 Rebecca M. Richards
Rebecca M. Richards Elena Sotillo2
Elena Sotillo2 Robbie G. Majzner
Robbie G. Majzner- 1Department of Pediatrics, Stanford University School of Medicine, Stanford, CA, United States
- 2Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, United States
Patients with high risk neuroblastoma have a poor prognosis and survivors are often left with debilitating long term sequelae from treatment. Even after integration of anti-GD2 monoclonal antibody therapy into standard, upftont protocols, 5-year overall survival rates are only about 50%. The success of anti-GD2 therapy has proven that immunotherapy can be effective in neuroblastoma. Adoptive transfer of chimeric antigen receptor (CAR) T cells has the potential to build on this success. In early phase clinical trials, CAR T cell therapy for neuroblastoma has proven safe and feasible, but significant barriers to efficacy remain. These include lack of T cell persistence and potency, difficulty in target identification, and an immunosuppressive tumor microenvironment. With recent advances in CAR T cell engineering, many of these issues are being addressed in the laboratory. In this review, we summarize the clinical trials that have been completed or are underway for CAR T cell therapy in neuroblastoma, discuss the conclusions and open questions derived from these trials, and consider potential strategies to improve CAR T cell therapy for patients with neuroblastoma.
Introduction
Neuroblastoma is a tumor of childhood arising from neural crest cells. Often diagnosed during the first 10 years of life, it is the most common extracranial solid tumor in childhood and is responsible for 11% of pediatric cancer deaths in patients younger than 15 years of age (1). Approximately 650 patients are diagnosed in the United States with neuroblastoma each year, which accounts for 7.5% of all cancer diagnoses for children younger than 15 years old (2, 3). Clinical presentation and outcomes are extremely variable. Newborns and infants are often incidentally found to have adrenal tumors that spontaneously regress without therapy, while toddlers and older children frequently present with widely metastatic disease that requires multimodal intensive therapy including surgery, chemotherapy, radiotherapy, autologous stem cell transplant, differentiation therapy, and monoclonal antibody-based immunotherapy. Patients with localized disease typically have excellent outcomes, with >90% event free survival (EFS) rates 5 years after diagnosis (4). In contrast, patients with high risk disease (defined by age >18 months, extent of metastases, and histologic and genetic factors such as N-MYC amplification) historically have had poor long term survival prospects, with 5-year EFS of about 50% (5–8). Patients who do survive often suffer long term sequelae from their intense treatment including hearing loss, growth retardation, and secondary malignancies (9). This population therefore has a desperate need for novel therapies to improve survival and to decrease morbidity.
Antibody-based immunotherapy was recently integrated into frontline protocols for patients with high risk neuroblastoma. A pivotal phase III clinical trial published in 2010 revealed an increase in 2 year EFS from 46 to 66% and overall survival (OS) from 75 to 86% for patients who received adjuvant anti-GD2 monoclonal antibody given with IL-2, GM-CSF, and retinoic acid compared to patients who received retinoic acid alone (6). Incorporation of anti-GD2 monoclonal antibodies into therapy for neuroblastoma has been one of the most successful interventions to improve survival for high risk patients (6, 10–13). This success has firmly established a new paradigm for the treatment of neuroblastoma that includes immunotherapy.
While survival rates have improved since the adoption of anti-GD2 antibodies, ~50% of patients will relapse and eventually die from their disease (6). Additionally, 20% of patients are refractory to induction therapy at diagnosis and may not ever receive anti-GD2 antibody (14). These patients are in need of more potent and targeted approaches. One such approach is adoptive transfer of chimeric antigen receptor (CAR) T cells, which combine the specificity of an antibody with the cytolytic capacity of T cells in an MHC independent manner (15). CD19 and CD22 CAR T cells have demonstrated remarkable success in children with relapsed and refractory leukemia and lymphoma (16–20). While anti-GD2 monoclonal antibodies have been successful in treating patients with neuroblastoma metastases in their bone marrow, they have generally not been useful as single agents against bulky disease (21). CAR T cells have the potential for increased potency and durability compared to monoclonal antibodies and thus could overcome this challenge. Additionally, while antibodies generally do not penetrate the central nervous system (CNS) (22), CAR T cells are able to cross the blood-brain barrier (23, 24). Relapsed neuroblastoma of the CNS has emerged as a clinical entity since the adoption of anti-GD2 monoclonal antibodies, and CARs could present an answer to this challenging clinical problem (25, 26).
CAR T cells have already shown promise in clinical trials for neuroblastoma with several objective responses seen in early phase studies (27–31). In general, however, CAR T cell activity has not been as robust in neuroblastoma as in hematologic malignancies. There are many challenges in designing CAR T cells against neuroblastoma including suboptimal T cell persistence and potency (27–29), a paucity of tumor specific targets (32, 33), and an immunosuppressive tumor microenvironment (34, 35). However, CAR T cell engineering is accelerating at a rapid pace, with the aim to improve potency and specificity of tumor targeting (36–41). Neuroblastoma is an excellent testing ground for these new therapeutics since immunotherapy has already been validated for these patients. In this review, we will discuss the clinical experience to date with neuroblastoma-directed CAR T cells and the challenges of applying these powerful therapeutics to neuroblastoma patients. As CAR T cell design becomes more sophisticated, these agents are primed to become part of the multimodal approach used to treat patients with high risk neuroblastoma.
Clinical Experience
Much of the early clinical experience treating children with CAR T cells has been in hematologic malignancies, but neuroblastoma has also been an area of intense investigation, with a steady stream of clinical trials of CAR T cells for patients with relapsed or refractory disease since the early 2000s. Despite preclinical development of CAR T cells against a variety of neuroblastoma associated antigens, only those directed against GD2 and L1-CAM (CD171) have reached clinical trials. Table 1 summarizes completed and ongoing clinical trials.
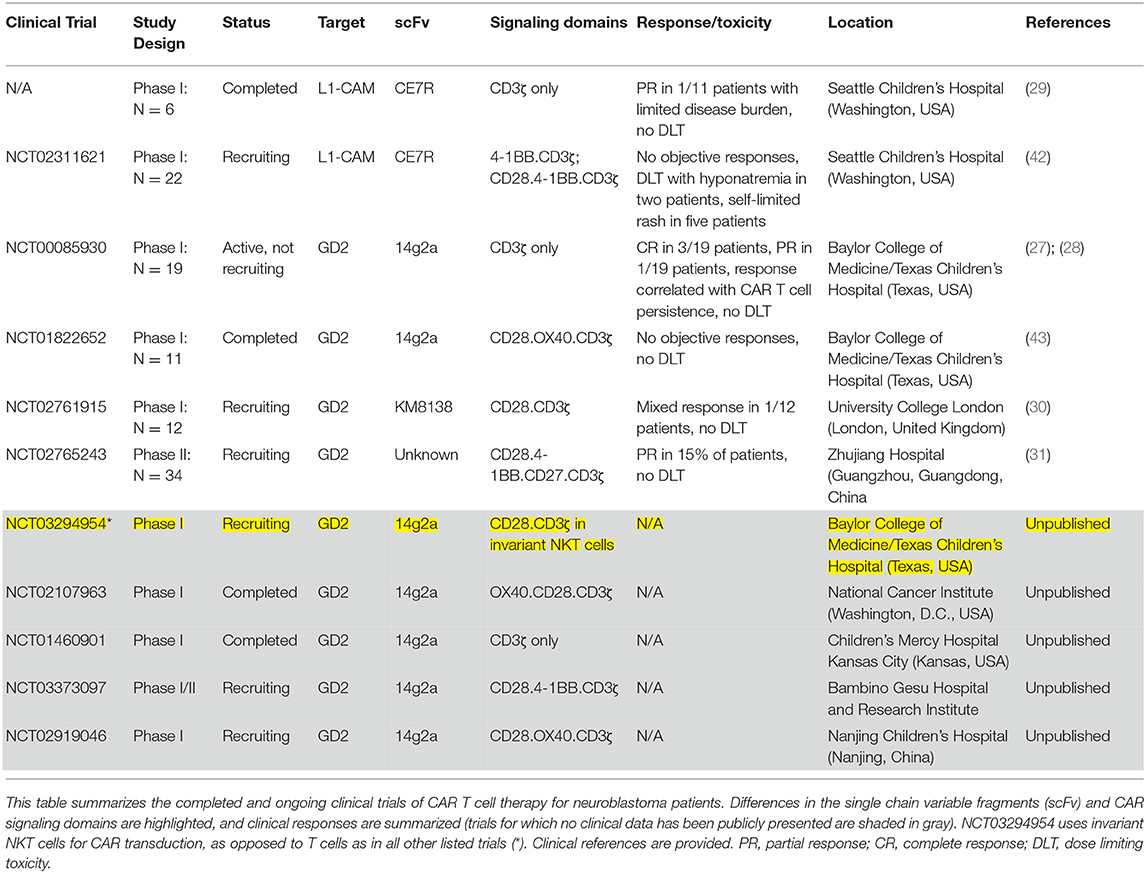
Table 1. Summary of CAR T cell clinical trials for neuroblastoma.
GD2
The most-studied tumor associated antigen in neuroblastoma is GD2. GD2 is a disialoganglioside that is highly and nearly universally expressed on neuroblastoma tissue (44) and likely plays a role in tumor immune evasion (45). It is a natural choice as a target for CAR T cell therapy in neuroblastoma based on the success of anti-GD2 monoclonal antibody therapy (6, 10–12).
One of the first CAR T cells products tested in children was a first generation anti-GD2 CAR (containing only the CD3ζ endodomain but no costimulatory domain). In preclinical models, Rossig et al. demonstrated that GD2 was a viable CAR T cell target for neuroblastoma (46). To translate the preclinical promise of anti-GD2 CAR T cells into patients, Pule et al. aimed to treat patients in a manner that could enhance CAR persistence. CAR T cells with first generation signaling domains (CD3ζ only) had previously demonstrated limited persistence in human trials for other indications, indicating that the CD3ζ only intracellular domain was not sufficient for optimal activity (47–49). Rather than endowing the CAR with embedded costimulation, Pule and colleagues generated a T cell product that could receive physiologic costimulation through engagement of a native T cell receptor (TCR).
These researchers drew on experience from clinical trials in which Epstein Barr Virus-specific cytotoxic T lymphocytes (EBV-CTLs) were adoptively transferred to patients with EBV-associated malignancies (50–53). In those trials, T cell persistence of at least 3 months was seen even with relatively low doses of EBV-CTLs. Adding tumor specificity with a CAR construct was a logical next step to take advantage of the longevity of EBV-CTLs. A Phase I trial (NCT00085930) tested this approach by infusing EBV-CTLs co-expressing a first generation anti-GD2 CAR into relapsed and refractory neuroblastoma patients who were seropositive for EBV viral capsid antigen (27).
In this trial, EBV-specific lymphocytes were extracted from eleven patients with refractory or recurrent neuroblastoma, transduced with retrovirus encoding a GD2 CAR molecule (containing the single chain variable fragment (scFv) derived from Dinutuximab, 14g2a), and stimulated ex vivo with autologous EBV-transformed lymphoblastoid cell lines (LCLs). This product was called GD2 CAR-CTL. Concurrently, bulk T cells were transduced with the same GD2 CAR but activated through the native TCR with anti-CD3 antibodies (GD2 CAR-ATC). Each patient received between 2 × 107 and 1 × 108 cells/m2 of both GD2 CAR-CTL and GD2 CAR-ATC. A 12-base pair mutation between the receptor stop codon and the 3′ LTR allowed for comparison of in vivo durability of the two cell types by RT-PCR. There was little to no detection of GD2 CAR-ATCs after 2 weeks, but clear persistence of the EBV specific GD2 CAR-CTLs until on average 6 weeks, demonstrating that costimulation is vital for CAR T cell persistence. Four of the eight patients (50%) with evaluable tumors had a partial or complete response, though all later progressed. Responses included one patient with a complete response of an extradural parietal lesion as measured by MIBG, one patient with a complete response of extensive bone marrow disease, and two patients with significant tumor necrosis confirmed by imaging and biopsies. These data support the hypothesis that ongoing costimulation increases persistence in vivo and results in increased efficacy and durability of response. A subsequent study with longer follow up determined that even low levels of persistent cells correlated strongly with slower time to disease progression (28).
While using viral specific CTLs takes advantage of the native TCR machinery with physiologic stimulation, there is some evidence that co-engagement of a CAR and TCR can result in T cell exhaustion and decreased CAR persistence (54). Most CAR constructs now rely on embedded costimulation. The same group from Baylor produced a third generation CAR containing both the CD28 and OX40 costimulatory domains. Preclinical studies demonstrated that incorporation of tandem costimulation domains increased expansion of the engineered T cell product and augmented cytokine release (55, 56), which prompted testing this construct in clinical trials.
The third generation anti-GD2 CAR was administered to eleven patients with relapsed or refractory neuroblastoma. Patients were treated in one of three cohorts: GD2 CAR T cells alone, GD2 CAR T cells after lymphodepleting chemotherapy, or GD2 CAR T cells after lymphodepleting chemotherapy given with the PD-1 inhibitor pembrolizumab. Patients who received lymphodepletion with or without checkpoint blockade had increased expansion of their CAR T cells and longer CAR T cell persistence. Anti-PD-1 therapy did not appear to dramatically affect these parameters or efficacy. Unfortunately, even after patients received proper lymphodepletion, this CAR was found to have minimal activity with no measurable responses (43). One explanation for the lack of long-term persistence seen in this trial is tonic signaling of the CAR T cell caused by aggregation of the 14g2a anti-GD2 scFv, leading to T cell exhaustion and limited anti-tumor efficacy (57). T cell exhaustion, which will be further discussed below, has emerged as an important factor that can limit CAR efficacy and is highly dependent on costimulation molecules (57, 58).
Another Phase I trial of anti-GD2 CARs is underway in the United Kingdom (NCT02761915) utilizing an scFv based on a previously described humanized murine antibody KM8138 (59) that is fused to a CD28 costimulatory domain and CD3ζ. Based on promising preclinical data (60), this trial is enrolling children with relapsed or refractory neuroblastoma and evaluable disease in a dose escalation model. Preliminary results presented in abstract form demonstrate minor clinical response by imaging criteria and cytokine release syndrome (CRS) in at least one patient at higher dose levels, but CAR T cell persistence also appears to be limited (30). A fourth generation GD2 CAR (including CD28, 4-1BB, and CD27 costimulatory domains in addition to CD3ζ) is also being tested in a multi-institutional Chinese Phase II trial for high-risk neuroblastoma patients. An abstract presented in 2017 reported 15% of 34 patients with a partial response and no dose limiting toxicities. Two patients had significant tumor regression, one with two bulky lesions that regressed by >90% each and one with a reduction in retroperitoneal tumor dimensions and standardized uptake value (SUV) by PET scan measured 2 months after CAR T cell therapy (31).
Despite mixed results in the early GD2 CAR clinical trials, this target remains an area of intense focus. There are currently many ongoing preclinical studies focused on targeting GD2 as well as five open clinical trials of CAR T cells directed against GD2 for neuroblastoma patients (NCT03373097, NCT02761915, NCT02765243, NCT03294954, NCT02919046). While the experience thus far with GD2 CARs in clinical trials has established safety and feasibility, limited T cell persistence has emerged as a major hurdle to success.
L1-CAM/CD171
Another target of interest in neuroblastoma is L1-CAM, an adhesion molecule that is overexpressed on neuroblastoma. Monoclonal antibody CE7 preferentially binds to a tumor-specific epitope of L1-CAM (61). The mechanism of tumor specificity has not been elucidated, but appears to be glycosylation-dependent (62–64). A first generation CAR containing the CE7 scFv, a CD4 transmembrane domain, and the CD3ζ intracellular signaling domain (CE7R CAR) demonstrated preclinical activity in xenograft models of neuroblastoma (65). A clinical construct was designed to include a selection-suicide fusion protein composed of hygromycin phosphotransferase and thymidine kinase (HyTK), allowing for CAR ablation with ganciclovir in the case of unforeseen toxicity. In a Phase I clinical trial of escalating doses of CE7R HyTK CD8+ CAR T cells, the authors demonstrated safety and observed no off-tumor, on-target toxicity. However, only one of six patients had a significant clinical response. That patient had limited disease burden, whereas the patients with higher disease burden had progressive disease. All patients ultimately died of their disease (29). Similar to GD2, lack of persistence of CAR T cells was also a major limiting factor in this study, which may have been related to the lack of costimulation in the CAR or to immunogenicity of the suicide HyTK protein (66).
To enhance the activity and persistence of L1-CAM directed CARs, the researchers then generated a second generation CAR (2G CE7 CAR) containing a 4-1BB costimulation domain and a truncated extracellular epidermal growth factor receptor (EGFRt) domain in place of the HyTK suicide switch (allowing for an alternative ablation strategy with cetuximab) (67, 68). Reassuringly, there was no significant clinical toxicity in non-human primates treated with 2G CE7 CAR T cells at doses 10–100 times higher than the doses employed in the clinical trial, though these primates did not have antigen positive malignancies (69).
A Phase I trial with the 2G CE7 CAR in rotation with a similar third generation product that also includes a CD28 endodomain is currently underway at Seattle Children's Hospital for recurrent or refractory high risk neuroblastoma patients (NCT02311621). Patients receive anti-L1-CAM CAR T cells in a defined ratio of 1:1 CD4:CD8 T cells. This strategy is based on previous successes of this controlled strategy for CAR T cell treatment of B-ALL and non-Hodgkin lymphoma at the same institution (70–72). Further study is required to determine the utility of a defined CD4:CD8 T cell product as this has not been tested in a randomized clinical trial, and equally impressive response rates have been obtained using non-selected populations of T cells or PBMCs after transduction (16, 17, 19, 20).
In a recently presented abstract, the researchers reported that L1-CAM CAR T cells infiltrate sites of disease in patients but appear to be causing off-tumor toxicity with transient skin rash (where the CAR T cells may colocalize with L1-CAM expressing normal cells) and poorly understood hyponatremia in some patients. Although these toxicities have all been transient and the trial is ongoing (42), the early finding of possible off-tumor, on-target toxicity is a reminder of the difficulty of identifying appropriate CAR-T cell targets (discussed further below).
Challenges in Targeting Neuroblastoma with CAR T Cells
Clinical experience thus far with CAR T cells for neuroblastoma indicates that T cell persistence is emerging as a major impediment for the success of these therapeutics. Outcomes have been encouraging but modest, with only a fraction of patients achieving measurable responses and very few patients demonstrating long term persistence of CAR T cells. In order to achieve the level of success that has been seen in hematologic malignancies, the field will have to address this challenge. Additionally, target selection is equally important, as many neuroblastoma targets are also expressed on normal tissues, creating the potential for off-tumor, on-target toxicity as may have been seen with L1-CAM CARs (albeit transiently). There may be a therapeutic window for CAR T cells against highly expressed tumor antigens that exhibit lower levels of expression on normal tissue, so this does not necessarily preclude these molecules as targets. Finally, as with other solid tumors, a complex, immunosuppressive microenvironment in neuroblastoma tumors presents a barrier for efficacious CAR T cell therapy.
T Cell Persistence and Exhaustion
CAR T cell persistence is essential for durable clinical responses (16, 47, 73–75). Long term follow-up of Baylor's first generation anti-GD2 CAR T cell trial demonstrated that time to disease progression was significantly delayed in patients whose T cells were detectable for longer (27, 28). In the trial of a first generation L1-CAM CAR, the only patient of six with a clinical response had detectable CAR T cells in the blood 56 days after treatment, while patients without objective response had shorter persistence (29).
CAR T cell persistence may be diminished due to T cell exhaustion. T cell exhaustion has primarily been studied in the setting of chronic antigen exposure including for viral infections (76, 77) and cancer (78–81). Exhausted T cells upregulate inhibitory receptors after excessive and continuous stimulation over a matter of days to weeks and exhibit diminished effector functions. T cell exhaustion appears to be partially reversible. This is fundamentally different from T cell senescence, which typically occurs over months to years, is associated with telomere shortening, and represents a terminally differentiated state without potential for reversibility or proliferation (82).
An exhausted CAR T cell phenotype has recently been described in GD2 CAR T cells, driven by antigen-independent tonic signaling (57). Long et al. explored why GD2 CAR T cells containing the 14g2a scFv appeared to be less functional than CD19 CAR T cells. The authors found that unlike the CD19 CAR, the GD2 CAR aggregated on the surface of T cells and subsequently triggered low level tonic signaling in the absence of antigen, which ultimately resulted in T cell exhaustion. Additionally, they demonstrated that integration of the CD28 costimulatory domain into tonically signaling CAR T cells amplified this phenotype, while inclusion of a 4-1BB costimulatory domain protected against T cell exhaustion (57). This finding is in line with clinical studies of CD19 CAR T cells, as those with 4-1BB costimulatory domains demonstrate long term persistence while those with CD28 costimulatory domains do not (16, 18, 19). Our group plans to open a clinical trial of GD2 CAR T cells with a 4-1BB costimulatory domain in early 2019, which will be the first such trial in North America.
Persistence can be affected by factors extrinsic to the CAR molecule. Early CAR T cell trials did not incorporate lymphodepletion prior to CAR T cell infusion, which may have compromised expansion of the engineered T cells (18, 43, 71). Lymphodepleting chemotherapy improves engraftment and efficacy and has become a standard part of CAR T cell regimens (83, 84). The mechanism of increased activity after lymphodepletion is thought to be depletion of regulatory immune cells and/or a reflexive increase in homeostatic cytokines IL-7 and IL-15 that drive CAR T cell proliferation (84–86). Given that endogenous cytokines may increase CAR efficacy, some groups have focused on increasing CAR potency by programming CAR T cells to secrete immunostimulatory cytokines locally (87, 88). Systemic infusion of cytokines is often associated with unacceptable toxicity (89–91), and overexpression of the cytokine receptor does not overcome a dearth of cytokines in the tumor microenvironment (92). Therefore, providing local and inducible cytokine release by the CAR T cells themselves is an attractive strategy. Initial reports have demonstrated improved potency of CD19 CAR when co-expressed with IL-7 (93), IL-12 (94), IL-15 (95), membrane bound chimeric IL-15 (88), and IL-21 (93). Further studies will be required to translate these results clinically and to see if this can be generalized to solid tumors and to neuroblastoma specifically.
Anti-carcinoembryonic antigen (CEA) CAR T cells were engineered to produce IL-12 only after engagement with target antigen by placing IL-12 under the control of a nuclear factor of activated T cells (NFAT) promoter. In a colon cancer model, CEA CAR T cells that expressed inducible IL-12 mediated greater tumor regression and abrogation of antigen negative tumor outgrowth. This effect was likely enhanced by activated macrophages that infiltrated the tumors in response to the locally secreted IL-12. (96). An alternative system combines oncolytic viruses that secrete cytokines IL-15 and CCL5 with anti-GD2 CAR T cell therapy in xenograft models of neuroblastoma in order to increase T cell infiltration and persistence (97).
Shum et al. recently described a system in which a constitutively active IL-7 receptor was co-expressed with a second generation GD2 CAR. This resulted in improved efficacy of GD2 CAR T cells in vitro and in a murine xenograft model of neuroblastoma (98). This modification did not lead to malignant transformation in short term assays, an important safety consideration as the IL-7 receptor was derived from a patient with T cell acute lymphoblastic leukemia (99). However, implementation of such a strategy into clinical trials will require caution due to the potential for delayed malignant transformation. These approaches to increase potency and persistence of CAR T cells are beginning to undergo testing in early clinical trials (NCT03635632), and may help to improve efficacy, durability, and ultimately clinical outcomes.
Target Selection and Potential for Toxicity
Choosing an optimal CAR T cell target in neuroblastoma and more generally in solid tumors is a daunting task. Much of the success of CD19 and CD22 CAR T cells hinges on the restriction of these targets to lymphoblasts and normal B cells, which are in large part dispensable with appropriate supportive measures (16–20). An ideal CAR target antigen is highly and homogeneously expressed on tumor cells with minimal expression on vital tissues. Fulfillment of these criteria is difficult for solid tumor antigens, as many antigens are expressed in cells of related origin.
Many antigens overexpressed on neuroblastoma are often present at lower levels in peripheral nerves and/or on other neural tissue (100–102), so an important consideration in the development of anti-GD2 CAR T cells is the potential for off-tumor, on-target toxicity. Anti-GD2 monoclonal antibodies cause pain requiring continuous infusion of narcotics for analgesia (103–106) due to their interaction with peripheral nerves and possibly engagement of the complement system (107). However, clinical trials of CAR T cells targeting GD2 have not resulted in toxicity despite clear signs of on-tumor efficacy (27, 28, 30, 43). Still, due to toxicity concerns, most anti-GD2 CAR T cells for clinical trials have been designed to include a “suicide switch” to allow for rapid ablation.
Oncologists have approached adoptive T cell therapy for solid tumors cautiously due to the overlap of antigen expression with normal tissues. There have been several incidents of off-tumor toxicity in human trials using engineered high affinity TCRs against MAGE-A3 (108) and MAGE-A12 (109) that cross reacted with normal tissue. Additionally, one patient with metastatic colon cancer died after treatment with a HER2-targeted CAR (110). The initial case report of this incident noted that there was pulmonary infiltration by CAR T cells that could be due off-tumor, on-target toxicity. However, that patient was administered a dose of CAR T cells that was found to be 100 times the maximum tolerated dose of CD19 CAR T cells as well as exogenous IL-2. She was found to have very high levels of circulating cytokines and our recent understanding of the toxicities associated with CAR T cells indicates that this was more likely to be caused by CRS than off-tumor, on-target toxicity (111). This is further supported by recent efforts at Baylor College of Medicine to target HER2 on pediatric sarcomas using CAR T cells. In carefully designed dose escalation trials conducted without and with lymphodepletion, anti-HER2 CAR T cells elicited no off-tumor, on-target toxicity but resulted in clinically significant responses including a complete response in a patient with metastatic rhabdomyosarcoma (112, 113).
One possible explanation for the lack of toxicity for both GD2 and HER2 CARs is the differential in antigen density between tumor cells and normal tissue. Antigen density is emerging as an important consideration for CAR efficacy. When our group engineered a CAR against Anaplastic Lymphoma Kinase (ALK) on neuroblastoma, there was a clear correlation between the number of surface molecules of target antigen and ALK CAR T cell efficacy. A threshold number of target molecules was required to elicit effector functions (114). In vivo, ALK CAR T cell efficacy was only seen when ALK expression was high on tumor cells. Similarly, in a Phase I trial of CD22 CAR T cells of children with ALL, after initially achieving a complete response, most patients relapsed with leukemia expressing lower levels of CD22 than their pre-treatment samples, apparently below the threshold for CAR efficacy (20). Others have found a similar relationship of CAR efficacy and antigen density in preclinical studies of CARs for targets including CD123, CD20, HER2, EGFR, and CD30 (115–121). This represents a paradigm shift in the field as it opens up potential therapeutic windows for targets expressed at low levels on normal tissue as long as expression on tumor is high (111).
As CARs are engineered to become more potent, they could also become more toxic due to recognition of lower levels of target. While several clinical trials of GD2 CAR T cells containing the 14g2a binder have been carried out without any reports of central or peripheral neurotoxicity (27, 28, 30, 43), one preclinical study of a high affinity GD2 CAR reported neurotoxicity and T cell infiltration in the brains of mice (122). However, studies of a CAR with the same high affinity binder in our laboratory do not cause neurotoxicity (123), calling into question whether the findings were truly due to off-tumor, on-target toxicity. The point remains, however, that as CAR T cells are better engineered to target low target antigen density tumor cells, there will be potential for increased toxicity and clinical trials must be conducted carefully.
Novel Targets
In addition to GD2 and L1-CAM, researchers are investigating several novel target antigens for CAR T cell therapy in neuroblastoma, and preclinical data are summarized below. Figure 1 depicts the targets currently under investigation for CAR T cell therapy for neuroblastoma.
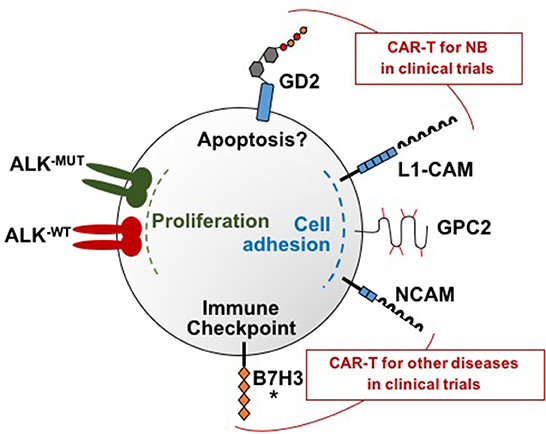
Figure 1. Molecular targets under investigation for CAR T cell therapy for neuroblastoma. There are a total of six neuroblastoma surface targets for which CAR T cells have been developed: GD2, L1-CAM, GPC2, B7H3, and ALK, and NCAM. These targets each have distinct functions that are depicted in this figure. Note that both wild type and mutated ALK are overexpressed on neuroblastoma samples and both can be targeted by the ALK CARs. GD2 and L1-CAM are the two targets currently in clinical trials for neuroblastoma. Clinical trials that include CAR T cells targeting NCAM are ongoing for multiple myeloma and AML but not yet for neuroblastoma. An asterisk marks B7H3 because clinical trials are currently being planned.
Glypican 2 (GPC2)
GPC2 is a member of the glypican family of proteins (124), and is instrumental for growth and differentiation of axons in the developing nervous system (125, 126). Gene expression-based exploration of the surfaceome of neuroblastoma cells identified GPC2 as a cell surface molecule that is highly expressed in neuroblastoma with low expression on normal tissue, indicating that it may be an ideal candidate for CAR T cell based immunotherapy (32, 33). Retrospective review demonstrated significantly decreased survival in neuroblastoma patients with tumors expressing high levels of GPC2. Bosse et al. generated an anti-GPC2 antibody drug conjugate (ADC) that demonstrated strong antitumor activity in a patient derived xenograft (PDX) mouse model (32).
Concurrently, another group developed CARs containing heavy chain only scFvs against GPC2 with 4-1BB and CD3ζ endodomains. Anti-GPC2 CAR T cells demonstrated in vitro activity and in vivo clearance of human neuroblastoma xenografts (127). Though this study needs to be expanded to include a broader array of neuroblastoma cell lines and primary human samples, preliminary data suggest that GPC2 should be further evaluated as a clinical target for CAR T cell therapy in neuroblastoma. These studies also demonstrate the importance and power of a surfaceome approach to identify new targets for CAR T cell immunotherapy, with a <5-year turn-around time from target identification to development of therapeutics with potential for clinical application.
B7-H3/CD276
B7-H3 (CD276) is a checkpoint molecule expressed at high levels on many pediatric solid tumors including neuroblastoma (128–131). It plays a role in immune evasion (132) and metastatic potential (133), and overexpression correlates with poor prognosis in many cancers (134). These characteristics have made B7-H3 an attractive target for immunotherapeutic strategies, and early phase clinical trials with monoclonal antibodies have demonstrated encouraging results in both neuroblastoma and other malignancies (135–137). 8H9, a monoclonal antibody recognizing B7-H3, has been in clinical trials for more than 10 years; an 8H9 radioconjugate is an important element of a regimen for relapsed CNS neuroblastoma (NCT00089245) (135, 136). More recently, early phase clinical trials with a tumor specific anti-B7-H3 monoclonal antibody (MGA271) demonstrated safety and efficacy in adult malignancies (138). Our group has developed an active CAR targeting B7-H3 containing the scFv derived from MGA271 and efficacy is currently being explored in neuroblastoma (139, 140).
Anaplastic Lymphoma Kinase (ALK)
Several groups have identified anaplastic lymphoma kinase (ALK) as a potential oncogene in neuroblastoma (141–143). ALK is a receptor tyrosine kinase and, similar to GPC2, its expression is primarily restricted to the central and peripheral nervous system during fetal development (144). ALK regulates cell proliferation, differentiation, and apoptosis and has been implicated in many signaling pathways including PI3K/AKT, RAS/MAPK, and STAT3 (145). Activating mutations occur almost universally in familial neuroblastoma but also occur in a sizable percentage of sporadic neuroblastoma cases. Additionally, 15–20% of neuroblastoma patients overexpress wild type ALK in the absence of an activating mutation (146).
Anti-ALK CARs with a 4-1BB costimulatory domain were generated using previously described monoclonal ALK antibodies (147). ALK CAR T cells demonstrated in vitro activity but had limited efficacy in vivo in xenograft models of neuroblastoma (114). Investigations into the reasons for limited CAR efficacy demonstrated that ALK expression on the neuroblastoma cell lines used was below the threshold of antigen expression required for CAR activity. This finding demonstrates the importance of antigen density for CAR T cell efficacy (115–121).
Neural Cell Adhesion Molecule (NCAM/CD56)
NCAM (CD56) is another glycoprotein that is important in neural development and is overexpressed on neuroblastoma (148). Similar to ALK and GPC2, it is overexpressed on tumors of neuroendocrine origin (149). It is also expressed on normal tissues, including most prominently on natural killer (NK) cells. Phase I and II clinical trials had demonstrated a favorable safety profile of anti-CD56 ADCs in solid tumors such as small cell lung cancer (150). The high and homogeneous expression on neuroblastoma and the limited toxicity of antibody-based therapy led one group to develop a CAR directed against CD56. This second generation CAR with a CD28 costimulation domain controlled tumor burden in a xenograft neuroblastoma model, but had only modest effects on survival (151). CD56 CAR T cells are being studied in clinical trials for relapsed multiple myeloma and for relapsed AML (NCT03473496, NCT03473457), though there are not yet published reports of any patient treated. Further investigation into CD56 as a target in neuroblastoma is warranted but off-tumor toxicity will need to be carefully monitored given significant normal tissue expression.
Natural Killer (NK) Cell Adoptive Therapy
NK cells have long been recognized as important in neuroblastoma and killer cell immunoglobulin-like receptors (KIR) haplotypes are strongly correlated to survival (152–154). NK cells lack the specificity of T cells, but they have the capacity to kill infected and malignant cells without the prerequisite priming and sensitization to peptide-MHC complexes on the target cell surface. Instead, NK activity is regulated by a balance of activating and inhibitory receptors (155). Several trials are underway in which neuroblastoma patients receive adoptively transferred ex vivo expanded but unmanipulated NK cells (NCT02573896, NCT01857934, NCT02650648, NCT03209869).
Given their importance in control of neuroblastoma, researchers have attempted to augment the anti-tumor effects of NK cells in by imparting them with tumor antigen specific CARs. One group generated patient-derived NK cells expressing a second generation GD2-specific CAR, and demonstrated significant improvement in cytotoxicity against primary patient neuroblastoma cells compared to NK cells without a CAR (156). Similarly, expressing the GD2-CAR in an NK-92 cell line promoted in vitro cytotoxicity against neuroblastoma cell lines that were resistant to killing by the parental NK-92 cell line (157). NK cells do not have the same proliferative capacity as T cells, and clinical trials of adoptively transferred NK cells are often marked by short persistence and disappointing anti-tumor effect (158). The persistence of NK cells and invariant NK T cells can be increased by constitutive secretion of IL-15, an approach being studied in clinical trials for children with neuroblastoma at Baylor College of Medicine (NCT03294954) (159, 160).
Engineering a Successful CAR T Cell Product
In contrast to standard chemotherapy or “off the shelf” immunotherapies such as monoclonal antibodies, an important consideration for CAR T cell therapy is the ability to manufacture adequate quantities of a viable, maximally efficacious T cell product. Some patients have poor expansion and inadequate production of CAR T cells. One group hypothesized that myeloid derived suppressor cells (MDSC) in the apheresis product may interfere with T cell expansion, and found higher proportions of monocytes in PBMC concentrates to inversely correlate with fold expansion of CD19 and GD2 CAR T cells (161). CAR T cell quality is of particular concern for patients who have undergone chemotherapy, radiation, and/or stem cell transplant, all important elements of upfront neuroblastoma therapy. Data presented in abstract form describe T cell fitness in PBMC samples collected at diagnosis and after each cycle of chemotherapy from children with a wide variety of cancers including neuroblastoma. These data suggest that after chemotherapy, patients develop poor CAR T cell potential, defined by a low proportion of naïve T cells, mitochondrial dysfunction, and poor spare respiratory capacity (162). Further study is warranted to understand this phenomenon and whether it ultimately impacts CAR T cell efficacy in patients, as highly active CD19 CAR T cells have been successfully generated from most patients with heavily pretreated ALL (18, 19).
Overcoming Immunosuppressive Tumor Microenvironment (TME)
The immunosuppressive tumor microenvironment (TME) presents a significant barrier to successful CAR T cell therapy for neuroblastoma. Neuroblastoma tumors are intermixed with a suppressive cell population that includes tumor associated macrophages (TAMs) and regulatory T cells (Tregs). Presence of these cells predicts poor outcomes (34, 163). Tumors also express inhibitory ligands such as PD-L1 that dampen T cell responses (164–167). Furthermore, the TME contains an array of soluble factors such as TGF-β and IL-10 that act to directly inhibit T cells (34, 168–171). Finally, physical barriers such as stroma, extracellular matrix (ECM) and tumor associated vasculature prevent tumor infiltrating T cells from easily accessing their target (172–175).
Enhancing Trafficking to Neuroblastoma
For CAR T cell therapy of hematologic malignancies, the majority of malignant cells are located within the hematopoietic system. Solid tumors are not as readily accessible, a fact supported by data from early clinical trials in which GD2 CAR T cells were easily detectable in peripheral blood but rarely seen in post-treatment tumor biopsies (27). Optimal trafficking of T cells occurs when the effector T cells express a chemokine receptor that is complementary to chemokines that are rich in the tumor microenvironment, either excreted by tumor cells or surrounding tumor stroma. Expression of chemokine CCL2 has long been associated with more effective immune responses against neuroblastoma and it is secreted by neuroblastoma cell lines and primary tumor cells (176, 177). However, CAR T cells generated from neuroblastoma patients were found to have very low expression of the corresponding chemokine receptor, CCR2, despite expressing high levels of other chemokine receptors. Transgenic expression of CCR2b on GD2 CAR T cells in a neuroblastoma xenograft model improved kinetics of CAR T cell chemotaxis and greater anti-tumor efficacy (177).
Depleting Suppressive Immune Cells
Assuming adoptively transferred T cells migrate appropriately to a solid tumor, they must circumvent many immunosuppressive factors within the TME. Many researchers are working to overcome this barrier. One strategy involves depleting suppressive immune cells. In a xenograft model of osteosarcoma, Long et al. observed that MDSCs decreased GD2 CAR T cell efficacy. When mice were treated with ATRA, which can induce differentiation of immature myeloid cells to a non-suppressive subtype (178), they had fewer suppressive MDSCs and there was a modest improvement in tumor control and survival (35). Alternatively, CARs themselves can be redirected against TAMs and regulatory T cells. One group took advantage of the dual specificity of CD123 CAR T cells against both Hodgkin lymphoma cells and TAMs. They found that with this strategy, they could target and eliminate TAMs and achieve durable remissions in Hodgkin lymphoma xenograft models (179).
Overcoming Inhibitory Signals
To evade the immune system, tumors express PD-L1, the ligand for PD-1, an inhibitory receptor on T cells. Engagement of this receptor dampens the native immune response (180) and blocking antibodies can “remove the brakes” and prompt an anti-tumor response, leading to success in early phase clinical trials (181–184). Neuroblastoma in particular was found to more frequently express PD-L1 than most other pediatric solid tumors. Additionally, PD-L1 expression [defined as >1% positive in tumor cells by immunohistochemistry, in line with some adult carcinoma scoring systems (185)] in neuroblastoma is associated with inferior survival (167).
PD-L1 upregulation on solid tumors can limit the efficacy of tumor-specific CAR T cells (186). Liu et al. postulated that they could improve anti-tumor control by combining CAR T cell therapy with a “switch-receptor” that would interrupt PD-1 inhibitory signaling. They endowed multiple CAR T cells with an additional chimeric receptor with a PD-1 extracellular domain directly connected to an intracellular CD28 co-receptor to provide costimulation and activation of T cells upon engagement with PD-L1. In all models, the switch receptor augmented CAR T cell function, and importantly, to a greater degree than anti-PD-1 monoclonal antibodies (39).
Interfering With Inhibitory Soluble Factors
When neuroblastoma directed CAR T cells penetrate the suppressive immune milieu, they inevitably encounter suppressive factors including soluble cytokines that can suppress T cell function. These factors can be secreted by tumor cells or by surrounding stromal cells and include TGF-β, IL-10, galectin-1, and galectin-3 (34, 168–171); they represent potential targets to enhance CAR T cell efficacy. TGF-β in particular has importance in the neuroblastoma TME. Elevated levels of TGF-β transcripts in primary neuroblastoma samples were associated with shorter EFS (187), and blockade of TGF-β induced a more potent NK cell response in conjunction with anti-GD2 monoclonal antibody in a neuroblastoma xenograft model (188). T cells engineered to express dominant negative TGF-β receptors have been shown in a number of settings to improve efficacy of adoptive T cell therapy (189–191). This strategy was recently corroborated in a preclinical CAR model using an anti-prostate-specific membrane antigen (PSMA) CAR (192). PSMA CAR T cells coexpressed with the dominant negative receptor demonstrated increased proliferation, cytokine secretion, exhaustion resistance, persistence, and anti-tumor efficacy. With such pre-clinical promise, this construct has been incorporated into a clinical trial (NCT03089203).
Targeting Tumor Stroma
CAR T cells must penetrate physical barriers within the tumor stromal compartment that augment tumor growth and prevent infiltration of surveilling immune cells. Cancer associated fibroblasts (CAF) are the dominant cell type in the tumor stroma and express fibroblast activating protein-α (FAP) at high levels (173, 174, 193, 194). In a murine model of lung cancer, the efficacy of CAR T cells targeting the Ephrin Receptor tyrosine kinase EphA2 was enhanced by coadministration of anti FAP CAR T cells (195), providing proof of principle that anti-stromal CAR T cells can contribute to successful CAR T cell therapy in the solid tumor setting. Though this CAR has not yet been tested in neuroblastoma models, CAFs derived from primary neuroblastoma samples universally express FAP and enhance tumor engraftment and growth, and thus represent a potential target within the neuroblastoma TME (196).
T cell infiltration into tumors requires degradation of ECM proteins, including heparan sulfate proteoglycans (HSPG) (172). HSPGs are expressed on neuronal tissue during development and neuroblastoma cells are known to express some HSPGs at high levels (127). Activated T cells secrete heparanase to actively break down HSPG (197), but ex vivo culture of T cells causes downregulation of heparanase and abrogates their ability to degrade ECM (198). Expression of heparanase in a GD2 CAR T cell significantly improved tumor infiltration and antitumor activity in a neuroblastoma xenograft model (198), validating this as a potential method to improve CAR T cell therapy in stromal-rich tumors.
The immunosuppressive tumor vasculature presents a third physical barrier that may be a viable target to improve CAR T cell therapy. Vascular endothelial growth factor (VEGF) is a proangiogenic factor secreted by tumors, and can directly suppress immune cell infiltration of tumors (175). In a neuroblastoma xenograft model, anti-GD2 CAR T cells co-administered with the anti-VEGF antibody bevacizumab had superior anti-tumor activity over GD2 CAR T cells alone, thought to be primarily related to increased tumor infiltration by T cells (140).
Conclusions
Immunotherapy with anti-GD2 antibodies has revolutionized the care of neuroblastoma patients, but there is still a great need for novel therapies for the patients with refractory or relapsed high risk disease. Early clinical trials with CAR T cells in neuroblastoma have demonstrated safety and shown some objective clinical responses. They have also provided insight into reasons for limited success, including lack of T cell persistence, difficulty in target antigen selection, and a suppressive tumor microenvironment. These challenges are universal in the CAR T cell field, in particular for solid tumors like neuroblastoma, and there are significant efforts underway to improve upon each of these domains. Successful CAR T cell therapy in neuroblastoma will require rational engineering approaches that address each of the above-mentioned barriers. Many studies presented in this review have encouraging pre-clinical results and thoughtful incorporation of some of these strategies into clinical trials will ultimately validate CAR T cells to treat neuroblastoma and improve patient outcomes.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a St. Baldrick's-Stand Up to Cancer Dream Team Translational Research Grant (SU2C-AACR-DT-27-17). Stand Up to Cancer is a division of the Entertainment Industry Foundation. Research Grants are administered by the American Association for Cancer Research. RM is supported by a Be Brooks Brave Fund St. Baldrick's Scholar Award, a Sarcoma Alliance for Research Through Collaboration (SARC) Career Development Award, and a Hyundai Hope on Wheels Young Investigator Award. RR is the Rachleff Pediatric Hematology/Oncology fellow and the Tashia and John Morgridge endowed postdoctoral fellow, supported by a grant from the Stanford University Children's Health Research Institute (CHRI).
Conflict of Interest Statement
RM has a pending patent application for the use of GD2 CAR T cells in H3K27M mutant gliomas.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Smith MA, Altekruse SF, Adamson PC, Reaman GH, Seibel NL. Declining childhood and adolescent cancer mortality. Cancer (2014) 120:2497–506. doi: 10.1002/cncr.28748
2. Matthay KK, Maris JM, Schleiermacher G, Nakagawara A, Mackall CL, Diller L, et al. Neuroblastoma. Nat Rev Dis Primers (2016) 2:16078. doi: 10.1038/nrdp.2016.78
3. Ries LAG SMA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR (eds). Cancer Incidence and Survival amoung Children and Adolescents: United States SEER Program 1975-1995. Bethesda, MD: National Cancer Institute, NIH (1999).
4. Strother DR, London WB, Schmidt ML, Brodeur GM, Shimada H, Thorner P, et al. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol. (2012) 30:1842–8. doi: 10.1200/JCO.2011.37.9990
5. Kreissman SG, Seeger RC, Matthay KK, London WB, Sposto R, Grupp SA, et al. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. Lancet Oncol. (2013) 14:999–1008. doi: 10.1016/S1470-2045(13)70309-7
6. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. (2010) 363:1324–34. doi: 10.1056/NEJMoa0911123
7. Park JR, Kreissman SG, London WB, Naranjo A, Cohn SL, Hogarty MD, et al. A phase III randomized clinical trial (RCT) of tandem myeloablative autologous stem cell transplant (ASCT) using peripheral blood stem cell (PBSC) as consolidation therapy for high-risk neuroblastoma (HR-NB): a Children's Oncology Group (COG) study. J Clin Oncol. (2016) 34(Suppl. 18):LBA3-LBA. doi: 10.1200/JCO.2016.34.15_suppl.LBA3
8. Pinto NR, Applebaum MA, Volchenboum SL, Matthay KK, London WB, Ambros PF, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. (2015) 33:3008–17. doi: 10.1200/JCO.2014.59.4648
9. Laverdiere C, Liu Q, Yasui Y, Nathan PC, Gurney JG, Stovall M, et al. Long-term outcomes in survivors of neuroblastoma: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. (2009) 101:1131–40. doi: 10.1093/jnci/djp230
10. Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol. (2001) 19:4189–94. doi: 10.1200/JCO.2001.19.22.4189
11. Cheung NK, Cheung IY, Kramer K, Modak S, Kuk D, Pandit-Taskar N, et al. Key role for myeloid cells: phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. Int J Cancer (2014) 135:2199–205. doi: 10.1002/ijc.28851
12. Gilman AL, Ozkaynak MF, Matthay KK, Krailo M, Yu AL, Gan J, et al. Phase I study of ch14.18 with granulocyte-macrophage colony-stimulating factor and interleukin-2 in children with neuroblastoma after autologous bone marrow transplantation or stem-cell rescue: a report from the Children's Oncology Group. J Clin Oncol. (2009) 27:85–91. doi: 10.1200/JCO.2006.10.3564
13. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol. (2012) 30:3264–70. doi: 10.1200/JCO.2011.41.3807
14. Kushner BH, Cheung NK. Induction for high-risk neuroblastoma. Pediatr Blood Cancer (2007) 49:221–3. doi: 10.1002/pbc.21206
15. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
16. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
17. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
18. Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood (2017) 129:3322–31. doi: 10.1182/blood-2017-02-769208
19. Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (2015) 385:517–28. doi: 10.1016/S0140-6736(14)61403-3
20. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. (2018) 24:20–8. doi: 10.1038/nm.4441
21. Shusterman S, London WB, Gillies SD, Hank JA, Voss SD, Seeger RC, et al. Antitumor activity of hu14.18-IL2 in patients with relapsed/refractory neuroblastoma: a Children's Oncology Group (COG) phase II study. J Clin Oncol. (2010) 28:4969–75. doi: 10.1200/JCO.2009.27.8861
22. Pardridge WM. The blood-brain barrier: bottleneck in brain drug development. NeuroRx (2005) 2:3–14. doi: 10.1602/neurorx.2.1.3
23. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
24. Abramson JS, McGree B, Noyes S, Plummer S, Wong C, Chen YB, et al. Anti-CD19 CAR T cells in CNS diffuse large-B-cell lymphoma. N Engl J Med. (2017) 377:783–4. doi: 10.1056/NEJMc1704610
25. Kushner BH, Ostrovnaya I, Cheung IY, Kuk D, Kramer K, Modak S, et al. Prolonged progression-free survival after consolidating second or later remissions of neuroblastoma with Anti-GD2 immunotherapy and isotretinoin: a prospective Phase II study. Oncoimmunology (2015) 4:e1016704. doi: 10.1080/2162402X.2015.1016704
26. Matthay KK, Brisse H, Couanet D, Couturier J, Benard J, Mosseri V, et al. Central nervous system metastases in neuroblastoma: radiologic, clinical, and biologic features in 23 patients. Cancer (2003) 98:155–65. doi: 10.1002/cncr.11448
27. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. (2008) 14:1264–70. doi: 10.1038/nm.1882
28. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
29. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. (2007) 15:825–33. doi: 10.1038/sj.mt.6300104
30. Straathof K, Flutter B, Wallace R, Thomas S, Cheung G, Collura A, et al. (eds). A Cancer Research UK Phase I Trial of Anti-GD2 Chimeric Antigen Receptor (CAR) Transduced T-Cells (1RG-CART) in Patients With Relapsed or Refractory Neuroblastoma. Chicago, IL: American Association for Cancer Research (2018).
31. Yang L, Ma X, Liu Y-C, Zhao W, Yu L, Qin M, et al. Chimeric antigen receptor 4SCAR-GD2-modified T cells targeting high-risk and recurrent neuroblastoma: a phase II multi-center trial in China. Blood (2017) 130(Suppl. 1):3335.
32. Bosse KR, Raman P, Zhu Z, Lane M, Martinez D, Heitzeneder S, et al. Identification of GPC2 as an oncoprotein and candidate immunotherapeutic target in high-risk neuroblastoma. Cancer Cell (2017) 32:295–309 e12. doi: 10.1016/j.ccell.2017.08.003
33. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol. (2012) 2:194. doi: 10.3389/fonc.2012.00194
34. Asgharzadeh S, Salo JA, Ji L, Oberthuer A, Fischer M, Berthold F, et al. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J Clin Oncol. (2012) 30:3525–32. doi: 10.1200/JCO.2011.40.9169
35. Long AH, Highfill SL, Cui Y, Smith JP, Walker AJ, Ramakrishna S, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. (2016) 4:869–80. doi: 10.1158/2326-6066.CIR-15-0230
36. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167:419–32 e16. doi: 10.1016/j.cell.2016.09.011
37. Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA. (2016) 113:E459–68. doi: 10.1073/pnas.1524155113
38. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. (2018) 24:352–9. doi: 10.1038/nm.4478
39. Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T Cells in Advanced Solid Tumors. Cancer Res. (2016) 76:1578–90. doi: 10.1158/0008-5472.CAN-15-2524
40. Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. (2017) 7:737. doi: 10.1038/s41598-017-00462-8
41. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci Rep. (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
42. Pinto N, Kuenkele A, Gardner RA, Finney O, Brakke H, Brown C, et al., (eds). ENCIT-01: A Phase 1 Study of Autologous T-Cells Lentivirally Transduced to Express CD171-Specific chimeric antigen receptors for recurrent/refractory high-risk neuroblastoma. San Francisco, CA: Advances in Neuroblastoma Research (2018).
43. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. (2017) 25:2214–24. doi: 10.1016/j.ymthe.2017.05.012
44. Mujoo K, Cheresh DA, Yang HM, Reisfeld RA. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. (1987) 47:1098–104.
45. Ladisch S, Li R, Olson E. Ceramide structure predicts tumor ganglioside immunosuppressive activity. Proc Natl Acad Sci USA. (1994) 91:1974–8. doi: 10.1073/pnas.91.5.1974
46. Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK. Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer (2001) 94:228–36. doi: 10.1002/ijc.1457
47. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. (2006) 12(20 Pt 1):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
48. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. (2011) 121:1822–6. doi: 10.1172/JCI46110
49. Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood (2008) 112:2261–71. doi: 10.1182/blood-2007-12-128843
50. Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. (1996) 2:551–5. doi: 10.1038/nm0596-551
51. O'Reilly RJ, Doubrovina E, Trivedi D, Hasan A, Kollen W, Koehne G. Adoptive transfer of antigen-specific T-cells of donor type for immunotherapy of viral infections following allogeneic hematopoietic cell transplants. Immunol Res. (2007) 38:237–50. doi: 10.1007/s12026-007-0059-2
52. Bollard CM, Gottschalk S, Leen AM, Weiss H, Straathof KC, Carrum G, et al. Complete responses of relapsed lymphoma following genetic modification of tumor-antigen presenting cells and T-lymphocyte transfer. Blood (2007) 110:2838–45. doi: 10.1182/blood-2007-05-091280
53. Straathof KC, Bollard CM, Popat U, Huls MH, Lopez T, Morriss MC, et al. Treatment of nasopharyngeal carcinoma with Epstein-Barr virus–specific T lymphocytes. Blood (2005) 105:1898–904. doi: 10.1182/blood-2004-07-2975
54. Yang Y, Kohler ME, Chien CD, Sauter CT, Jacoby E, Yan C, et al. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Sci Transl Med. (2017) 9:eaag1209. doi: 10.1126/scitranslmed.aag1209
55. Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. (2005) 12:933–41. doi: 10.1016/j.ymthe.2005.04.016
56. Hombach AA, Abken H. Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int J Cancer (2011) 129:2935–44. doi: 10.1002/ijc.25960
57. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. (2015) 21:581–90. doi: 10.1038/nm.3838
58. Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology (2018) 7:e1433518. doi: 10.1080/2162402X.2018.1433518
59. Nakamura K, Tanaka Y, Shitara K, Hanai N. Construction of humanized anti-ganglioside monoclonal antibodies with potent immune effector functions. Cancer Immunol Immunother. (2001) 50:275–84. doi: 10.1007/PL00006689
60. Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLoS ONE (2016) 11:e0152196. doi: 10.1371/journal.pone.0152196
61. Novak-Hofer I, Amstutz HP, Haldemann A, Blaser K, Morgenthaler JJ, Blauenstein P, et al. Radioimmunolocalization of neuroblastoma xenografts with chimeric antibody chCE7. J Nucl Med. (1992) 33:231–6.
62. Hong H, Stastny M, Brown C, Chang WC, Ostberg JR, Forman SJ, et al. Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J Immunother. (2014) 37:93–104. doi: 10.1097/CJI.0000000000000018
63. Meli ML, Carrel F, Waibel R, Amstutz H, Crompton N, Jaussi R, et al. Anti-neuroblastoma antibody chCE7 binds to an isoform of L1-CAM present in renal carcinoma cells. Int J Cancer (1999) 83:401–8. doi: 10.1002/(SICI)1097-0215(19991029)83:3<401::AID-IJC17>3.0.CO;2-A
64. Novak-Hofer I, Amstutz HP, Morgenthaler JJ, Schubiger PA. Internalization and degradation of monoclonal antibody chCE7 by human neuroblastoma cells. Int J Cancer (1994) 57:427–32. doi: 10.1002/ijc.2910570322
65. Gonzalez S, Naranjo A, Serrano LM, Chang WC, Wright CL, Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J Gene Med. (2004) 6:704–11. doi: 10.1002/jgm.489
66. Berger C, Flowers ME, Warren EH, Riddell SR. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood (2006) 107:2294–302. doi: 10.1182/blood-2005-08-3503
67. Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood (2011) 118:1255–63. doi: 10.1182/blood-2011-02-337360
68. Paszkiewicz PJ, Frassle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. (2016) 126:4262–72. doi: 10.1172/JCI84813
69. Kunkele A, Taraseviciute A, Finn LS, Johnson AJ, Berger C, Finney O, et al. Preclinical assessment of CD171-directed CAR T-cell adoptive therapy for childhood neuroblastoma: CE7 epitope target safety and product manufacturing feasibility. Clin Cancer Res. (2017) 23:466–77. doi: 10.1158/1078-0432.CCR-16-0354
70. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30:492–500. doi: 10.1038/leu.2015.247
71. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
72. Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. (2016) 8:355ra116. doi: 10.1126/scitranslmed.aaf8621
73. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. (2010) 16:1245–56. doi: 10.1016/j.bbmt.2010.03.014
74. Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. (2004) 173:7125–30. doi: 10.4049/jimmunol.173.12.7125
75. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. (2006) 66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160
76. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell (2009) 138:30–50. doi: 10.1016/j.cell.2009.06.036
77. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature (1993) 362:758–61. doi: 10.1038/362758a0
78. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
79. Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. (2011) 121:2350–60. doi: 10.1172/JCI46102
80. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
81. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
82. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
83. Yao X, Ahmadzadeh M, Lu YC, Liewehr DJ, Dudley ME, Liu F, et al. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood (2012) 119:5688–96. doi: 10.1182/blood-2011-10-386482
84. Cui Y, Zhang H, Meadors J, Poon R, Guimond M, Mackall CL. Harnessing the physiology of lymphopenia to support adoptive immunotherapy in lymphoreplete hosts. Blood (2009) 114:3831–40. doi: 10.1182/blood-2009-03-212134
85. Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. (2005) 202:907–12. doi: 10.1084/jem.20050732
86. Fry TJ, Connick E, Falloon J, Lederman MM, Liewehr DJ, Spritzler J, et al. A potential role for interleukin-7 in T-cell homeostasis. Blood (2001) 97:2983–90. doi: 10.1182/blood.V97.10.2983
87. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia (2018) 32:520–31. doi: 10.1038/leu.2017.226
88. Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci USA. (2016) 113:E7788–97. doi: 10.1073/pnas.1610544113
89. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
90. Sportes C, Babb RR, Krumlauf MC, Hakim FT, Steinberg SM, Chow CK, et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin Cancer Res. (2010) 16:727–35. doi: 10.1158/1078-0432.CCR-09-1303
91. Lacy MQ, Jacobus S, Blood EA, Kay NE, Rajkumar SV, Greipp PR. Phase II study of interleukin-12 for treatment of plateau phase multiple myeloma (E1A96): a trial of the Eastern Cooperative Oncology Group. Leuk Res. (2009) 33:1485–9. doi: 10.1016/j.leukres.2009.01.020
92. Vera JF, Hoyos V, Savoldo B, Quintarelli C, Giordano Attianese GM, Leen AM, et al. Genetic manipulation of tumor-specific cytotoxic T lymphocytes to restore responsiveness to IL-7. Mol Ther. (2009) 17:880–8. doi: 10.1038/mt.2009.34
93. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood (2010) 115:3508–19. doi: 10.1182/blood-2009-09-241398
94. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood (2012) 119:4133–41. doi: 10.1182/blood-2011-12-400044
95. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia (2010) 24:1160–70. doi: 10.1038/leu.2010.75
96. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
97. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. (2014) 74:5195–205. doi: 10.1158/0008-5472.CAN-14-0697
98. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
99. Zenatti PP, Ribeiro D, Li W, Zuurbier L, Silva MC, Paganin M, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. (2011) 43:932–9. doi: 10.1038/ng.924
100. Lammie G, Cheung N, Gerald W, Rosenblum M, Cordoncardo C. Ganglioside gd expression in the human nervous-system and in neuroblastomas - an immunohistochemical study. Int J Oncol. (1993) 3:909–15. doi: 10.3892/ijo.3.5.909
101. Schulz G, Cheresh DA, Varki NM, Yu A, Staffileno LK, Reisfeld RA. Detection of ganglioside GD2 in tumor tissues and sera of neuroblastoma patients. Cancer Res. (1984) 44(12 Pt 1):5914–20.
102. Svennerholm L, Bostrom K, Fredman P, Jungbjer B, Lekman A, Mansson JE, et al. Gangliosides and allied glycosphingolipids in human peripheral nerve and spinal cord. Biochim Biophys Acta (1994) 1214:115–23. doi: 10.1016/0005-2760(94)90034-5
103. Mody R, Naranjo A, Van Ryn C, Yu AL, London WB, Shulkin BL, et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): an open-label, randomised, phase 2 trial. Lancet Oncol. (2017) 18:946–57. doi: 10.1016/S1470-2045(17)30355-8
104. Navid F, Sondel PM, Barfield R, Shulkin BL, Kaufman RA, Allay JA, et al. Phase I trial of a novel anti-GD2 monoclonal antibody, Hu14.18K322A, designed to decrease toxicity in children with refractory or recurrent neuroblastoma. J Clin Oncol. (2014) 32:1445–52. doi: 10.1200/JCO.2013.50.4423
105. Kushner BH, Kramer K, Modak S, Cheung NK. Successful multifold dose escalation of anti-GD2 monoclonal antibody 3F8 in patients with neuroblastoma: a phase I study. J Clin Oncol. (2011) 29:1168–74. doi: 10.1200/JCO.2010.28.3317
106. Osenga KL, Hank JA, Albertini MR, Gan J, Sternberg AG, Eickhoff J, et al. A phase I clinical trial of the hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the Children's Oncology Group. Clin Cancer Res. (2006) 12:1750–9. doi: 10.1158/1078-0432.CCR-05-2000
107. Isaacs JD, Greenwood J, Waldmann H. Therapy with monoclonal antibodies. II. The contribution of Fc gamma receptor binding and the influence of C(H)1 and C(H)3 domains on in vivo effector function. J Immunol. (1998) 161:3862–9.
108. Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood (2013) 122:863–71. doi: 10.1182/blood-2013-03-490565
109. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. (2013) 36:133–51. doi: 10.1097/CJI.0b013e3182829903
110. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
111. Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
112. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive Sarcoma. J Clin Oncol. (2015) 33:1688–96. doi: 10.1200/JCO.2014.58.0225
113. Hegde M, DeRenzo CC, Zhang H, Mata M, Gerken C, Shree A, et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J Clin Oncol. (2017) 35(Suppl. 15):10508. doi: 10.1200/JCO.2017.35.15_suppl.10508
114. Walker AJ, Majzner RG, Zhang L, Wanhainen K, Long AH, Nguyen SM, et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol Ther. (2017) 25:2189–201. doi: 10.1016/j.ymthe.2017.06.008
115. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, et al. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther. (2017) 25:1933–45. doi: 10.1016/j.ymthe.2017.04.017
116. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol. (2004) 173:7647–53. doi: 10.4049/jimmunol.173.12.7647
117. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. (2015) 194:911–20. doi: 10.4049/jimmunol.1402346
118. Hombach AA, Gorgens A, Chmielewski M, Murke F, Kimpel J, Giebel B, et al. Superior therapeutic index in lymphoma therapy: CD30(+) CD34(+) hematopoietic stem cells resist a chimeric antigen receptor T-cell attack. Mol Ther. (2016) 24:1423–34. doi: 10.1038/mt.2016.82
119. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. (2015) 75:3505–18. doi: 10.1158/0008-5472.CAN-15-0139
120. Turatti F, Figini M, Balladore E, Alberti P, Casalini P, Marks JD, et al. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J Immunother. (2007) 30:684–93. doi: 10.1097/CJI.0b013e3180de5d90
121. Weijtens ME, Hart EH, Bolhuis RL. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. (2000) 7:35–42. doi: 10.1038/sj.gt.3301051
122. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
123. Majzner RG, Weber EW, Lynn RC, Xu P, Mackall CL. Neurotoxicity associated with a high-affinity GD2 CAR-letter. Cancer Immunol Res. (2018) 6:494–5. doi: 10.1158/2326-6066.CIR-18-0089
124. Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. (2008) 9:224. doi: 10.1186/gb-2008-9-5-224
125. Stipp CS, Litwack ED, Lander AD. Cerebroglycan: an integral membrane heparan sulfate proteoglycan that is unique to the developing nervous system and expressed specifically during neuronal differentiation. J Cell Biol. (1994) 124:149–60. doi: 10.1083/jcb.124.1.149
126. Ivins JK, Litwack ED, Kumbasar A, Stipp CS, Lander AD. Cerebroglycan, a developmentally regulated cell-surface heparan sulfate proteoglycan, is expressed on developing axons and growth cones. Dev Biol. (1997) 184:320–32. doi: 10.1006/dbio.1997.8532
127. Li N, Fu H, Hewitt SM, Dimitrov DS, Ho M. Therapeutically targeting glypican-2 via single-domain antibody-based chimeric antigen receptors and immunotoxins in neuroblastoma. Proc Natl Acad Sci USA. (2017) 114:E6623–31. doi: 10.1073/pnas.1706055114
128. Baral A, Ye HX, Jiang PC, Yao Y, Mao Y. B7-H3 and B7-H1 expression in cerebral spinal fluid and tumor tissue correlates with the malignancy grade of glioma patients. Oncol Lett. (2014) 8:1195–201. doi: 10.3892/ol.2014.2268
129. Gregorio A, Corrias MV, Castriconi R, Dondero A, Mosconi M, Gambini C, et al. Small round blue cell tumours: diagnostic and prognostic usefulness of the expression of B7-H3 surface molecule. Histopathology (2008) 53:73–80. doi: 10.1111/j.1365-2559.2008.03070.x
130. Zhao JL, Chen FL, Zhou Q, Pan W, Wang XH, Xu J, et al. B7-H3 protein expression in a murine model of osteosarcoma. Oncol Lett. (2016) 12:383–6. doi: 10.3892/ol.2016.4675
131. Zhou Z, Luther N, Ibrahim GM, Hawkins C, Vibhakar R, Handler MH, et al. B7-H3, a potential therapeutic target, is expressed in diffuse intrinsic pontine glioma. J Neurooncol. (2013) 111:257–64. doi: 10.1007/s11060-012-1021-2
132. Picarda E, Ohaegbulam KC, Zang X. Molecular pathways: targeting B7-H3 (CD276) for human cancer immunotherapy. Clin Cancer Res. (2016) 22:3425–31. doi: 10.1158/1078-0432.CCR-15-2428
133. Tekle C, Nygren MK, Chen YW, Dybsjord I, Nesland JM, Maelandsmo GM, et al. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int J Cancer (2012) 130:2282–90. doi: 10.1002/ijc.26238
134. Ye Z, Zheng Z, Li X, Zhu Y, Zhong Z, Peng L, et al. B7-H3 overexpression predicts poor survival of cancer patients: a meta-analysis. Cell Physiol Biochem. (2016) 39:1568–80. doi: 10.1159/000447859
135. Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, et al. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. J Neurooncol. (2010) 97:409–18. doi: 10.1007/s11060-009-0038-7
136. Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Tomlinson U, Wolden SL, et al. A curative approach to central nervous system metastases of neuroblastoma. J Clin Oncol. (2017) 35(Suppl. 15):10545. doi: 10.1200/JCO.2017.35.15_suppl.10545
137. Souweidane MM, Kramer K, Pandit-Taskar N, Zhou Z, Haque S, Zanzonico P, et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: a single-centre, dose-escalation, phase 1 trial. Lancet Oncol. (2018). 19:1040–50. doi: 10.1016/S1470-2045(18)30322-X
138. Powderly J, Cote G, Flaherty K, Szmulewitz RZ, Ribas A, Weber J, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J Immunother Cancer (2015) 3(Suppl. 2):O8. doi: 10.1186/2051-1426-3-S2-O8
139. Majzner RG, Nellan A, Heitzeneder S, Theruvath J, Mackall CL. (eds). CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Pittsburgh, PA: American Society of Pediatric Hematology/Oncology (2018).
140. Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology (2017) 7:e1378843. doi: 10.1080/2162402X.2017.1378843
141. Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature (2008) 455:971–4. doi: 10.1038/nature07399
142. George RE, Sanda T, Hanna M, Frohling S, Luther W 2nd, Zhang J, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature (2008) 455:975–8. doi: 10.1038/nature07397
143. Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature (2008) 455:930–5. doi: 10.1038/nature07261
144. Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene (1997) 14:439–49. doi: 10.1038/sj.onc.1200849
145. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer (2008) 8:11–23. doi: 10.1038/nrc2291
146. De Brouwer S, De Preter K, Kumps C, Zabrocki P, Porcu M, Westerhout EM, et al. Meta-analysis of neuroblastomas reveals a skewed ALK mutation spectrum in tumors with MYCN amplification. Clin Cancer Res. (2010) 16:4353–62. doi: 10.1158/1078-0432.CCR-09-2660
147. Moog-Lutz C, Degoutin J, Gouzi JY, Frobert Y, Brunet-de Carvalho N, Bureau J, et al. Activation and inhibition of anaplastic lymphoma kinase receptor tyrosine kinase by monoclonal antibodies and absence of agonist activity of pleiotrophin. J Biol Chem. (2005) 280:26039–48. doi: 10.1074/jbc.M501972200
148. Zeromski J, Nyczak E, Dyszkiewicz W. Significance of cell adhesion molecules, CD56/NCAM in particular, in human tumor growth and spreading. Folia Histochem Cytobiol. (2001) 39 (Suppl. 2):36–7.
149. Wachowiak R, Rawnaq T, Metzger R, Quaas A, Fiegel H, Kahler N, et al. Universal expression of cell adhesion molecule NCAM in neuroblastoma in contrast to L1: implications for different roles in tumor biology of neuroblastoma? Pediatr Surg Int. (2008) 24:1361–4. doi: 10.1007/s00383-008-2264-z
150. Shah MH, Lorigan P, O'Brien ME, Fossella FV, Moore KN, Bhatia S, et al. Phase I study of IMGN901, a CD56-targeting antibody-drug conjugate, in patients with CD56-positive solid tumors. Invest New Drugs (2016) 34:290–9. doi: 10.1007/s10637-016-0336-9
151. Crossland DL, Denning WL, Ang S, Olivares S, Mi T, Switzer K, et al. Antitumor activity of CD56-chimeric antigen receptor T cells in neuroblastoma and SCLC models. Oncogene (2018) 37:3686–97. doi: 10.1038/s41388-018-0187-2
152. Bottino C, Dondero A, Bellora F, Moretta L, Locatelli F, Pistoia V, et al. Natural killer cells and neuroblastoma: tumor recognition, escape mechanisms, and possible novel immunotherapeutic approaches. Front Immunol. (2014) 5:56. doi: 10.3389/fimmu.2014.00056
153. Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer Res. (2010) 70:9554–61. doi: 10.1158/0008-5472.CAN-10-2211
154. Erbe AK, Wang W, Carmichael L, Kim K, Mendonca EA, Song Y, et al. Neuroblastoma patients' KIR and KIR-ligand genotypes influence clinical outcome for dinutuximab-based immunotherapy: a report from the children's oncology group. Clin Cancer Res. (2017) 24:189–96. doi: 10.1158/1078-0432.CCR-17-1767
155. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
156. Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. (2009) 15:4857–66. doi: 10.1158/1078-0432.CCR-08-2810
157. Esser R, Muller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. (2012) 16:569–81. doi: 10.1111/j.1582-4934.2011.01343.x
158. Burns LJ, Weisdorf DJ, DeFor TE, Vesole DH, Repka TL, Blazar BR, et al. IL-2-based immunotherapy after autologous transplantation for lymphoma and breast cancer induces immune activation and cytokine release: a phase I/II trial. Bone Marrow Transplant. (2003) 32:177–86. doi: 10.1038/sj.bmt.1704086
159. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood (2014) 124:2824–33. doi: 10.1182/blood-2013-11-541235
160. Heczey A, Huang W, Liu D, Guo L, Marinova E, Wood M, et al., (eds). IND-Enabling Preclinical Evaluation of NKT Cells Expressing a GD2-Specific Chimeric Antigen Receptor and IL-15 for Treating Neuroblastoma. San Francisco, CA: Advances in Neuroblastoma Research (2018).
161. Stroncek DF, Ren J, Lee DW, Tran M, Frodigh SE, Sabatino M, et al. Myeloid cells in peripheral blood mononuclear cell concentrates inhibit the expansion of chimeric antigen receptor T cells. Cytotherapy (2016) 18:893–901. doi: 10.1016/j.jcyt.2016.04.003
162. Das RK, Storm J, Barrett DM. Abstract 1631: T cell dysfunction in pediatric cancer patients at diagnosis and after chemotherapy can limit chimeric antigen receptor potential. Cancer Res. (2018) 78(Suppl. 13):1631 doi: 10.1158/1538-7445.AM2018-1631
163. Mina M, Boldrini R, Citti A, Romania P, D'Alicandro V, De Ioris M, et al. Tumor-infiltrating T lymphocytes improve clinical outcome of therapy-resistant neuroblastoma. Oncoimmunology. (2015) 4:e1019981. doi: 10.1080/2162402X.2015.1019981
164. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
165. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
166. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. (2008) 8:467–77. doi: 10.1038/nri2326
167. Majzner RG, Simon JS, Grosso JF, Martinez D, Pawel BR, Santi M, et al. Assessment of programmed death-ligand 1 expression and tumor-associated immune cells in pediatric cancer tissues. Cancer (2017) 123:3807–15. doi: 10.1002/cncr.30724
168. Morandi F, Croce M, Cangemi G, Barco S, Rigo V, Carlini B, et al. IL-10 and ARG-1 concentrations in bone marrow and peripheral blood of metastatic neuroblastoma patients do not associate with clinical outcome. J Immunol Res. (2015) 2015:718975. doi: 10.1155/2015/718975
169. Scarpa S, Coppa A, Ragano-Caracciolo M, Mincione G, Giuffrida A, Modesti A, et al. Transforming growth factor β regulates differentiation and proliferation of human neuroblastoma. Exp Cell Res. (1996) 229:147–54. doi: 10.1006/excr.1996.0352
170. Seo N, Hayakawa S, Takigawa M, Tokura Y. Interleukin-10 expressed at early tumour sites induces subsequent generation of CD4(+) T-regulatory cells and systemic collapse of antitumour immunity. Immunology (2001) 103:449–57. doi: 10.1046/j.1365-2567.2001.01279.x
171. Soldati R, Berger E, Zenclussen AC, Jorch G, Lode HN, Salatino M, et al. Neuroblastoma triggers an immunoevasive program involving galectin-1-dependent modulation of T cell and dendritic cell compartments. Int J Cancer (2012) 131:1131–41. doi: 10.1002/ijc.26498
172. Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. (1999) 68:729–77. doi: 10.1146/annurev.biochem.68.1.729
173. Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. (1986) 315:1650–9. doi: 10.1056/NEJM198612253152606
174. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
175. Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunol Res. (2001) 23:263–72. doi: 10.1385/IR:23:2-3:263
176. Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi Z, Asgharzadeh S, et al. Natural killer T cells infiltrate neuroblastomas expressing the chemokine CCL2. J Exp Med. (2004) 199:1213–21. doi: 10.1084/jem.20031462
177. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
178. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. (2006) 66:9299–307. doi: 10.1158/0008-5472.CAN-06-1690
179. Ruella M, Klichinsky M, Kenderian SS, Shestova O, Ziober A, Kraft DO, et al. Overcoming the immunosuppressive tumor microenvironment of Hodgkin lymphoma using chimeric antigen receptor T cells. Cancer Discov. (2017) 7:1154–67. doi: 10.1158/2159-8290.CD-16-0850
180. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. (2002) 99:12293–7. doi: 10.1073/pnas.192461099
181. Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. (2015) 372:311–9. doi: 10.1056/NEJMoa1411087
182. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. (2010) 363:711–23. doi: 10.1056/NEJMoa1003466
183. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515:558–62. doi: 10.1038/nature13904
184. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. (2013) 369:122–33. doi: 10.1056/NEJMoa1302369
185. Brahmer JR, Govindan R, Anders RA, Antonia SJ, Sagorsky S, Davies MJ, et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of non-small cell lung cancer (NSCLC). J Immunother Cancer (2018) 6:75. doi: 10.1186/s40425-018-0382-2
186. Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. (2014) 20:4262–73. doi: 10.1158/1078-0432.CCR-13-2627
187. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Vα24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
188. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβR1 blockade with galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
189. Wallace A, Kapoor V, Sun J, Mrass P, Weninger W, Heitjan DF, et al. Transforming growth factor-β receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res. (2008) 14:3966–74. doi: 10.1158/1078-0432.CCR-08-0356
190. Foster AE, Dotti G, Lu A, Khalil M, Brenner MK, Heslop HE, et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J Immunother. (2008) 31:500–5. doi: 10.1097/CJI.0b013e318177092b
191. Zhang L, Yu Z, Muranski P, Palmer DC, Restifo NP, Rosenberg SA, et al. Inhibition of TGF-β signaling in genetically engineered tumor antigen-reactive T cells significantly enhances tumor treatment efficacy. Gene Ther. (2013) 20:575–80. doi: 10.1038/gt.2012.75
192. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
193. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-α. Science (2010) 330:827–30. doi: 10.1126/science.1195300
194. Rettig WJ, Garin-Chesa P, Beresford HR, Oettgen HF, Melamed MR, Old LJ. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc Natl Acad Sci USA. (1988) 85:3110–4. doi: 10.1073/pnas.85.9.3110
195. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
196. Borriello L, Nakata R, Sheard MA, Fernandez GE, Sposto R, Malvar J, et al. Cancer-associated fibroblasts share characteristics and protumorigenic activity with mesenchymal stromal cells. Cancer Res. (2017) 77:5142–57. doi: 10.1158/0008-5472.CAN-16-2586
197. Naparstek Y, Cohen IR, Fuks Z, Vlodavsky I. Activated T lymphocytes produce a matrix-degrading heparan sulphate endoglycosidase. Nature (1984) 310:241–4. doi: 10.1038/310241a0
Keywords: neuroblastoma, pediatric oncology, immunotherapy, CAR T cells, adoptive T cell therapy, clinical trials
Citation: Richards RM, Sotillo E and Majzner RG (2018) CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 9:2380. doi: 10.3389/fimmu.2018.02380
Received: 02 August 2018; Accepted: 25 September 2018;
Published: 16 October 2018.
Edited by:
Avery Dexter Posey Jr., University of Pennsylvania, United StatesReviewed by:
Andras Heczey, Baylor College of Medicine, United StatesDavid Barrett, Children's Hospital of Philadelphia, United States
Copyright © 2018 Richards, Sotillo and Majzner. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robbie G. Majzner, cm1hanpuZXJAc3RhbmZvcmQuZWR1