 Pere Soler-Palacín1,2
Pere Soler-Palacín1,2 Marina Garcia-Prat1,2
Marina Garcia-Prat1,2 Andrea Martín-Nalda1,2
Andrea Martín-Nalda1,2 Clara Franco-Jarava2,3
Clara Franco-Jarava2,3 Jacques G. Rivière1,2Alberto Plaja4
Jacques G. Rivière1,2Alberto Plaja4 Daniela Bezdan5,6Mattia Bosio5,6
Daniela Bezdan5,6Mattia Bosio5,6 Mónica Martínez-Gallo2,3
Mónica Martínez-Gallo2,3 Stephan Ossowski5,6,7
Stephan Ossowski5,6,7 Roger Colobran2,3,4*
Roger Colobran2,3,4*- 1Pediatric Infectious Diseases and Immunodeficiencies Unit, Hospital Universitari Vall d'Hebron, Vall d'Hebron Research Institute, Universitat Autònoma de Barcelona, Barcelona, Spain
- 2Jeffrey Modell Foundation Excellence Center, Barcelona, Spain
- 3Immunology Division, Department of Cell Biology, Physiology and Immunology, Hospital Universitari Vall d'Hebron, Vall d'Hebron Research Institute, Autonomous University of Barcelona, Barcelona, Spain
- 4Department of Clinical and Molecular Genetics, Hospital Universitari Vall d'Hebron, Barcelona, Spain
- 5Centre for Genomic Regulation, The Barcelona Institute of Science and Technology, Barcelona, Spain
- 6Universitat Pompeu Fabra, Barcelona, Spain
- 7Institute of Medical Genetics and Applied Genomics, University of Tübingen, Tübingen, Germany
LRBA deficiency was first described in 2012 as an autosomal recessive disorder caused by biallelic mutations in the LRBA gene (OMIM #614700). It was initially characterized as producing early-onset hypogammaglobulinemia, autoimmune manifestations, susceptibility to inflammatory bowel disease, and recurrent infection. However, further reports expanded this phenotype (including patients without hypogammaglobulinemia) and described LRBA deficiency as a clinically variable syndrome with a wide spectrum of clinical manifestations. We present the case of a female patient who presented with type 1 diabetes, psoriasis, oral thrush, and enlarged liver and spleen at the age of 8 months. She later experienced recurrent bacterial and viral infections, including pneumococcal meningitis and Epstein Barr viremia. She underwent two consecutive stem cell transplants at the age of 8 and 9 years, and ultimately died. Samples from the patient and her parents were subjected to whole exome sequencing, which revealed a homozygous 1-bp insertion in exon 23 of the patient's LRBA gene, resulting in frameshift and premature stop codon. The patient's healthy mother was heterozygous for the mutation and her father tested wild-type. This finding suggested that either one copy of the paternal chromosome 4 bore a deletion including the LRBA locus, or the patient inherited two copies of the mutant maternal LRBA allele. The patient's sequencing data showed a 1-Mb loss of heterozygosity region in chromosome 4, including the LRBA gene. Comparative genomic hybridization array of the patient's and father's genomic DNA yielded normal findings, ruling out genomic copy number abnormalities. Here, we present the first case of LRBA deficiency due to a uniparental disomy (UPD). In contrast to classical Mendelian inheritance, UPD involves inheritance of 2 copies of a chromosomal region from only 1 parent. Specifically, our patient carried a small segmental isodisomy of maternal origin affecting 1 Mb of chromosome 4.
Introduction
Primary immunodeficiencies (PIDs) comprise a range of genetically determined diseases characterized by partial or complete loss of immune system function. Patients with PIDs typically have increased susceptibility to infections and many show evidence of immune dysregulation, manifesting as autoimmunity, allergy, or malignancy (1). In most cases, PIDs are inherited as monogenic disorders, although common variable immunodeficiency (CVID) is a clear exception. CVID is a primary antibody deficiency characterized by hypogammaglobulinaemia and increased susceptibility to infections, particularly of the respiratory tract. CVID shows considerable phenotypic and genetic heterogeneity, and monogenic forms account for < 20% of all cases (2). In the last few years, next-generation sequencing (NGS) technology has accelerated the discovery of autosomal recessive and dominant genes causing various conditions previously included in the clinical diagnosis of CVID (3, 4).
One such condition is LPS-responsive beige-like anchor (LRBA) protein deficiency (OMIM #614700). LRBA deficiency was first described in 2012 and initially characterized as producing early-onset hypogammaglobulinemia, autoimmune manifestations, susceptibility to inflammatory bowel disease, and recurrent infection (5). However, later reports expanded this phenotype to include patients without hypogammaglobulinaemia and described LRBA deficiency as a clinically variable syndrome with a wide spectrum of manifestations (6, 7).
LRBA is a cytosolic protein located in the endoplasmic reticulum, trans-Golgi apparatus, endocytosis vesicles, and lysosomes. It is ubiquitously expressed by almost all cell types, but shows higher expression levels in immune effector cells. LRBA participates in polarized vesicle trafficking and is probably involved in other critical cellular processes (5).
LRBA deficiency is an autosomal recessive disorder caused by biallelic mutations in the LRBA gene. Concordantly, all reported patients with a molecular diagnosis of this condition carry homozygous or compound heterozygous mutations. These mutations are distributed throughout the gene and essentially include missense mutations, splice site mutations, small indels, and nonsense mutations (6). Large structural rearrangements affecting several exons of LRBA have also been described in a few patients (7, 8).
Here, we present the case of a patient with LRBA deficiency due to a uniparental disomy (UPD), the first such case described. In contrast to classical Mendelian inheritance, UPD involves inheritance of two copies of a region of a chromosome from only one parent and can be heterodisomic (inheritance of a pair of homologous chromosomes from one parent) or isodisomic (inheritance of two copies of a single chromosome homolog from one parent). UPD can affect the entire chromosome or only a segment (this latter situation is termed partial or segmental UPD). The case reported here involves a cytogenetically normal female with a lethal LRBA deficiency resulting from a novel homozygous mutation. We demonstrated that the homozygosity was the result of a small segmental isodisomy of maternal origin affecting 1 Mb of chromosome 4.
Materials and Methods
Patient
Clinical data were obtained from the patient's medical records. Written informed consent was provided by the patient's parents for the studies reported here and for the publication of this case report, in accordance with the procedures of the Ethics Review Board of Hospital Universitari Vall d'Hebron, Barcelona (Spain).
Whole Exome Sequencing
Sample Preparation and Sequencing
DNA from the parent-child trio underwent whole exome sequencing (WES) as described previously (9, 10). DNA from blood was sheared using Covaris with the following settings: 300–400 bp (target size), C = 35 ng/μL, V = 45 μL, DC = 10, I = 5, C/b = 200, t = 040 s, water level = 12.5. WES libraries were prepared using the Illumina TruSeq DNA kit (Illumina, San Diego, CA, USA), NEXTflex Rapid DNA-Seq Kit, and NEXTflex Rapid Pre-Capture Combo Kit (BIOO Scientific, Austin, TX, USA) following the supplier's instructions. Pools of 6 samples were subjected to whole exome capture, applying Nimblegene SeqCap EZ Human Exome, version 3 (Roche NimbleGen, Madison, WI, USA). Libraries were sequenced to an average coverage of 50 × on an Illumina HiSeq 2500 sequencer using the 2 × 100 bp paired-end read protocol.
Computational Analysis
We used the eDiVA pipeline (http://ediva.crg.eu) to analyse the sequencing data of the parent-child trio. eDiVA performs read alignment using bwa-mem followed by GATK indel realignment, variant prediction by GATK HaplotypeCaller, as well as variant annotation and variant pathogenicity classification using customized methods. Finally, causal variant prioritization was performed according to four inheritance patterns (autosomal recessive homozygous, autosomal recessive compound heterozygous, autosomal dominant de novo, X-linked), as described (9, 10). Furthermore, we identified regions affected by loss of heterozygosity using Bcfttols v1.3 (11), and detected copy number variants using Conifer (12).
Microarray-Based Comparative Genomic Hybridization
DNA copy-number variations were investigated with an array comparative genomic hybridization assay using the CytoSure Constitutional v3 array 8 × 60 K (Oxford Gene Technology, UK) according to manufacturer recommendations on DNA extracted from peripheral blood with the GENTRA Puregene Kit (Qiagen, Hilden, Germany). DNA from the patient and her father were compared with a reference DNA sample (healthy control). Data analysis was performed by Cytogenomics 2.1 software with the ADM-2 algorithm and a minimum of three consecutive probes to detect an anomaly.
Results
The patient was a Caucasian girl with clinical onset at 8 months of age. She was the only daughter of a non-consanguineous couple; her mother had psoriasis and her father vitiligo. Her grandfather was affected with type 1 insulin-dependent diabetes mellitus, diagnosed at the age of 30. Her mother had no previous miscarriages and the prenatal history was uneventful. No allergies were demonstrated and the vaccination schedule, including live vaccines, had been updated to the age of 12 months with no significant adverse events.
At the age of 8 months the patient presented with type 1 diabetes, psoriasis, oral thrush, and enlarged liver and spleen. Two months later, lower right lobe pneumonia caused by Streptococcus pneumoniae was diagnosed. At that point, splenomegaly persisted and Epstein Barr virus (EBV) viremia was detected (viral load 9,298 copies/mL). When she was 2 years old, pneumococcal meningitis and immune thrombocytopenia were diagnosed and she experienced recurrent pneumonia despite immunoglobulin replacement therapy and antibiotic prophylaxis. These episodes led to the development of bronchiectasis in the left lower lobe and lingula, documented on CT scanning when she was 4 years old. Immunological studies performed at that time showed essentially normal lymphocyte counts, although T cells with effector phenotype were expanded and B and NK cell numbers were slightly decreased (Table 1). Despite low/normal immunoglobulin values, specific antibody production was impaired. NK cytotoxicity was repeatedly normal. When she was 28 months old, an EBV-related lymphoproliferative syndrome (mature B cell lymphoma) was diagnosed and appropriate chemotherapy was started with a good clinical and radiological response. Subsequently, she had varicella zoster meningitis at the age of 3 and severe inflammatory enteropathy starting at the age of 5.
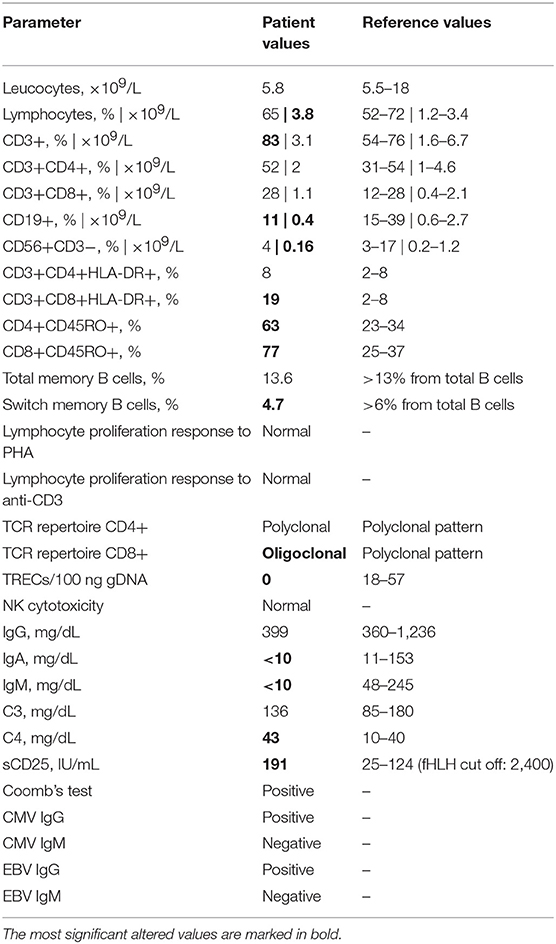
Table 1. Immunological parameters.
The patient underwent two consecutive stem cell transplants (SCT) due to loss of the first graft, at the age of 7 and 8 years, respectively, to treat persistent, refractory enteropathy and bone marrow failure. One month after the second transplant, she was admitted to the intensive care unit because of respiratory distress and sadly, she died. Disseminated toxoplasmosis was demonstrated in post-mortem studies.
Whole exome sequencing revealed a homozygous 1-bp insertion in exon 23 of the patient's LRBA gene, causing a frameshift and introduction of a premature stop codon resulting in a predicted truncated protein of 1,123 aa (wild-type LRBA protein: 2,863 aa). This mutation had not been reported in the literature or databases, and, following the recommendations of the Human Genome Variation Society (HGVS), we named it c.3366insA (p.Ala1123SerfsX1). WES data from the parents showed that the patient's healthy mother was heterozygous for the mutation and her father did not carry the mutation. Considering the autosomal recessive inheritance of LRBA deficiency, this was not consistent with the homozygous occurrence in our patient. We confirmed the genotypes by Sanger sequencing (Figure 1), and paternity was confirmed by high-resolution HLA typing. These findings suggested that either the paternal chromosome 4 bore a deletion including the LRBA locus (at least exon 23), or the patient inherited two copies of the mutant maternal LRBA allele. The patient's WES data showed a loss of heterozygosity (LOH) region of 1.013 Mb in chromosome 4 including the LRBA gene (Figure 1). LOH regions can be copy number-neutral or show copy number loss. Comparative genomic hybridization (CGH) array and WES-based copy number analysis of the patient's and father's genomic DNA yielded normal results, thereby ruling out genomic copy number abnormalities. We carefully checked the 10 CGH array probes located along the LRBA gene, which displayed a normal signal (Figure 1). In this 1.013-Mb region, the patient was homozygous for a haplotype present in the mother. Specifically, within the LOH region we detected 5 common single-nucleotide polymorphisms (SNPs) surrounding the LRBA mutation, in which the patient had not inherited any allele from her father. The genotypes of these SNPs were confirmed by Sanger sequencing (Figure 1). Collectively, these results indicate that the homozygous state of the patient's c.3366insA LRBA mutation was due to a segmental maternal isodisomy (Figure 1).
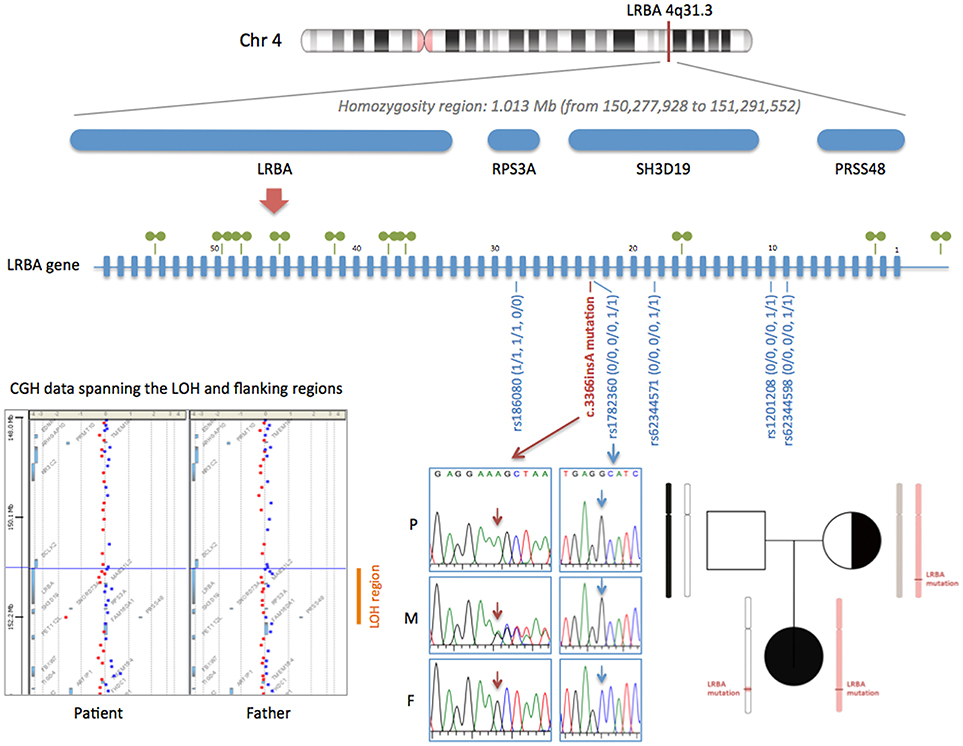
Figure 1. Segmental isodisomy of maternal origin containing the c.3366insA LRBA mutation. The LOH region in chromosome 4 of the patient is indicated by a red line. The LOH region, detected by analyzing WES data, includes four genes (LRBA, RPS3A, SH3D19, and PRSS48). A diagram of the LRBA gene with its 58 exons is shown. The 10 CGH array probes located along the LRBA gene are depicted with green symbols. All LRBA probes displayed a normal signal, confirming a copy number-neutral region. The c.3366insA mutation is shown in red (electropherograms of the patient and her parents). Five common SNPs surrounding the LRBA mutation are also shown in blue. The genotype of the patient (P) and her mother (M) and father (F) are indicated using the numbers 0 and 1 for the 2 possible alleles. Array-CGH data spanning the LOH and flanking regions is shown. gDNA from patient and father was labeled with Cy3 (red) and control gDNA with Cy5 (blue). The pedigree of the family shows the inherited chromosome 4, with the segmental isodisomy of maternal origin.
Discussion
Mutations in the LRBA gene encoding the LRBA protein are associated with an autosomal recessive monogenic disorder called LRBA deficiency and Autoimmunity, regulatory T (Treg) cell defects, Autoimmune Infiltration and Enteropathy (LATAIE) syndrome or, more commonly, merely LRBA deficiency (5). Why LRBA deficiency leads to this immune dysregulation syndrome was elucidated in 2015 when a study demonstrated that LRBA interacts with the cytoplasmic tail of CTLA4 and protects it from being sorted to lysosomes for degradation (13). Therefore, LRBA deficiency triggers a de facto CTLA4 deficiency. Not surprisingly, deleterious heterozygous mutations in the CTLA4 gene cause an autosomal dominant disorder called CTLA-4 Haploinsufficiency with Autoimmune Infiltration (CHAI) (or simply CTLA4 deficiency) which has similar symptoms (14, 15). Remarkably, LRBA deficiency results in lower CTLA4 levels than those of CTLA4-deficient patients, explaining the earlier disease onset, the nearly complete penetrance, and the greater severity of LATAIE compared to CHAI disease (16).
More than 70 patients with LRBA deficiency have been reported to date, with a range of diverse mutations identified and with highly variable clinical and immunologic characteristics including chronic diarrhea, hypogammaglobulinaemia, autoimmune disorders, organomegaly, recurrent infection, and combinations of these phenotypes (5–7). From the clinical viewpoint, our patient presented a severe LRBA-deficient phenotype when compared to patients in previous studies (6). She showed immune dysregulation, organomegaly, recurrent infection, and failure to thrive, thus fulfilling nearly the entire clinical spectrum of the disease. Due to immune dysregulation, the patient was treated with steroids and rapamycin but the clinical response was suboptimal and she required HSCT, which has been proposed as an appropriate treatment option (17, 18), but unfortunately, she died. Of note, and similar to what has been seen in previous cases, our patient had a lymphoid malignancy, suggesting that proliferative disease may be another feature of LRBA deficiency (5, 13, 19).
Using massive parallel sequencing (WES) we found a homozygous loss of function mutation in the LRBA gene, c.3366insA (p.Ala1123SerfsX1), which perfectly fit the patient's clinical phenotype. In the analysis of WES data, no additional PID related mutations were found. The most interesting part of this study is the uncommon genetic mechanism of heredity that led to this homozygous mutation (considering that the father did not carry the mutation). By combining data from a CGH array with WES computational analysis, we found a copy number-neutral LOH region of 1 Mb in chromosome 4 including the LRBA gene. These results, together with the marker segregation pattern, indicate that the homozygous state of the patient's c.3366insA LRBA mutation was due to segmental maternal isodisomy.
Uniparental disomy is a condition in which both copies of an entire chromosome or a specific chromosomal region are inherited from the same progenitor. UPD can be pathogenic, because of either imprinting defects or inheritance of a recessive condition from a single carrier parent (20). Specifically, our patient had segmental isodisomy of maternal origin, which means that she inherited 2 copies of a fragment (~1 Mb) of a single chromosome 4 from her mother. Segmental isodisomy is considered the result of post-zygotic mitotic recombination after normal fertilization (21), a well-recognized major mechanism of the LOH of tumor suppressor genes in cancer (22). Segmental UPD is a fairly common event, found in up to 15% of control populations (23). Usually, segmental UDPs have a relatively small size (median, 8.7 Mb), are primarily interstitial (22), and typically do not exert any adverse effect on the individual. The probability that a segmental UPD reveals an autosomal recessive disease is very low. Consequently, UPD as the cause of a PID is extremely rare.
To our knowledge, only 10 cases of PID due to UPD (including the patient described here) have been reported to date. We have summarized them Table 2. Nine of the 10 cases have been reported in the last 10 years. This is not surprising, as recent technological advances, such as SNP arrays and NGS, have facilitated detection of UPD. All reported cases are isodisomies or mixed forms, according to the recessive inheritance of the diseases, and 7 of the 10 involve the entire chromosome. Our study reports the smallest segmental UPD described in PIDs, with a fragment of only 1 Mb. Most UPDs in PIDs are of maternal origin, which concurs with the observation that maternal UPD is 1.6 to 3-fold more common than paternal UPD of an entire chromosome (33).
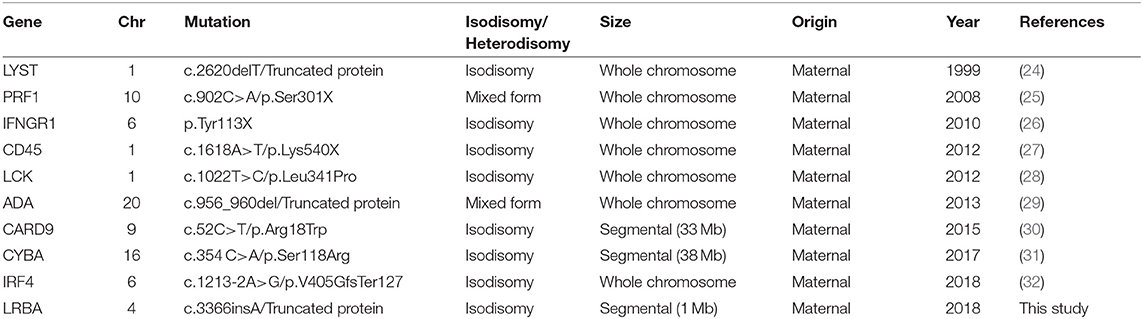
Table 2. Reported cases of uniparental disomy in primary immunodeficiencies.
A final relevant aspect of this case are the consequences in terms of genetic counseling: as segmental UPD is basically a rare, accidental mitotic error, the recurrence risk for siblings is not 25% as in general autosomal recessive diseases, but can be considered negligible or, at the least, much lower.
Author Contributions
PS-P was the principal clinician in charge of the patient's care and wrote part of the manuscript. MG-P, CF-J, and MM-G performed the immunological analysis. AM-N and JR were involved in management of the patient. AP performed the comparative genomic hybridization array and data analysis. DB, MB, and SO performed whole exome sequencing and the data analysis. RC performed the genetic analysis and was responsible for designing the study, writing the manuscript, and approving the final draft. All authors reviewed the manuscript and contributed to the final draft.
Funding
This study was funded by Instituto de Salud Carlos III, grants PI11/01086, PI14/00405 and PI17/00660, cofinanced by the European Regional Development Fund (ERDF). We acknowledge support of the Spanish Ministry of Economy and Competitiveness, Centro de Excelencia Severo Ochoa 2013–2017, the CERCA Programme/Generalitat de Catalunya, the CRG emergent translational research award and the la Caixa Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are deeply grateful to the affected individual and her family who participated in this study. We acknowledge Celine Cavallo for English language support.
References
1. Bousfiha A, Jeddane L, Al-Herz W, Ailal F, Casanova J-L, Chatila T, et al. The 2015 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2015) 35:727–38. doi: 10.1007/s10875-015-0198-5
2. Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. (2016) 53:575–90. doi: 10.1136/jmedgenet-2015-103690
3. Kienzler A-K, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol. (2017) 188:326–32. doi: 10.1111/cei.12947
4. Ameratunga R, Lehnert K, Woon S-T, Gillis D, Bryant VL, Slade CA, et al. Review: diagnosing common variable immunodeficiency disorder in the era of genome sequencing. Clin Rev Allergy Immunol. (2018) 54:261–8. doi: 10.1007/s12016-017-8645-0
5. Lopez-Herrera G, Tampella G, Pan-Hammarström Q, Herholz P, Trujillo-Vargas CM, Phadwal K, et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am J Hum Genet. (2012) 90:986–1001. doi: 10.1016/j.ajhg.2012.04.015
6. Gámez-Díaz L, August D, Stepensky P, Revel-Vilk S, Seidel MG, Noriko M, et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J Allergy Clin Immunol. (2016) 137:223–30. doi: 10.1016/j.jaci.2015.09.025
7. Alkhairy OK, Abolhassani H, Rezaei N, Fang M, Andersen KK, Chavoshzadeh Z, et al. Spectrum of phenotypes associated with mutations in LRBA. J Clin Immunol. (2016) 36:33–45. doi: 10.1007/s10875-015-0224-7
8. Burns SO, Zenner HL, Plagnol V, Curtis J, Mok K, Eisenhut M, et al. LRBA gene deletion in a patient presenting with autoimmunity without hypogammaglobulinemia. J Allergy Clin Immunol. (2012) 130:1428–32. doi: 10.1016/j.jaci.2012.07.035
9. Bahamonde MI, Serra SA, Drechsel O, Rahman R, Marcé-Grau A, Prieto M, et al. A single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS ONE (2015) 10:e0146035. doi: 10.1371/journal.pone.0146035
10. Wambach JA, Stettner GM, Haack TB, Writzl K, Škofljanec A, Maver A, et al. Survival among children with “Lethal” congenital contracture syndrome 11 caused by novel mutations in the gliomedin gene (GLDN). Hum Mutat. (2017) 38:1477–84. doi: 10.1002/humu.23297
11. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics (2009) 25:2078–9. doi: 10.1093/bioinformatics/btp352
12. Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature (2012) 485:237–41. doi: 10.1038/nature10945
13. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. Autoimmune Disease. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349:436–40. doi: 10.1126/science.aaa1663
14. Lo B, Abdel-Motal UM. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr Opin Immunol. (2017) 49:14–9. doi: 10.1016/j.coi.2017.07.014
15. Schwab C, Gabrysch A, Olbrich P, Patiño V, Warnatz K, Wolff D, et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J Allergy Clin Immunol. (2018) doi: 10.1016/j.jaci.2018.02.055. [Epub ahead of print].
16. Hou TZ, Verma N, Wanders J, Kennedy A, Soskic B, Janman D, et al. Identifying functional defects in patients with immune dysregulation due to LRBA and CTLA-4 mutations. Blood (2017) 129:1458–68. doi: 10.1182/blood-2016-10-745174
17. Seidel MG, Hirschmugl T, Gamez-Diaz L, Schwinger W, Serwas N, Deutschmann A, et al. Long-term remission after allogeneic hematopoietic stem cell transplantation in LPS-responsive beige-like anchor (LRBA) deficiency. J Allergy Clin Immunol. (2015) 135:1384–1390.e1–8. doi: 10.1016/j.jaci.2014.10.048
18. Tesi B, Priftakis P, Lindgren F, Chiang SCC, Kartalis N, Löfstedt A, et al. Successful hematopoietic stem cell transplantation in a patient with LPS-responsive beige-like anchor (LRBA) gene mutation. J Clin Immunol. (2016) 36:480–9. doi: 10.1007/s10875-016-0289-y
19. Bratanič N, Kovač J, Pohar K, Trebušak Podkrajšek K, Ihan A, Battelino T, et al. Multifocal gastric adenocarcinoma in a patient with LRBA deficiency. Orphanet J Rare Dis. (2017) 12:131. doi: 10.1186/s13023-017-0682-5
20. Yamazawa K, Ogata T, Ferguson-Smith AC. Uniparental disomy and human disease: an overview. Am J Med Genet C Semin Med Genet. (2010) 154C:329–34. doi: 10.1002/ajmg.c.30270
21. Niida Y, Ozaki M, Shimizu M, Ueno K, Tanaka T. Classification of uniparental isodisomy patterns that cause autosomal recessive disorders: proposed mechanisms of different proportions and parental origin in each pattern. Cytogenet Genome Res. (2018) 154:137–46. doi: 10.1159/000488572
22. O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood (2010) 115:2731–9. doi: 10.1182/blood-2009-10-201848
23. Mohamedali A, Gäken J, Twine NA, Ingram W, Westwood N, Lea NC, et al. Prevalence and prognostic significance of allelic imbalance by single-nucleotide polymorphism analysis in low-risk myelodysplastic syndromes. Blood (2007) 110:3365–73. doi: 10.1182/blood-2007-03-079673
24. Dufourcq-Lagelouse R, Lambert N, Duval M, Viot G, Vilmer E, Fischer A, et al. Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur J Hum Genet. (1999) 7:633–7. doi: 10.1038/sj.ejhg.5200355
25. Al-Jasmi F, Abdelhaleem M, Stockley T, Lee K-S, Clarke JTR. Novel mutation of the perforin gene and maternal uniparental disomy 10 in a patient with familial hemophagocytic lymphohistiocytosis. J Pediatr Hematol Oncol. (2008) 30:621–4. doi: 10.1097/MPH.0b013e31817580fd
26. Prando C, Boisson-Dupuis S, Grant AV, Kong X-F, Bustamante J, Feinberg J, et al. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-gamma receptor 1 deficiency. Am J Med Genet A (2010) 152A:622–9. doi: 10.1002/ajmg.a.33291
27. Roberts JL, Buckley RH, Luo B, Pei J, Lapidus A, Peri S, et al. CD45-deficient severe combined immunodeficiency caused by uniparental disomy. Proc Natl Acad Sci USA. (2012) 109:10456–61. doi: 10.1073/pnas.1202249109
28. Hauck F, Randriamampita C, Martin E, Gerart S, Lambert N, Lim A, et al. Primary T-cell immunodeficiency with immunodysregulation caused by autosomal recessive LCK deficiency. J Allergy Clin Immunol. (2012) 130:1144–1152.e11. doi: 10.1016/j.jaci.2012.07.029
29. Geelen J, Pfundt R, Meijer J, Verheijen FW, van Kuilenburg ABP, Warris A, et al. Severe phenotype of severe combined immunodeficiency caused by adenosine deaminase deficiency in a patient with a homozygous mutation due to uniparental disomy. J Allergy Clin Immunol. (2013) 132:222–3. doi: 10.1016/j.jaci.2012.11.006
30. Lanternier F, Barbati E, Meinzer U, Liu L, Pedergnana V, Migaud M, et al. Inherited CARD9 deficiency in 2 unrelated patients with invasive Exophiala infection. J Infect Dis. (2015) 211:1241–50. doi: 10.1093/infdis/jiu412
31. Bravo García-Morato M, Nevado J, González-Granado LI, Sastre Urgelles A, Rodríguez Pena R, Ferreira Cerdán A. Chronic granulomatous disease caused by maternal uniparental isodisomy of chromosome 16. J Allergy Clin Immunol Pract. (2017) 5:1146–8. doi: 10.1016/j.jaip.2017.01.018
32. Bravo García-Morato M, Aracil Santos FJ, Briones AC, Blázquez Moreno A, Del Pozo Maté Á, Domínguez-Soto Á, et al. New human combined immunodeficiency caused by interferon regulatory factor 4 (IRF4) deficiency inherited by uniparental isodisomy. J Allergy Clin Immunol. (2018) 141:1924–7.e18. doi: 10.1016/j.jaci.2017.12.995
Keywords: primary immunodeficiency, LRBA deficiency, uniparental disomy, whole exome sequencing, comparative genomic hybridization array
Citation: Soler-Palacín P, Garcia-Prat M, Martín-Nalda A, Franco-Jarava C, Rivière JG, Plaja A, Bezdan D, Bosio M, Martínez-Gallo M, Ossowski S and Colobran R (2018) LRBA Deficiency in a Patient With a Novel Homozygous Mutation Due to Chromosome 4 Segmental Uniparental Isodisomy. Front. Immunol. 9:2397. doi: 10.3389/fimmu.2018.02397
Received: 07 August 2018; Accepted: 27 September 2018;
Published: 16 October 2018.
Edited by:
Shen-Ying Zhang, Rockefeller University, United StatesReviewed by:
Satoshi Okada, School of Medicine, Hiroshima University, JapanBernice Lo, Sidra Medical and Research Center, Qatar
Copyright © 2018 Soler-Palacín, Garcia-Prat, Martín-Nalda, Franco-Jarava, Rivière, Plaja, Bezdan, Bosio, Martínez-Gallo, Ossowski and Colobran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roger Colobran, cmNvbG9icmFuQHZoZWJyb24ubmV0