully lead us to a long-term solution that can deal with both human and livestock infections, as well as the ever surviving wildlife trypanosome reservoir.
Author Contributions
All authors contributed to the writing of the paper. In addition all artwork was prepared by JP.
Funding
We acknowledge the financial support grants from the FWO (FWO G015016N and G.0.028.10.N.10) and the Bill and Melinda Gates Foundation, grant 641K760.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Desquesnes M, Holzmuller P, Lai D, Dargantes A, Lun Z, Jittaplapong S. Trypanosoma evansi and surra: a review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. Biomed Res Int. (2013) 2013:194176. doi: 10.1155/2013/194176
2. Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet (2017) 390:2397–409. doi: 10.1016/S0140-6736(17)31510-6
3. Diall O, Cecchi G, Wanda G, Argilés-Herrero R, Vreysen MJB, Cattoli G, et al. Developing a progressive control pathway for African animal trypanosomosis. Trends Parasitol. (2017) 33:499–509. doi: 10.1016/j.pt.2017.02.005
4. Molinari J, Moreno SA. Trypanosoma brucei Plimmer & Bradford, 1899 is a synonym of T. evansi (Steel, 1885) according to current knowledge and by application of nomenclature rules. Syst Parasitol. (2018) 95:249–56. doi: 10.1007/s11230-018-9779-z
5. Gibson W. Liaisons dangereuses: sexual recombination among pathogenic trypanosomes. Res Microbiol. (2015) 166:459–66. doi: 10.1016/j.resmic.2015.05.005
6. Peacock L, Ferris V, Sharma R, Sunter J, Bailey M, Carrington M, et al. Identification of the meiotic life cycle stage of Trypanosoma brucei in the tsetse fly. Proc Natl Acad Sci USA. (2011) 108:3671–6. doi: 10.1073/pnas.1019423108
7. Lugli EB, Pouliot M, Portela MDPM, Loomis MR, Raper J. Characterization of primate trypanosome lytic factors. Mol Biochem Parasitol. (2004) 138:9–20. doi: 10.1016/j.molbiopara.2004.07.004
8. Capewell P, Cooper A, Clucas C, Weir W, MacLeod A. A co-evolutionary arms race: trypanosomes shaping the human genome, humans shaping the trypanosome genome. Parasitology (2015) 142:S108–19. doi: 10.1017/S0031182014000602
9. Guilliams M, Bosschaerts T, Hérin M, Hünig T, Loi P, Flamand V, et al. Experimental expansion of the regulatory T cell population increases resistance to African trypanosomiasis. J Infect Dis. (2008) 198:781–91. doi: 10.1086/590439
10. Bakhiet M, Mix E, Kristensson K, Wigzell H, Olsson T. T cell activation by aTrypanosoma brucei brucei-deriyed lymphocyte triggering factor is dependent on tyrosine protein kinases but not on protein kinase C and A. Eur J Immunol. (1993) 23:1535–1539. doi: 10.1002/eji.1830230721
11. Truc P, Büscher P, Cuny G, Gonzatti MI, Jannin J, Joshi P, et al. A typical human infections by animal trypanosomes. PLoS Negl Trop Dis. (2013) 7:e2256. doi: 10.1371/journal.pntd.0002256
12. Overath P, Haag J, Mameza MG, Lischke A. Freshwater fish trypanosomes: definition of two types, host control by antibodies and lack of antigenic variation. Parasitology (1999) 119(Pt 6):591–601. doi: 10.1017/S0031182099005089
13. Zídková L, Cepicka I, Szabová J, Svobodová M. Biodiversity of avian trypanosomes. Infect Genet Evol. (2012) 12:102–12. doi: 10.1016/j.meegid.2011.10.022
14. Haag J, O'hUigin C, Overath P. The molecular phylogeny of trypanosomes: evidence for an early divergence of the Salivaria. Mol Biochem Parasitol. (1998) 91:37–49. doi: 10.1016/S0166-6851(97)00185-0
15. Pandey A, Galvani A. Strategies for Trypanosoma brucei gambiense elimination. Lancet Glob Health (2017) 5:e10–1. doi: 10.1016/S2214-109X(16)30284-4
16. Holmes P. On the road to elimination of rhodesiense human African trypanosomiasis: first WHO meeting of stakeholders. PLoS Negl Trop Dis. (2015) 9:10–12. doi: 10.1371/journal.pntd.0003571
17. Informal Expert Group on Gambiense HAT Reservoirs, Büscher P, Bart J-M, Boelaert M, Bucheton B, Cecchi G, et al. Do cryptic reservoirs threaten gambiense-sleeping sickness elimination? Trends Parasitol. (2018) 34:197–207. doi: 10.1016/j.pt.2017.11.008
18. Desquesnes M, Dargantes A, Lai D-H, Lun Z-R, Holzmuller P, Jittapalapong S. Trypanosoma evansi and Surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed Res Int. (2013) 1–20. doi: 10.1155/2013/321237
19. Morrison LJ, Vezza L, Rowan T, Hope JC. Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends Parasitol. (2016) 32:599–607. doi: 10.1016/j.pt.2016.04.012
20. Auty H, Torr SJ, Michoel T, Jayaraman S, Morrison LJ. Cattle trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev Sci Tech. (2015) 34:587–98.
21. Giordani F, Morrison LJ, Rowan TG, DE Koning HP, Barrett MP. The animal trypanosomiases and their chemotherapy: a review. Parasitology (2016) 143:1862–89. doi: 10.1017/S0031182016001268
22. P. Finelle. African animal trypanosomiasis transmission of trypanosomes. World Anim Rev. (1973) 7:1–6.
23. Lun ZR, Lai DH, Li FJ, Lukeš J, Ayala FJ. Trypanosoma brucei: two steps to spread out from Africa. Trends Parasitol. (2010) 26:424–7. doi: 10.1016/j.pt.2010.05.007
24. Baldacchino F, Desquesnes M, Mihok S, Foil LD, Duvallet G, Jittapalapong S. Tabanids: neglected subjects of research, but important vectors of disease agents! Infect Genet Evol. (2014) 28:596–615. doi: 10.1016/j.meegid.2014.03.029
25. Banerjee D, Kumar V, Maity A, Ghosh B, Tyagi K, Singha D, et al. Identification through DNA barcoding of Tabanidae (Diptera) vectors of surra disease in India. Acta Trop. (2015) 150:52–8. doi: 10.1016/j.actatropica.2015.06.023
26. Taioe MO, Motloang MY, Namangala B, Chota A, Molefe NI, Musinguzi SP, et al. Characterization of tabanid flies (Diptera: Tabanidae) in South Africa and Zambia and detection of protozoan parasites they are harboring. Parasitology (2017) 144:1162–78. doi: 10.1017/S0031182017000440
27. Misra KK, Roy S, Choudhury A. Biology of Trypanosoma (Trypanozoon) evansi in experimental heterologous mammalian hosts. J Parasit Dis. (2016) 40:1047–61. doi: 10.1007/s12639-014-0633-1
28. Borst P, Fase-Fowler F, Gibson WC. Kinetoplast DNA of Trypanosoma evansi. Mol Biochem Parasitol. (1987) 23:31–8.
29. Paris Z, Hashimi H, Lun S, Alfonzo JD, Lukeš J. Futile import of tRNAs and proteins into the mitochondrion of Trypanosoma brucei evansi. Mol Biochem Parasitol. (2011) 176:116–20. doi: 10.1016/j.molbiopara.2010.12.010
30. Carnes J, Anupama A, Balmer O, Jackson A, Lewis M, Brown R, et al. Genome and phylogenetic analyzes of Trypanosoma evansi reveal extensive similarity to T. brucei and multiple independent origins for dyskinetoplasty. PLoS Negl Trop Dis. (2015) 9:e3404. doi: 10.1371/journal.pntd.0003404
31. Cuypers B, Van den Broeck F, Van Reet N, Meehan CJ, Cauchard J, Wilkes JM, et al. Genome-Wide SNP analysis reveals distinct origins of Trypanosoma evansi and Trypanosoma equiperdum. Genome Biol Evol. (2017) 9:1990–7. doi: 10.1093/gbe/evx102
32. Richardson JB, Lee K-Y, Mireji P, Enyaru J, Sistrom M, Aksoy S, et al. Genomic analyses of African Trypanozoon strains to assess evolutionary relationships and identify markers for strain identification. PLoS Negl Trop Dis. (2017) 11:e0005949. doi: 10.1371/journal.pntd.0005949
33. Gibson W. Molecular characterization of field isolates of human pathogenic trypanosomes. Trop Med Int Health (2001) 6:401–6. doi: 10.1046/j.1365-3156.2001.00711.x
34. Radwanska M, Chamekh M, Vanhamme L, Claes F, Magez S, Magnus E, et al. The serum resistance-associated gene as a diagnostic tool for the detection of Trypanosoma brucei rhodesiense. Am J Trop Med Hyg. (2002) 67:684–90. doi: 10.4269/ajtmh.2002.67.684
35. Welburn SC, Picozzi K, Fèvre EM, Coleman PG, Odiit M, Carrington M, et al. Identification of human-infective trypanosomes in animal reservoir of sleeping sickness in Uganda by means of serum-resistance-associated (SRA) gene. Lancet (2001) 358:2017–9. doi: 10.1016/S0140-6736(01)07096-9
36. Radwanska M, Claes F, Magez S, Magnus E, Perez-Morga D, Pays E, et al. Novel primer sequences for polymerase chain reaction-based detection of Trypanosoma brucei gambiense. Am J Trop Med Hyg. (2002) 67:289–95. doi: 10.4269/ajtmh.2002.67.289
37. Claes F, Büscher P, Touratier L, Goddeeris BM. Trypanosoma equiperdum: master of disguise or historical mistake? Trends Parasitol. (2005) 21:316–21. doi: 10.1016/j.pt.2005.05.010
38. Jones TW, Dávila AMR. Trypanosoma vivax–out of Africa. Trends Parasitol. (2001) 17:99–101. doi: 10.1016/S1471-4922(00)01777-3
39. Moloo SK, Sabwa CL, Kabata JM. Vector competence of Glossina pallidipes and G. morsitans centralis for Trypanosoma vivax, T. congolense and T. b. brucei. Acta Trop. (1992) 51:271–80. doi: 10.1016/0001-706X(92)90045-Y
40. Croft SL, Kuzoe FA, Ryan L, Molyneux DH. Trypanosome infection rates of Glossina spp. (Diptera: Glossinidae) in transitional forest-savanna near Bouaflé, Ivory Coast. Tropenmed Parasitol. (1984) 35:247–50.
41. Gibson W. The origins of the trypanosome genome strains Trypanosoma brucei brucei TREU 927, T. b. gambiense DAL 972, T. vivax Y486 and T. congolense IL3000. Parasit Vectors (2012) 5:71. doi: 10.1186/1756-3305-5-71
42. Truc P, Jamonneau V, N'Guessan P, N'Dri L, Diallo PB, Cuny G. Trypanosoma brucei ssp. and T congolense: mixed human infection in Côte d'Ivoire. Trans R Soc Trop Med Hyg. (1996) 92:537–8.
43. Van Xong H, De Baetselier P, Pays E, Magez S. Selective pressure can influence the resistance of Trypanosoma congolense to normal human serum. Exp Parasitol. (2002) 102:61–5. doi: 10.1016/S0014-4894(03)00032-8
44. Wiesenhütter E. Research into the relative importance of tabanidae (Diptera) in mechanical disease transmission I. the seasonal occurrence and relative abundance of tabanidae in a dar es salaam dairy farm. J Nat Hist. (1975) 9:377–84. doi: 10.1080/00222937500770271
45. Gerold P, Striepen B, Reitter B, Geyer H, Geyer R, Reinwald E, et al. Glycosyl-phosphatidylinositols of Trypanosoma congolense: two common precursors but a new protein-anchor. J Mol Biol. (1996) 261:181–94. doi: 10.1006/jmbi.1996.0451
46. Okwor I, Onyilagha C, Kuriakose S, Mou Z, Jia P, Uzonna JE. Regulatory T cells enhance susceptibility to experimental Trypanosoma Congolense infection independent of mouse genetic background. PLoS Negl Trop Dis. (2012) 6:e1761. doi: 10.1371/journal.pntd.0001761
47. Inverso JA, De Gee AL, Mansfield JM. Genetics of resistance to the African trypanosomes. VII. Trypanosome virulence is not linked to variable surface glycoprotein expression. J Immunol. (1988) 140:289–93.
48. Magez S, Stijlemans B, Radwanska M, Pays E, Ferguson MA, De Baetselier P. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J Immunol. (1998) 160:1949–56. doi: 10.4049/jimmunol.164.4.2070
49. Sileghem M, Flynn JN. Suppression of interleukin 2 secretion and interleukin 2 receptor expression during tsetse-transmitted trypanosomiasis in cattle. Eur J Immunol. (1992) 22:767–73. doi: 10.1002/eji.1830220321
50. Obishakin E, de Trez C, Magez S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. (2014) 36:187–98. doi: 10.1111/pim.12099
51. Garside LH, Gibson WC. Molecular characterization of trypanosome species and subgroups within subgenus Nannomonas. Parasitology (1995) 111(Pt 3):301–12. doi: 10.1017/S0031182000081853
52. Vickerman K. Developmental cycles and biology of pathogenic trypanosomes. Br Med Bull. (1985) 41:105–14. doi: 10.1093/oxfordjournals.bmb.a072036
53. Peacock L, Bailey M, Gibson W. Dynamics of gamete production and mating in the parasitic protist Trypanosoma brucei. Parasit Vectors (2016) 9:404. doi: 10.1186/s13071-016-1689-9
54. Maclean L, Chisi JE, Odiit M, Gibson WC, Ferris V, Picozzi K, et al. Severity of human African trypanosomiasis in East Africa is associated with geographic location, parasite genotype, and host in ammatory cytokine response pro le. Infect Immun. (2004) 72:7040–4. doi: 10.1128/IAI.72.12.7040
55. Murphy WJ, Brentano ST, Rice-Ficht AC, Dorfman DM, Donelson JE. DNA rearrangements of the variable surface antigen genes of the trypanosomes1. J Protozool (1984) 31:65–73. doi: 10.1111/j.1550-7408.1984.tb04291.x
56. Hall JPJ, Wang H, David Barry J. Mosaic VSGs and the scale of Trypanosoma brucei antigenic variation. PLoS Pathog. (2013) 9:e1003502. doi: 10.1371/journal.ppat.1003502
57. Levine RF, Mansfield JM. Genetics of resistance to the African trypanosomes. III. Variant-specific antibody responses of H-2-compatible resistant and susceptible mice. J Immunol. (1984) 133:1564–9.
58. Mugnier MR, Stebbins CE, Papavasiliou FN. Masters of disguise: antigenic variation and the VSG coat in Trypanosoma brucei. PLoS Pathog. (2016) 12:e1005784. doi: 10.1371/journal.ppat.1005784
59. Pays E. Regulation of antigen gene expression in Trypanosoma brucei. Trends Parasitol. (2005) 21:517–20. doi: 10.1016/j.pt.2005.08.016
60. Horn D. Antigenic variation in African trypanosomes. Mol Biochem Parasito (2014) 195:123–9. doi: 10.1016/j.molbiopara.2014.05.001
61. Weiss BL, Wang J, Aksoy S. Tsetse immune system maturation requires the presence of obligate symbionts in larvae. PLoS Biol. (2011) 9:e1000619. doi: 10.1371/journal.pbio.1000619
62. Weiss BL, Maltz M, Aksoy S. Obligate symbionts activate immune system development in the tsetse fly. J Immunol. (2012) 188:3395–403. doi: 10.4049/jimmunol.1103691
63. Weiss BL, Wang J, Maltz MA, Wu Y, Aksoy S. Trypanosome infection establishment in the tsetse fly gut is influenced by microbiome-regulated host immune barriers. PLoS Pathog. (2013) 9:e1003318. doi: 10.1371/journal.ppat.1003318
64. Wang J, Wu Y, Yang G, Aksoy S. Interactions between mutualist Wigglesworthia and tsetse peptidoglycan recognition protein (PGRP-LB) influence trypanosome transmission. Proc Natl Acad Sci USA. (2009) 106:12133–8. doi: 10.1073/pnas.0901226106
65. Weiss BL, Savage AF, Griffith BC, Wu Y, Aksoy S. The peritrophic matrix mediates differential infection outcomes in the tsetse fly gut following challenge with commensal, pathogenic, and parasitic microbes. J Immunol. (2014) 193:773–82. doi: 10.4049/jimmunol.1400163
66. Hao Z, Kasumba I, Lehane MJ, Gibson WC, Kwon J, Aksoy S. Tsetse immune responses and trypanosome transmission: implications for the development of tsetse-based strategies to reduce trypanosomiasis. Proc Natl Acad Sci USA. (2001) 98:12648–53. doi: 10.1073/pnas.221363798
67. Hu C, Aksoy S. Innate immune responses regulate trypanosome parasite infection of the tsetse fly glossina morsitans morsitans. Mol Microbiol. (2006) 60:1194–204. doi: 10.1111/j.1365-2958.2006.05180.x
68. Hao Z, Kasumba I, Aksoy S. Proventriculus (cardia) plays a crucial role in immunity in tsetse fly (Diptera: Glossinidiae). Insect Biochem Mol Biol. (2003) 33:1155–64. doi: 10.1016/j.ibmb.2003.07.001
69. Haines LR, Lehane SM, Pearson TW, Lehane MJ. Tsetse EP protein protects the fly midgut from trypanosome establishment. PLoS Pathog. (2010) 6:e1000793. doi: 10.1371/journal.ppat.1000793
70. Akoda K, Van Den Abbeele J, Marcotty T, De Deken R, Sidibe I, Van den Bossche P. Nutritional stress of adult female tsetse flies (Diptera: Glossinidae) affects the susceptibility of their offspring to trypanosomal infections. Acta Trop. (2009) 111:263–7. doi: 10.1016/j.actatropica.2009.05.005
71. Akoda K, Van den Bossche P, Marcotty T, Kubi C, Coosemans M, De Deken R, et al. Nutritional stress affects the tsetse fly's immune gene expression. Med Vet Entomol. (2009) 23:195–201. doi: 10.1111/j.1365-2915.2009.00799.x
72. Akoda K, Van den Bossche P, Lyaruu E A, De Deken R, Marcotty T, Coosemans M, et al. Maturation of a Trypanosoma Brucei infection to the infectious metacyclic stage is enhanced in nutritionally stressed tsetse flies : table 1. J Med Entomol. (2009) 46:1446–9. doi: 10.1603/033.046.0629
73. Peacock L, Cook S, Ferris V, Bailey M, Gibson W. The life cycle of Trypanosoma (Nannomonas) congolense in the tsetse fly. Parasit Vectors (2012) 5:109. doi: 10.1186/1756-3305-5-109
74. Gibson W, Peacock L, Hutchinson R. Microarchitecture of the tsetse fly proboscis. Parasit Vectors (2017) 10:430. doi: 10.1186/s13071-017-2367-2
75. Morrison LJ, Tweedie A, Black A, Pinchbeck GL, Christley RM, Schoenefeld A, et al. Discovery of mating in the major African livestock pathogen trypanosoma congolense. PLoS ONE (2009) 4:e5564. doi: 10.1371/journal.pone.0005564
76. Caljon G, Van Den Abbeele J, Stijlemans B, Coosemans M, De Baetselier P, Magez S. Tsetse fly saliva accelerates the onset of Trypanosoma brucei infection in a mouse model associated with a reduced host inflammatory response. Infect Immun. (2006) 74:6324–30. doi: 10.1128/IAI.01046-06
77. Caljon G, De Ridder K, De Baetselier P, Coosemans M, Van Den Abbeele J. Identification of a tsetse fly salivary protein with dual inhibitory action on human platelet aggregation. PLoS ONE (2010) 5:e9671. doi: 10.1371/journal.pone.0009671
78. Sutherland CS, Stone CM, Steinmann P, Tanner M, Tediosi F. Seeing beyond 2020: an economic evaluation of contemporary and emerging strategies for elimination of Trypanosoma brucei gambiense. Lancet Glob Health (2017) 5:e69–79. doi: 10.1016/S2214-109X(16)30237-6
79. Simarro PP, Diarra A, Postigo JAR, Franco JR, Jannin JG. The human african trypanosomiasis control and surveillance program of the World Health Organization 2000-2009: the way forward. PLoS Negl Trop Dis. (2011) 5:e1007. doi: 10.1371/journal.pntd.0001007
80. Joshi PP, Shegokar VR, Powar RM, Herder S, Katti R, Salkar HR, et al. Human trypanosomiasis caused by Trypanosoma evansi in India: the first case report. Am J Trop Med Hyg. (2005) 73:491–5. doi: 10.4269/ajtmh.2005.73.491
81. Vanhollebeke B, Truc P, Poelvoorde P, Pays A, Joshi PP, Katti R, et al. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med. (2006) 355:2752–6. doi: 10.1056/NEJMoa063265
82. Truc P, Gibson W, Herder S. Genetic characterization of Trypanosoma evansi isolated from a patient in India. Infect Genet Evol. (2007) 7:305–7. doi: 10.1016/j.meegid.2006.07.004
83. Haridy FM, El-Metwally MT, Khalil HHM, Morsy TA. Trypanosoma evansi in dromedary camel: with a case report of zoonosis in greater Cairo, Egypt. J Egypt Soc Parasitol. (2011) 41:65–76.
84. Duffy CW, Morrison LJ, Black A, Pinchbeck GL, Christley RM, Schoenefeld A, et al. Trypanosoma vivax displays a clonal population structure. Int J Parasitol. (2009) 39:1475–83. doi: 10.1016/j.ijpara.2009.05.012
85. Jackson AP, Berry A, Aslett M, Allison HC, Burton P, Vavrova-Anderson J, et al. Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proc Natl Acad Sci USA. (2012) 109:3416–21. doi: 10.1073/pnas.1117313109
86. Biteau N, Asencio C, Izotte J, Rousseau B, Fèvre M, Pillay D, et al. Trypanosoma brucei gambiense infections in mice lead to tropism to the reproductive organs, and horizontal and vertical transmission. PLoS Negl Trop Dis. (2016) 10:1–15. doi: 10.1371/journal.pntd.0004350
87. Rocha G, Martins A, Gama G, Brandão F, Atouguia J. Possible cases of sexual and congenital transmission of sleeping sickness [5]. Lancet (2004) 363:247. doi: 10.1016/S0140-6736(03)15345-7
88. Seed JR, Sechelski JB, Ortiz JC, Chapman JF. Relationship between human serum trypanocidal activity and host resistance to the African trypanosomes. J Parasitol. (1993) 79:226–32. doi: 10.1016/j.molbiopara.2015.03.007
89. Raper J, Fung R, Ghiso J, Nussenzweig V, Tomlinson S. Characterization of a novel trypanosome lytic factor from human serum. Infect Immun. (1999) 67:1910–6.
90. Raper J, Portela MP, Lugli E, Frevert U, Tomlinson S. Trypanosome lytic factors: novel mediators of human innate immunity. Curr Opin Microbiol. (2001) 4:402–8. doi: 10.1016/S1369-5274(00)00226-5
91. De Greef C, Hamers R. The serum resistance-associated (SRA) gene of Trypanosoma brucei rhodesiense encodes a variant surface glycoprotein-like protein. Mol Biochem Parasitol. (1994) 68:277–84. doi: 10.1016/0166-6851(94)90172-4
92. Xong H V, Vanhamme L, Chamekh M, Chimfwembe CE, Van Den Abbeele J, Pays A, et al. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell (1998) 95:839–46.
93. Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Abbeele J Van Den, et al. Apolipoprotein L-1 is the trypanosome lytic factor of human serum. Nature (2003) 422:83–7. doi: 10.1038/nature01457.1.
94. Vanwalleghem G, Fontaine F, Lecordier L, Tebabi P, Klewe K, Nolan DP, et al. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat Commun. (2015) 6:8078. doi: 10.1038/ncomms9078
95. Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, Nolan DP, Lins L, Homblé F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science (2005) 309:469–72. doi: 10.1126/science.1114566
96. Vanhollebeke B, Lecordier L, Perez-Morga D, Amiguet-Vercher A, Pays E. Human serum lyses Trypanosoma brucei by triggering uncontrolled swelling of the parasite lysosome. J Eukaryot Microbiol. (2007) 54:448–51. doi: 10.1111/j.1550-7408.2007.00285.x
97. Oli MW, Cotlin LF, Shiflett AM, Hajduk SL. Serum resistance-associated protein blocks lysosomal targeting of trypanosome lytic factor in Trypanosoma brucei. Eukaryot Cell (2006) 5:132–9. doi: 10.1128/EC.5.1.132-139.2006
98. Greene AS, Hajduk SL. Trypanosome lytic factor-1 initiates oxidation-stimulated osmotic lysis of Trypanosoma brucei brucei. J Biol Chem. (2016) 291:3063–75. doi: 10.1074/jbc.M115.680371
99. DeJesus E, Kieft R, Albright B, Stephens NA, Hajduk SL. A single amino acid substitution in the group 1 Trypanosoma brucei gambiense haptoglobin-hemoglobin receptor abolishes TLF-1 binding. PLoS Pathog. (2013) 9:1–10. doi: 10.1371/journal.ppat.1003317
100. Bullard W, Kieft R, Capewell P, Veitch NJ, Macleod A, Hajduk SL. Haptoglobin-hemoglobin receptor independent killing of African trypanosomes by human serum and trypanosome lytic factors. Virulence (2012) 3:72–6. doi: 10.4161/viru.3.1.18295
101. Vanhollebeke B, De Muylder G, Nielsen MJ, Pays A, Tebabi P, Dieu M, et al. A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans. Science (2008) 320:677–81. doi: 10.1126/science.1156296
102. Enyaru JCK, Matovu E, Nerima B, Akol M, Sebikali C. Detection of T.b. rhodesiense trypanosomes in humans and domestic animals in South East Uganda by amplification of serum resistance-associated gene. Ann N Y Acad Sci. (2006) 1081:311–9. doi: 10.1196/annals.1373.041
103. Gibson W, Peacock L, Ferris V, Fischer K, Livingstone J, Thomas J, et al. Genetic recombination between human and animal parasites creates novel strains of human pathogen. PLoS Negl Trop Dis. (2015) 9:1–16. doi: 10.1371/journal.pntd.0003665
104. Echodu R, Sistrom M, Bateta R, Murilla G, Okedi L, Aksoy S, et al. Genetic diversity and population structure of <italic>Trypanosoma brucei </italic> in uganda: implications for the epidemiology of sleeping sickness and nagana. PLoS Negl Trop Dis. (2015) 9:e0003353. doi: 10.5061/dryad.m7q4c
105. Capewell P, Veitch NJ, Turner CMR, Raper J, Berriman M, Hajduk SL, et al. Differences between Trypanosoma brucei gambiense groups 1 and 2 in their resistance to killing by trypanolytic factor 1. PLoS Negl Trop Dis. (2011) 5:e1287. doi: 10.1371/journal.pntd.0001287
106. Higgins MK, Tkachenko O, Brown A, Reed J, Raper J, Carrington M. Structure of the trypanosome haptoglobin-hemoglobin receptor and implications for nutrient uptake and innate immunity. Proc Natl Acad Sci USA. (2013) 110:1905–10. doi: 10.1073/pnas.1214943110
107. Kieft R, Capewell P, Turner CMR, Veitch NJ, MacLeod A, Hajduk S. Mechanism of Trypanosoma brucei gambiense (group 1) resistance to human trypanosome lytic factor. Proc Natl Acad Sci USA. (2010) 107:16137–41. doi: 10.1073/pnas.1007074107
108. Berberof M, Pérez-Morga D, Pays E. A receptor-like flagellar pocket glycoprotein specific to Trypanosoma brucei gambiense. Mol Biochem Parasitol. (2001) 113:127–38. doi: 10.1016/S0166-6851(01)00208-0
109. Capewell P, Clucas C, DeJesus E, Kieft R, Hajduk S, Veitch N, et al. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLoS Pathog. (2013) 9:2–7. doi: 10.1371/journal.ppat.1003686
110. Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homblé F, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature (2013) 501:430–4. doi: 10.1038/nature12516
111. Radwanska M, Bockstal V, Brombacher F, Magez S. Parasite-induced B-cell apoptosis results in loss of specific protective anti-trypanosome antibody responses, and abolishment of vaccine induced protective memory. In: XII INTERNATIONAL CONGRESS OF PARASITOLOGY (ICOPA) (Melbourne, VIC). (2010). p. 31–38.
112. Lai D-H, Wang Q-P, Li Z, Luckins AG, Reid SA, Lun Z-R. Investigations into human serum sensitivity expressed by stocks of Trypanosoma brucei evansi. Int J Parasitol. (2010) 40:705–10. doi: 10.1016/j.ijpara.2009.11.009
113. Faulkner SD, Oli MW, Kieft R, Cotlin L, Widener J, Shiflett A, et al. In vitro generation of human high-density-lipoprotein-resistant Trypanosoma brucei brucei. Eukaryot Cell (2006) 5:1276–86. doi: 10.1128/EC.00116-06
114. Vanhamme L, Renauld H, Lecordier L, Poelvoorde P, Van Den Abbeele J, Pays E. The Trypanosoma brucei reference strain TREU927/4 contains T. brucei rhodesiense-specific SRA sequences, but displays a distinct phenotype of relative resistance to human serum. Mol Biochem Parasitol. (2004) 135:39–47. doi: 10.1016/j.molbiopara.2004.01.004
115. Lai D, Wang Q, Li Z, Julius L, Lun Z. Evolution of the serum resistance-associated SRA gene in African trypanosomes. Sci Bull. (2009) 54:1275–8. doi: 10.1007/s11434-009-0137-z
116. Van Vinh Chau N, Buu Chau L, Desquesnes M, Herder S, Phu Huong Lan N, Campbell JI, et al. A clinical and epidemiological investigation of the first reported human infection with the zoonotic parasite Trypanosoma evansi in Southeast Asia. Clin Infect Dis. (2016) 62:1002–8. doi: 10.1093/cid/ciw052
117. Wabale V, Nalage P, Joshi A, Bharadwaj R, Deshpande K, Chowdhary A. Human Asian Trypanosomiasis due to Trypanosoma evansi: a rare case. J Adv Parasitol. (2015) 3:65–8. doi: 10.14737/journal.jap/2015/2.3.65.68
118. Baral TN, De Baetselier P, Brombacher F, Magez S. Control of Trypanosoma evansi infection is IgM mediated and does not require a type I inflammatory response. J Infect Dis. (2007) 195:1513–20. doi: 10.1086/515577
119. Desquesnes M, Yangtara S, Kunphukhieo P, Chalermwong P, Jittapalapong S, Herder S. Zoonotic trypanosomes in South East Asia: attempts to control Trypanosoma lewisi using veterinary drugs. Exp Parasitol. (2016) 165:35–42. doi: 10.1016/j.exppara.2016.03.009
120. Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA. (2014) 111:E2130–9. doi: 10.1073/pnas.1400699111
121. Smith EE, Malik HS. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. (2009) 19:850–8. doi: 10.1101/gr.085647.108
122. Thomson R, Molina-Portela P, Mott H, Carrington M, Raper J. Hydrodynamic gene delivery of baboon trypanosome lytic factor eliminates both animal and human-infective African trypanosomes. Proc Natl Acad Sci USA. (2009) 106:19509–14. doi: 10.1073/pnas.0905669106
123. Cooper A, Capewell P, Clucas C, Veitch N, Weir W, Thomson R, et al. A primate APOL1 variant that kills Trypanosoma brucei gambiense. PLoS Negl Trop Dis. (2016) 10:e0004903. doi: 10.1371/journal.pntd.0004903
124. Borst P. Antigenic variation and allelic exclusion. Cell (2002) 109:5–8. doi: 10.1016/S0092-8674(02)00711-0
125. Pays E, Vanhamme L, Pérez-Morga D. Antigenic variation in Trypanosoma brucei: facts, challenges and mysteries. Curr Opin Microbiol. (2004) 7:369–74. doi: 10.1016/j.mib.2004.05.001
126. Radwanska M, Magez S, Michel A, Stijlemans B, Geuskens M, Pays E. Comparative analysis of antibody responses against HSP60, invariant surface glycoprotein 70, and variant surface glycoprotein reveals a complex antigen-specific pattern of immunoglobulin isotype switching during infection by Trypanosoma brucei. Infect Immun. (2000) 68:848–60. doi: 10.1128/IAI.68.2.848-860.2000
127. Bockstal V, Guirnalda P, Caljon G, Goenka R, Telfer JC, Frenkel D, et al. T. brucei infection reduces B lymphopoiesis in bone marrow and truncates compensatory splenic lymphopoiesis through transitional B-cell apoptosis. PLoS Pathog. (2011) 7:e1002089. doi: 10.1371/journal.ppat.1002089
128. Engstler M, Pfohl T, Herminghaus S, Boshart M, Wiegertjes G, Heddergott N, et al. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell (2007) 131:505–15. doi: 10.1016/j.cell.2007.08.046
129. La Greca F, Haynes C, Stijlemans B, De Trez C, Magez S. Antibody-mediated control of Trypanosoma vivax infection fails in the absence of tumor necrosis factor. Parasite Immunol. (2014) 36:271–6. doi: 10.1111/pim.12106
130. Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. (2008) 4:e1000078. doi: 10.1371/journal.ppat.1000078
131. Cnops J, Kauffmann F, De Trez C, Baltz T, Keirsse J, Radwanska M, et al. Maintenance of B cells during chronic murine Trypanosoma brucei gambiense infection. Parasite Immunol. (2016) 38:642–7. doi: 10.1111/pim.12344
132. Cnops J, De Trez C, Bulte D, Radwanska M, Ryffel B, Magez S. IFN-γ mediates early B-cell loss in experimental African trypanosomosis. Parasite Immunol. (2015) 37:479–84. doi: 10.1111/pim.12208
133. Frenkel D, Zhang F, Guirnalda P, Haynes C, Bockstal V, Radwanska M, et al. Trypanosoma brucei Co-opts NK cells to kill splenic B2 B cells. PLoS Pathog. (2016) 12:e1005733. doi: 10.1371/journal.ppat.1005733
134. Cnops J, Magez S, De Trez C. Escape mechanisms of African trypanosomes: why trypanosomosis is keeping us awake. Parasitology (2015) 142:417–27. doi: 10.1017/S0031182014001838
135. Tizard IR, Mittal KR, Nielsen K. Depressed immunoconglutinin responses in calves experimentally infected with Trypanosoma congolense. Res Vet Sci. (1980) 28:203–6.
136. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Cytokines and antibody responses during Trypanosoma congolense infections in two inbred mouse strains that differ in resistance. Parasite Immunol. (1999) 21:57–71. doi: 10.1046/j.1365-3024.1999.00202.x
137. Schopf LR, Filutowicz H, Bi XJ, Mansfield JM. Interleukin-4-dependent immunoglobulin G1 isotype switch in the presence of a polarized antigen-specific Th1-cell response to the trypanosome variant surface glycoprotein. Infect Immun. (1998) 66:451–61.
138. Lejon V, Mumba Ngoyi D, Kestens L, Boel L, Barbé B, Kande Betu V, et al. Gambiense human african trypanosomiasis and immunological memory: effect on phenotypic lymphocyte profiles and humoral immunity. PLoS Pathog. (2014) 10:e1003947. doi: 10.1371/journal.ppat.1003947
139. Holland WG, Do TT, Huong NT, Dung NT, Thanh NG, Vercruysse J, et al. The effect of Trypanosoma evansi infection on pig performance and vaccination against classical swine fever. Vet Parasitol. (2003) 111:115–23. doi: 10.1016/S0304-4017(02)00363-1
140. Holland WG, My LN, Dung TV, Thanh NG, Tam PT, Vercruysse J, et al. The influence of T. evansi infection on the immuno-responsiveness of experimentally infected water buffaloes. Vet Parasitol. (2001) 102:225–34. doi: 10.1016/S0304-4017(01)00534-9
141. Rurangirwa FR, Musoke AJ, Nantulya VM, Tabel H. Immune depression in bovine trypanosomiasis: effects of acute and chronic Trypanosoma congolense and chronic Trypanosoma vivax infections on antibody response to Brucella abortus vaccine. Parasite Immunol. (1983) 5:267–76. doi: 10.1111/j.1365-3024.1983.tb00743.x
142. Rurangirwa FR, Tabel H, Losos GJ, Tizard IR. Suppression of antibody response to Leptospira biflexa and Brucella abortus and recovery from immunosuppression after Berenil treatment. Infect Immun. (1979) 26:822–6.
143. Ilemobade AA, Adegboye DS, Onoviran O, Chima JC. Immunodepressive effects of trypanosomal infection in cattle immunized against contagious bovine pleuropneumonia. Parasite Immunol. (1982) 4:273–82. doi: 10.1111/j.1365-3024.1982.tb00438.x
144. Mwangi DM, Munyua WK, Nyaga PN. Immunosuppression in caprine trypanosomiasis: effects of acute Trypanosoma congolense infection on antibody response to anthrax spore vaccine. Trop Anim Health Prod. (1990) 22:95–100. doi: 10.1007/BF02239832
145. Sharpe RT, Langley AM, Mowat GN, Macaskill JA, Holmes PH. Immunosuppression in bovine trypanosomiasis: response of cattle infected with Trypanosoma congolense to foot-and-mouth disease vaccination and subsequent live virus challenge. Res Vet Sci. (1982) 32:289–293.
146. De Geea L, Levine RF, Mansfield JM. Genetics of resistance to the African trypanosomes. VI. Heredity of resistance and variable surface glycoprotein-specific immune responses. J Immunol. (1988) 140:283–8.
147. Dagenais TR, Demick KP, Bangs JD, Forest KT, Paulnock DM, Mansfield JM. T-cell responses to the trypanosome variant surface glycoprotein are not limited to hypervariable subregions. Infect Immun. (2009) 77:141–51. doi: 10.1128/IAI.00729-08
148. Dagenais TR, Freeman BE, Demick KP, Paulnock DM, Mansfield JM. Processing and presentation of variant surface glycoprotein molecules to T cells in African trypanosomiasis. J Immunol. (2009) 183:3344–55. doi: 10.4049/jimmunol.0802005
149. Schleifer KW, Mansfield JM. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J Immunol. (1993) 151:5492–503.
150. Hertz CJ, Filutowicz H, Mansfield JM. Resistance to the African trypanosomes is IFN-gamma dependent. J Immunol. (1998) 161:6775–83.
151. Hertz CJ, Mansfield JM. IFN-gamma-dependent nitric oxide production is not linked to resistance in experimental African trypanosomiasis. Cell Immunol. (1999) 192:24–32. doi: 10.1006/cimm.1998.1429
152. Magez S, Radwanska M, Drennan M, Fick L, Baral TN, Brombacher F, et al. Interferon-gamma and nitric oxide in combination with antibodies are key protective host immune factors during Trypanosoma congolense Tc13 infections. J Infect Dis. (2006) 193:1575–83. doi: 10.1086/503808
153. Uzonna JE, Kaushik RS, Gordon JR, Tabel H. Experimental murine Trypanosoma congolense infections. I. Administration of anti-IFN-gamma antibodies alters trypanosome-susceptible mice to a resistant-like phenotype. J Immunol. (1998) 161:5507–15.
154. Cnops J, De Trez C, Stijlemans B, Keirsse J, Kauffmann F, Barkhuizen M, et al. NK-, NKT- and CD8-Derived IFNγ drives myeloid cell activation and erythrophagocytosis, resulting in trypanosomosis-associated acute anemia. PLoS Pathog. (2015) 11:e1004964. doi: 10.1371/journal.ppat.1004964
155. Wu H, Liu G, Shi M. Interferon gamma in African Trypanosome infections: friends or foes? Front Immunol. (2017) 8:1105. doi: 10.3389/fimmu.2017.01105
156. Bakhiet M, Büscher P, Harris RA, Kristensson K, Wigzell H, Olsson T. Different trypanozoan species possess CD8 dependent lymphocyte triggering factor-like activity. Immunol Lett. (1996) 50:71–80. doi: 10.1016/0165-2478(96)02521-7
157. Bakhiet M, Hamadien M, Tjernlund A, Mousal A, Seiger A. African trypanosomes activate human fetal brain cells to proliferation and IFN-gamma production. Neuroreport (2002) 13:53–6. doi: 10.1097/00001756-200201210-00015
158. Masocha W, Robertson B, Rottenberg ME, Mhlanga J, Sorokin L, Kristensson K. Cerebral vessel laminins and IFN-gamma define Trypanosoma brucei brucei penetration of the blood-brain barrier. J Clin Invest. (2004) 114:689–94. doi: 10.1172/JCI22104
159. Liu G, Sun D, Wu H, Zhang M, Huan H, Xu J, et al. Distinct contributions of CD4+ and CD8+ T cells to pathogenesis of Trypanosoma brucei infection in the context of gamma interferon and interleukin-10. Infect Immun. (2015) 83:2785–95. doi: 10.1128/IAI.00357-15
160. Olsson T, Bakhiet M, Edlund C, Höjeberg B, Van der Meide PH, Kristensson K. Bidirectional activating signals between Trypanosoma brucei and CD8+ T cells: a trypanosome-released factor triggers interferon-gamma production that stimulates parasite growth. Eur J Immunol. (1991) 21:2447–54. doi: 10.1002/eji.1830211022
161. Sternberg JM, Mabbott NA. Nitric oxide-mediated suppression of T cell responses duringTrypanosoma brucei infection: soluble trypanosome products and interferon-γ are synergistic inducers of nitric oxide synthase. Eur J Immunol. (1996) 26:539–43. doi: 10.1002/eji.1830260306
162. Lopez R, Demick KP, Mansfield JM, Paulnock DM. Type I IFNs play a role in early resistance, but subsequent susceptibility, to the African trypanosomes. J Immunol. (2008) 181:4908–17. doi: 10.4049/jimmunol.181.7.4908
163. Magez S, Radwanska M, Beschin A, Sekikawa K, De Baetselier P. Tumor necrosis factor alpha is a key mediator in the regulation of experimental Trypanosoma brucei infections. Infect Immun. (1999) 67:3128–32.
164. Iraqi F, Sekikawa K, Rowlands J, Teale A. Susceptibility of tumor necrosis factor-alpha genetically deficient mice to Trypanosoma congolense infection. Parasite Immunol. (2001) 23:445–51. doi: 10.1046/j.1365-3024.2001.00401.x
165. Magez S, Lucas R, Darji A, Songa EB, Hamers R, De Baetselier P. Murine tumor necrosis factor plays a protective role during the initial phase of the experimental infection with Trypanosoma brucei brucei. Parasite Immunol. (1993) 15:635–41. doi: 10.1111/j.1365-3024.1993.tb00577.x
166. Magez S, Truyens C, Merimi M, Radwanska M, Stijlemans B, Brouckaert P, et al. P75 tumor necrosis factor-receptor shedding occurs as a protective host response during African trypanosomiasis. J Infect Dis. (2004) 189:527–39. doi: 10.1086/381151
167. Stijlemans B, De Baetselier P, Magez S, Van Ginderachter JA, De Trez C. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol. (2018) 9:218. doi: 10.3389/fimmu.2018.00218
168. Lucas R, Magez S, De Leys R, Fransen L, Scheerlinck JP, Rampelberg M, et al. Mapping the lectin-like activity of tumor necrosis factor. Science (1994) 263:814–7. doi: 10.1126/science.8303299
169. Magez S, Radwanska M, Stijlemans B, Xong H V, Pays E, De Baetselier P. A conserved flagellar pocket exposed high mannose moiety is used by African trypanosomes as a host cytokine binding molecule. J Biol Chem. (2001) 276:33458–64. doi: 10.1074/jbc.M103412200
170. Daulouède S, Bouteille B, Moynet D, De Baetselier P, Courtois P, Lemesre JL, et al. Human macrophage tumor necrosis factor (TNF)-alpha production induced by Trypanosoma brucei gambiense and the role of TNF-alpha in parasite control. J Infect Dis. (2001) 183:988–91. doi: 10.1086/319257
171. Paulnock DM, Freeman BE, Mansfield JM. Modulation of innate immunity by African trypanosomes. Parasitology (2010) 137:2051–63. doi: 10.1017/S0031182010001460
172. O'Gorman GM, Park SDE, Hill EW, Meade KG, Mitchell LC, Agaba M, et al. Cytokine mRNA profiling of peripheral blood mononuclear cells from trypanotolerant and trypanosusceptible cattle infected with Trypanosoma congolense. Physiol Genomics (2006) 28:53–61. doi: 10.1152/physiolgenomics.00100.2006
173. Magez S, Stijlemans B, Baral T, De Baetselier P. VSG-GPI anchors of African trypanosomes: their role in macrophage activation and induction of infection-associated immunopathology. Microbes Infect. (2002) 4:999–1006. doi: 10.1016/S1286-4579(02)01617-9
174. Leppert BJ, Mansfield JM, Paulnock DM. The soluble variant surface glycoprotein of African trypanosomes is recognized by a macrophage scavenger receptor and induces I kappa B alpha degradation independently of TRAF6-mediated TLR signaling. J Immunol. (2007) 179:548–56. doi: 10.4049/jimmunol.179.1.548
175. Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN- -induced nitric oxide production via reduction in STAT1 phosphorylation in African Trypanosomiasis. J Immunol. (2003) 171:1466–72. doi: 10.4049/jimmunol.171.3.1466
176. Stijlemans B, Baral TN, Guilliams M, Brys L, Korf J, Drennan M, et al. A glycosylphosphatidylinositol-based treatment alleviates trypanosomiasis-associated immunopathology. J Immunol. (2007) 179:4003–14. doi: 10.4049/jimmunol.179.6.4003
177. Stijlemans B, Vankrunkelsven A, Brys L, Raes G, Magez S, De Baetselier P. Scrutinizing the mechanisms underlying the induction of anemia of inflammation through GPI-mediated modulation of macrophage activation in a model of African trypanosomiasis. Microbes Infect. (2010) 12:389–99. doi: 10.1016/j.micinf.2010.02.006
178. Harris TH, Cooney NM, Mansfield JM, Paulnock DM. Signal transduction, gene transcription, and cytokine production triggered in macrophages by exposure to trypanosome DNA. Infect Immun. (2006) 74:4530–7. doi: 10.1128/IAI.01938-05
179. Barkhuizen M, Magez S, Ryffel B, Brombacher F. Interleukin-12p70 deficiency increases survival and diminishes pathology in Trypanosoma congolense infection. J Infect Dis. (2008) 198:1284–91. doi: 10.1086/592048
180. Krishnamoorthy P, Sengupta PP, Das S, Ligi M, Shome BR, Rahman H. Cytokine gene expression and pathology in mice experimentally infected with different isolates of Trypanosoma evansi. Exp Parasitol. (2016) 170:168–76. doi: 10.1016/j.exppara.2016.09.019
181. Flynn JN, Sileghem M. The role of the macrophage in induction of immunosuppression in Trypanosoma congolense-infected cattle. Immunology (1991) 74:310–6.
182. Maclean L, Odiit M, Macleod A, Morrison L, Sweeney L, Cooper A, et al. Spatially and genetically distinct African Trypanosome virulence variants defined by host interferon-gamma response. J Infect Dis. (2007) 196:1620–8. doi: 10.1086/522011
183. Kennedy PGE. Cytokines in central nervous system trypanosomiasis: cause, effect or both? Trans R Soc Trop Med Hyg. (2009) 103:213–4. doi: 10.1016/j.trstmh.2008.08.013
184. Murray M, Morrison WI, Whitelaw DD. Host susceptibility to African trypanosomiasis: trypanotolerance. Adv Parasitol. (1982) 21:1–68. doi: 10.1016/S0065-308X(08)60274-2
185. Naessens J. Bovine trypanotolerance: a natural ability to prevent severe anemia and haemophagocytic syndrome? Int J Parasitol. (2006) 36:521–8. doi: 10.1016/j.ijpara.2006.02.012
186. Rifkin MR, Landsberger FR. Trypanosome variant surface glycoprotein transfer to target membranes: a model for the pathogenesis of trypanosomiasis. Proc Natl Acad Sci USA. (1990) 87:801–5. doi: 10.1073/pnas.87.2.801
187. Guegan F, Plazolles N, Baltz T, Coustou V. Erythrophagocytosis of desialylated red blood cells is responsible for anemia during Trypanosoma vivax infection. Cell Microbiol. (2013) 15:1285–303. doi: 10.1111/cmi.12123
188. Suzuki T, Ueta YY, Inoue N, Xuan X, Saitoh H, Suzuki H. Beneficial effect of erythropoietin administration on murine infection with Trypanosoma congolense. Am J Trop Med Hyg. (2006) 74:1020–5. doi: 10.4269/ajtmh.2006.74.1020
189. Authié E, Boulangé A, Muteti D, Lalmanach G, Gauthier F, Musoke AJ. Immunisation of cattle with cysteine proteinases of Trypanosoma congolense: targetting the disease rather than the parasite. Int J Parasitol. (2001) 31:1429–33. doi: 10.1016/S0020-7519(01)00266-1
190. Stijlemans B, Brys L, Korf H, Bieniasz-Krzywiec P, Sparkes A, Vansintjan L, et al. MIF-mediated hemodilution promotes pathogenic anemia in experimental African trypanosomosis. PLOS Pathog. (2016) 12:e1005862. doi: 10.1371/journal.ppat.1005862
191. Thompson PD, Tipney H, Brass A, Noyes H, Kemp S, Naessens J, et al. Claudin 13, a member of the claudin family regulated in mouse stress induced erythropoiesis. PLoS ONE (2010) 5:e12667. doi: 10.1371/journal.pone.0012667
192. Blom-Potar MC, Chamond N, Cosson A, Jouvion G, Droin-Bergère S, Huerre M, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. II. Immunobiological dysfunctions. PLoS Negl Trop Dis. (2010) 4:e793. doi: 10.1371/journal.pntd.0000793
193. Chamond N, Cosson A, Blom-Potar MC, Jouvion G, D'Archivio S, Medina M, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. I. Parasitological, hematological and pathological parameters. PLoS Negl Trop Dis. (2010) 4:e792. doi: 10.1371/journal.pntd.0000792
194. Magez S, Stijlemans B, Caljon G, Eugster H-P, De Baetselier P. Control of experimental Trypanosoma brucei infections occurs independently of lymphotoxin-alpha induction. Infect Immun. (2002) 70:1342–51. doi: 10.1128/IAI.70.3.1342
195. Bosschaerts T, Guilliams M, Noel W, Hérin M, Burk RF, Hill KE, et al. Alternatively activated myeloid cells limit pathogenicity associated with African trypanosomiasis through the IL-10 inducible gene selenoprotein P. J Immunol. (2008) 180:6168–75. doi: 10.1186/1756-3305-4-74
196. Vankrunkelsven A, De Ceulaer K, Hsu D, Liu F-T, De Baetselier P, Stijlemans B. Lack of galectin-3 alleviates trypanosomiasis-associated anemia of inflammation. Immunobiology (2016) 215:833–41. doi: 10.1016/j.imbio.2010.05.028
197. Katunguka-Rwakishaya E, Murray M, Holmes PH. Pathophysiology of Trypanosoma congolense infection in two breeds of sheep, Scottish blackface and Finn dorset. Vet Parasitol. (1997) 68:215–25. doi: 10.1016/S0304-4017(96)01075-8
198. Katunguka-Rwakishaya E, Murray M, Holmes PH. Pathophysiology of ovine trypanosomiasis: ferrokinetics and erythrocyte survival studies. Res Vet Sci. (1992) 53:80–6. doi: 10.1016/0034-5288(92)90089-K
199. da Silva CB, Wolkmer P, Paim FC, Da Silva AS, Siqueira LC, de Souza CL, et al. Iron metabolism and its relationship to anemia and immune system in Trypanosoma evansi infected rats. Exp Parasitol. (2013) 133:357–64. doi: 10.1016/j.exppara.2012.12.010
200. Mackie JT, Stenner R, Gillett AK, Barbosa A, Ryan U, Irwin PJ. Trypanosomiasis in an Australian little red flying fox (Pteropus scapulatus). Aust Vet J. (2017) 95:259–61. doi: 10.1111/avj.12597
201. Doua F, Yapo FB. Human trypanosomiasis in the Ivory Coast: therapy and problems. Acta Trop. (1993) 54:163–8. doi: 10.1016/0001-706X(93)90090-X
202. Doua F, Boa FY, Schechter PJ, Miézan TW, Diai D, Sanon SR, et al. Treatment of human late stage gambiense trypanosomiasis with alpha-difluoromethylornithine (eflornithine): efficacy and tolerance in 14 cases in Côte d'Ivoire. Am J Trop Med Hyg. (1987) 37:525–33.
203. Burri C, Brun R. Eflornithine for the treatment of human African trypanosomiasis. Parasitol Res. (2003) 90(Supp 1):S49–52. doi: 10.1007/s00436-002-0766-5
204. Milord F, Pépin J, Loko L, Ethier L, Mpia B. Efficacy and toxicity of eflornithine for treatment of Trypanosoma brucei gambiense sleeping sickness. Lancet (1992) 340:652–5. doi: 10.1016/0140-6736(92)92180-N
205. Paul M, Stefaniak J, Smuszkiewicz P, Van Esbroeck M, Geysen D, Clerinx J. Outcome of acute East African trypanosomiasis in a polish traveler treated with pentamidine. BMC Infect Dis. (2014) 14:111. doi: 10.1186/1471-2334-14-111
206. Ngotho M, Kagira JM, Kariuki C, Maina N, Thuita JK, Mwangangi DM, et al. Influence of trypanocidal therapy on the hematology of vervet monkeys experimentally infected with Trypanosoma brucei rhodesiense. Acta Trop. (2011) 119:14–8. doi: 10.1016/j.actatropica.2011.02.013
207. Chisi JE, Misiri H, Zverev Y, Nkhoma A, Sternberg JM. Anaemia in human African trypanosomiasis caused by Trypanosoma brucei rhodesiense. East Afr Med J. (2004) 81:505–8. doi: 10.4314/eamj.v81i10.9232
208. MacLean L, Chisi JE, Odiit M, Gibson WC, Ferris V, Picozzi K, et al. Severity of human African trypanosomiasis in east Africa is associated with geographic location, parasite genotype, and host inflammatory cytokine response profile. Infect Immun. (2004) 72:7040–4. doi: 10.1128/IAI.72.12.7040-7044.2004
Keywords: trypanosomosis, immunology, pathology, anemia, transmission
Citation: Radwanska M, Vereecke N, Deleeuw V, Pinto J and Magez S (2018) Salivarian Trypanosomosis: A Review of Parasites Involved, Their Global Distribution and Their Interaction With the Innate and Adaptive Mammalian Host Immune System. Front. Immunol. 9:2253. doi: 10.3389/fimmu.2018.02253
Received: 31 May 2018; Accepted: 11 September 2018;
Published: 02 October 2018.
Edited by:
Xun Suo, China Agricultural University, ChinaReviewed by:
Debora Decote-Ricardo, Universidade Federal Rural do Rio de Janeiro, BrazilDolores Correa, Instituto Nacional de Pediatria, Mexico
Xin Zhao, Institute of Microbiology (CAS), China
Copyright © 2018 Radwanska, Vereecke, Deleeuw, Pinto and Magez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Magez, c3RlZmFuLm1hZ2V6QHZ1Yi5iZQ==