 Sebastian Peer1
Sebastian Peer1 Giuseppe Cappellano1,2
Giuseppe Cappellano1,2 Natascha Hermann-Kleiter1
Natascha Hermann-Kleiter1 Karin Albrecht-Schgoer1
Karin Albrecht-Schgoer1 Reinhard Hinterleitner1,3‡
Reinhard Hinterleitner1,3‡ Gottfried Baier1†
Gottfried Baier1† Thomas Gruber1*†
Thomas Gruber1*†- 1Division of Translational Cell Genetics, Department for Medical Genetics, Molecular and Clinical Pharmacology, Medical University of Innsbruck, Innsbruck, Austria
- 2Department of Dermatology, Venereology and Allergology, Medical University of Innsbruck, Innsbruck, Austria
- 3Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
Genome-wide association studies as well as lymphatic expression analyses have linked both Cbl-b and GM-CSF to human multiple sclerosis as well as other autoimmune diseases. Both Cbl-b and GM-CSF have been shown to play a prominent role in the development of murine encephalomyelitis; however, no functional connection between the two has yet been established. In this study, we show that Cblb knockout mice demonstrated significantly exacerbated severity of experimental autoimmune encephalomyelitis (EAE), augmented T cell infiltration into the central nervous system (CNS) and strongly increased production of GM-CSF in T cells in vitro and in vivo.GM-CSF neutralization demonstrated that the increased susceptibility of Cblb−/− mice to EAE was dependent on GM-CSF. Mechanistically, p50 binding to the GM-CSF promoter and the IL-3/GM-CSF enhancer element “CNSa” was strongly increased in nuclear extracts from Cbl-b-deficient T cells. This study suggests that Cbl-b limits autoimmunity by preventing the pathogenic effects of GM-CSF overproduction in T cells.
Introduction
The E3 ubiquitin ligase Cbl-b regulates T cell activation thresholds by mediating the requirement for CD28 costimulation (1, 2), thus playing an essential role in immunotolerance and limiting autoimmunity via CD28 (1, 3). Mechanistically, Cbl-b restricts activation of the T cell antigen receptor (TCR) by controlling key molecules involved in T cell stimulation. It has been proposed that Cbl-b inhibits activation of the p85 subunit of phosphoinositide 3-kinase (PI3K) (4, 5), protein kinase C theta (PKCθ), and phospholipase C-γ1 (PLC-γ1) (3, 6) and acts in concert with c-Cbl to promote antigen-induced downregulation of the TCR (7). In addition, Cbl-b represses transactivation of the transcription factor nuclear factor-κB (NF-κB) (8). Furthermore, Cbl-b has been shown to mediate the suppressive effects of TGF-β, leading to reduced T cell sensitivity toward TGF-β and to inhibition by regulatory T cells (9–15).
Cbl-b-deficient mice are prone to develop autoimmunity (1) and two (2, 16) out of three (13) studies showed such mice to have a high susceptibility to experimental autoimmune encephalomyelitis (EAE), the animal model for multiple sclerosis (MS). Along these lines, various genome-wide association studies showed that variants of the CBLB gene are associated with MS (17–19). Furthermore, Cbl-b in T cells was reported to be decreased in patients with MS, and low expression of mRNA was associated with increased risk of relapse (20). The underlying molecular functions of Cbl-b, however, have remained elusive.
In MS and EAE, the central nervous system (CNS) becomes inflamed, and the insulating myelin sheath of axons gets attacked by autoreactive immune cells (21–23). In EAE, the CNS inflammation and subsequent paralysis is induced by active immunization of mice with self-antigens such as myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG) or proteolipid protein (PLP) (24). Single epitopes of such proteins, such as MOG35–55 and PLP139–151 peptides, are also sufficient to induce disease (25–27). Onset of MS and EAE is believed to be dependent on autoreactive CD4+ T cells which migrate into the CNS to activate and recruit other immune cells (28, 29). Cytokines derived from these CNS antigen-specific CD4+ T cells and their corresponding transcription factors are of significant interest in autoimmune research.
Somewhat surprisingly, it has been shown that many proinflammatory cytokines such as IFNγ, IL-12, IL-17A, IL-17F, IL-21, or IL-22 are not required for EAE development (30–38). Instead, it has been shown in several studies that GM-CSF, mainly produced by T cells in an inflammatory setting (39), is an essential cytokine in the pathogenesis of autoimmune neuroinflammation such as EAE (28, 40–42). Its elevated expression in T cells is also implicated in MS (43, 44), and blocking GM-CSF is being tested as a potential treatment option (45).
GM-CSF is a monomeric glycoprotein (46), recognized by a heterodimeric receptor whose β-chain is shared with the IL-3 and IL-5 receptors (47, 48). Signalling at low GM-CSF concentrations occurs via the PI-3 kinase pathway, while higher concentrations further activate JAK2/STAT5 (49) and can lead to proliferation, protection from apoptosis, early commitment to myelopoiesis, differentiation/maturation of committed progenitors and multiple activation and motility functions in mature cells (50). Its ability to promote macrophage polarization and subsequent inflammatory mediator production (51, 52) is of particular interest in the EAE setting.
Along this line of argumentation, IL-3, a cytokine closely related to GM-CSF (47, 48, 53–55), has also been associated with autoimmune neuroinflammation. IL-3 was shown to exacerbate EAE (56) and to be upregulated in MS lesions (57), whereas another study describes it to be a marker of encephalitogenic T helper 1 (Th1) and Th17 cells but redundant for the severeness of EAE symptoms (58).
Because of the apparent importance of GM-CSF and IL-3 for CNS autoimmunity, their deregulation in MS-patients (44, 57, 59, 60), their functional relationship (47, 48, 55) and the correlation of Cbl-b with autoimmunity as well as the hyper-responsive Cblb−/− T cell phenotype (1–3, 17–19), we were interested to find out whether Cbl-b suppresses EAE through the regulation of GM-CSF and IL-3. We show that Cbl-b deficiency leads to massive upregulation of GM-CSF and IL-3 in CD4+ T cells, a potentially causative factor in increased neuroinflammation.
Results
Cbl-b-Deficient T Cells Produce Significantly More GM-CSF and IL-3 in vitro
Since Th17 cells were implicated in EAE pathogenesis and reported to be an important source of GM-CSF (41, 42, 61, 62), we isolated naïve CD4+ T cells from wt and Cblb−/− mice, differentiated them into the Th17 subset in vitro (Figure 1C) and determined their cytokine production. As a result, unskewed Th0 cells lacking Cbl-b produced significantly more GM-CSF and IL-3 than their wt counterparts both at protein and mRNA levels (Figures 1A,B,D,E). Interestingly, however, Th17 differentiation repressed rather than upregulated GM-CSF and IL-3, and this repression was also pronounced in Cbl-b-deficient Th17 cells (Figures 1A,B). Since the downregulation of GM-CSF and IL-3 is dependent on IL-6 [(63) and data not shown], this indicates that the IL-6/STAT3 pathway is not affected by the loss of Cbl-b.
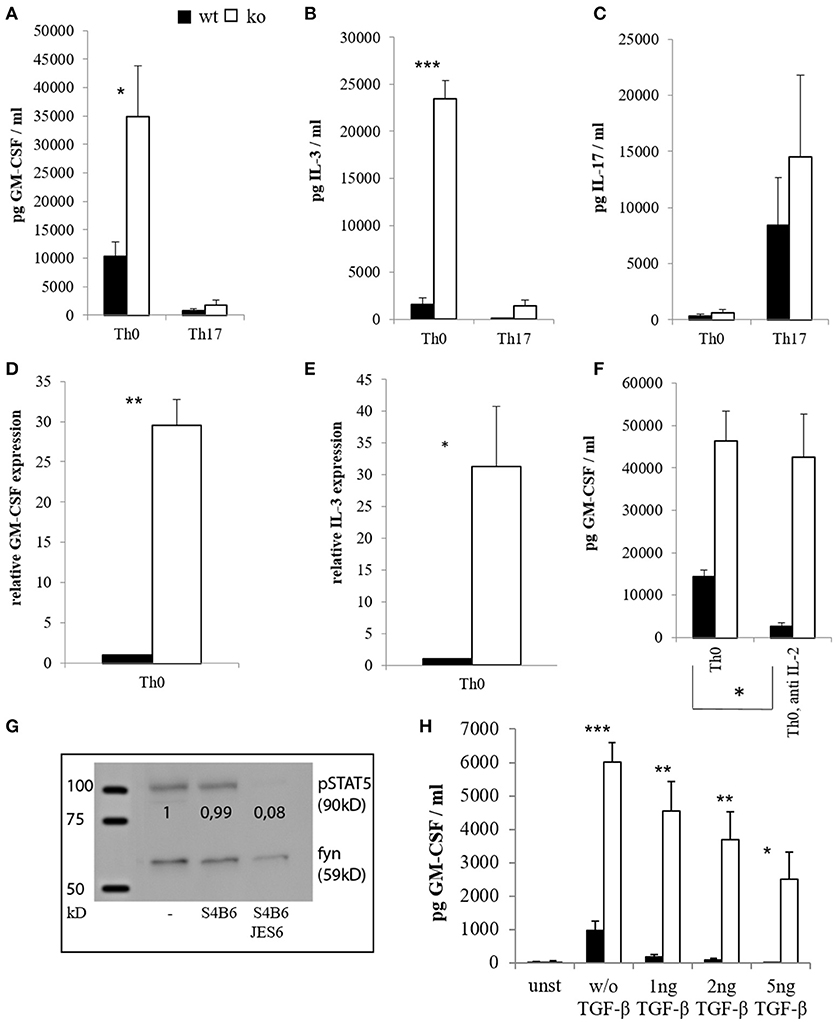
Figure 1. Cytokine expression of CD4+ cells in vitro. CD4+ CD62L+ cells of wt and Cblb−/− mice were kept in Iscove's Modified Dulbecco's Medium (IMDM) supplemented with 10% FCS, 2 mM l-glutamine and penicillin-streptomycin [50 U/ml] and stimulated with 1 μg/ml anti-CD28 and 3 μg/ml platebound anti-CD3, with or without Th17 differentiation conditions (5 ng/ml TGF-β, 20 ng/ml IL-6, 10 ng/ml IL-23, 2 μg/ml antiIFN-γ, 2 μg/ml antiIL-4). GM-CSF [(A), n = 4–8; 3–6 independent experiments] and IL-3 levels [(B), n = 4–8; 3–6 independent experiments] were measured on day 3 in the cell culture supernatants. To validate Th17 differentiation, IL-17 was measured as well [(C), n = 4; 4 independent experiments]. RNA was extracted on day 2, and qRT-PCR for GM-CSF [(D), n = 6; 5 independent experiments] and IL-3 [(E), n = 6; 5 independent experiments] was performed. In some experiments, IL-2 blocking antibodies JES6 (30 μg/ml) and S4B6 (40 μg/ml) were added in combination (= anti IL-2), and GM-CSF levels were measured in the supernatants on day 3 [(F), n = 4; 3 independent experiments]. To validate the antibody function, cells were lysed, submitted to western blot and pSTAT5 was detected [(G), 1 out of 2 experiments]. The loading control was considered in the quantification. (H) (n = 4; 2 independent experiments) shows GM-CSF amounts in supernatants on day 3 of unstimulated (= unst) or stimulated CD4+ cells treated with different amounts of TGF-β.
It has been shown that activation of STAT5 induces the expression of GM-CSF and IL-3 (63, 64). It is also known that Cbl-b-deficient T cells produce enhanced amounts of IL-2 (1, 2, 16). To exclude the possibility that increased GM-CSF expression by Cblb−/− T cells was just due to auto- and paracrine IL-2/STAT5 signaling, we neutralized IL-2. Blocking IL-2 with the combination of two different antibodies efficiently inhibited STAT5 activation, thus showing this approach to be valid (Figure 1G). As shown in Figure 1F, increased GM-CSF secretion by Cbl-b-deficient T cells was conserved during IL-2 blockade, indicating that this effect is not dependent on IL-2.
The immunosuppressive cytokine TGF-β is implicated in the suppression of EAE (65, 66), and Cblb−/− cells have been shown to be relatively resistant against TGF-β-mediated inhibition (9, 10, 15). Therefore, we wanted to find out whether this would also apply to GM-CSF expression. As shown in Figure 1H, TGF-β efficiently suppressed GM-CSF secretion of wt T cells in a dose dependent manner (99% inhibition at 5 ng/ml TGF-β), whereas Cblb−/− T cells were partially resistant to TGF-β inhibition (58% inhibition at 5 ng/ml TGF-β). Notably, even with the highest concentration of TGF-β used, Cbl-b-deficient T cells still produced more GM-CSF than wt T cells without addition of TGF-β (Figure 1H).
The proximal promoter of the GM-CSF gene, encompassing about 120 bp upstream of the transcription start site, contains a CD28 response region that consists of one SP1 and two NF-κB binding sites. The proximal NF-κB site is responsive to TCR signals and binds RelA/p50 heterodimers. Mutation of this site has been shown to reduce the activation of a GM-CSF promoter reporter by at least 50% (67, 68). A distal enhancer element “CNSa” in the IL-3/GM-CSF gene cluster (69) also shows potential NF-κB binding sites. Since our results showed deregulated GM-CSF and IL-3 expression in Cbl-b-deficient T cells, we were interested to find out whether this was reflected by changes in NF-κB binding to the GM-CSF promoter and CNSa. Therefore, we prepared nuclear extracts from anti-CD3/anti-CD28-stimulated wt and Cblb−/− CD4+ T cells and performed electrophoretic mobility shift assays (EMSA). As DNA oligonucleotides, we used the wt sequence of the proximal NF-κB binding site of the GM-CSF promoter (−110 to −125; Figure 2A) and the sequences of two predicted NF-κB binding sites within CNSa (Figures 2B,C) as well as mutated forms of the respective oligos to confirm the specificity of the interaction. As shown in Figure 2, binding of NF-κB p50 was well inducible by T cell stimulation (validated by a p50 super-shift) at all investigated sites. Notably, p50 binding to the NF-κB binding site was strongly enhanced in nuclear extracts from Cbl-b-deficient T cells at the GM-CSF promoter as well as the CNSa enhancer. Using the mutant oligonucleotide sequences, either labeled (not shown) or as cold competitors, validated the specificity of the NF-κB p50 binding site.
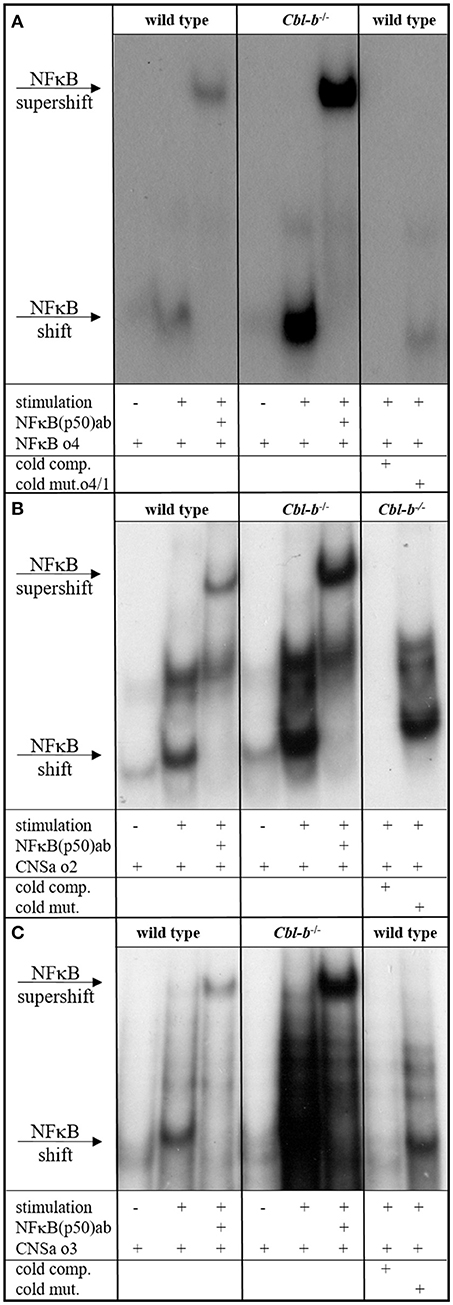
Figure 2. NFκB EMSA. EMSA of nuclear extracts from wt and Cblb−/− CD4+ T cells, unstimulated or stimulated overnight with 3 μg/ml platebound anti-CD3 and 1 μg/ml anti-CD28. As wt oligo, the NFκB consensus sequence within the minimal GM-CSF promoter (A) or predicted NFκB sites in the distal enhancer element of the IL-3/GM-CSF gene cluster “CNSa” (B,C) were used. To validate binding specificity, mutated oligos were used instead. Wt and mutated sequences were added in excess as unlabeled competition oligos (cold comp., cold mut.). Where indicated, an anti-p50 antibody was added. One representative experiment out of three (A) or two (B,C) is shown.
Cbl-b-Deficient Mice are Hyper-Susceptible to EAE Which is Correlated With Dysregulated GM-CSF Expression
In several models, Cbl-b has been shown to be crucial for tolerance induction and prevention of autoimmunity (1, 3, 70, 71). However, the studies on Cbl-b in EAE so far did not yield consistent results (2, 13, 16). Given the fact that Cbl-b is a threshold regulator in T cells, these divergent results could be due to different EAE protocols (1, 2, 72). To address this issue, we used an EAE protocol that leads to only mild signs of disease in wt mice. Applying this protocol, Cbl-b-deficient animals demonstrated significantly enhanced disease severity (Figure 3A), which was accompanied by significantly increased T cell infiltration into the CNS (Figure 3B). The frequency of regulatory T cells (Tregs) was increased as well but was not sufficient to impair EAE progression in Cblb−/− animals (Figure 3B, right) likely due to the increased resistance of Cblb−/− effector T cells to Treg suppression in vivo (9, 13).
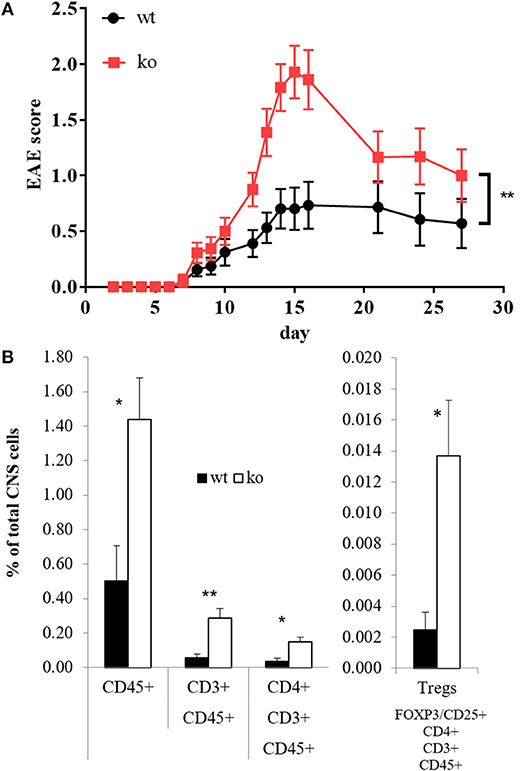
Figure 3. EAE score and CNS-infiltrating T cells. EAE was induced in wt and Cblb−/− mice and disease progression was monitored [(A), n = 16–18; 5 independent experiments]. At the peak of disease (day 14), FACS analysis of brain and spinal cord was performed [(B), n = 6; 2 independent experiments].
On the peak of disease, restimulation of mononuclear CNS cells with CD3 crosslinking led to strongly enhanced GM-CSF and IL-3 secretion in the absence of Cbl-b (Figures 4A,B). Consistent with this observation, restimulation of draining lymph node cells with the MOG peptide showed the same effect (Figures 4C,D). Importantly, cells isolated from non-MOG-challenged control mice did not express any of these cytokines upon stimulation (not shown).
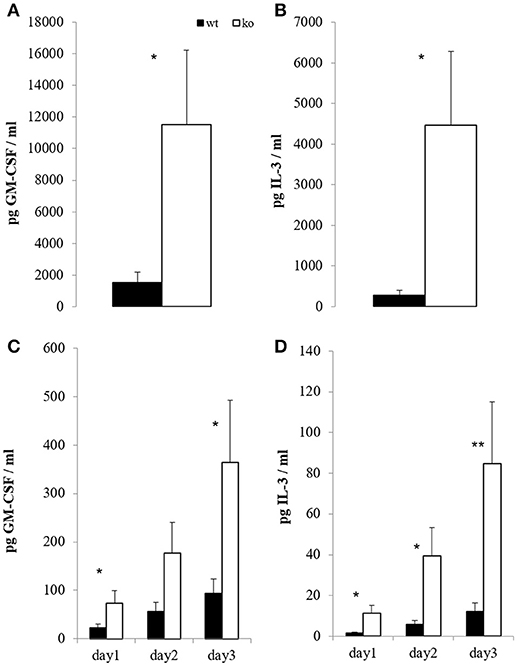
Figure 4. In vitro T cell recall. At the peak of EAE (day 14), mononuclear cells from brain and spinal cord were restimulated overnight with 5 μg/ml platebound anti-CD3; GM-CSF [(A), n = 6; 2 independent experiments] and IL-3 [(B), n = 6; 2 independent experiments] were measured in the supernatants. In parallel, draining inguinal lymph node cells were restimulated with 100 μg/ml MOG for the indicated time periods, and GM-CSF [(C), n = 8–10; 3 independent experiments] and IL-3 [(D), n = 8–10; 3 independent experiments] were measured in the supernatants.
To test the relevance of GM-CSF for the increased EAE disease severity of Cblb−/− mice we therapeutically administered GM-CSF blocking antibodies to Cblb−/− and wt mice during the course of an EAE experiment.
Strikingly, neutralization of GM-CSF led to an exorbitant amelioration of EAE symptoms in Cblb−/− mice down to wt levels (Figure 5).
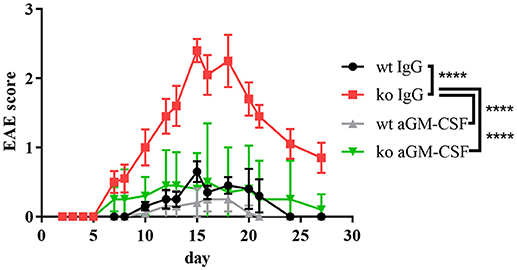
Figure 5. aGM-CSF therapy in EAE. EAE was induced in wt and Cblb−/− mice (n = 5 per group) and 400 μg aGM-CSF or IgG control antibody were injected i.p. on day 6, 9, 12, 15, 17, and 20. Disease progression was monitored.
Overall, our findings demonstrate that the increased susceptibility of Cblb−/− mice to a MOG-induced neuroinflammatory autoimmune reaction in vivo is mediated via GM-CSF and suggests that Cbl-b limits autoimmunity by preventing the pathogenic effects of GM-CSF overproduction in T cells.
Discussion
Initially, GM-CSF was not considered as a prominent cytokine in neuroinflammatory diseases such as EAE or MS. Instead, CNS autoimmunity was long thought to be mediated by Th1 and Th17 cells through the production of IFNγ and IL-17 until the non-redundant function of T cell-derived GM-CSF in EAE was demonstrated in a number of publications (28, 41, 42); this led to the further discovery that T cells in MS show elevated GM-CSF production (43, 44).
While there is agreement on T cells being the main source for GM-CSF, there is no consensus on the T cell subset that is primarily responsible for the increased production of this cytokine. There are many reports describing Th1 or Th17 cells as main GM-CSF producers (28, 42), some claim that there is a GM-CSF-producing T cell subset (63) while others do not link GM-CSF production to T cell polarization (41). In this study, stimulation of naïve CD4+ T cells with CD3 and CD28 crosslinking without any skewing conditions (Th0) yielded the highest levels of GM-CSF, while Th17 differentiation clearly repressed GM-CSF production (Figure 1A). This was mainly due to the action of IL-6 (not shown), which is in agreement with previous reports (42, 63). Thus, we show that GM-CSF is not a Th17-specific cytokine under in vitro differentiation conditions.
The E3 ubiquitin ligase Cbl-b is an established negative regulator of T cell activation, providing a “safety net” against autoimmunity. Here we provide strong experimental evidence that Cbl-b regulates lymphatic GM-CSF expression in vitro and in vivo during EAE pathogenesis (Figures 1, 4).
We and others have shown that Cbl-b indirectly controls TGF-β-mediated inhibition in T cells (9, 10, 15). The reduced effectiveness of TGF-β in Cblb−/− mice and the fact that TGF-β ameliorates EAE (65, 66, 73, 74) potentially explain why Cblb−/− mice develop such severe EAE (Figure 3A), probably secondary to increased GM-CSF expression (Figures 1, 4).
Interestingly and along this line of argumentation, GM-CSF expression itself is inhibited by TGF-β and this effect is considerably dampened in Cblb−/− CD4+ T cells (Figure 1H).
Additionally, as described previously, GM-CSF production is induced by NF-κB binding to the GM-CSF promoter (67, 68, 75). Furthermore, it has been shown that Cbl-b negatively regulates NF-κB (76) and that NF-κB binding to the IL-2 promoter is enhanced in Cbl-b-deficient mice (16). In keeping with these reports, we show that NF-κB binding to the GM-CFS promoter as well as to a distal enhancer element in the IL-3/GM-CSF gene cluster is enhanced in Cbl-b-deficient mice (Figure 2), providing further mechanistic insight into the observed robust upregulation of GM-CSF and IL-3 in Cbl-b-deficient T cells.
The discovery of IL-3 involvement in the development of EAE (56) is even more recent but not unexpected due to its close relationship to GM-CSF (54, 55). Similar to the effect on GM-CSF, the dysregulation of IL-3 in Cbl-b-deficient T cells is also drastic, an observation of potential interest, especially once more research on IL-3 in CNS autoimmunity has been conducted.
Since MS has a family recurrence rate of 20% (77), genetic factors are evolving to be of increasing interest in MS research. Cbl-b seems to be one such genetic factor, as it has emerged to be a potential MS risk gene (17–19). This fits well to our results, which show that Cbl-b-deficient mice develop drastically increased EAE symptoms and CNS infiltration by T cells (Figure 3). As the frequency of Tregs is also increased (Figure 3B) we hereby prove that the higher EAE susceptibility of Cblb−/− mice is not due to diminished Treg numbers. We showed not only that more T cells infiltrate the CNS, but also that they produce more GM-CSF and IL-3 upon restimulation with CD3 crosslinking (Figures 4A,B). Furthermore, restimulation of draining lymph node cells with MOG peptide resulted in significantly increased GM-CSF and IL-3 levels in supernatants from Cblb−/− cells (Figures 4C,D). As treatment with MOG would only trigger MOG-experienced cells, and CD3 crosslinking alone would only stimulate cytokine production in effector T cells, this shows that in the Cblb−/− mice more effector cells produce more GM-CSF and IL-3. This finding may prove to be a useful gauge: low lymphatic Cbl-b expression in MS patients could be an indication for higher levels of GM-CSF in MS lesions and better responses could be expected from antibody therapies. This speculation is strongly supported by our blocking experiments, which showed that neutralization of GM-CSF was particularly effective in Cbl-b-deficient mice (Figure 5). Research on such therapeutic antibodies has already yielded promising results: Blockade of GM-CSF receptor has been reported to ameliorate chronic as well as relapsing-remitting EAE (78), and a monoclonal antibody against human GM-CSF (MOR103) has already been tested in a phase Ib trial for MS (45).
Taken together, we identified the Cbl-b/GM-CSF signaling axis as a potentially causative actor in neuroinflammatory patho-mechanism.
Materials and Methods
Mice
Cbl-b knockout mice on a C57Bl/6 background have been described previously (1). Mice were maintained under SPF conditions. All animal experiments were performed in accordance with the Austrian “Tierversuchsgesetz” (BGBI. Nr.501/1989 i.d.g.F. and BMWF-66.011/0061-II/3b/2013) and were approved by the Bundesministerium für Wissenschaft und Forschung (bm:wf).
EAE Induction
Wt and Cblb−/− mice received s.c. injection of 300 μl emulsion containing 200 μg MOG35-55 (GenScript) in incomplete Freund's adjuvant (ThermoFisherScientific) supplemented with killed Mycobacterium tuberculosis (BD; 2 mg/ml). Pertussis toxin (Sigma; 500 μg in 0.1 ml PBS) was injected intraperitoneally on days 0 and 2. The mice were observed from day 0, and the EAE clinical scores were evaluated every day as follows: 0.5, partial tail limpness; 1, tail limpness; 1.5, reversible impaired righting reflex; 2, impaired righting reflex and hindlimb weakness; 2.5, paralysis of one hindlimb; 3, paralysis of both hindlimbs; 3.5, paralysis of both hindlimbs and one forelimb; 4, hind- and forelimb paralysis; 5, death (79).
Antibody Therapy
Four hundred microgram aGM-CSF (BioXcell BE-0259; clone MP1-22E9) or IgG2a (BioXcell BE0089, clone 2A3) in 400 μl PBS were injected intraperitoneally to all mice, as soon as the first mice showed EAE symptoms on day 6. Treatment was administered on day 6, 9, 12, 15, 17, and 20 after EAE induction.
Brain and Spinal Cord Homogenization
Mice were perfused with PBS 14 days after EAE induction, and brain and spinal cords were taken and cut into small pieces in 5 ml PBS supplemented with 2.5 mg/ml collagenase (Sigma), 1.8 mg/ml DNAse (Sigma), 2.5 mM MgCl and 2% fetal calf serum (FCS, S1810). After 45 min in a humid, 37° incubator, EDTA was added (final concentration 1 mM) and incubated for another 5 min. The pieces were transferred onto a 70 μm cell strainer and pushed through with the back of a 10 ml syringe. The suspension was further passed through a 40 μm cell strainer and then kept on ice for further analysis.
Isolation of CNS Leukocytes
Mononuclear cells were isolated by Percoll (Sigma) gradient centrifugation from homogenized combined brain and spinal cords as described previously (80).
Restimulation of Draining Lymph Node Cells
The draining inguinal lymph nodes were prepared 14 days after EAE induction, pressed through a metal sieve and restimulated in RPMI+++ (Roswell Park Memorial Institute 1640 medium [PAA; E15-039] supplemented with 10% FCS, 2 mM l-glutamine and penicillin-streptomycin [50 U/ml]) with 100 μg/ml MOG.
Isolation of T Cells
Spleen and lymph nodes (axillary, brachial, inguinal) were mashed through a metal sieve, depleted of erythrocytes using the mouse erythrocyte lysing kit (R&D, WL2000) and CD4+ T cells were isolated using the “CD4+ T Cell Isolation Kit, mouse” (Miltenyi Biotech 130-104-454) according to the manufacturer's protocol. For Th0 and Th17, cells were further subjected to positive CD62L selection (Miltenyi, Cat.no.130-093-227).
Flow Cytometry
Homogenized brain and spinal cord cells were incubated with FC-block (BD 553141) for 15 min at 4°, then stained for 20 min at 4°C with specific antibodies (CD8 PerCPCy5.5 [eBiosciences 45-0081-80], CD45 APC [eBiosciences 17-0451-81], CD4 V500 [BD 560782], CD3 PB [Biolegend 100214]) diluted 1:200 in PBS/2% FCS. Data acquisition was performed on a FACSVerse and data analysis was conducted using the FlowJo 10.0.8r1 software.
Western Blot
A 48-well plate was coated with PBS containing 3 μg/ml anti-CD3 (2C11, in-house made) and incubated for 3 h at 37°C. After washing with PBS, 1.5 × 106 CD4+ cells per well were cultured overnight in 0.5 ml RPMI+++ and costimulated with 1 μg/ml anti-CD28 (BD 553294) and antibodies against IL-2 (JES-6, 30 μg/ml; S4B6, 40 μg/ml).
Cells were collected, washed once in ice-cold PBS and lysed in 30 μl lysis buffer (5 mM NaP2P, 5 mM NaF, 1 mM Na3VO4, 5 mM EDTA, 150 mM NaCl, 50 mM Tris [pH 7.3], 1% NP-40, aprotinin and leupeptin [50 μg/ml each]) for 30 min on ice. After centrifugation (15,000 g, 4°C), protein lysates were subjected to Western blotting analysis with antibodies against pSTAT5 (cell signaling 9351; 1:1000) and fyn (Santa Cruz-16; 1:1,000). Densitometric analysis was performed using ImageJ.
Nuclear Extracts
5 × 106 CD4+ cells were harvested, washed once with cold PBS, (1 min, 9,300 rcf, 4°C) resuspended in 300 μl BufferA (10 mM Hepes pH 7.9; 10 mM KCl; 0.1 mM EDTA; 0.1 mM EGTA; 1 mM DTT; 0.5 mM PMSF) and kept on ice for 15 min. 20 μl 10%NP-40 (final concentration 0.63%) were added, the pellet vortexed (10 s) and washed two times with 300 μl BufferA (5 min, 2300 rcf, 4°C). The supernatant was discarded, 30 μl BufferC (20 mM Hepes pH 7.9; 0.4 M NaCl; 1 mM EDTA; 1 mM EGTA; 1 mM DTT; 1 mM PMSF) added and put on a shaker for 30 min at 4°C. After centrifugation (10 min, 13,000 rcf, 4°C) supernatants were taken and stored at −80°C. Protein concentrations were determined by Bradford assay.
Gel Mobility Shift Assay
Single-stranded oligonucleotides were synthesized by Eurofins MWG Operon and annealed.
Overnight resting and anti-CD3/anti-CD28 stimulated CD4+ T cells from wt or Cbl-b- deficient mice were lysed and nuclear extracts were prepared. The following oligonucleotides were used, and the core binding motifs are underlined:
NFκB o4:5′ - TCCACAACTCAGGTAGTTCCCCCGC-3′ (spanning −125 to −110 relative to the transcription start of the mouse GM-CSF gene)
mutated o4/1:5′ -TCCACAACTCTCTTAGTTCCCCCGC-3′
CNSa o2:5′ -GAGAAATACAGGGAATTCCCTACTCTGAGGATAATGG-3′ (predicted by TRANSFAC analysis of CNSa)
CNSa mut o2:5′ - GAGAAATACAGTTAATTCCATACTCTGAGGATGGTGG-3′
CNSa o3:5′ -TGGATCTTGATGGGAAATTAAGTGAAGT-3′ (predicted, published NFκB binding site within CNSa) (69)
CNSa mut o3:5′ - TGGATCTTGATGGGCGCTTAAGTGAAGT-3′.
Binding reactions and supershifts were performed for 30 min at 4–8°C using the Binding Buffer B-1 (Active Motif 37480) together with Stabilizing Solution D (Active Motif 37488) containing 4 μg mouse T cell nuclear extracts (Active Motif 36042) and 2 μg of anti NF-κB (p50) (Santa Cruz-1190) antibody.
Binding reactions with the 3 × 105 cpm labeled probe were performed for 20 min at 4°-8°C using Binding Buffer C-1 (Active Motif 37484) together with Stabilizing Solution D (Active Motif 37488). Samples were run on a 4% native polyacrylamide gel in 0.5 × TBE for 3 h at 250 V. For competition assays, 10-fold unlabeled oligonucleotides identical to the radioactive-labeled probes were added to the binding reaction.
qRT-PCR
5 × 105 CD4+ cells were cultured in a 96-flat bottom well for 2 days. RNA was extracted using the RNeasy Mini Kit (Qiagen, Cat.no.74106) according to the manufacturer's protocol. For cDNA synthesis, Omniscript RT Kit (Qiagen, Cat.no.205111), Oligo-dT 15 primer (Promega, Cat.no.C1101) and RNase inhibitor (Promega, Cat.no.N2111) were used according to the manufacturer's protocol.
Real-time PCR was performed using Bio&SELL 5x QPCR MixII (Rox) (BS76.520.5000) and TaqMan probes for the relevant genes on a 7500 FAST Real-Time PCR system. Results were normalized to GAPDH.
Cytokine Measurements
Cell culture supernatants were analyzed via Bio-Plex multianalyte technology (BioRad).
Statistics
Results are expressed as mean ± standard error of the mean (SEM). Groups were compared using the paired Student's t-test. For EAE, two-way Anova with Sidak's post hoc test was performed. Data analysis was performed using GraphPad Prism 7.00. Significant differences are indicated as *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, ****p ≤ 0.0001.
Author Contributions
TG and GB conceptualization and supervision. SP performed most of the experimental work and data analysis along with TG, GC, NH-K, KA-S, and RH. SP, TG, and GB writing. All coauthors have read and revised the manuscript.
Funding
This work was supported by grants from the Austrian Science Fund [MCBO PhD 641 program W1101, P 25440-B21, P31383-B30, 30324-B21 (GB), P 26892-B13 (TG)] as well as the Christian Doppler (CD) Society (GB; CD Laboratory I643 CARE).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Prof. Hermann Dietrich, Mag.med.vet. Anja Beierfuss and the caretakers of the animal facility of Innsbruck Medical University, and our colleagues in the Division of Translational Cell Genetics for valuable discussions.
References
1. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-Dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature (2000) 403:211–6. doi: 10.1038/35003228
2. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature (2000) 403:216–20. doi: 10.1038/35003235
3. Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity (2004) 21:167–77. doi: 10.1016/j.immuni.2004.07.013
4. Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol. (2001) 2:870–5. doi: 10.1038/ni0901-870
5. Fang D, Wang HY, Fang N, Altman Y, Elly C, Liu YC. Cbl-b, a RING-type E3 ubiquitin ligase, targets phosphatidylinositol 3-kinase for ubiquitination in T cells. J Biol Chem. (2001) 276:4872–8. doi: 10.1074/jbc.M008901200
6. Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. (2004) 5:255–65. doi: 10.1038/ni1047
7. Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. (2002) 3:1192–9. doi: 10.1038/ni855
8. Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. (2008) 28:2470–80. doi: 10.1128/MCB.01505-07
9. Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF-beta in Cbl-b−−/−− mice. J Immunol. (2004) 173:1059–65. doi: 10.4049/jimmunol.173.2.1059
10. Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J Immunol. (2006) 176:1316–20. doi: 10.4049/jimmunol.176.3.1316
11. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. (2007) 117:1029–36. doi: 10.1172/JCI29472
12. Loeser S, Loser K, Bijker MS, Rangachari M, Van Der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. (2007) 204:879–91. doi: 10.1084/jem.20061699
13. Adams CO, Housley WJ, Bhowmick S, Cone RE, Rajan TV, Forouhar F, et al. Cbl-b(−/−) T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-beta. J Immunol. (2010) 185:2051–8. doi: 10.4049/jimmunol.1001171
14. Lutz-Nicoladoni C, Wallner S, Stoitzner P, Pircher M, Gruber T, Wolf AM, et al. Reinforcement of cancer immunotherapy by adoptive transfer of cblb-deficient CD8(+) T cells combined with a DC vaccine. Immunol Cell Biol. (2011) 90:130–4. doi: 10.1038/icb.2011.11
15. Gruber T, Hinterleitner R, Hermann-Kleiter N, Meisel M, Kleiter I, Wang CM, et al. Cbl-b mediates TGFbeta sensitivity by downregulating inhibitory SMAD7 in primary T cells. J Mol Cell Biol. (2013) 5:358–68. doi: 10.1093/jmcb/mjt017
16. Gruber T, Hermann-Kleiter N, Hinterleitner R, Fresser F, Schneider R, Gastl G, et al. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Sci Signal. (2009) 2:ra30. doi: 10.1126/scisignal.2000046
17. Sanna S, Pitzalis M, Zoledziewska M, Zara I, Sidore C, Murru R, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet. (2010) 42:495–7. doi: 10.1038/ng.584
18. Corrado L, Bergamaschi L, Barizzone N, Fasano ME, Guerini FR, Salvetti M, et al. Association of the CBLB gene with multiple sclerosis: new evidence from a replication study in an Italian population. J Med Genet. (2011) 48:210–1. doi: 10.1136/jmg.2010.081380
19. Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J Immunol. (2014) 193:4439–47. doi: 10.4049/jimmunol.1303077
20. Zhou WB, Wang R, Deng YN, Ji XB, Huang GX, Xu YZ. Study of Cbl-b dynamics in peripheral blood lymphocytes isolated from patients with multiple sclerosis. Neurosci Lett. (2008) 440:336–9. doi: 10.1016/j.neulet.2008.05.089
21. Loma I, Heyman R. Multiple sclerosis: pathogenesis and treatment. Curr Neuropharmacol. (2011) 9:409–16. doi: 10.2174/157015911796557911
23. Kister I, Bacon TE, Chamot E, Salter AR, Cutter GR, Kalina JT, et al. Natural history of multiple sclerosis symptoms. Int J MS Care (2013) 15:146–58. doi: 10.7224/1537-2073.2012-053
24. Rangachari M, Kuchroo VK. Using EAE to better understand principles of immune function and autoimmune pathology. J Autoimmun. (2013) 45:31–9. doi: 10.1016/j.jaut.2013.06.008
25. Whitham RH, Bourdette DN, Hashim GA, Herndon RM, Ilg RC, Vandenbark AA, et al. Lymphocytes from SJL/J mice immunized with spinal cord respond selectively to a peptide of proteolipid protein and transfer relapsing demyelinating experimental autoimmune encephalomyelitis. J Immunol. (1991) 146:101–7.
26. Mcrae BL, Vanderlugt CL, Dal Canto MC, Miller SD. Functional evidence for epitope spreading in the relapsing pathology of experimental autoimmune encephalomyelitis. J Exp Med. (1995) 182:75–85. doi: 10.1084/jem.182.1.75
27. Mendel I, Kerlero De Rosbo N, Ben-Nun A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V beta expression of encephalitogenic T cells. Eur J Immunol. (1995) 25:1951–9. doi: 10.1002/eji.1830250723
28. Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. (2007) 178:39–48. doi: 10.4049/jimmunol.178.1.39
29. Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol. (2017) 133:223–44. doi: 10.1007/s00401-016-1631-4
30. Krakowski M, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. (1996) 26:1641–6. doi: 10.1002/eji.1830260735
31. Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. (2000) 164:2759–68. doi: 10.4049/jimmunol.164.5.2759
32. Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, et al. IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol. (2002) 169:7104–10. doi: 10.4049/jimmunol.169.12.7104
33. Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, et al. Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol. (2003) 170:2153–60. doi: 10.4049/jimmunol.170.4.2153
34. Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, et al. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol. (2007) 179:8098–104. doi: 10.4049/jimmunol.179.12.8098
35. Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI. Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J Immunol. (2008) 180:7097–101. doi: 10.4049/jimmunol.180.11.7097
36. Sonderegger I, Kisielow J, Meier R, King C, Kopf M. IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol. (2008) 38:1833–8. doi: 10.1002/eji.200838511
37. Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. (2008) 205:1063–75. doi: 10.1084/jem.20071978
38. Haak S, Croxford AL, Kreymborg K, Heppner FL, Pouly S, Becher B, et al. IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice. J Clin Invest. (2009) 119:61–9.
39. Becher B, Tugues S, Greter M. GM-CSF: From growth factor to central mediator of tissue inflammation. Immunity (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
40. Mcqualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. (2001) 194:873–82. doi: 10.1084/jem.194.7.873
41. Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. (2011) 12:560–7. doi: 10.1038/ni.2027
42. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. (2011) 12:568–75. doi: 10.1038/ni.2031
43. Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. (2014) 5:5056. doi: 10.1038/ncomms6056
44. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in multiple sclerosis and suppressed by IFN-beta therapy. J Immunol. (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
45. Constantinescu CS, Asher A, Fryze W, Kozubski W, Wagner F, Aram J, et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. (2015) 2:e117. doi: 10.1212/NXI.0000000000000117
46. Souza LM, Boone TC, Lai PH, Zsebo KM, Murdock DC, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science (1986) 232, 61–65. doi: 10.1126/science.2420009
47. Kitamura T, Sato N, Arai K, Miyajima A. Expression cloning of the human IL-3 receptor cDNA reveals a shared beta subunit for the human IL-3 and GM-CSF receptors. Cell (1991) 66:1165–74. doi: 10.1016/0092-8674(91)90039-2
48. Tavernier J, Devos R, Cornelis S, Tuypens T, Vanderheyden J, Fiers W, et al. A human high-affinity Interleukin-5 receptor (Il5r) is composed of an Il5-specific alpha-chain and a beta-chain shared with the receptor for Gm-Csf. Cell (1991) 66:1175–84. doi: 10.1016/0092-8674(91)90040-6
49. Shiomi A, Usui T. Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediators Inflamm. (2015) 2015:568543. doi: 10.1155/2015/568543
50. Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, et al. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood (2009) 114:1289–98. doi: 10.1182/blood-2008-12-164004
51. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. (2008) 8:533–44. doi: 10.1038/nri2356
52. Van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, Van Den Berg W, Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol. (2013) 56:675–82. doi: 10.1016/j.molimm.2013.05.002
53. Cockerill PN. Mechanisms of transcriptional regulation of the human IL-3/GM-CSF locus by inducible tissue-specific promoters and enhancers. Crit Rev Immunol. (2004) 24:385–408. doi: 10.1615/CritRevImmunol.v24.i6.10
54. Mirabella F, Baxter EW, Boissinot M, James SR, Cockerill PN. The human IL-3/granulocyte-macrophage colony-stimulating factor locus is epigenetically silent in immature thymocytes and is progressively activated during T cell development. J Immunol. (2010) 184:3043–54. doi: 10.4049/jimmunol.0901364
55. Broughton SE, Dhagat U, Hercus TR, Nero TL, Grimbaldeston MA, Bonder CS, et al. The GM-CSF/IL-3/IL-5 cytokine receptor family: from ligand recognition to initiation of signaling. Immunol Rev. (2012) 250:277–302. doi: 10.1111/j.1600-065X.2012.01164.x
56. Renner K, Hellerbrand S, Hermann F, Riedhammer C, Talke Y, Schiechl G, et al. IL-3 promotes the development of experimental autoimmune encephalitis. JCI Insight (2016) 1:e87157. doi: 10.1172/jci.insight.87157
57. Baranzini SE, Elfstrom C, Chang SY, Butunoi C, Murray R, Higuchi R, et al. Transcriptional analysis of multiple sclerosis brain lesions reveals a complex pattern of cytokine expression. J Immunol. (2000) 165:6576–82. doi: 10.4049/jimmunol.165.11.6576
58. Lee PW, Xin MK, Pei W, Yang YH, Lovett-Racke AE. IL-3 is a marker of encephalitogenic T cells, but not essential for CNS autoimmunity. Front Immunol. (2018) 9:1255. doi: 10.3389/fimmu.2018.01255
59. Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga, F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. (1998) 20:373–82. doi: 10.3109/08923979809034820
60. Imitola J, Rasouli J, Watanabe F, Mahajan K, Sharan AD, Ciric B, et al. Elevated expression of granulocyte-macrophage colony-stimulating factor receptor in multiple sclerosis lesions. J Neuroimmunol. (2017) 317:45–54. doi: 10.1016/j.jneuroim.2017.12.017
61. Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature (2003) 421:744–8. doi: 10.1038/nature01355
62. Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. (2005) 201:233–40. doi: 10.1084/jem.20041257
63. Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. (2014) 24:1387–402. doi: 10.1038/cr.2014.154
64. Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci. Transl. Med. (2014) 6:241ra280. doi: 10.1126/scitranslmed.3008706
65. Cautain B, Damoiseaux J, Bernard I, Van Straaten H, Van Breda Vriesman P, Boneu B, et al. Essential role of TGF-beta in the natural resistance to experimental allergic encephalomyelitis in rats. Eur J Immunol. (2001) 31:1132–40. doi: 10.1002/1521-4141(200104)31:4<1132::AID-IMMU1132>3.0.CO;2-N
66. Gonzalez-Garcia I, Zhao Y, Ju S, Gu Q, Liu L, Kolls JK, et al. IL-17 signaling-independent central nervous system autoimmunity is negatively regulated by TGF-beta. J Immunol. (2009) 182:2665–71. doi: 10.4049/jimmunol.0802221
67. Jenkins F, Cockerill PN, Bohmann D, Shannon MF. Multiple signals are required for function of the human granulocyte-macrophage colony-stimulating factor gene promoter in T cells. J Immunol. (1995) 155:1240–51.
68. Thomas RS, Tymms MJ, Mckinlay LH, Shannon MF, Seth A, Kola I. ETS1, NF-κB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene (1997) 14:2845–55. doi: 10.1038/sj.onc.1201125
69. Wurster AL, Precht P, Pazin MJ. NF-kappa B and BRG1 bind a distal regulatory element in the IL-3/GM-CSF locus. Mol Immunol. (2011) 48:2178–88. doi: 10.1016/j.molimm.2011.07.016
70. Gronski MA, Boulter JM, Moskophidis D, Nguyen LT, Holmberg K, Elford AR, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. (2004) 10:1234–9. doi: 10.1038/nm1114
71. Gomez-Martin D, Ibarra-Sanchez M, Romo-Tena J, Cruz-Ruiz J, Esparza-Lopez J, Galindo-Campos M, et al. Casitas B lineage lymphoma b is a key regulator of peripheral tolerance in systemic lupus erythematosus. Arthritis Rheum. (2013) 65:1032–42. doi: 10.1002/art.37833
72. Rudd CE, Schneider H. Lymphocyte signaling: Cbl sets the threshold for autoimmunity. Curr Biol. (2000) 10:R344–347. doi: 10.1016/S0960-9822(00)00463-2
73. Johns LD, Flanders KC, Ranges GE, Sriram S. Successful treatment of experimental allergic encephalomyelitis with transforming growth factor-beta 1. J Immunol. (1991) 147:1792–6.
74. Johns LD, Sriram S. Experimental allergic encephalomyelitis: neutralizing antibody to TGF beta 1 enhances the clinical severity of the disease. J Neuroimmunol. (1993) 47:1–7. doi: 10.1016/0165-5728(93)90278-7
75. Schreck R, Baeuerle PA. NF-kappa B as inducible transcriptional activator of the granulocyte-macrophage colony-stimulating factor gene. Mol Cell Biol. (1990) 10:1281–6. doi: 10.1128/MCB.10.3.1281
76. Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol. (2008) 28:2470–80.
77. Compston A, Coles A. Multiple sclerosis. Lancet (2008) 372:1502–17. doi: 10.1016/S0140-6736(08)61620-7
78. Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J Autoimmun. (2017) 84:1–11. doi: 10.1016/j.jaut.2017.06.005
79. Inoue M, Chen PH, Siecinski S, Li QJ, Liu C, Steinman L, et al. An interferon-beta-resistant and NLRP3 inflammasome-independent subtype of EAE with neuronal damage. Nat Neurosci. (2016) 19:1599–609. doi: 10.1038/nn.4421
Keywords: adaptive immunity, multiple sclerosis, experimental autoimmune encephalomyelitis, Cbl-b, GM-CSF
Citation: Peer S, Cappellano G, Hermann-Kleiter N, Albrecht-Schgoer K, Hinterleitner R, Baier G and Gruber T (2018) Regulation of Lymphatic GM-CSF Expression by the E3 Ubiquitin Ligase Cbl-b. Front. Immunol. 9:2311. doi: 10.3389/fimmu.2018.02311
Received: 13 April 2018; Accepted: 17 September 2018;
Published: 08 October 2018.
Edited by:
Yun-Cai Liu, Tsinghua University, ChinaReviewed by:
Margot Thome, Université de Lausanne, SwitzerlandDominik Wolf, Universitätsklinikum Bonn, Germany
Copyright © 2018 Peer, Cappellano, Hermann-Kleiter, Albrecht-Schgoer, Hinterleitner, Baier and Gruber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Gruber, dGhvbWFzLmdydWJlckBpLW1lZC5hYy5hdA==
† These authors have contributed equally to this work
‡ Present Address: Reinhard Hinterleitner, Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States