 Amarshi Mukherjee1†
Amarshi Mukherjee1† Vanessa Jantsch2†Rida Khan1Wolfgang Hartung2René Fischer3
Vanessa Jantsch2†Rida Khan1Wolfgang Hartung2René Fischer3 Jonathan Jantsch4Boris Ehrenstein2*
Jonathan Jantsch4Boris Ehrenstein2* Maximilian F. Konig5*
Maximilian F. Konig5* Felipe Andrade1*
Felipe Andrade1*- 1Division of Rheumatology, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 2Klinik und Poliklinik für Rheumatologie, Klinische Immunologie, Asklepios Klinikum Bad Abbach, Bad Abbach, Germany
- 3Department of Otorhinolaryngology, University Hospital Regensburg, Regensburg, Germany
- 4Institute of Clinical Microbiology and Hygiene, University Hospital Regensburg and University of Regensburg, Regensburg, Germany
- 5Department of Medicine, Massachusetts General Hospital, Boston, MA, United States
Background: Aggregatibacter actinomycetemcomitans (Aa) is a Gram-negative coccobacillus recognized as a pathogen in periodontitis and infective endocarditis. By producing a toxin (leukotoxin A, LtxA) that triggers global hypercitrullination in neutrophils, Aa has been recently linked to rheumatoid arthritis (RA) pathogenesis. Although mechanistic and clinical association studies implicate Aa infection in the initiation of autoimmunity in RA, direct evidence in humans is lacking.
Case:We describe a 59-year-old man with anti-citrullinated protein antibody (ACPA)-positive RA who presented for evaluation of refractory disease. He was found to have Aa endocarditis. Following antibiotic treatment, joint symptoms resolved and ACPAs normalized. Given the implications for RA immunopathogenesis, we further investigated the bacterial, genetic and immune factors that may have contributed to the patient's clinical and autoimmune phenotypes.
Methods:DNA was extracted from serum and used to amplify the Aa leukotoxin (ltx) promoter region by PCR, which was further analyzed by Sanger sequencing. High-resolution identification of HLA alleles was performed by sequenced based typing (SBT). TNF-α, IFN-γ, GM-CSF, IL-1β, IL-6, IL-8, IL-17A, IL-18, IL-21, and IL-22 were quantified in serum by a multiplex immunoassay. IgG and IgA antibodies to Aa LtxA were assayed by ELISA.
Results:Aa genotyping confirmed infection with a highly leukotoxic strain carrying a 530-bp ltx promoter deletion, shown to result in 10- to 20-fold higher bacterial expression of LtxA. Immuno-phenotyping showed high anti-LtxA antibodies, elevated cytokines implicated in RA pathogenesis (Th1/Th17), and specific host susceptibility conferred by three HLA alleles strongly linked to ACPAs and RA (DRB1*04:04, DRB1*15:01, and DPB1*04:01). One year after eradication of Aa, the patient remained free of arthritis and anti-CCP antibodies.
Conclusion: In the context of genetic risk for RA, systemic subacute infection with a leukotoxic strain of Aa can drive ACPA production and a clinical phenotype similar to RA.
Introduction
Aggregatibacter actinomycetemcomitans (Aa) is a Gram-negative coccobacillus first described in 1912 as a co-isolate from actinomycosis lesions (“Bacterium actinomycetem comitans”) (1). Aa has since been recognized as a pathogen in periodontitis and, as part of the HACEK group, in rare cases of infective endocarditis (IE) (2–4). Recently, Aa has been proposed as a link between periodontitis and autoimmunity in rheumatoid arthritis (RA) due to its ability to induce citrullinated autoantigens targeted by anti-citrullinated protein antibodies (ACPAs) (5).
Leukotoxin A (LtxA) is an acylated protein toxin secreted by Aa and a major virulence factor in periodontitis (4). By acting as a pore-forming toxin, LtxA induces membranolysis and cell death in host immune cells, thus permitting escape from immune surveillance (4). This pathway has been shown to drive hypercitrullination of RA autoantigens in human neutrophils, thus linking Aa leukotoxicity to RA immunopathogenesis (5). Leukotoxic strains of Aa (as measured by antibodies to LtxA) are highly prevalent in RA. Exposure to Aa is strongly associated with ACPAs and rheumatoid factor (RF) in individuals carrying HLA-DRB1 shared epitope (SE) alleles, which confer genetic susceptibility to RA. Together, these findings have implicated Aa as a candidate trigger of autoimmunity in individuals at risk for RA (5). However, experimental evidence to demonstrate a causative effect is missing. Here, we report a case of early RA associated with Aa endocarditis and its resolution with antibiotic therapy. We believe that this case provides direct evidence that in the setting of genetic susceptibility, Aa is an etiologic agent that can induce ACPA production and arthritis in humans.
Case Report
A 59-year-old Caucasian man with a history of severe mitral regurgitation and recent diagnosis of seropositive RA was admitted to the hospital for evaluation of refractory joint pain and swelling. Four years prior to admission, the patient had undergone prosthetic mitral valve replacement. Since then, he had received deep dental cleanings twice a year. The patient was in his usual health until 11 months prior to admission, when he developed intermittent pain and swelling of his knees, right hip, right elbow, and wrists bilaterally that was associated with morning stiffness of >1 h. He endorsed 11 lbs. weight loss and night sweats, but no fevers. Following 6 months of persistent symptoms, the patient saw a local rheumatologist who noted synovitis of the 2nd left metacarpophalangeal joint and tenosynovitis of the extensor tendons of his left hand. Laboratory workup showed evidence of systemic inflammation [C-reactive protein (CRP) 100 mg/L, erythrocyte sedimentation rate (ESR) 84 mm/h] and positive ACPAs (measured by the anti-CCP antibody assay). Testing for RF was negative. The patient was diagnosed with early seropositive RA, and he was started on immunosuppression with prednisolone and methotrexate. Given lack of clinical improvement, leflunomide was added. Due to persistent joint pain and swelling, the patient was hospitalized 2 months later for evaluation.
At the time of hospital admission, laboratory evaluation showed CRP 112 mg/L, ESR 79 mm/h, and high-titer anti-CCP IgG antibodies (262 U/mL; reference range <17 U/mL). Musculoskeletal ultrasound (US) showed effusion of the 2nd and 3rd right proximal interphalangeal joints as well as 1st and 4th right metatarsophalangeal joints. There was evidence of tenosynovitis of the right wrist extensor tendons, and inflammation of the flexor tendons of the right ankle and right Achilles tendon. Radiographs of the hands and feet showed no erosions. Prednisolone was increased. The patient was started on etanercept, and leflunomide was discontinued. Following a brief period of improvement, the pain around the right Achilles tendon and right wrist flexor tendons worsened within 3 weeks. US revealed new abscess formation along the right Achilles tendon. Incision and drainage was performed with wound cultures demonstrating Aa by PCR and sequence analysis. Blood cultures grew Aa in 2/3 sets of bottles, and echocardiography confirmed prosthetic mitral valve endocarditis. All immunosuppressive medications were discontinued, and antibiotic therapy with ceftriaxone 2 g IV daily was started. CRP levels decreased, and the joint pain improved. After completing a 6 week course of intravenous antibiotics, the patient's joint pain and swelling had resolved. Thereafter, anti-CCP antibody levels started to decline (Figure 1A). A non-ulcerated squamous cell carcinoma of the tongue was subsequently diagnosed and treated with radiotherapy. At ~1 year follow-up, the patient remained free of joint symptoms, and anti-CCP antibodies and CRP levels had normalized (anti-CCP 13.5 U/mL; CRP 5 mg/L) (Figure 1A).
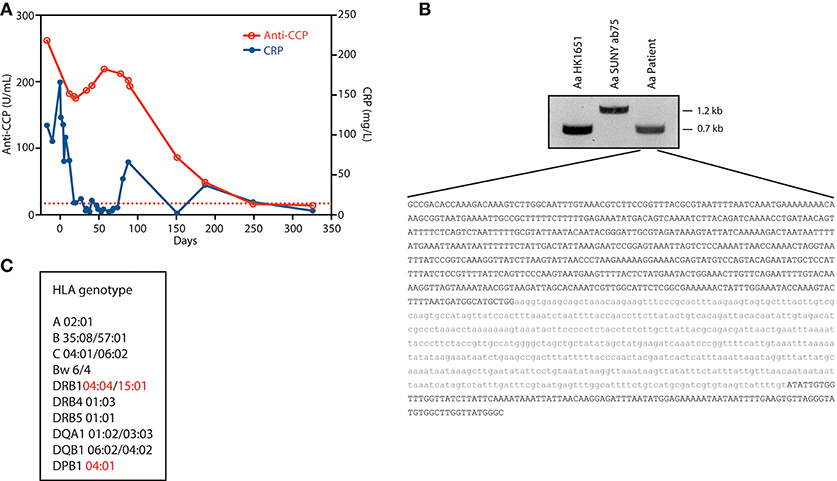
Figure 1. Autoimmune, genetic, and microbial factors associated with Aa-induced early RA. (A) Serial measurements of C-reactive protein (CRP) and anti-cyclic citrullinated peptide (CCP) antibodies. Day 0 marks the diagnosis of Aa endocarditis. Anti-CCP was determined in serum using an automated electrochemiluminescence immunoassay platform (Elecsys, Roche). The dotted line marks the cut-off for anti-CCP antibody positivity (17 U/mL). (B) DNA extracted from patient serum was used to amplify the Aa ltx promoter region by PCR. PCR products of strains Aa HK1651 and Aa SUNY ab75 were used as a reference for JP2 and non-JP2 clones, respectively. The PCR product amplified from patient serum was analyzed by Sanger sequencing. The resulting nucleotide sequence is shown in black. The missing nucleotide sequence corresponding to the 530-bp deletion (Δ530) previously described in Aa JP2 clones is shown in gray. (C) HLA genotype. HLA risk alleles linked to ACPA production and RA are marked in red.
Materials and Methods
Aa Ltx Promoter Genotyping
DNA extracted from patient serum (QIAamp DNA kit, Qiagen), and DNA from strains Aa HK1651 (serotype b, ATCC 700685) and Aa SUNY ab75 (serotype a, ATCC 43717) were used to amplify the Aa ltx promoter region by PCR using primers ltx3/ltx4 (ltx3: 5′-GCCGACACCAAAGACAAAGTCT-3′ and ltx4: 5′-GCCCATAACCAAGCCACATAC-3′) as previously described (6). Aa strains HK1651 and SUNY ab75 were used as a reference for JP2 and non-JP2 clones, respectively. The PCR product amplified from patient serum was analyzed by Sanger sequencing in both 5′ and 3′ directions using the ltx3 and ltx4 primers, respectively.
HLA Typing
Sequenced based typing (SBT) was used for high-resolution identification of alleles of HLA-A, -B, -C, - DRB1, -DQB1, and -DPB1. SBT uses PCR to amplify the locus of interest and Sanger sequencing to determine the nucleotide sequence. HLA-typing was performed at the Johns Hopkins University Immunogenetics Laboratory.
Quantification of Cytokines and Anti-LtxA Antibodies in Serum
Serial measurements of TNF-α, IFN-γ, GM-CSF, IL-1β, IL-6, IL-8, IL-17A, IL-18, IL-21, and IL-22 were quantified in patient serum by multiplex immunoassay (Meso Scale Diagnostics). IgG and IgA antibodies to LtxA were assayed in serial serum samples by ELISA as previously described (5) using a recombinant immunodominant peptide of LtxA (amino acids 730-1055) as antigen (5).
Results
Bacterial Virulence of Aa
The virulence factor LtxA generates citrullinated autoantigens in human neutrophils (5). Expression of LtxA varies considerably among Aa strains and is regulated by both environmental factors and genetic variation within the ltx promoter (7, 8). To define potential variations in the ltx promoter region associated with highly virulent strains, bacterial DNA from patient serum was amplified by PCR (Figure 1B). The amplified ltx promoter contained a 530-bp deletion (Δ530), which has been shown to result in 10- to 20-fold higher expression of LtxA compared to wild-type Aa strains due to deletion of a transcriptional terminator (6, 8, 9). These data indicate that the patient was infected with a highly leukotoxic strain of Aa.
Genetic Susceptibility
Since the presence of autoantibodies in RA patients carrying HLA-DRB1 susceptibility alleles is strongly linked to Aa exposure (5), HLA-typing was performed at high resolution (Figure 1C). The patient's haplotypes revealed at least 3 HLA alleles strongly associated with seropositive RA: two heterozygous DRB1 risk alleles (*04:04 and *15:01) and one homozygous DPB1 risk allele (*04:01) (10, 11). DRB1*04:04 confers strong disease susceptibility by the presence of residues Val, Arg and Ala at positions 11, 71, and 74, respectively. In DRB1*15:01 and DPB1*04:01, risk is defined by amino acid positions 74 (Ala) and 9 (Phe), respectively. These residues are located within the antigen-binding groove of the HLA class II molecule (10), and may facilitate presentation of citrullinated self-peptides (12).
Inflammatory Milieu
To define the inflammatory milieu that may have contributed to loss of immune tolerance and arthritis, a panel of cytokines was quantified in samples collected longitudinally after diagnosis of Aa infection (Figures 2A–J). The cytokine profile induced by Aa was characterized by expression of several cytokines implicated in autoimmunity and RA (13). These included TNF-α, IFN-γ, IL-1β, IL-17A, and IL-18 (Figures 2A–E). Levels decreased progressively within weeks to months after initiation of antibiotic treatment, which coincided with clinical improvement. Other cytokines did not show similar dynamics, including GM-CSF, IL-6, IL-8, IL-21, and IL-22 (Figures 2F–J).
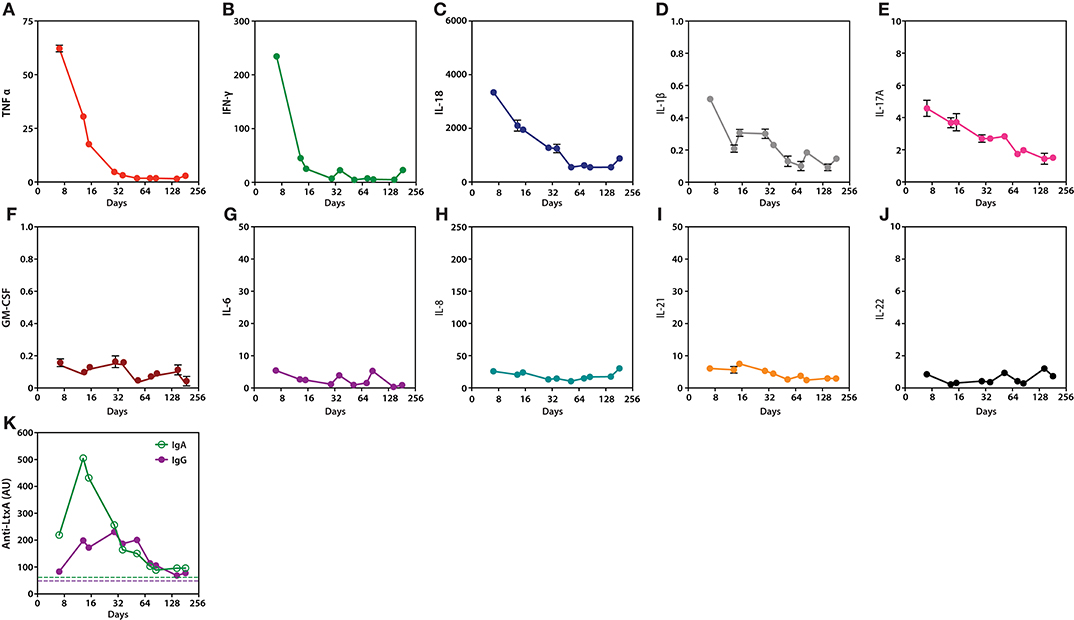
Figure 2. Serum cytokines and anti-LtxA antibodies during Aa infection and after initiation of antibiotic treatment. (A–J), Serial measurements of TNF-α, IFN-γ, IL-18, IL-1β, IL-17A, GM-CSF, IL-6, IL-8, IL-21, and IL-22 (pg/mL), respectively. (K) Serial quantification of IgG and IgA antibodies to LtxA. Dotted lines mark cut-offs for anti-LtxA positivity, which were established using a cohort of healthy individuals without periodontitis (5). Cytokine and anti-LtxA antibody levels were assayed in triplicate and duplicate, respectively. Mean values are shown. Day 0 marks the diagnosis of Aa endocarditis.
Humoral Immunity to Aa
Although IgA and IgG antibodies to LtxA were positive at the time of IE diagnosis, levels markedly increased several days after initiation of antibiotic treatment (Figure 2K). This coincided with a decline in peripheral cytokine levels (Figures 2A–E). The increase in anti-LtxA antibody levels suggests a more efficient humoral immune response against Aa, which may have aided pathogen clearance. Anti-LtxA antibody levels decreased with time, but remained positive at lower titers. Similar dynamics of the anti-Aa antibody response have previously been reported with initiation of periodontal treatment (14).
Discussion
Autoimmune diseases result from the interplay of genetic susceptibility, environmental factors, and stochastic events that together determine an individual's risk of developing disease. Loss of tolerance to peptidylcitrulline is an immune hallmark of RA (15), and pathogens with the potential to provoke autoantigen citrullination have emerged as putative agents that may trigger or sustain the anti-CCP response in RA (16). An association between RA and Aa is supported by mechanistic and clinical evidence (5). We believe this case provides proof of concept that in the genetically predisposed individual, Aa can induce the autoimmune responses associated with RA. The resolution of RA-associated symptoms (i.e., morning stiffness, polyarthritis, tenosynovitis) and anti-CCP antibodies with antibiotic therapy further supports an etiological role of Aa in driving autoimmunity in this patient.
Aa endocarditis is an insidious disease with a subacute or chronic course, which can have a prolonged period of symptoms before diagnosis (up to 60 weeks) (17). The presence of prosthetic heart valves, oral infection and dental procedures are among the risk factors associated with this illness (17). Since periodontal infection was not documented in the case presented here, it is possible to suggest that Aa may have reached the vascular compartment as result of inoculation during deep dental cleaning. Different to other cases of Aa endocarditis that present with prominent systemic features (fever, weight loss, rash, hepatosplenomegaly, hematological abnormalities, and hematuria) and often severe complications such as heart failure, renal failure, mycotic aneurysms, and septic embolization (17), the clinical course in this patient was exceptionally defined by arthritis and anti-CCP antibodies.
It is possible that several factors may have contributed to the patient's risk of developing loss of tolerance to peptidylcitrulline and RA-associated symptoms in the setting of (sub-)acute Aa infection. First, we identified a strong genetic susceptibility for seropositive RA as conferred by 3 distinct HLA-class II risk alleles in 2 loci (DRB1*04:04, DRB1*15:01, and DPB1*04:01). Distinct alleles are thought to confer additional disease risk by expanding the repertoire of possible RA autoantigens presented on HLA-class II molecules (18). In this patient, the different risk alleles likely conferred independent susceptibility to RA through the presence of distinct amino acid residues within the HLA class II binding groove (10).
Second, the patient was found to be infected with a highly leukotoxic Aa strain that carries a Δ530 ltx promoter deletion (8). This genotype of Aa, first described in the highly leukotoxic strain JP2 (8), is endemic in various populations originating in North and Central Africa and associated with aggressive periodontitis (4). The prevalence of JP2-like strains in Aa endocarditis is unknown, but to our knowledge, no cases are reported to date. In a study of 35 blood isolates of Aa, no JP2 promoter deletions were identified (19). As the generation of citrullinated autoantigens by Aa is dependent on LtxA alone (5), the identification of the highly leukotoxic JP2 genotype may explain why loss of immunologic tolerance to citrullinated proteins and clinical autoimmunity occurred in this patient. Although certain autoantibodies including RF are common in IE, anti-CCP antibodies have only been reported in one case of IE in which the pathogen was not identified (20). Indeed, the unlikely co-incidence of infection with a rare, highly virulent genotype of Aa in a patient uniquely predisposed to make ACPAs may explain why anti-CCPs and RA have not been reported in other cases of Aa endocarditis.
A third factor that may have facilitated the induction of arthritis is the unique pro-inflammatory milieu associated with Aa JP2 infection. Aa serotype b, which includes JP2 clones (8), is a potent inducer of Th1 (TNF-α and IFN-γ) and Th17 (IL-17A) immune phenotypes (21). LtxA also induces production of IL-1β and IL-18 by macrophages (22). These cytokines, which are central mediators in the immunopathogenesis of RA (13), also characterized the systemic cytokine profile identified in this patient during active Aa infection (Figures 2A–E). Although it is difficult to define whether these cytokines played a role in the patient's clinical presentation, it is interesting that although the addition of etanercept transiently improved the joint symptoms, it may also have allowed for abscess formation which help to unmask the underlying infection with Aa. Thus, it is possible that the cytokine milieu induced by the Aa JP2 strain helped to limit bacterial dissemination and the development of common complications associated with Aa endocarditis, but also contributed (together with Aa-induced anti-CCPs) to the development of arthritis in this patient. TNFα blockade may indeed have changed the clinical course of the disease from an indolent infection with RA phenotype to a more typical presentation of Aa endocarditis (e.g., septic emboli).
Although the patient fulfilled the 2010 ACR/EULAR classification criteria for RA [8/10 points based on synovitis of at least 4 small joints (3 pt.); ACPA >3xULN (3 pt.); duration >6 weeks [1 pt.]; and abnormal CRP and ESR (1 pt.)] (23), his disease course is not classic. In RA, the transition from pre-clinical autoimmunity to overt arthritis often takes several years (24). This is consistent with a model of indolent infection such as periodontitis or bronchiectasis leading to chronic antigenic simulation (25). In cases of RA potentially associated with Aa periodontitis, the production of citrullinated autoantigens would represent a chronic, localized process which may result in long preclinical phase. During this phase, amplification pathways may be established that maintain disease. In this case of systemic Aa infection, widespread production of RA autoantigens triggered by a highly leukotoxic strain of Aa may have resulted in acute autoantibody production and arthritis, which rapidly resolved after the driver of autoantigen production (i.e., Aa) was eradicated.
It is now appreciated that the relationship between infectious agents and autoimmune arthritis is more complex than the one pathogen-one disease model that built the conceptual framework for Koch's postulates. The existence of a single pathogen that acts as a driver of autoimmunity in all patients with RA is unlikely. Similarly, only a fraction of individuals infected with a microbial species of arthritogenic potential will develop RA. This case underscores the importance of both genetic susceptibility and environmental agents for the induction of autoimmune pathology, and in a human model provides direct evidence that leukotoxic strains of Aa can trigger autoimmune features found in RA. Moreover, it suggests that in some cases, this pathogen can be a reversible cause of RA. Although the idea of definitive treatment of early autoimmunity in RA is enticing, it remains uncertain whether established RA associated with Aa periodontitis may be modifiable by antibiotic therapy.
Ethics Statement
The patient gave written informed consent prior presentation of the case. The case report is not part of a clinical trial.
Author Contributions
AM, JJ, and RK performed in vitro studies. VJ, WH, RF, and BE clinically followed the patient. FA supervised experiments. MK and FA wrote the manuscript and prepared the figures. All authors contributed to the preparation of the final manuscript.
Funding
Funding for this project was provided by the Jerome L. Greene Foundation, and National Institute of Arthritis, and Musculoskeletal and Skin Diseases (NIAMS)/National Institutes of Health (NIH) grant R01 AR069569. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIAMS or the NIH.
Conflict of Interest Statement
FA received a grant from Medimmune and is author on issued Patent No. 8,975,033 entitled “Human Autoantibodies Specific for PAD3 which are Cross-reactive with PAD4 and their Use in the Diagnosis and Treatment of Rheumatoid Arthritis and Related Diseases.” FA has served as consultant for Bristol-Myers Squibb Company and Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer YP and handling Editor declared their shared affiliation at the time of the review.
References
2. Geraci JE, Wilson WR. Symposium on infective endocarditis. III. Endocarditis due to gram-negative bacteria. Report of 56 cases. Mayo Clin Proc. (1982) 57:145–8.
3. Tanner AC, Haffer C, Bratthall GT, Visconti RA, Socransky SS. A study of the bacteria associated with advancing periodontitis in man. J Clin Periodontol. (1979) 6:278–307. doi: 10.1111/j.1600-051X.1979.tb01931.x
4. Johansson A. Aggregatibacter actinomycetemcomitans leukotoxin: a powerful tool with capacity to cause imbalance in the host inflammatory response. Toxins (2011) 3:242–59. doi: 10.3390/toxins3030242
5. Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med. (2016) 8:369ra176. doi: 10.1126/scitranslmed.aaj1921
6. Poulsen K, Ennibi OK, Haubek D. Improved PCR for detection of the highly leukotoxic JP2 clone of Actinobacillus actinomycetemcomitans in subgingival plaque samples. J Clin Microbiol. (2003) 41:4829–32. doi: 10.1128/JCM.41.10.4829-4832.2003
7. Hoglund AC, Haubek D, Kwamin F, Johansson A, Claesson R. Leukotoxic activity of Aggregatibacter actinomycetemcomitans and periodontal attachment loss. PLoS ONE (2014) 9:e104095. doi: 10.1371/journal.pone.0104095
8. Brogan JM, Lally ET, Poulsen K, Kilian M, Demuth DR. Regulation of Actinobacillus actinomycetemcomitans leukotoxin expression: analysis of the promoter regions of leukotoxic and minimally leukotoxic strains. Infect Immun. (1994) 62:501–8.
9. Sampathkumar V, Velusamy SK, Godboley D, Fine DH. Increased leukotoxin production: Characterization of 100 base pairs within the 530 base pair leukotoxin promoter region of Aggregatibacter actinomycetemcomitans. Sci Rep. (2017) 7:1887. doi: 10.1038/s41598-017-01692-6
10. Raychaudhuri S, Sandor C, Stahl EA, Freudenberg J, Lee HS, Jia X, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. (2012) 44:291–6. doi: 10.1038/ng.1076
11. Terao C, Suzuki A, Ikari K, Kochi Y, Ohmura K, Katayama M, et al. An association between amino acid position 74 of HLA-DRB1 and anti-citrullinated protein antibody levels in Japanese patients with anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. (2015) 67:2038–45. doi: 10.1002/art.39133
12. Scally SW, Petersen J, Law SC, Dudek NL, Nel HJ, Loh KL, et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J Exp Med. (2013) 210:2569–82. doi: 10.1084/jem.20131241
13. McInnes IB, Buckley CD, Isaacs JD. Cytokines in rheumatoid arthritis - shaping the immunological landscape. Nat Rev Rheumatol. (2016) 12:63–8. doi: 10.1038/nrrheum.2015.171
14. Sjostrom K, Ou J, Whitney C, Johnson B, Darveau R, Engel D, et al. Effect of treatment on titer, function, and antigen recognition of serum antibodies to Actinobacillus actinomycetemcomitans in patients with rapidly progressive periodontitis. Infect Immun. (1994) 62:145–51.
15. Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. (1998) 101:273–81. doi: 10.1172/JCI1316
16. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
17. Paturel L, Casalta JP, Habib G, Nezri M, Raoult D. Actinobacillus actinomycetemcomitans endocarditis. Clin Microbiol Infect. (2004) 10:98–118. doi: 10.1111/j.1469-0691.2004.00794.x
18. Lenz TL, Deutsch AJ, Han B, Hu X, Okada Y, Eyre S, et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet. (2015) 47:1085–90. doi: 10.1038/ng.3379
19. Paju S, Carlson P, Jousimies-Somer H, Asikainen S. Heterogeneity of Actinobacillus actinomycetemcomitans strains in various human infections and relationships between serotype, genotype, and antimicrobial susceptibility. J Clin Microbiol. (2000) 38:79–84.
20. Bojalil R, Mazon-Gonzalez B, Carrillo-Cordova JR, Springall R, Amezcua-Guerra LM. Frequency and clinical significance of a variety of autoantibodies in patients with definite infective endocarditis. J Clin Rheumatol. (2012) 18:67–70. doi: 10.1097/RHU.0b013e318247caf0
21. Diaz-Zuniga J, Melgar-Rodriguez S, Alvarez C, Monasterio G, Benitez A, Ciuchi P, et al. T-lymphocyte phenotype and function triggered by Aggregatibacter actinomycetemcomitans is serotype-dependent. J Periodontal Res. (2015) 50:824–35. doi: 10.1111/jre.12270
22. Kelk P, Abd H, Claesson R, Sandstrom G, Sjostedt A, Johansson A. Cellular and molecular response of human macrophages exposed to Aggregatibacter actinomycetemcomitans leukotoxin. Cell Death Dis. (2011) 2:e126. doi: 10.1038/cddis.2011.6
23. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. (2010) 62:2569–81. doi: 10.1002/art.27584
24. Nielen MM, van SD, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum. (2004) 50:380–6. doi: 10.1002/art.20018
Keywords: rheumatoid arthritis, ACPA, anti-CCP, Aggregatibacter actinomycetemcomitans, autoantibodies
Citation: Mukherjee A, Jantsch V, Khan R, Hartung W, Fischer R, Jantsch J, Ehrenstein B, Konig MF and Andrade F (2018) Rheumatoid Arthritis-Associated Autoimmunity Due to Aggregatibacter actinomycetemcomitans and Its Resolution With Antibiotic Therapy. Front. Immunol. 9:2352. doi: 10.3389/fimmu.2018.02352
Received: 03 July 2018; Accepted: 24 September 2018;
Published: 16 October 2018.
Edited by:
Juan-Manuel Anaya, Universidad del Rosario, ColombiaReviewed by:
Yovana Pacheco, Universidad del Rosario, ColombiaGabriel J. Tobón, Fundacion Valle del Lili, Colombia
Copyright © 2018 Mukherjee, Jantsch, Khan, Hartung, Fischer, Jantsch, Ehrenstein, Konig and Andrade. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Boris Ehrenstein, Yi5laHJlbnN0ZWluQGFza2xlcGlvcy5jb20=
Maximilian F. Konig, a29uaWdAamhtaS5lZHU=
Felipe Andrade, YW5kcmFkZUBqaG1pLmVkdQ==
†These authors have contributed equally to this work