 Arjan van Caam
Arjan van Caam Madelon Vonk2
Madelon Vonk2- 1Experimental Rheumatology, Radboudumc, Nijmegen, Netherlands
- 2Department of Rheumatology, Radboudumc, Nijmegen, Netherlands
Systemic sclerosis (SSc) is a severe auto-immune disease, characterized by vasculopathy and fibrosis of connective tissues. SSc has a high morbidity and mortality and unfortunately no disease modifying therapy is currently available. A key cell in the pathophysiology of SSc is the myofibroblast. Myofibroblasts are fibroblasts with contractile properties that produce a large amount of pro-fibrotic extracellular matrix molecules such as collagen type I. In this narrative review we will discuss the presence, formation, and role of myofibroblasts in SSc, and how these processes are stimulated and mediated by cells of the (innate) immune system such as mast cells and T helper 2 lymphocytes. Furthermore, current novel therapeutic approaches to target myofibroblasts will be highlighted for future perspective.
Introduction
Systemic sclerosis (SSc) is a rare but severe auto-immune disease characterized by inflammation, vasculopathy and excessive fibrosis of connective tissues. Its incidence worldwide is on average an estimated 13 people per 1 million per year, with a prevalence of ~200 people per 1 million (1). Risk factors include genetic predisposition (2), female sex (3), and exposure to environmental cues such as chemicals like silica or solvents (4), but its etiology remains poorly understood. The excessive fibrosis characteristic for SSc typically starts distally in the skin of the extremities and moves upwards toward and trough the trunk until it greatly negatively affects the function of many organs like the gastro-intestinal tract and lungs. SSc is therefore accompanied by a high morbidity and patients often require extensive medical care with a (severely) reduced quality of life (5). Mortality is also increased in SSc patients. On average, the standard mortality rate of all causes is 2.7, with lung involvement being the major cause of death (6). Furthermore, the estimated loss of life-expectancy for patients is more than 15 years (7). Unfortunately, to date, no targeted disease-modifying therapy is available, resulting in a large unmet medical need. Because of this need, SSc has been designated an orphan disease to support research and development of a treatment.
The lack of targeted therapy for SSc is partly due to a lack of understanding of its pathophysiology. Its pathophysiology is a complex interplay between endothelium, the innate and acquired immune system, target organs and connective tissue which culminates in excessive fibrosis of e.g., skin and internal organs. A key cellular player in many fibrotic conditions such as keloid formation, Dupuytren's contracture and post-operative scarring is the myofibroblast, which is a special type of fibroblast. In this review we will discuss the role of myofibroblasts in SSc, their formation and how these cells are at the center of SSc pathophysiology, by regulating many of this disease's aspects.
On the Myofibroblast and Its Biological Function
Myofibroblasts were first identified in granulation tissue during open wound healing, as cells that resembled fibroblasts but contained microfilaments in their cytoplasm similar to those of smooth muscle cells (8, 9). Subsequently, it was demonstrated that these cells have contractile properties and are key in open wound closure (9). Myofibroblasts facilitate wound healing in several ways (Figure 1); First, they are capable of producing large amounts of extra cellular matrix (ECM) molecules such as collagen type I, collagen type III and fibronectin to replace lost ECM. Secondly, myofibroblasts are contractile. Their microfilaments (also known as stress fibers) consist of alpha smooth muscle actin (αSMA) and non-muscle myosin type II (10) and can contract in typical actin-myosin fashion, albeit rather slowly compared to muscle actin myosin filaments. Thirdly, myofibroblasts strongly connect physically to their environment; via integrin-mediated focal adhesions and cadherin-mediated adherens junctions their actin cytoskeleton is strongly anchored to their surrounding ECM and neighboring cells, respectively (11). The combination of this strong connection to the environment with their ability to contract allows myofibroblasts to exert tension on their surroundings and contract (damaged) tissue. This contraction decreases wound size and is crucial for open wound healing. Long term wound healing is further supported by myofibroblasts via their ability to strengthen the ECM; myofibroblasts express several protein and collagen crosslinking enzymes such as protein-glutamine gamma-glutamyltransferase 2 (=transglutaminase 2), protein-lysine 6-oxidase (LOX), and procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) (12). These enzymes help strengthen e.g., fibrillar collagen bundles by post-translationally modifying collagen molecules, which results in increased crosslinking of these molecules in collagen networks during the maturation phase of wound healing. These crosslinks increase this networks' strength and prevents enzymatic degradation and thus strengthen the (scar) tissue.
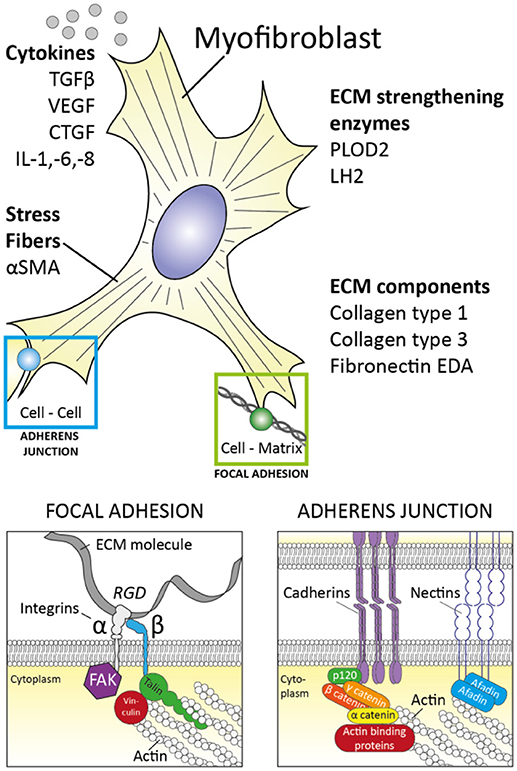
Figure 1. The myofibroblast and its properties. Myofibroblasts are characterized by stress fibers containing αSMA, production of extracellular matrix (ECM) components and ECM strengthening enzymes. Furthermore, myofibroblast are closely linked to their environment via focal adhesions and adherens junctions. Cytokines which are produced by myofibroblasts include TGFβ, VEGF, CTGF, IL-1, IL-6, and IL-8. These characteristics help myofibroblasts fulfill their role in wound healing.
Myofibroblasts also secrete and/or activate various autocrine and paracrine mediators to facilitate wound healing. For example, myofibroblasts produce vascular endothelial growth factor (VEGF) (13). This polypeptide growth factor is key in the formation of new blood vessels. Furthermore, myofibroblasts produce endothelin 1, a potent vasoconstrictor but also a factor which stimulates the formation of new myofibroblasts (14) and enhances their function in regard to collagen production and contractile properties (15). Myofibroblast function is also enhanced by their production of connective tissue growth factor (CTGF), a matricellular protein which stimulates e.g., their formation and collagen type I production. A key growth factor which is produced (13) and potently activated by myofibroblasts is transforming growth factor β (TGFβ) (16). This polypeptide growth factor is strongly pro-fibrotic and stimulates myofibroblast formation and activity. TGFβ is produced in latent form [bound by latency associated peptide (LAP) and latent TGFβ binding proteins (LTBP)] but can efficiently be activated by myofibroblasts via an integrin-mediated process (16, 17). Of note, TGFβ induces the expression of ET-1, CTGF, and VEGF in myofibroblasts, indicating that this growth factor lays at the heart of the expression of these factors. In addition, myofibroblasts can produce a range of various cytokines and chemokines to aid in the recruitment and facilitate the function of (innate) immune cells (13). Most notably, they produce interleukin 1 (IL-1), interleukin 6 (IL-6), interleukin 8 (IL-8), and monocyte chemoattractive protein 1 (MCP-1) (13).
Together these abilities make myofibroblasts well suited to facilitate wound healing.
On the Presence of Myofibroblasts in SSc
Myofibroblasts have long been associated with SSc pathophysiology (18). Already in 1972 it was identified that fibroblasts obtained from SSc skin have a pro-fibrotic phenotype and produce more collagens than control fibroblasts (19). In 1990 it was confirmed using immunohistochemistry that fibroblasts of SSc patients near lesional areas in skin, esophagus, and lungs contain alpha smooth muscle actin (20) and are thus myofibroblasts.
In skin, the presence of myofibroblasts correlates with the amount of (hyalinized) collagen and skin parameters related to fibrosis such as tightness, hardness and stiffness, and does so more significantly than inflammation (21–23), supporting for a role of myofibroblasts in the pathogenesis of these clinical signs. This skin thickening and hardening can occur to such extent that it impairs movement of e.g., fingers. Furthermore, excessive matrix deposition leads to loss of tissue architecture such as sweat glands and hair follicles.
In lungs of SSc patients, the presence of myofibroblasts in the interstitial space can already be observed early during the fibrotic process (24), and with progression of interstitial lung disease they can ultimately also be observed in bronchoalveolar lavage liquid of SSc patients (25). The presence of pathological myofibroblasts greatly negatively affects lung function. Their matrix producing ability destroys alveolar architecture and increases interstitial space thickness, which both hamper respiration. Furthermore, the presence of myofibroblasts can induce stenosis; the abnormal narrowing of bloodvessels, and blood vessel narrowing is further enhanced by myofibroblasts' expression of ET-1, a potent vasoconstrictor. This hampers pulmonary blood flow, and as a consequence induces strain on the right heart ventricule.
Another location where myofibroblasts can be detected in SSc is in the esophagus and gastric wall of patients with severe fibrosis (26). Here, myofibroblast presence results in loss of muscle function, making these tissues unable to contract. As a consequence, gastric acid can flow into the esophagus, causing gastro-oesophageal reflux disease.
Together, these observations place myofibroblasts in the various organs that can be affected by SSc. In addtion, organs such as kidney, intestine and myocard can also be affected by myofibroblast-driven fibrosis in SSc (18). However, of note, in late stage fibrotic atrophic SSc skin these cells can no longer be detected (27). Figure 2 gives an overview of the location of myofibroblasts in SSc.
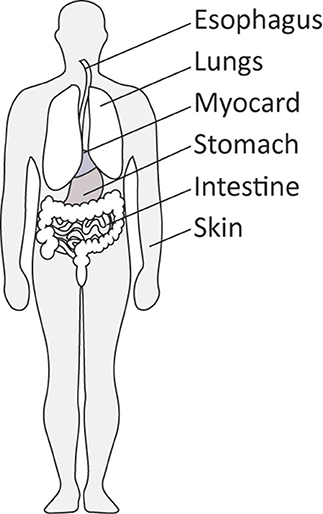
Figure 2. Organs commonly affected by diffuse cutaneous SSc.
In healthy tissues, the presence of myofibroblasts is (very) rare due to the tendency of myofibroblasts to undergo apoptosis when they are no longer needed for the healing process (28, 29). However, a putative resident type of myofibroblast can be found in lung alveolar ducts, where they help regulate alveolar function. In contrast, in SSc their presence is unwanted and attributed to a lowered susceptibility of myofibroblasts to undergo apoptosis and to increased formation.
Decreased Apoptosis of Myofibroblasts in SSc
Two major pathways govern cellular apoptosis; the intrinsic and extrinsic pathway. The extrinsic pathway is induced by activation of fas cell surface death receptor (Fas). Fas is a membrane spanning receptor of the TNF receptor superfamily and can, upon binding of Fas ligand, trigger the formation of a death-inducing signaling complex (DISC). This complex subsequently activates apoptosis-initiator caspase 8 to start a caspase pathway ultimately culminating in activation of caspase-3 and apoptosis (Figure 3). The intrinsic pathway is triggered by release of cytochrome c from mitochondria, which is subsequently incorporated into apoptosomes, cellular structures which activate the apoptosis-initiator caspase-9 to initiate apoptosis (30). A key protein in release of cytochrome c from mitochondria is BCL2-associated X protein (BAX), which, upon oligomerization, forms pores in the mitochondrial membrane through which cytochrome c can leak (31). Two important inhibitors of BAX are BCL2 and BCL2-XL (also known as BCL2L1), which both prevent oligomerization of BAX and are thus anti-apoptotic. Of note, the extrinsic and intrinsic pathways are not fully discrete but linked, for example via BH3 interacting domain death agonist (BID), a protein which is activated by caspase 8 and subsequently forms mitochondrial membrane pores in cooperation with BAX (32). Ultimately, whether cells like myofibroblasts undergo apoptosis is determined by the ratio of activity between pro-apoptotic mitochondrial membrane pore forming proteins (e.g., BAX) and their anti-apoptotic inhibitors (e.g., BCL2). Pro-survival signaling can skew this balance in favor of anti-apoptotic proteins.
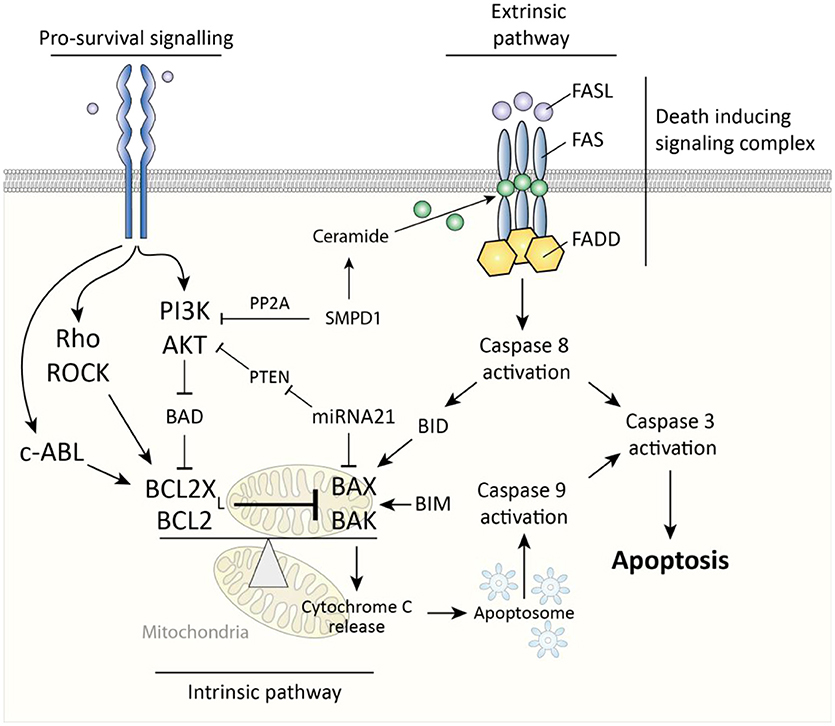
Figure 3. Caspase-dependent apoptosis pathways in myofibroblasts. The extrinsic pathway is activated via death inducing signaling complex and results in caspase 8-mediated caspase 3 activity which results in apoptosis. The intrinsic pathway is triggered by cytochrome c release from mitochondria which results in caspase 9-mediated caspase 3 activity. This cytochrome c release is governed by the ratio between pro-apoptotic BAX/BAK and BCL2(XL). Pro-survival signaling affects this ratio in favor of BCL2(XL).
In systemic sclerosis, myofibroblasts are less prone to undergo apoptosis for several reasons. To begin, it has been observed that, in quiescent state, SSc myofibroblasts express less pro-apoptotic BAX compared to myofibroblasts of control subjects (33). A possible cause for this is increased activity of tyrosine-protein kinase ABL1 (c-Abl). Silencing of c-ABL enhances apoptosis in both healthy and SSc skin fibroblasts by increasing the BAX/BCL2 ratio toward pro-apoptotic BAX (34). An example of how c-ABL can be activated is via TGFβ signaling; in idiopathic pulmonary fibrosis, c-Abl is activated by TGFβ (35), and silencing of c-Abl inhibits the pro-survival effects of TGFβ on myofibroblast apoptosis (34).
Secondly, in fibrotic tissues, extracellular matrix stiffness is increased compared to healthy tissue. This increased stiffness is an important survival signal for myofibroblasts; via mechanosensing such stiffness results in intracellular activation of Rho and Rho-associated kinase (ROCK) whose activity increases BCL2-XL expression (36). Importantly, this increased, stiffness-induced, BCL2-XL expression is needed to counteract the function of the pro-apoptotic protein BIM (36). BIM is an activator of BAX and accumulates in myofibroblasts exposed to a stiff matrix. This accumulation primes the cells to undergo apoptosis (36), and only the continued presence of BCL2-XL prevents this. This balance between BCL-2 and BIM serves a role during normal wound healing; once the matrix softens during the final wound remodeling stage, pro-surivival ROCK signaling drops, resulting in loss of BCL-2 expression, and rapid BIM-mediated apoptosis of myofibroblasts (36). Recently, it has been shown that pharmacological inhibition of BCL2-XL can mimic this process and induce targeted BIM-mediated apoptosis in myofibroblasts and even revert established (murine) fibrosis (36).
In addition, in SSc skin, phosphatidylinositol 3-kinase (PI3K)/AKT serine/threonine kinase (AKT) signaling (37) is increased. This pathway facilitates myofibroblasts survival by inhibiting the activity of BAX. It does so by inactivating bcl2-associated agonist of cell death (BAD) via phosphorylation, after which this protein can no longer inhibit the function of anti-apoptotic proteins such as BCL2-XL. Many growth factors can induce PI3K/AKT signaling, including TGFβ. TGFβ signaling is increased in skin of SSc patients, and TGFβ has been demonstrated to induce AKT signaling in dermal fibroblasts to lower myofibroblasts' sensitivity for Fas-mediated apoptosis (34, 37, 38). Furthermore, TGFβ signaling also lowers expression of acid sphingomyelinase (SMPD1) (39). This enzyme induces the activation of protein phosphatase 2 (PP2A), i.e., an inhibitor of AKT signaling, and a reduction in SMPD1 thus enhances pro-survival AKT signaling. Additionaly, SMPD1 facilitates Fas-dependent apoptosis via its product; i.e., the lipid ceramide, which helps cluster Fas at the cell membrane, thus facilitating the formation of death inducing signaling complexes (40). In SSc fibroblasts, it has been shown that TGFβ lowers Fas-mediated apoptosis and that overexpression of SMPD1 prevented this effect, indicating its importance (39).
Finally, a role for micro RNAs (miRNA) in protecting myofibroblasts against apoptosis has been described in SSc. miRNAs are small non coding RNA molecules that can bind messenger RNAs and induce their degradation via an RNA-induced silencing complex (RISC). In SSc skin, expression of miRNA21 is increased, and this miRNA targets and degrades pro-apoptotic BAX mRNA (41). Additionally, miRNA21 targets phosphatase and tensin homolog (PTEN), which is an inhibitor of AKT signaling, as this phosphatase lowers intracellular PIP3 levels, the activator of AKT signaling (38). Via these mechanisms, presence of this miRNA lowers cellular sensitivity to apoptosis. Notably, TGFβ induces expression of miRNA21 in fibroblasts (38).
Together these mechanisms protect myofibroblasts from apoptosis in SSc which, in contrast to their final loss during wound healing, ensures their continued presence (long) after their formation.
On the Formation of Myofibroblasts in SSc: Pathways
In SSc, not only the apoptosis of myofibroblasts is decreased but also their formation is increased. Myofibroblasts can originate in several ways, including the differentiation of fibroblasts toward myofibroblasts. This process is key in normal wound healing and facilitated by growth factors such as TGFβ, Wnts, damage associated molecular patterns such as fibronectin cloths, and tissue stiffness; the stiffer the matrix the more prone fibroblasts are to become myofibroblasts (42). In Figure 4 several intracellular pathways are listed that are involved in the transition of fibroblasts to myofibroblasts.
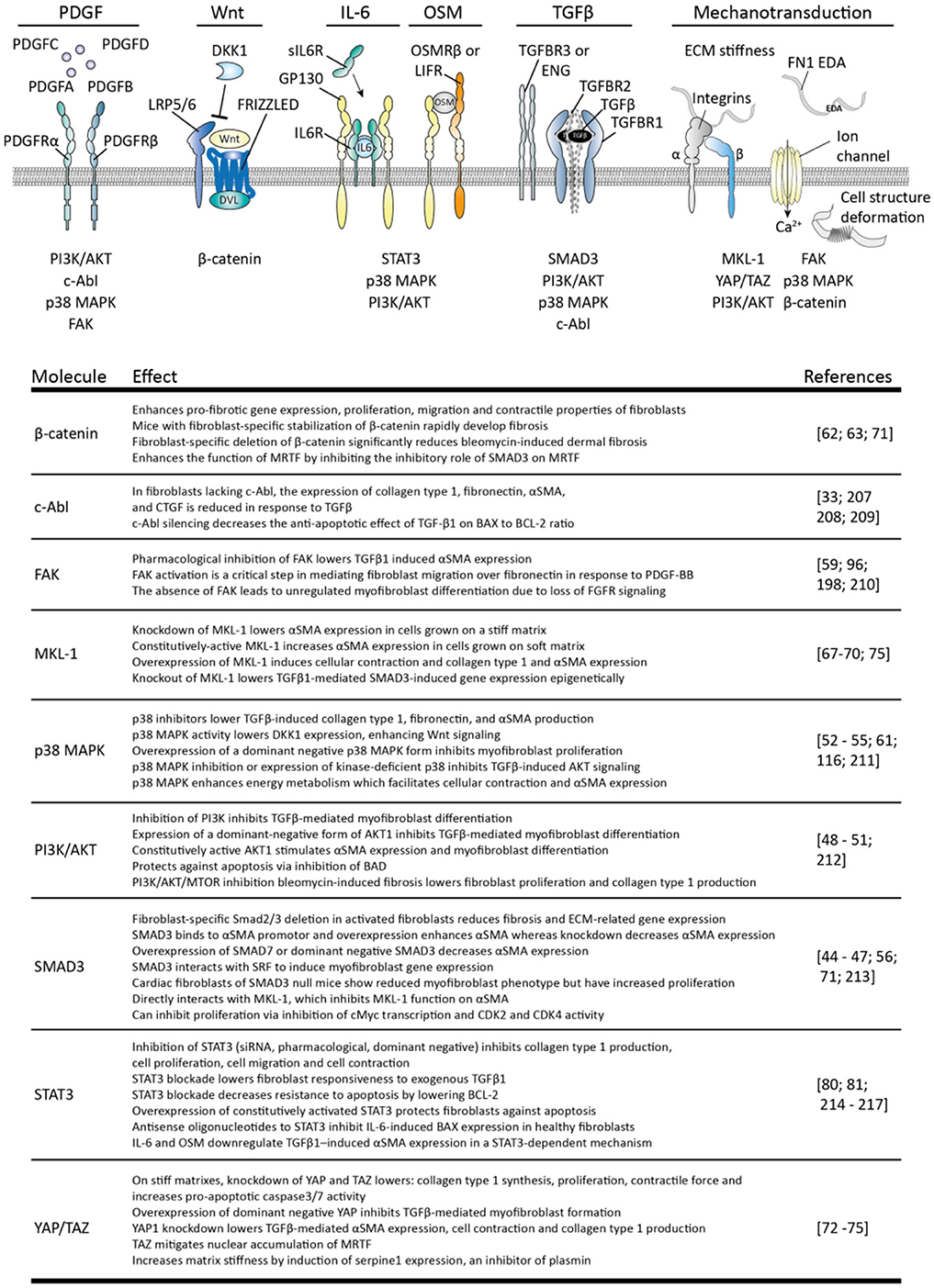
Figure 4. Stimuli for myofibroblast formation and their intracellular pathways. The four variants of platelet-derived growth factor (PDGF) can interact with homo- or heterodimers of PDGF receptor alpha (PDGFRα) and beta (PDGFRβ) to induce signaling by: phosphoinositide 3-kinase (PI3K)/AKT serine/threonine kinase (AKT), p38 mitogen-activated protein kinases (p38 MAPK), focal adhesion kinase (FAK) and tyrosine-protein kinase ABL1 (c-ABL). Canonical Wnt signaling is activated via formation of a wnt/frizzled/LRP5/6 complex which recruits disheveled (DVL) to the plasma membrane. This inhibits β-catenin degradation, leading to the accumulation of this protein and subsequent signaling. Interleukin 6 (IL-6) signaling uses a complex of membrane-bound or soluble IL-6 receptor (IL-6R) and glycoprotein 130 (gp130) to activate PI3K/AKT, p38MAPK and signal transducer and activator of transcription 3 (STAT3) signaling. Oncostatin M (OSM) also uses gp130, but together with oncostatin M receptor beta (OSMRβ) or leukemia inhibitory factor receptor (LIFR). Transforming growth factor beta (TGFβ) induces heterotetramerization of TGFβ-receptor type I (TGFBR1) and II (TGBR2) and results in intracellular activation of SMAD3, p38 MAPK, PI3K/AKT c-ABL. TGFβ-receptor type III receptors such as betaglycan (TGFBR3), and endoglin (ENG) guide TGFβ availability and receptor complex formation. Mechanotransduction can occur via mechanosensitive ion channels, leading to e.g., calcium ion (Ca2+) influx, integrin complexes and deformation of cellular structures, leading to activation of myocardin-like protein 1 (MLK1), β-catenin, FAK, p38 MAPK, PI3K/AKT, and yes-associated protein 1 (YAP)/WW domain-containing transcription regulator protein 1 (TAZ). The effects of each of these pathways are listed in the table. Note that not all intracellular pathways are listed for each stimulus, only those connected to myofibroblast formation.
To begin, a key growth factor for myofibroblast formation is TGFβ; this growth factor directly induces extracellular matrix production and αSMA expression in fibroblasts. TGFβ activity is increased in skin of SSc patients, just as expression of its activating integrin αVβ5 (43, 44). This integrin can recognize latent TGFβ via its RGD domain and can mechanically separate the latency conferring peptides from the active peptide (42). The importance of integrin-mediated TGFβ activation is illustrated by the observation that inhibition of integrin αVβ5 by the use of antibodies or antisense RNA inhibits myofibroblasts formation (43, 44). Various intracellular pathways play a role in establishing the effects of TGFβ, in particular: SMAD3, PI3K/AKT, p38 MAPK, and c-ABL. Overexpression of SMAD3 enhances, whereas knockdown inhibits αSMA and extracellular matrix production in fibroblasts (45–48). Furthermore, fibroblast-specific deletion of SMAD3 reduces αSMA production and myofibroblast phenotype (49–52), for example, loss of SMAD3 lowers the number of activated myofibroblasts in cardiac fibrosis in vivo and reduces extracellular matrix production by myofibroblasts (47). Inhibition of PI3K/AKT signaling inhibits TGFβ-mediated myofibroblast formation, whereas overexpression of a constitutively active form of AKT1 enhances myofibroblasts development. The use of p38 MAPK inhibitors also lowers TGFβ-induced collagen type I and αSMA production and prevents TGFβ-induced AKT signaling (53–55). Additionally, this pathway alters cellular energy metabolism in such a way that is facilitates cellular contraction (56). Finally, in fibroblasts lacking c-ABL the expression of extracellular matrix molecules and αSMA is reduced in response to TGFβ. Of note, TGFβ can also negatively affect myofibroblasts. For example, SMAD3 can inhibit cellular proliferation via lowering the expression of c-myc and preventing the progression of cell division from G1 to S phase (57). Furthermore, pre-treatment of granulation tissue (myo) fibroblasts with TGFβ enhances their sensitivity to undergo bFGF-mediated apoptosis (58). This last observation illustrates that cellular context, e.g., the presence of bFGF, can greatly impact TGFβ signaling outcome.
Importantly, TGFβ facilitates the function of various other growth factors in fibroblasts. In SSc skin fibroblasts, TGFβ makes fibroblasts more sensitive to anabolic stimulation with platelet derived growth factor (PDGF), via induction of its receptor (PDGFR) (59). This growth factor induces extracellular matrix production and proliferation via the activation of PI3K/AKT, p38 MAPK, c-ABL, and focal adhesion kinase (FAK) pathways. In addition, this last pathway regulates PDGF-induced migration of myofibroblasts which recruits myofibroblasts to fibrotic areas (60). TGFβ and PDGF can work in concert, for example, in mouse corneal stromal fibroblasts co-stimulation of fibroblasts with TGFβ and PDGF greatly enhances myofibroblast formation compared to TGFβ alone (61).
Another pathway enhanced by TGFβ in SSc is canonical Wnt signaling. TGFβ signaling via p38 MAPK lowers dickkopf-1 (DKK1) expression (62), which is an inhibitor of canonical Wnt signaling via β-catenin. In skin and fibroblasts of SSc patients, decreased DKK1 expression is observed (62), together with increased β-catenin accumulation (63), and increased expression of Wnt signaling-related genes (62, 64). Stimulation of fibroblasts with canonical Wnts such as Wnt-1 or Wnt3a upregulates collagen type 1 and αSMA expression, and does so to a similar extent as TGFβ. Furthermore, mice with continuous fibroblast-specific Wnt signaling by artificial β-catenin stabilization rapidly develop skin fibrosis whereas fibroblast-specific deletion of β-catenin protects mice from bleomycin-induced skin fibrosis (63). Notably, Wnt signaling can induce autocrine TGFβ signaling (64) and overexpression of DKK1 protects mice even against TGFβ-receptor mediated skin fibrosis, indicating that both pathways are closely interwoven and interdependent.
Apart from the abovementioned effects, TGFβ can induce the expression of fibronectin 1 extra domain A (FN1 EDA) in (myo) fibroblasts. FN1 EDA is a splice variant of fibronectin which contains the so-called EDA domain. Normally, FN1 EDA is not expressed in healthy tissues but its expression is induced during wound healing (65) Fibroblasts can detect FN1 EDA via membrane bound receptors such as α4 containing integrins or toll like receptor 4 (TLR4), and its presence is a prerequisite for TGFβ-mediated myofibroblast formation; its expression precedes αSMA expression, and mice that lack their FN1 EDA domain are unable to produce myofibroblasts during injury (65–67). In turn, FN1 EDA facilitates the mechanical activation of TGFβ because it binds the latent form of TGFβ and presents this to integrins.
Next to these aforementioned stimuli, cellular mechanosensing is another crucial element in the transition of fibroblasts to myofibroblasts. Via for example intergrins, mechanosensitive ion channels, and cell structure deformation, fibroblasts can sense mechanical cues such as matrix stiffness. This mechanosensing results in activation of various intracellular pathways such as FAK, PI3K/AKT, p38 MAPK, and β-catenin, and activation of transcription activators such as myocardin-like protein 1 (MKL-1) and transcriptional coactivator YAP1 (YAP1) and WW domain-containing transcription regulator protein 1 (TAZ). Both MKL-1 and YAP/TAZ directly regulate myofibroblast phenotype. Knockdown of MKL-1 lowers αSMA expression in cells grown on a stiff matrix whereas overexpression of a constitutively active form of MKL-1 increases αSMA expression in cells grown on a soft matrix (68, 69). MKL-1 also activates collagen type 1 expression in lung fibroblasts (70). Furthermore, MKL-1 interacts with SMAD3 to bind the promoters of collagen type I and ASMA, and knockdown of MKL-1 lowers SMAD3-dependent gene expression (71). However, this interaction with SMAD3 can result in more rapid degradation of MKL-1, leading to repression of MKL-1-dependent genes (72). β-catenin has been shown to counteract this effect of SMAD3 (72), indicating that MKL-1 function depends on the integration of various pathways. Knockdown of YAP/TAZ in fibroblasts that are grown on stiff matrixes lowers proliferation, collagen type 1 synthesis, contractile force and increases pro-apoptotic caspase3 and caspase 7 activity. Furthermore, knockdown of YAP or overexpression of a dominant negative form lowers TGFβ-mediated myofibroblast formation (73–76). Notably, YAP/TAZ influence matrix stiffness by directly inducing serpine1 expression (73). Serpine1 inhibits the activation of plasmin, a protease which degrades extracellular matrix molecules such as fibrin and fibronection and can activate collagenases. Plasmin activity thus degrades and softens the extracellular matrix, but YAP/TAZ activity counteracts this (73) of note, serpine1 expression can also be rapidly and highly induced by TGFβ (77), and mechanical activation of TGFβ is enhanced in stiffer matrixes (42). Both YAP/TAZ and TGFβ activity can thus result in a feed forward loop in which tissue stiffness results in tissue stiffness-enhancing activity. Such a mechanism can explain continued fibrosis in absence of a exogenous stimulus.
Finally, the transition of fibroblasts to myofibroblasts is also facilitated by intracellular STAT3 signaling. STAT3 is induced by various cytokines such as interleukin 6 (IL-6) and oncostatin M (OSM). IL-6 expression is strongly expressed in SSc skin fibroblasts (78), and in vitro, stimulation of SSc skin fibroblasts with IL-6 results in collagen and αSMA expression (78–80). Furthermore, in the murine bleomycin model for skin fibrosis, knockout of IL-6 reduces skin pathology, as does administration of an anti-IL-6 receptor antibody (MR16-1) (79). In SSc skin, STAT3 signaling is activated (81) resulting in pro-fibrotic gene expression in fibroblasts; for example, STAT3 regulates collagen type I expression in SSc skin fibroblasts (82). However, of note, in lungs of SSc patients no enhanced STAT3 activation can be observed (82). Importantly, in both bleomycin induced skin and lung fibrosis in mice, knockout or pharmacological inhibition of STAT3 ameliorates fibrosis (83) (81). Furthermore, in both models, STAT3 was shown to be downstream of TGFβ signaling, as inhibition of STAT3 prevented TGFβ-induced myofibroblasts formation (81, 83).
Together these pathways can mediate the transition of fibroblasts to myofibroblasts and direct myofibroblasts activity after formation but cellular context plays an important role in guiding the outcome.
On the Formation of Myofibroblasts in SSc: Cells
Apart from the transition of fibroblasts to myofibroblasts, an important source of myofibroblasts in SSc is the transdifferentiation of other cell types (Figure 5).
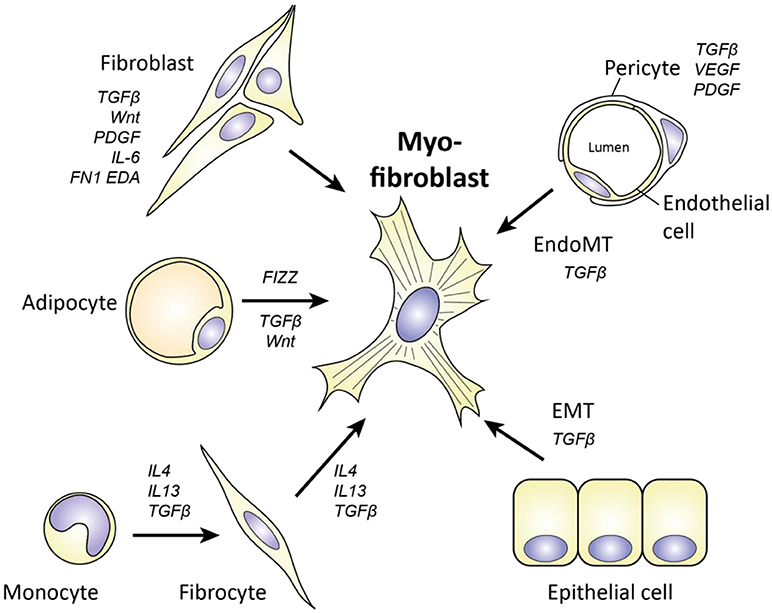
Figure 5. Cellular origins of myofibroblasts in SSc. Myofibroblasts can originate from various cell types, including fibroblasts, adipocytes, monocytes/fibrocytes, pericytes, endothelial cells, and epithelial cells. Key molecules for each transition are depicted. For epithelial cells to become myofibroblasts, they have to undergo epithelial to mesenchymal transition (EMT). For endothelial cells a similar process is needed, called endothelial to mesenchymal transition (EndoMT).
To begin, one cell type that can function as a source of myofibroblasts is the pericyte. These contractile cells surround endothelial cells in the microvasculature and regulate blood flow. Pericytes already express αSMA, and can become myofibroblasts if they leave their cellular niche and start to express proteins such as collagen type I and FN1-EDA. That this process occurs in SSc is suggested by a study that shows that pericytes in SSc skin, but not in healthy skin, express FN1-EDA and other myofibroblast markers (27). Furthermore, using lineage tracing it has elegantly been demonstrated that perivascular cells end up in skin scars as myofibroblasts (84). In addition, this transition is also observed in lung, liver, and kidney fibrosis (85), indicating that pericyte to myofibroblast transition is a common aspect of many fibrotic disorders. Putative drivers of this transition are VEGF, PDGF, and TGFβ.
Another cell type which can give rise to myofibroblasts is the fibrocyte. Fibrocytes are circulating cells of myeloid origin with stem cell like characteristics. These cells were first identified as the myeloid cells that rapidly invade wounds and, in contrast to other myeloid cells, produce ECM molecules. Their migration to wounds is guided by damage associated molecular patterns (DAMPs) and chemokines such as Chemokine (C-C motif) ligand 21 (CCL21) (86), and after arrival, these cells start differentiating into a myofibroblast-like phenotype under the influence of factors such as TGFβ (86). Of note, fibrocytes can originate from monocytes, and, importantly, SSc monocytes display increased maturation toward myofibroblasts as indicated by αSMA expression when compared to monocytes from healthy controls (87). Furthermore, fibrocyte presence and involvement in pulmonary fibrosis can readily be detected in SSc (87). Paradoxically, fibrocyte numbers in blood are lower in SSc patients than in healthy controls. Possibly, these cells are recruited out of the blood compartment into affected areas which would explain their lower numbers in blood.
In addition to the abovementioned cells, adipocytes, i.e., fat cells, are another source of myofibroblasts in SSc. Via the process of adipocyte to myofibroblast transition these cells can become myofibroblasts. In SSc skin, subcutaneous fat disappears over the course of the disease (88). With the use of adiponectin-lineage tracking, it has been demonstrated in the murine bleomycin model of skin fibrosis that adipocytes can lose their adipocyte-related gene expression and start expressing αSMA to become myofibroblasts (88). Importantly, in this model of skin fibrosis the loss of fat tissue precedes fibrosis (88) indicating that this process can underlie the fibrotic process. Adipocyte to myofibroblast transition is strongly driven by TGFβ (88), Found in inflammatory zone 1 (FIZZ1) and possibly Wnt signaling (89). In vitro, FIZZ1 suppresses adipogenesis and stimulates myofibroblast differentiation via Notch1 signaling. Furthermore, mice lacking FIZZ1 retain more fat and develop less fibrosis in response to bleomycin skin injury (90). Of note, FIZZ1 has also been attributed a role in lung fibrosis, by recruiting bone marrow derived stem like cells like to damaged lung tissue (91), and its levels are increased in serum of SSc patients (90).
Finally, two important sources of myofibroblasts in SSc are epithelial to mesenchymal transition (EMT) and endothelial to mesenchymal transition (EndoMT). In both processes, respectively epithelial and endothelial cells lose their phenotype and become myofibroblasts. Both processes can be observed in SSc. EndoMT can be identified using immunohistochemistry by observing endothelial cells with both endothelial (cluster of differentiation (CD31, and VE-cadherin) and myofibroblast markers (αSMA), and has been observed in skin and in lungs of SSc patients (92, 93). Furthermore, EndoMT has been linked to endothelial dysfunction as a cause for pulmonary arterial hypertension, a major complication in SSc (94). Notably, endothelial cells that undergo EndoMT produce more IL-6, IL-8 and TNFα compared to normal endothelial cells (94). EMT is an important driver of lung fibrosis, in which alveolar epithelial cells become myofibroblasts (95). This was demonstrated using alveolar specific lineage tracking, which visualized that alveolar cells started to express αSMA upon overexpression of TGFβ1 (95). The role of EMT in skin fibrosis is less clear. In SSc skin, expression of the key EMT inducing transcription factor SNAI1 can be observed in keratinocytes, but not loss of their epithelial E-Cadherin marker (96). Possibly, the EMT process is therefore only partially evoked here.
In conclusion, myofibroblasts can originate from many sources in SSc. Possibly, their origin has an effect on their phenotype and function, yet little is known if this is the case.
On Increased Activity of Myofibroblasts in SSc
Because of reduced apoptosis and increased formation, myofibroblasts numbers are increased in SSc. However, also their activity is markedly increased in SSc. For example, skin (myo) fibroblasts of SSc patients show more activation of focal adhesion kinase (FAK) in vitro than those of controls (97). This focal adhesion kinase is a key component of integrin signaling, and regulates fibroblast migration, survival and growth. Furthermore, in vitro, (myo)fibroblasts obtained from SSc patients produce more extracellular matrix molecules such as collagen type I than those of healthy controls, and their migratory and contractile properties are also increased (19, 98). Because the activated phenotype of SSc (myo) fibroblasts persists ex vivo, e.g., during cell culture, epigenetic changes most likely play an important role in this phenotype. For example, recent research has shown that in SSc skin fibroblasts, expression of the histone demethylase Jumonji domain-containing protein 3 (JMJD3) is increased (99). This histone demethylase removes the so-called H3K27me3 mark from histones, and this mark can repress expression of pro-fibrotic genes such as collagen type I in fibroblasts (100). Furthermore, pharmacological inhibition of H3K27 trimethylation induces skin fibrosis and aggravates pathology in bleomcyin induced skin fibrosis (100). A key target which is activated by JMJD3 is Fos-related antigen 2 (Fra-2) (99). This transcription factor has been identified as an important regulator of extracellular matrix production in skin fibroblasts; transgenic overexpression of Fra-2 results in increased dermal thickness and myofibroblast formation and is a mouse model for SSc (101), whereas knockdown of Fra-2 reduces both TGFβ- and PDGF-induced collagen production in primary skin fibroblasts of SSc patients (102).
Next to epigenetic changes, several cytokines can enhance the formation and function of myofibroblasts. In Table 1 an overview is given of how various cytokines affect myofibroblasts activity. As already mentioned TGFβ, PDGF, Wnts, IL-6, and OSM are key cytokines for myofibroblasts formation and activity. In addition to these factors, both IL-4 and IL-13 are pro-fibrotic (150). Both cytokines induce αSMA expression in primary lung fibroblasts in a dose- and time-dependent manner (105, 150), and enhance the production of collagen type I in normal fibroblasts (108). IL-22 has been described to have similar effect (118). Less clear is the role of IL-1 and Tumor necrosis factor α (TNFα). Of these factors both inhibitory and stimulatory effects on (myo) fibroblasts have been described. In atrial and intestinal myofibroblasts TNFα induces proliferation and collagen synthesis (119, 120). However, in dermal fibroblasts TNFα can inhibit αSMA expression by inhibiting TGFβ signaling (124). Interleukin 1 can not only induce, but also inhibit, collagen production, proliferation and myofibroblasts formation in dermal and lung fibroblasts by inhibition of TGFβ signaling (103, 104). Apart from these stimulatory cytokines, several signaling molecules inhibit myofibroblast formation and activity. For example, interferon γ (IFNγ) inhibits collagen synthesis, sensitizes dermal fibroblast to Fas-mediated apoptosis (125, 126) and inhibits IL-4 effects (125). Prostaglandin E2 has similar effects on formation and apoptosis in lung and keloid fibroblasts (145–147). The role of basic fibroblast growth factor (FGF2) is less clear, as it can inhibit TGFβ-mediated myofibroblast formation (140), but can also increase myofibroblast proliferation (151).
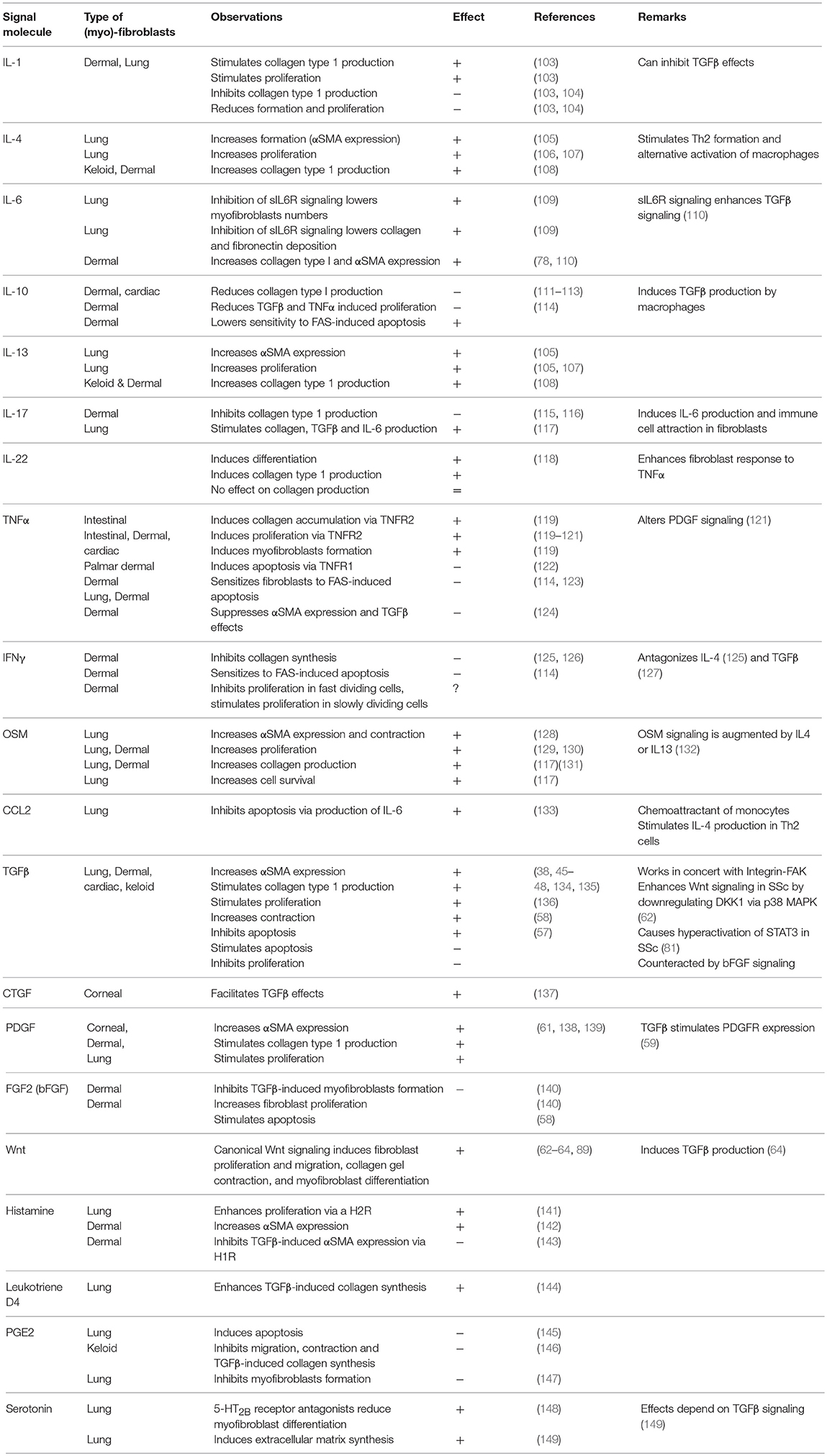
Table 1. Influence of various cytokines on myofibroblast biology.
The increased presence and activity of myofibroblasts in SSc results in various deleterious effects. First of all, their excessive matrix production and remodeling capabilities can destruct organ architecture leading to loss of function like in lung fibrosis. Furthermore, deposition of extracellular matrix molecules such as collagens in the interstitial space of lung tissue inhibits gas exchange, greatly lowering lung function and resulting in interstitial lung disease. In skin excessive matrix deposition increases stiffness, increases hardness, and leads to loss of cutaneous tissues like, fat tissue, sweat glands, hair follicles, and sebaceous glands (152). In the gastro-intestinal tract, myofibroblast-induced fibrosis negatively affect motility, digestion, absorption, and excretion (153).
Blood vessel function is also impacted by myofibroblasts. To begin, myofibroblasts produce endothelin-1 (15). Endothelin 1 is a potent vasoconstrictor, leading to increased blood pressure. Notably, endothelin 1 also stimulates the formation of new myofibroblasts. Furthermore, myofibroblasts also produce VEGF (154), e.g., during wound healing, and can also express angiopoietin 1 and 2, both of which stimulate the formation of new blood vessels (155). As mentioned, myofibroblasts also produce and activate TGFβ. VEGF, angiopoietins, and TGFβ are all key regulators of endothelial homeostasis, and normally these factors are well balanced to maintain this homeostasis. However, this balance can be disturbed by the myofibroblast's production of these factors, leading to aberrant vascular remodeling. For example, uncontrolled VEGF signaling has been suggested to be a cause for capillary malformations in SSc (154).
Myofibroblast also have an immunomodulatory role. As mentioned, they express for example interleukin 1 (IL-1), interleukin 6 (IL-6), interleukin 8 (IL-8), monocyte chemoattractive protein 1 (MCP-1) (13). Both IL-8 and MCP-1, also known as CCL2, are chemokines, attracting neutrophils, monocytes and T cells and in this way facilitate inflammation. Both IL-1 and IL-6 can enhances pro-inflammatory gene expression in immune cells. Furthermore, both factors can participate in the differentiation of monocytes toward macrophages and play a role in the differentiation of naive T-cells toward an effector subtype (156).
On the Role of the (Innate) Immune System in Myofibroblast Formation and Function
Myofibroblast survival, formation, and function are all increased in SSc. The (innate) immune system plays an important role in this. In Figure 6 an overview is given of how.
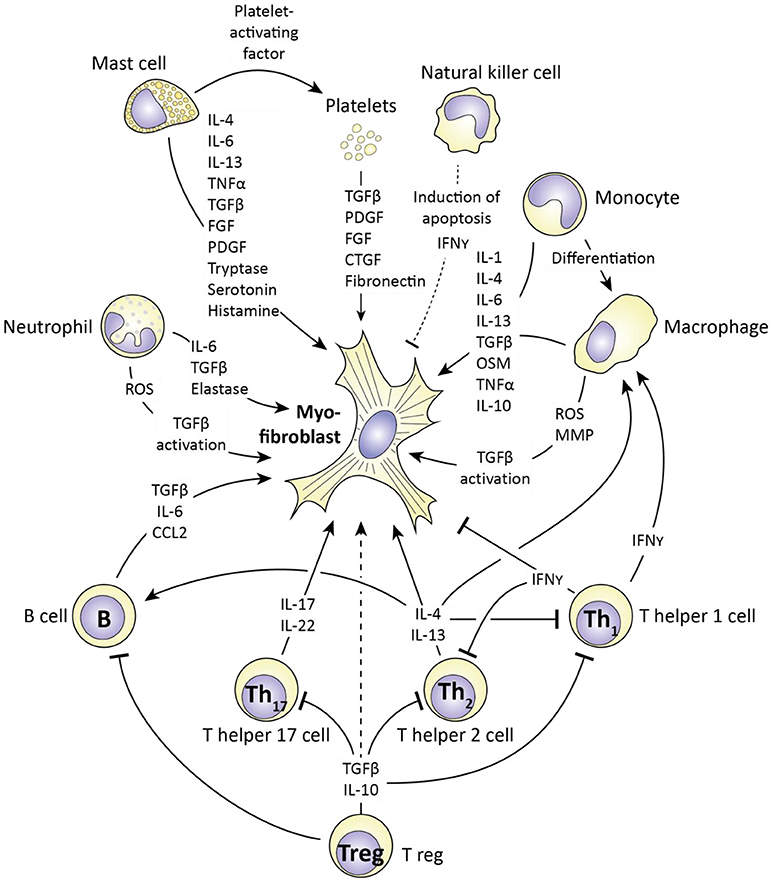
Figure 6. The influence of immune cells on myofibroblast formation and function. Immune cells produce various mediators (also see Table 1) that influence myofibroblast formation and function. For each cell type (and platelets) the corresponding mediators are depicted. Cells which stimulate myofibroblast function include mast cells, monocytes/macrophages and T helper 2 lymphocytes via e.g. production of IL-4, IL-13, and TGFβ. In contrast, T helper 1 cells can negatively affect myofibroblast function via production of interferon gamma (IFNγ). Importantly, the ultimate outcome of an immune response on myofibroblast function depends on the interplay between immune cells, as this interplay regulates the production of the mediators the affect myofibroblast function.
One immune cell which can induce myofibroblasts formation and activity is the mast cell. Mast cells are part of the innate immune system and well known for their role in allergy. However, they have already been implicated in SSc pathophysiology for a long time (157), because they can produce several mediators which stimulate fibrosis (158). One such factor is Platelet-activating factor, which stimulates platelet aggregation and degranulation. Platelet degranulation releases many (growth) factors, including TGFβ, PDGF, and fibronectin, all of which are factors which stimulate myofibroblasts formation and function. Another product of mast cells and platelets is serotonin. Serotonin has long been implicated in fibrotic disorders; already in 1958 it was demonstrated that subcutaneous injections of serotonin induce skin fibrosis (159). More recently, it was demonstrated that serotonin directly increases extracellular matrix production in primary skin fibroblasts (149). This effect runs via the 5H-T2b receptor; inhibition of this receptor with terguride decreases collagen and fibronectin production by fibroblasts. Importantly, mice that lack this receptor () are protected against bleomycin-induced skin fibrosis, just as mice in which the 5H-T2b, receptor is pharmacologically inhibited (149). Mast cells also produce tryptase, a serine proteinase, which, remarkably, stimulates fibroblast proliferation and collagen production (142, 160, 161), and histamine, which also induces (lung) fibroblast proliferation (141). Next to these factors, mast cells also produce a large array of pro-fibrotic cytokines; IL-4, IL-6, IL-13 TNF-α, TGFβ, and PDGF (158) which directly stimulate the formation and activity of myofibroblasts. Interestingly, mast cells can directly interact with skin (myo) fibroblasts, and this facilitates their role in fibrosis. This interaction was shown to be serpine1 dependent. Apart from the aforementioned role as inhibitor of plasmin activation, this protein is a chemotactic for mast cells and induces the expression of intercellular adhesion molecule 1 (ICAM1) in fibroblasts, which is needed for mast cells to adhere to fibroblasts (162). Of note, serpine1 is a downstream target of TGFβ signaling in many cell types, including fibroblasts.
Another innate immune cell which can have a pro-fibrotic role is the neutrophil. Like mast cells, neutrophils produce various pro-fibrotic cytokines including: TGFβ, IL-6, and VEGF (163). Furthermore, activated neutrophils release reactive oxygen species (ROS) (164). Reactive oxygen species activate fibroblasts and stimulate fibrosis (165). In part, this effect is due to the activation of TGFβ. Chemical reaction of reactive oxygen species with latent TGFβ disrupts the quaternary protein structure of latent TGFβ, and results in release of active TGFβ (165). Of note, neutrophils of SSc patients release more ROS than neutrophils of healthy controls when challenged with TNFα (164). Recently, it was also demonstrated that neutrophil elastase, a serine proteinase, can induce myofibroblasts formation (166). Mice lacking this enzyme are protected against asbestos-induced lung fibrosis, and in vitro neutrophil elastase directly stimulates myofibroblasts formation, proliferation, and contractility (166). Furthermore, pharmacological inhibition of neutrophil elastase by sivelestat protects mice from bleomycin induced lung fibrosis (167), demonstrating that at least in lungs, neutrophil elastase is pro-fibrotic.
Next to mast cells and neutrophils, also macrophages can stimulate the formation and activity of myofibroblasts. To begin, macrophages, and their precursor the monocyte, can produce large amounts of TGFβ, for example during bleomycin induced lung fibrosis in rats (168). Apart from TGFβ, macrophages produce many cytokines with pro-fibrotic effects, including IL-4, IL-6, and IL-13 (156). Especially alternatively activated macrophages, also known as M2 macrophages, are associated with production of pro-fibrotic cytokines. These cells have a less pro-inflammatory and more repair oriented phenotype than classically activated macrophages, i.e., M1 macrophages (156). Macrophages, like neutrophils, also produce reactive oxygen species which enhance fibrosis. The importance of macrophages in regulating fibrosis is demonstrated by the observation that in mice, deletion of lung macrophages using liposomal chlodronate reduces bleomycin induced lung fibrosis, and a similar effect is obtained if circulating monocytes are depleted using liposomal chlodronate (169).
A cell of the innate immune system with a possible anti-fibrotic role is the natural killer (NK) cell. In liver fibrosis, this cell type can recognize myofibroblasts and stimulate them to undergo apoptosis (170). Furthermore, NK cells produce IFNγ a strong inhibitor of myofibroblasts formation and function (171). However, in SSc, both the killing ability and stimulation-dependent IFNγ production of NK cells has been reported to be reduced (171).
In addition to the cells of the innate immune system, cells of the acquired immune system also play a role in fibrosis. A cell type particularly associated with fibrosis in SSc is the T helper 2 cell (Th2). These cells produce the pro-fibrotic cytokines IL-4, IL-5, and IL-13, which directly stimulate fibroblasts but also induce the formation of alternatively activated macrophages (172, 173). SSc is characterized by Th2 polarization, i.e., a Th2 cytokine profile in blood, and importantly, in SSc, the extent of Th2 polarization directly positively correlates with active interstitial lung disease (i.e., lung fibrosis), supporting for a role of Th2 cells in this process (132). Also T helper 17 cells (Th17) can play a role in fibrosis, in part via their production of IL-17 and IL-22, which can stimulate collagen, TGFβ and IL-6 production in pulmonary fibroblasts (117, 118). In contrast to these two T helper subtypes, T helper 1 (Th1) cells are more associated with inhibition of myofibroblast function, for example in pulmonary fibrosis (174). This effect of Th1 cells is attributed to their production of IFNγ, which directly inhibits myofibroblast formation and function, but also directs macrophage polarization away from the pro-fibrotic, alternatively activated (M2) phenotype. The role of regulatory T cells (Treg) in fibrosis and myofibroblast activity is less clear. These cells produce TGFβ and IL-10 which can directly regulate myofibroblast function, but also affect the activity of Th1, Th2, and Th17 cells. Because these effector T cells have different functions on myofibroblasts, the end effect of Treg activity is difficult to predict. Finally, B lymphocytes have also been demonstrated to be able to promote fibrosis; co-culture of B cells with skin fibroblasts induced the expression of collagen and αSMA by the latter (175). For this effect cell-cell contact was required, as the use of a transwell system negated the stimulatory effect of B cells on fibroblast activity (175). However, B lymphocytes are able to produce TGFβ, CCL2, and IL-6, which enhance myofibroblast activity without the need for cell contact.
On Therapeutic Targeting of Myofibroblasts in SSc
In view of the role of myofibroblasts in SSc, inhibiting their formation or function makes an excellent option for targeted therapy. Several compounds that have been investigated in, or are currently under investigation in clinical trials are listed in Tables 2, 3, respectively. Whether these compounds truly target myofibroblasts is up for debate, yet they do target cellular processes important for myofibroblast formation and function.
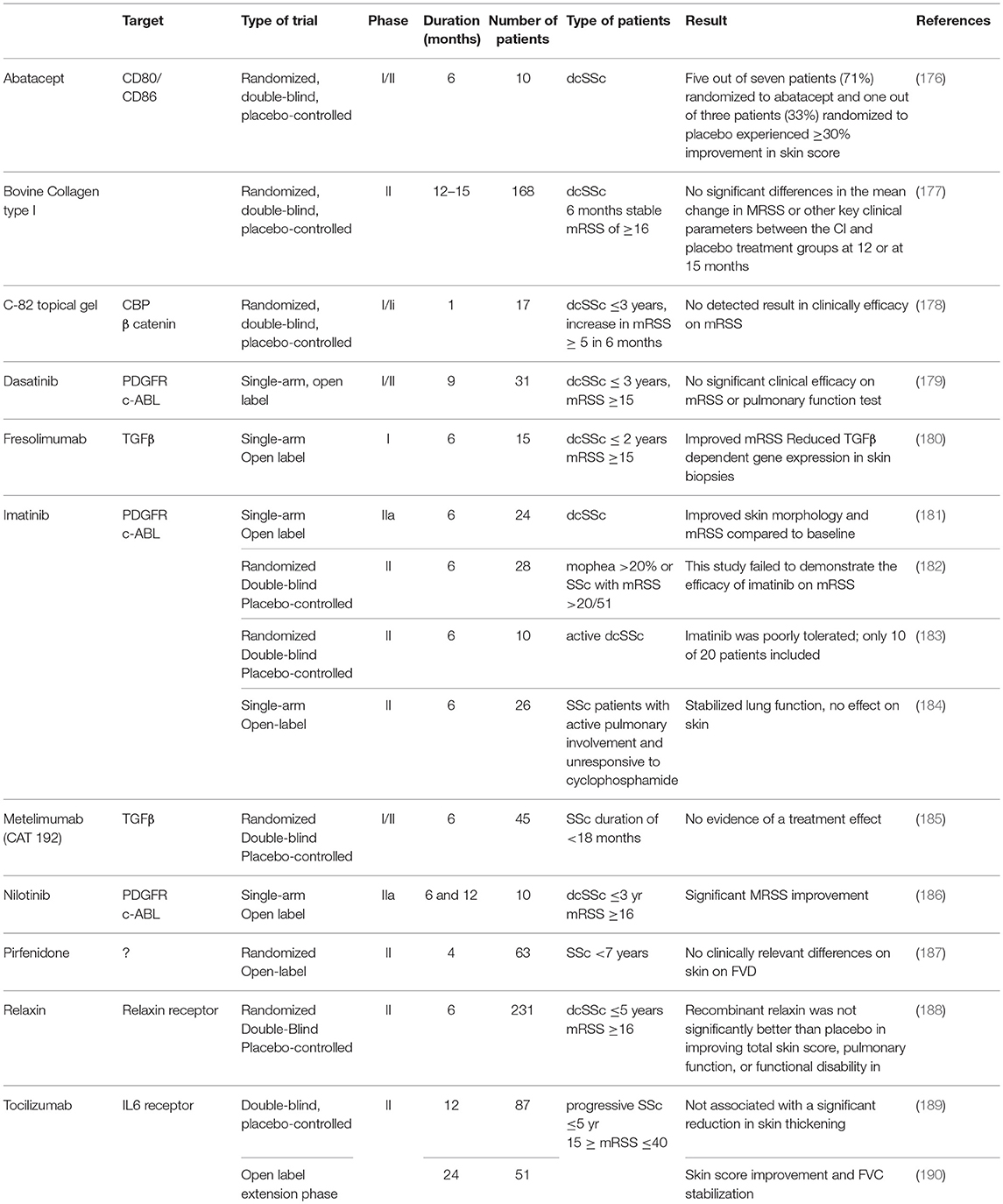
Table 2. Clinical trials conducted with putative anti-fibrotic agents in SSc.
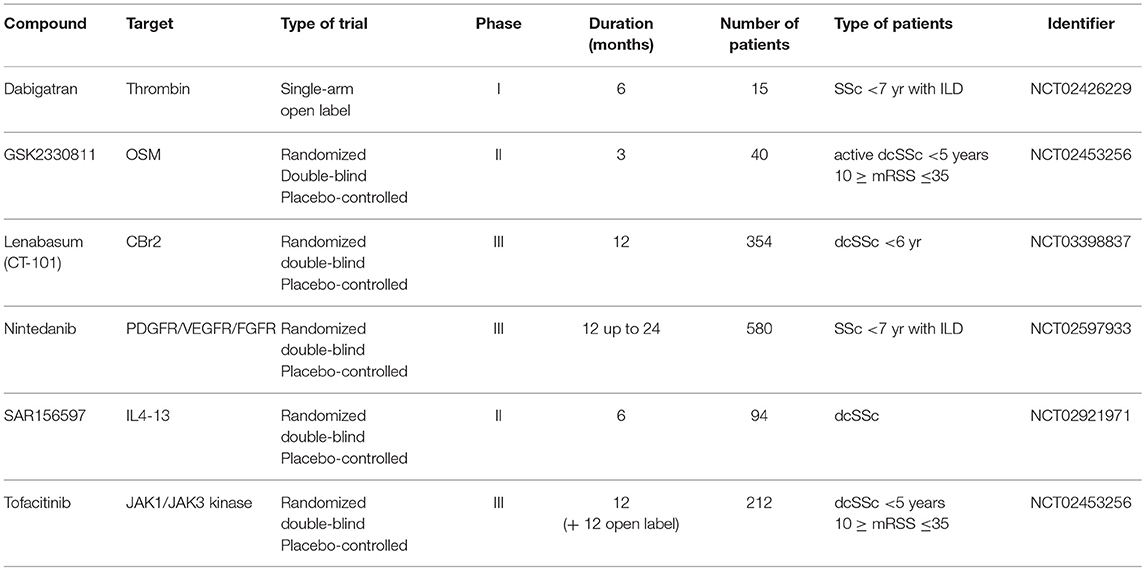
Table 3. Clinical trials currently underway with putative anti-fibrotic agents in SSc.
To begin, one compound that is currently under investigation is tocilizumab. Tocilizumab is a humanized antibody directed against the IL-6 receptor and currently enrolled in a phase III trial for SSc therapy (98). In an initial phase 2 double-blind, placebo controlled study tocilizumab did not significant reduce skin thickening (189), but the open label extension phase of this study did show encouraging protective effects on skin thickening and loss of forced vital capacity in SSc patients (190). Interestingly, skin biopsies were collected from enrolled patients before and after treatment and analyzed for fibroblast phenotype. Treatment with tocilizumab for 24 weeks decreased fibroblast protein production, migration and contractility compared to baseline (98). Furthermore, a large effect between the tocilizumab and placebo treated groups was observed on gene expression profile; in the placebo group, gene expression was not significantly altered over 24 weeks, whereas in the tocilizumab treated group 2,136 genes were significantly differentially expressed. Strikingly, many TGFβ signaling related genes, together with cell contractility pathways, were downregulated to a level similar to normal expression levels (98). This study thus demonstrates that tocilizumab is a serious candidate for targeting (myo-) fibroblasts in SSc.
In view of these results with tocilizumab, the results of tofacitinib in SSc treatment will be of interest. Tofacitinib is a small molecule JAK1 and JAK3 kinase inhibitor downstream of IL-6 signaling which can be used for the treatment of rheumatoid arthritis. Because JAK1 and JAK3 both activate STAT3 this compound can be expected to inhibit myofibroblast function. Currently, tofacitinib is under investigation in a small double-blinded phase I/II trial for safety and efficacy in SSc.
Another compound of interest for treatment of fibrosis in SSc is pirfenidone. Pirfenidone is used for the treatment of idiopathic pulmonary fibrosis and is a pyridone derivative. Dietary intake of this compound was shown to inhibit bleomycin-induced lung fibrosis in hamsters (191). Furthermore, this compound reduces fibroblast proliferation and attenuates TGFβ-induced αSMA and collagen production in primary skin fibroblast (192, 193). In lung fibroblast of SSc patients with interstitial lung disease (ILD), treatment with pirfenidone lowered αSMA and fibronectin expression (194). However, in an open label phase 2 study with 63 SSc patients with ILD, no beneficial effects of pirfenidone were observed on disease outcomes (187).
Nintedanib is a small molecule kinase inhibitor of platelet derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and fibroblast growth factor receptor (FGFR), which has been approved for the treatment of interstitial lung disease, and which can possibly be used for the treatment of (ILD in) SSc. For this latter application, it was recently granted a fast track designation by the U.S. Food and Drug Administration (FDA). In lung fibroblasts in vitro, nintedanib inhibits proliferation and motility as induced by FGF and PDGF, but also inhibits TGFβ-induced collagen deposition (195). In vivo, nintedanib protects mice and rats against bleomycin-induced lung fibrosis (195, 196), and lowers the amount of lymphocytes and neutrophils but not macrophages in bronchoalveolar lavage liquid in this model. In SSc skin fibroblasts, nintedanib also prevents proliferation and motility and lowers the expression of αSMA (197). Furthermore, it lowers the myofibroblasts count and skin fibrosis in bleomycin induced skin fibrosis in mice (197). In two phase 3 trials with patients suffering from interstitial lung disease, nintedanib profoundly lowered the decline in forced vital capacity (195, 198). Currently a phase 3 trial is underway to test its safety and efficacy in SSc.
PDGFR signaling is also targeted by nilotinib. This small molecule kinase inhibitor inhibits both PDGFR signaling and c-ABL signaling. In dermal fibroblasts, nilotinib inhibits TGFβ- or PDGF-induced collagen production in a dose dependent manner (199). Furthemore, this compound strongly lowered myofibroblasts formation and dermal thickness in bleomycin induced skin fibrosis in mice (199). Nilotinib has been tested in a small open label trial with 10 SSc patients (186), and has shown promising results on the modified Rodnan skin score after 6 and 12 months of treatment. A compound similar to nilotinib is imatinib, which also targets PDGFR and c-ABL signaling (200) This compound also reduces collagen production in skin fibroblasts, and also protects mice against bleomycin induced fibrosis via reduction of myofibroblasts formation and matrix deposition (200). Several clinical phase 2 trials have been conducted with imatinib in SSc. In an open label, single-arm clinical trial a positive effect of imatinib on skin thickening was reported after 6 months of treatment (181). However, in another placebo controlled, double blinded phase 2 study no efficacy on modified Rodnan skin score was reported after 6 months of treatment (182). Furthermore, a single center randomized, double-blind, placebo-controlled phase 2 trial warned for poor drug tolerance in SSc patients (183). A low dose of imatinib has also been tested in an open label study for treatment of intestitial lung disease in SSc patients unresponsive to cyclophosphamide (184), and was reported to stabilize lung function but again had no effect on skin.
In view of its pro-fibrotic effects, TGFβ has also been targeted in SSc. Currently, several TGFβ signaling targeting drugs are under clinical development for e.g., cancer treatment (201), but no trials for SSc are currently reported. In a small open label single center study, Fresolimumab (180), a high affinity TGFβ inactivating monoclonal antibody was recently tested in SSc patients, and reduced TGFβ dependent gene expression in skin biopsies and improved modified Rodnan skin score (180). In the past, a low affinity antibody had no such effect (185). Interestingly, several compounds that inhibit TGFβ activation by integrins are under development for various diseases. In a genetic mouse model for SSc (characterized by a mutation in fibrilin) antibodies against integrin α5β1 and integrin α5β3 inhibit skin fibrosis (202). The effects of these antibodies are mimicked by a TGFβ inhibiting antibody, illustrating that these effects possibly run via inhibition of TGFβ activation. In addition, a monoclonal antibody targeting integrin αVβ6 has been shown to protect mice from radiation induced fibrosis (203).
Currently also under development for treatment of SSc is lenabasum (CT-101). Lenabasum is a canabinoid type 2 receptor (CBr2) agonist and is currently being tested in a phase 3 trial for its efficacy and safety in treatment of SSc. Skin fibroblasts express CBr2, and this expression is increased in SSc (204). Stimulation of SSc skin fibroblasts with the synthetic cannabinoid WIN55,212-2 lowers matrix production, myofibroblast formation, and production of TGFβ, CTGF, and IL-6 (204). Furthermore, addition of this compound to mice inhibits bleomycin induced skin fibrosis by lowering fibroblast to myofibroblast transition and TGFβ, CTGF, and PDGF production (205). In addition, in bleomycin lung fibrosis activation of canabinoid receptor type 2 signaling by JWH133 lowered both the inflammatory response and extracellular collagen deposition, which was accompanied by reduced levels of TGFβ in blood (206). These observations make lenabasum a promising compound.
Finally, a therapy currently under investigation in a phase 2 trial as targeted therapy for SSc is the use of abatacept. Abatacept is a fusion protein consisting out of an IgG1 Fc tail fused with the extracellular part of CTL4 and is currently in use for the treatment of rheumatoid arthritis. Abatacept targets and prevents the function of CD80/CD86 molecules of professional antigen producing cells. This prevents these antigen presenting cells from activating T cells, as CD80/CD86 provide the co-stimulatory signal required in addition to MHCII binding to initiate T (helper) cell differentiation. Early SSc skin is characterized by perivascular T cell infiltrates (172), and (late stage) SSc patients have increased Th2 cell activation (Th2 polarization) and these cells express the pro-fibrotic cytokines IL-4, IL-5, IL-6, and IL-13 (172). In bleomycin induced skin fibrosis in mice, abatacept lowers the influx of monocytes, T cells and B cells into lesional areas, lowers IL-6 and IL-10 levels and lowers skin fibrosis (207). Importantly, abatacept does not affect skin fibrosis in the murine Tsk1 model of SSc which is less dependent on inflammation nor in bleomycin induced skin fibrosis in SCID mice which lack T cells. In a very small double-blind placebo controlled trial of 10 patients, abatacept improved the mRSS of patients. Especially patients with an inflammatory gene expression profile in their blood responded well to abatacept (176). These results indicate that abatacept can possibly eleviate inflammation driven fibrosis, but not by directly targeting myofibroblasts.
Conclusion and Future Perspectives
In this review, we have addressed the role of myofibroblasts in SSc pathophysiology. The presence and formation of these cells are increased in SSc, giving rise to pathology due to their ability to produce excessive amounts of extracellular matrix molecules like collagen type I, their ability to affect vascular biology by production of e.g., VEGF and ET1, but also due to their immuno-modulatory effects via production of IL-6 and TGFβ. Targeting these cells is therefore a feasible strategy to get to a targeted therapy for SSc. Currently multiple drugs doing just that are in phase 3 trials, giving hope for the future of SSc treatment.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol. (2012) 24:165–70. doi: 10.1097/BOR.0b013e32834ff2e8
2. Ramos PS, Silver RM, Feghali-Bostwick CA. Genetics of systemic sclerosis: recent advances. Curr Opin Rheumatol. (2015) 27:521–9. doi: 10.1097/BOR.0000000000000214
3. Freire M, Rivera A, Sopena B, Tolosa Vilella C, Guillen-Del Castillo A, Colunga Arguelles D, et al. Clinical and epidemiological differences between men and women with systemic sclerosis: a study in a Spanish systemic sclerosis cohort and literature review. Clin Exp Rheumatol. (2017) 35(Suppl. 106):89–97.
4. Rubio-Rivas M, Moreno R, Corbella X. Occupational and environmental scleroderma. Systematic review and meta-analysis. Clin Rheumatol. (2017) 36:569–82. doi: 10.1007/s10067-016-3533-1
5. Almeida C, Almeida I, Vasconcelos C. Quality of life in systemic sclerosis. Autoimmun Rev. (2015) 14:1087–96. doi: 10.1016/j.autrev.2015.07.012
6. Rubio-Rivas M, Royo C, Simeon CP, Corbella X, Fonollosa V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum. (2014) 44:208–19. doi: 10.1016/j.semarthrit.2014.05.010
7. Mok CC, Kwok CL, Ho LY, Chan PT, Yip SF. Life expectancy, standardized mortality ratios, and causes of death in six rheumatic diseases in Hong Kong, China. Arthritis Rheum. (2011) 63:1182–9. doi: 10.1002/art.30277
8. Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia (1971) 27:549–50. doi: 10.1007/BF02147594
9. Gabbiani G, Hirschel BJ, Ryan GB, Statkov PR, Majno G. Granulation tissue as a contractile organ. A study of structure and function. J Exp Med. (1972) 135:719–34. doi: 10.1084/jem.135.4.719
10. Bond JE, Ho TQ, Selim MA, Hunter CL, Bowers EV, Levinson H. Temporal spatial expression and function of non-muscle myosin II isoforms IIA and IIB in scar remodeling. Laborat Invest. (2011) 91:499–508. doi: 10.1038/labinvest.2010.181
11. Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C. Alpha-smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts. Mol Biol Cell (2003) 14:2508–19. doi: 10.1091/mbc.E02-11-0729
12. Klingberg F, Hinz B, White ES. The myofibroblast matrix: implications for tissue repair and fibrosis. J Pathol. (2013) 229:298–309. doi: 10.1002/path.4104
13. Kendall RT, Feghali-Bostwick CA. Fibroblasts in fibrosis: novel roles and mediators. Front Pharmacol. (2014) 5:123. doi: 10.3389/fphar.2014.00123
14. Lagares D, Busnadiego O, Garcia-Fernandez RA, Lamas S, Rodriguez-Pascual F. Adenoviral gene transfer of endothelin-1 in the lung induces pulmonary fibrosis through the activation of focal adhesion kinase. Am J Res Cell Mol Biol. (2012) 47:834–42. doi: 10.1165/rcmb.2011-0446OC
15. Lagares D, Garcia-Fernandez RA, Jimenez CL, Magan-Marchal N, Busnadiego O, Lamas S, et al. Endothelin 1 contributes to the effect of transforming growth factor beta1 on wound repair and skin fibrosis. Arthritis Rheum. (2010) 62:878–89. doi: 10.1002/art.27307
16. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. (2007) 179:1311–23. doi: 10.1083/jcb.200704042
17. Hinz B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. (2015) 47:54–65. doi: 10.1016/j.matbio.2015.05.006
18. Gilbane AJ, Denton CP, Holmes AM. Scleroderma pathogenesis: a pivotal role for fibroblasts as effector cells. Arthritis Res Ther. (2013) 15:215. doi: 10.1186/ar4230
19. Leroy EC. Connective tissue synthesis by scleroderma skin fibroblasts in cell culture. J Exp Med. (1972) 135:1351–62. doi: 10.1084/jem.135.6.1351
20. Sappino AP, Masouye I, Saurat JH, Gabbiani G. Smooth muscle differentiation in scleroderma fibroblastic cells. Am J Pathol. (1990) 137:585–91.
21. Kissin EY, Merkel PA, Lafyatis R. Myofibroblasts and hyalinized collagen as markers of skin disease in systemic sclerosis. Arthritis Rheum. (2006) 54:3655–60. doi: 10.1002/art.22186
22. Ziemek J, Man A, Hinchcliff M, Varga J, Simms RW, Lafyatis R. The relationship between skin symptoms and the scleroderma modification of the health assessment questionnaire, the modified Rodnan skin score, and skin pathology in patients with systemic sclerosis. Rheumatology (2016) 55:911–7. doi: 10.1093/rheumatology/kew003
23. Van Praet JT, Smith V, Haspeslagh M, Degryse N, Elewaut D, De Keyser F. Histopathological cutaneous alterations in systemic sclerosis: a clinicopathological study. Arthrit Res Ther. (2011) 13:R35. doi: 10.1186/ar3267
24. Beon M, Harley RA, Wessels A, Silver RM, Ludwicka-Bradley A. Myofibroblast induction and microvascular alteration in scleroderma lung fibrosis. Clin Exp Rheumatol. (2004) 22:733–42.
25. Ludwicka A, Trojanowska M, Smith EA, Baumann M, Strange C, Korn JH, et al. Growth and characterization of fibroblasts obtained from bronchoalveolar lavage of patients with scleroderma. J Rheumatol. (1992) 19:1716–23.
26. Manetti M, Neumann E, Milia AF, Tarner IH, Bechi P, Matucci-Cerinic M, et al. Severe fibrosis and increased expression of fibrogenic cytokines in the gastric wall of systemic sclerosis patients. Arthritis Rheum. (2007) 56:3442–7. doi: 10.1002/art.22940
27. Rajkumar VS, Howell K, Csiszar K, Denton CP, Black CM, Abraham DJ. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res Ther. (2005) 7:R1113–23. doi: 10.1186/ar1790
28. Moulin V, Larochelle S, Langlois C, Thibault I, Lopez-Valle CA, Roy M. Normal skin wound and hypertrophic scar myofibroblasts have differential responses to apoptotic inductors. J Cell Physiol. (2004) 198:350–8. doi: 10.1002/jcp.10415
29. Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. (1995) 146:56–66.
30. Yuan S, Akey CW. Apoptosome structure, assembly, and procaspase activation. Structure (2013) 21:501–15. doi: 10.1016/j.str.2013.02.024
31. Zhang M, Zheng J, Nussinov R, Ma B. Release of cytochrome C from bax pores at the mitochondrial membrane. Sci Rep. (2017) 7:2635. doi: 10.1038/s41598-017-02825-7
32. Billen LP, Shamas-Din A, Andrews DW. Bid: a Bax-like BH3 protein. Oncogene (2008) 27(Suppl. 1):S93–104. doi: 10.1038/onc.2009.47
33. Santiago B, Galindo M, Rivero M, Pablos JL. Decreased susceptibility to Fas-induced apoptosis of systemic sclerosis dermal fibroblasts. Arthritis Rheum. (2001) 44:1667–76. doi: 10.1002/1529-0131(200107)44:7<1667::AID-ART291>3.0.CO;2-Y
34. Karimizadeh E, Gharibdoost F, Motamed N, Jafarinejad-Farsangi S, Jamshidi A, Mahmoudi M. c-Abl silencing reduced the inhibitory effects of TGF-beta1 on apoptosis in systemic sclerosis dermal fibroblasts. Mol Cell Biochem. (2015) 405:169–76. doi: 10.1007/s11010-015-2408-0
35. Daniels CE, Wilkes MC, Edens M, Kottom TJ, Murphy SJ, Limper AH, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest. (2004) 114:1308–16. doi: 10.1172/JCI200419603
36. Lagares D, Santos A, Grasberger PE, Liu F, Probst CK, Rahimi RA, et al. Targeted apoptosis of myofibroblasts with the BH3 mimetic ABT-263 reverses established fibrosis. Sci Trans Med. (2017) 9:aal3765. doi: 10.1126/scitranslmed.aal3765
37. Jelaska A, Korn JH. Role of apoptosis and transforming growth factor beta1 in fibroblast selection and activation in systemic sclerosis. Arthritis Rheum. (2000) 43:2230–9. doi: 10.1002/1529-0131(200010)43:10<2230::AID-ANR10>3.0.CO;2-8
38. Liu Y, Li Y, Li N, Teng W, Wang M, Zhang Y, et al. TGF-beta1 promotes scar fibroblasts proliferation and transdifferentiation via up-regulating MicroRNA-21. Sci Rep. (2016) 6:32231. doi: 10.1038/srep32231
39. Samuel GH, Lenna S, Bujor AM, Lafyatis R, Trojanowska M. Acid sphingomyelinase deficiency contributes to resistance of scleroderma fibroblasts to Fas-mediated apoptosis. J Dermatol Sci. (2012) 67:166–72. doi: 10.1016/j.jdermsci.2012.06.001
40. Smith EL, Schuchman EH. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. (2008) 22:3419–31. doi: 10.1096/fj.08-108043
41. Jafarinejad-Farsangi S, Farazmand A, Gharibdoost F, Karimizadeh E, Noorbakhsh F, Faridani H, et al. Inhibition of MicroRNA-21 induces apoptosis in dermal fibroblasts of patients with systemic sclerosis. Int J Dermatol. (2016) 55:1259–67. doi: 10.1111/ijd.13308
42. Hinz B. Tissue stiffness, latent TGF-beta1 activation, and mechanical signal transduction: implications for the pathogenesis and treatment of fibrosis. Curr Rheumatol Rep. (2009) 11:120–6. doi: 10.1007/s11926-009-0017-1
43. Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Involvement of alphavbeta5 integrin-mediated activation of latent transforming growth factor beta1 in autocrine transforming growth factor beta signaling in systemic sclerosis fibroblasts. Arthritis Rheum. (2005) 52:2897–905. doi: 10.1002/art.21246
44. Asano Y, Ihn H, Yamane K, Jinnin M, Tamaki K. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am J Pathol. (2006) 168:499–510. doi: 10.2353/ajpath.2006.041306
45. Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. (2003) 29:397–404. doi: 10.1165/rcmb.2003-0063OC
46. Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB, Tian XL. Effect of TGF-beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol Sin. (2007) 28:382–91. doi: 10.1111/j.1745-7254.2007.00468.x
47. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
48. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circul Res. (2010) 107:418–28. doi: 10.1161/CIRCRESAHA.109.216101
49. Kulkarni AA, Thatcher TH, Olsen KC, Maggirwar SB, Phipps RP, Sime PJ. PPAR-gamma ligands repress TGFbeta-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis. PLoS ONE (2011) 6:e15909. doi: 10.1371/journal.pone.0015909
50. Abdalla M, Goc A, Segar L, Somanath PR. Akt1 mediates alpha-smooth muscle actin expression and myofibroblast differentiation via myocardin and serum response factor. J Biol Chem. (2013) 288:33483–93. doi: 10.1074/jbc.M113.504290
51. Lu Y, Azad N, Wang L, Iyer AK, Castranova V, Jiang BH, et al. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. (2010) 42:432–41. doi: 10.1165/rcmb.2009-0002OC
52. Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, et al. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. (2017) 7:14272. doi: 10.1038/s41598-017-14612-5
53. Meyer-Ter-Vehn T, Gebhardt S, Sebald W, Buttmann M, Grehn F, Schlunck G, et al. p38 inhibitors prevent TGF-beta-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. (2006) 47:1500–9. doi: 10.1167/iovs.05-0361
54. Sanders YY, Cui Z, Le Saux CJ, Horowitz JC, Rangarajan S, Kurundkar A, et al. SMAD-independent down-regulation of caveolin-1 by TGF-beta: effects on proliferation and survival of myofibroblasts. PLoS ONE (2015) 10:e0116995. doi: 10.1371/journal.pone.0116995
55. Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, et al. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. (2004) 279:1359–67. doi: 10.1074/jbc.M306248200
56. Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem. (2015) 290:25427–38. doi: 10.1074/jbc.M115.646984
57. Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell (2002) 110:19–32. doi: 10.1016/S0092-8674(02)00801-2
58. Akasaka Y, Ono I, Kamiya T, Ishikawa Y, Kinoshita T, Ishiguro S, et al. The mechanisms underlying fibroblast apoptosis regulated by growth factors during wound healing. J Pathol. (2010) 221:285–99. doi: 10.1002/path.2710
59. Yamakage A, Kikuchi K, Smith EA, LeRoy EC, Trojanowska M. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J Exp Med. (1992) 175:1227–34. doi: 10.1084/jem.175.5.1227
60. Zhao XK, Cheng Y, Liang Cheng M, Yu L, Mu M, Li H, et al. Focal adhesion kinase regulates fibroblast migration via integrin beta-1 and plays a central role in fibrosis. Sci Rep. (2016) 6:19276. doi: 10.1038/srep19276
61. Singh V, Barbosa FL, Torricelli AA, Santhiago MR, Wilson SE. Transforming growth factor beta and platelet-derived growth factor modulation of myofibroblast development from corneal fibroblasts in vitro. Exp Eye Res. (2014) 120:152–60. doi: 10.1016/j.exer.2014.01.003
62. Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat Commun. (2012) 3:735. doi: 10.1038/ncomms1734
63. Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, et al. Beta-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. (2012) 71:761–7. doi: 10.1136/annrheumdis-2011-200568
64. Wei J, Fang F, Lam AP, Sargent JL, Hamburg E, Hinchcliff ME, et al. Wnt/beta-catenin signaling is hyperactivated in systemic sclerosis and induces Smad-dependent fibrotic responses in mesenchymal cells. Arthritis Rheum. (2012) 64:2734–45. doi: 10.1002/art.34424
65. White ES, Muro AF. Fibronectin splice variants: understanding their multiple roles in health and disease using engineered mouse models. IUBMB Life (2011) 63:538–46. doi: 10.1002/iub.493
66. Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. (1998) 142:873–81. doi: 10.1083/jcb.142.3.873
67. Muro AF, Moretti FA, Moore BB, Yan M, Atrasz RG, Wilke CA, et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am J Respir Critic Care Med. (2008) 177:638–45. doi: 10.1164/rccm.200708-1291OC
68. Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. (2012) 47:340–8. doi: 10.1165/rcmb.2012-0050OC
69. Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, et al. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci USA. (2013) 110:16850–5. doi: 10.1073/pnas.1316764110
70. Luchsinger LL, Patenaude CA, Smith BD, Layne MD. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J Biol Chem. (2011) 286:44116–25. doi: 10.1074/jbc.M111.276931
71. Fan Z, Hao C, Li M, Dai X, Qin H, Li J, et al. MKL1 is an epigenetic modulator of TGF-beta induced fibrogenesis. Biochim Biophys Acta (2015) 1849:1219–28. doi: 10.1016/j.bbagrm.2015.07.013
72. Charbonney E, Speight P, Masszi A, Nakano H, Kapus A. Beta-catenin and Smad3 regulate the activity and stability of myocardin-related transcription factor during epithelial-myofibroblast transition. Mol Biol Cell (2011) 22:4472–85. doi: 10.1091/mbc.e11-04-0335
73. Liu F, Lagares D, Choi KM, Stopfer L, Marinkovic A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol. (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
74. Liang M, Yu M, Xia R, Song K, Wang J, Luo J, et al. Yap/Taz Deletion in Gli(+) Cell-derived myofibroblasts attenuates fibrosis. J Am Soc Nephrol. (2017) 28:3278–90. doi: 10.1681/ASN.2015121354
75. Piersma B, de Rond S, Werker PM, Boo S, Hinz B, van Beuge MM, et al. YAP1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. (2015) 185:3326–37. doi: 10.1016/j.ajpath.2015.08.011
76. Speight P, Kofler M, Szaszi K, Kapus A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat Commun. (2016) 7:11642. doi: 10.1038/ncomms11642
77. Dennler S, Itoh S, Vivien D, Ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. (1998) 17:3091–100. doi: 10.1093/emboj/17.11.3091
78. Khan K, Xu S, Nihtyanova S, Derrett-Smith E, Abraham D, Denton CP, et al. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann Rheum Dis. (2012) 71:1235–42. doi: 10.1136/annrheumdis-2011-200955
79. Kitaba S, Murota H, Terao M, Azukizawa H, Terabe F, Shima Y, et al. Blockade of interleukin-6 receptor alleviates disease in mouse model of scleroderma. Am J Pathol. (2012) 180:165–76. doi: 10.1016/j.ajpath.2011.09.013
80. Gallucci RM, Lee EG, Tomasek JJ. IL-6 modulates alpha-smooth muscle actin expression in dermal fibroblasts from IL-6-deficient mice. J Invest Dermatol. (2006) 126:561–8. doi: 10.1038/sj.jid.5700109
81. Chakraborty D, Sumova B, Mallano T, Chen CW, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. (2017) 8:1130. doi: 10.1038/s41467-017-01236-6
82. Papaioannou I, Xu S, Denton CP, Abraham DJ, Ponticos M. STAT3 controls COL1A2 enhancer activation cooperatively with JunB, regulates type I collagen synthesis posttranscriptionally, and is essential for lung myofibroblast differentiation. Mol Biol Cell (2018) 29:84–95. doi: 10.1091/mbc.e17-06-0342
83. Pedroza M, Le TT, Lewis K, Karmouty-Quintana H, To S, George AT, et al. STAT-3 contributes to pulmonary fibrosis through epithelial injury and fibroblast-myofibroblast differentiation. FASEB J. (2016) 30:129–40. doi: 10.1096/fj.15-273953
84. Dulauroy S, Di Carlo SE, Langa F, Eberl G, Peduto L. Lineage tracing and genetic ablation of ADAM12(+) perivascular cells identify a major source of profibrotic cells during acute tissue injury. Nat Med. (2012) 18:1262–70. doi: 10.1038/nm.2848
85. Greenhalgh SN, Iredale JP, Henderson NC. Origins of fibrosis: pericytes take centre stage. F1000Prime Rep. (2013) 5:37. doi: 10.12703/P5-37
86. Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. (2001) 166:7556–62. doi: 10.4049/jimmunol.166.12.7556
87. Tourkina E, Bonner M, Oates J, Hofbauer A, Richard M, Znoyko S, et al. Altered monocyte and fibrocyte phenotype and function in scleroderma interstitial lung disease: reversal by caveolin-1 scaffolding domain peptide. Fibrogen Tissue Repair (2011) 4:15. doi: 10.1186/1755-1536-4-15
88. Marangoni RG, Korman BD, Wei J, Wood TA, Graham LV, Whitfield ML, et al. Myofibroblasts in murine cutaneous fibrosis originate from adiponectin-positive intradermal progenitors. Arthritis Rheumatol. (2015) 67:1062–73. doi: 10.1002/art.38990
89. Wei J, Melichian D, Komura K, Hinchcliff M, Lam AP, Lafyatis R, et al. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: a novel mouse model for scleroderma? Arthritis Rheum. (2011) 63:1707–17. doi: 10.1002/art.30312
90. Martins V, Gonzalez De Los Santos F, Wu Z, Capelozzi V, Phan SH, Liu T. FIZZ1-induced myofibroblast transdifferentiation from adipocytes and its potential role in dermal fibrosis and lipoatrophy. Am J Pathol. (2015) 185:2768–76. doi: 10.1016/j.ajpath.2015.06.005
91. Liu T, Yu H, Ullenbruch M, Jin H, Ito T, Wu Z, et al. The in vivo fibrotic role of FIZZ1 in pulmonary fibrosis. PLoS ONE (2014) 9:e88362. doi: 10.1371/journal.pone.0088362
92. Manetti M, Romano E, Rosa I, Guiducci S, Bellando-Randone S, De Paulis A, et al. Endothelial-to-mesenchymal transition contributes to endothelial dysfunction and dermal fibrosis in systemic sclerosis. Ann Rheum Dis. (2017) 76:924–34. doi: 10.1136/annrheumdis-2016-210229
93. Mendoza FA, Piera-Velazquez S, Farber JL, Feghali-Bostwick C, Jimenez SA. Endothelial cells expressing endothelial and mesenchymal cell gene products in Systemic Sclerosis-associated interstitial lung disease lung tissues. Arthritis Rheum. (2015) 68:210–217. doi: 10.1002/art.39421
94. Good RB, Gilbane AJ, Trinder SL, Denton CP, Coghlan G, Abraham DJ, et al. Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am J Pathol. (2015) 185:1850–8. doi: 10.1016/j.ajpath.2015.03.019
95. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. (2006) 103:13180–5. doi: 10.1073/pnas.0605669103
96. Nikitorowicz-Buniak J, Denton CP, Abraham D, Stratton R. Partially evoked epithelial-mesenchymal transition (EMT) is associated with increased TGFbeta signaling within lesional scleroderma skin. PLoS ONE (2015) 10:e0134092. doi: 10.1371/journal.pone.0134092
97. Mimura Y, Ihn H, Jinnin M, Asano Y, Yamane K, Tamaki K. Constitutive phosphorylation of focal adhesion kinase is involved in the myofibroblast differentiation of scleroderma fibroblasts. J Invest Dermatol. (2005) 124:886–92. doi: 10.1111/j.0022-202X.2005.23701.x
98. Denton CP, Ong VH, Xu S, Chen-Harris H, Modrusan Z, Lafyatis R, et al. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann Rheum Dis. (2018). doi: 10.1136/annrheumdis-2018-213031
99. Bergmann C, Brandt A, Merlevede B, Hallenberger L, Dees C, Wohlfahrt T, et al. The histone demethylase Jumonji domain-containing protein 3 (JMJD3) regulates fibroblast activation in systemic sclerosis. Ann Rheum Dis. (2018) 77:150–8. doi: 10.1136/annrheumdis-2017-211501
100. Kramer M, Dees C, Huang J, Schlottmann I, Palumbo-Zerr K, Zerr P, et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Ann Rheum Dis. (2013) 72:614–20. doi: 10.1136/annrheumdis-2012-201615
101. Maurer B, Distler JH, Distler O. The Fra-2 transgenic mouse model of systemic sclerosis. Vascul Pharmacol. (2013) 58:194–201. doi: 10.1016/j.vph.2012.12.001
102. Reich N, Maurer B, Akhmetshina A, Venalis P, Dees C, Zerr P, et al. The transcription factor Fra-2 regulates the production of extracellular matrix in systemic sclerosis. Arthritis Rheum. (2010) 62:280–90. doi: 10.1002/art.25056
103. Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. (2016) 38:517–34. doi: 10.1007/s00281-016-0559-z
104. Mia MM, Boersema M, Bank RA. Interleukin-1beta attenuates myofibroblast formation and extracellular matrix production in dermal and lung fibroblasts exposed to transforming growth factor-beta1. PLoS ONE (2014) 9:e91559. doi: 10.1371/journal.pone.0091559
105. Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol. (2003) 132:168–76. doi: 10.1159/000073718
106. Fukai A, Kawamura N, Saito T, Oshima Y, Ikeda T, Kugimiya F, et al. Akt1 in murine chondrocytes controls cartilage calcification during endochondral ossification under physiologic and pathologic conditions. Arthritis Rheum. (2010) 62:826–36. doi: 10.1002/art.27296
107. Kraft M, Lewis C, Pham D, Chu HW. IL-4, IL-13, and dexamethasone augment fibroblast proliferation in asthma. J Allergy Clin Immunol. (2001) 107:602–6. doi: 10.1067/mai.2001.113760
108. Oriente A, Fedarko NS, Pacocha SE, Huang SK, Lichtenstein LM, Essayan DM. Interleukin-13 modulates collagen homeostasis in human skin and keloid fibroblasts. J Pharmacol Exp Ther. (2000) 292:988–94.
109. Le TT, Karmouty-Quintana H, Melicoff E, Le TT, Weng T, Chen NY, et al. Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J Immunol. (2014) 193:3755–68. doi: 10.4049/jimmunol.1302470
110. O'Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. (2014) 289:9952–60. doi: 10.1074/jbc.M113.545822
111. Moroguchi A, Ishimura K, Okano K, Wakabayashi H, Maeba T, Maeta H. Interleukin-10 suppresses proliferation and remodeling of extracellular matrix of cultured human skin fibroblasts. Eur Surg Res. (2004) 36:39–44. doi: 10.1159/000075073
112. Reitamo S, Remitz A, Tamai K, Uitto J. Interleukin-10 modulates type I collagen and matrix metalloprotease gene expression in cultured human skin fibroblasts. J Clin Invest. (1994) 94:2489–92. doi: 10.1172/JCI117618
113. Jung M, Ma Y, Iyer RP, DeLeon-Pennell KY, Yabluchanskiy A, Garrett MR, et al. IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res Cardiol. (2017) 112:33. doi: 10.1007/s00395-017-0622-5
114. Santiago B, Galindo M, Palao G, Pablos JL. Intracellular regulation of Fas-induced apoptosis in human fibroblasts by extracellular factors and cycloheximide. J Immunol. (2004) 172:560–6.
115. Brembilla NC, Montanari E, Truchetet ME, Raschi E, Meroni P, Chizzolini C. Th17 cells favor inflammatory responses while inhibiting type I collagen deposition by dermal fibroblasts: differential effects in healthy and systemic sclerosis fibroblasts. Arthritis Res Ther. (2013) 15:R151. doi: 10.1186/ar4334
116. Nakashima T, Jinnin M, Yamane K, Honda N, Kajihara I, Makino T, et al. Impaired IL-17 signaling pathway contributes to the increased collagen expression in scleroderma fibroblasts. J Immunol. (2012) 188:3573–83. doi: 10.4049/jimmunol.1100591
117. Lei L, Zhao C, Qin F, He ZY, Wang X, Zhong XN. Th17 cells and IL-17 promote the skin and lung inflammation and fibrosis process in a bleomycin-induced murine model of systemic sclerosis. Clin Exp Rheumatol. (2016) 34(Suppl. 100):14–22.
118. McGee HM, Schmidt BA, Booth CJ, Yancopoulos GD, Valenzuela DM, Murphy AJ, et al. IL-22 promotes fibroblast-mediated wound repair in the skin. J Invest Dermatol. (2013) 133:1321–9. doi: 10.1038/jid.2012.463
119. Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J Biol Chem. (2005) 280:36099–109. doi: 10.1074/jbc.M505291200
120. Porter KE, Turner NA, O'Regan DJ, Ball SG. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: inhibition by simvastatin. Cardiovasc Res. (2004) 64:507–15. doi: 10.1016/j.cardiores.2004.07.020
121. Battegay EJ, Raines EW, Colbert T, Ross R. TNF-alpha stimulation of fibroblast proliferation. Dependence on platelet-derived growth factor (PDGF) secretion and alteration of PDGF receptor expression. J Immunol. (1995) 154:6040–7.
122. Alikhani M, Alikhani Z, Raptis M, Graves DT. TNF-alpha in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol. (2004) 201:341–8. doi: 10.1002/jcp.20067
123. Frankel SK, Cosgrove GP, Cha SI, Cool CD, Wynes MW, Edelman BL, et al. TNF-alpha sensitizes normal and fibrotic human lung fibroblasts to Fas-induced apoptosis. Am J Respir Cell Mol Biol. (2006) 34:293–304. doi: 10.1165/rcmb.2005-0155OC
124. Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: an implication for abnormal wound healing. J Invest Dermatol. (2007) 127:2645–55. doi: 10.1038/sj.jid.5700890
125. Serpier H, Gillery P, Salmon-Ehr V, Garnotel R, Georges N, Kalis B, et al. Antagonistic effects of interferon-gamma and interleukin-4 on fibroblast cultures. J Invest Dermatol. (1997) 109:158–62. doi: 10.1111/1523-1747.ep12319207
126. Rosenbloom J, Feldman G, Freundlich B, Jimenez SA. Inhibition of excessive scleroderma fibroblast collagen production by recombinant gamma-interferon. Association with a coordinate decrease in types I and III procollagen messenger RNA levels. Arthritis Rheum. (1986) 29:851–6.
127. Eickelberg O, Pansky A, Koehler E, Bihl M, Tamm M, Hildebrand P, et al. Molecular mechanisms of TGF-(beta) antagonism by interferon (gamma) and cyclosporine A in lung fibroblasts. FASEB J. (2001) 15:797–806. doi: 10.1096/fj.00-0233com
128. Nagahama KY, Togo S, Holz O, Magnussen H, Liu X, Seyama K, et al. Oncostatin M modulates fibroblast function via signal transducers and activators of transcription proteins-3. Am J Respir Cell Mol Biol. (2013) 49:582–91. doi: 10.1165/rcmb.2012-0460OC
129. Scaffidi AK, Mutsaers SE, Moodley YP, McAnulty RJ, Laurent GJ, Thompson PJ, et al. Oncostatin M stimulates proliferation, induces collagen production and inhibits apoptosis of human lung fibroblasts. Br J Pharmacol. (2002) 136:793–801. doi: 10.1038/sj.bjp.0704769
130. Ihn H, Tamaki K. Oncostatin M stimulates the growth of dermal fibroblasts via a mitogen-activated protein kinase-dependent pathway. J Immunol. (2000) 165:2149–55. doi: 10.4049/jimmunol.165.4.2149
131. Ihn H, LeRoy EC, Trojanowska M. Oncostatin M stimulates transcription of the human alpha2(I) collagen gene via the Sp1/Sp3-binding site. J Biol Chem. (1997) 272:24666–72. doi: 10.1074/jbc.272.39.24666
132. Boin F, De Fanis U, Bartlett SJ, Wigley FM, Rosen A, Casolaro V. T cell polarization identifies distinct clinical phenotypes in scleroderma lung disease. Arthritis Rheum. (2008) 58:1165–74. doi: 10.1002/art.23406
133. Liu X, Das AM, Seideman J, Griswold D, Afuh CN, Kobayashi T, et al. The CC chemokine ligand 2 (CCL2) mediates fibroblast survival through IL-6. Am J Respir Cell Mol Biol. (2007) 37:121–8. doi: 10.1165/rcmb.2005-0253OC
134. Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, et al. Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. (2003) 278:12384–9. doi: 10.1074/jbc.M208544200
135. Bettinger DA, Yager DR, Diegelmann RF, Cohen IK. The effect of TGF-beta on keloid fibroblast proliferation and collagen synthesis. Plast Reconstr Surg. (1996) 98:827–33. doi: 10.1097/00006534-199610000-00012
136. Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1). Am J Respir Cell Mol Biol. (1999) 21:658–65. doi: 10.1165/ajrcmb.21.6.3720
137. Garrett Q, Khaw PT, Blalock TD, Schultz GS, Grotendorst GR, Daniels JT. Involvement of CTGF in TGF-beta1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Invest Ophthalmol Vis Sci. (2004) 45:1109–16. doi: 10.1167/iovs.03-0660
138. Bostrom H, Willetts K, Pekny M, Leveen P, Lindahl P, Hedstrand H, et al. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell (1996) 85:863–73. doi: 10.1016/S0092-8674(00)81270-2
139. Donovan J, Shiwen X, Norman J, Abraham D. Platelet-derived growth factor alpha and beta receptors have overlapping functional activities towards fibroblasts. Fibrogenesis Tissue Repair (2013) 6:10. doi: 10.1186/1755-1536-6-10
140. Dolivo DM, Larson SA, Dominko T. FGF2-mediated attenuation of myofibroblast activation is modulated by distinct MAPK signaling pathways in human dermal fibroblasts. J Dermatol Sci. (2017) 88:339–48. doi: 10.1016/j.jdermsci.2017.08.013
141. Jordana M, Befus AD, Newhouse MT, Bienenstock J, Gauldie J. Effect of histamine on proliferation of normal human adult lung fibroblasts. Thorax (1988) 43:552–8. doi: 10.1136/thx.43.7.552
142. Gailit J, Marchese MJ, Kew RR, Gruber BL. The differentiation and function of myofibroblasts is regulated by mast cell mediators. J Invest Dermatol. (2001) 117:1113–9. doi: 10.1046/j.1523-1747.2001.15211.x
143. Lin L, Yamagata K, Nakayamada S, Sawamukai N, Yamaoka K, Sakata K, et al. Histamine inhibits differentiation of skin fibroblasts into myofibroblasts. Biochem Biophys Res Commun. (2015) 463:434–9. doi: 10.1016/j.bbrc.2015.05.094
144. Asakura T, Ishii Y, Chibana K, Fukuda T. Leukotriene D4 stimulates collagen production from myofibroblasts transformed by TGF-beta. J Allergy Clin Immunol. (2004) 114:310–5. doi: 10.1016/j.jaci.2004.04.037
145. Huang SK, White ES, Wettlaufer SH, Grifka H, Hogaboam CM, Thannickal VJ, et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. (2009) 23:4317–26. doi: 10.1096/fj.08-128801
146. Sandulache VC, Parekh A, Li-Korotky H, Dohar JE, Hebda PA. Prostaglandin E2 inhibition of keloid fibroblast migration, contraction, and transforming growth factor (TGF)-beta1-induced collagen synthesis. Wound Repair Regener. (2007) 15:122–33. doi: 10.1111/j.1524-475X.2006.00193.x
147. Kolodsick JE, Peters-Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. (2003) 29:537–44. doi: 10.1165/rcmb.2002-0243OC
148. Lofdahl A, Rydell-Tormanen K, Muller C, Martina Holst C, Thiman L, Ekstrom G, et al. 5-HT2B receptor antagonists attenuate myofibroblast differentiation and subsequent fibrotic responses in vitro and in vivo. Physiol Rep. (2016) 4:12873. doi: 10.14814/phy2.12873
149. Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, et al. Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. (2011) 208:961–72. doi: 10.1084/jem.20101629
150. Hashimoto S, Gon Y, Takeshita I, Maruoka S, Horie T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase-dependent pathway. J Allergy Clin Immunol. (2001) 107:1001–8. doi: 10.1067/mai.2001.114702
151. Perl AK, Gale E. FGF signaling is required for myofibroblast differentiation during alveolar regeneration. Am J Physiol. (2009) 297:L299–308. doi: 10.1152/ajplung.00008.2009
152. Krieg T, Takehara K. Skin disease: a cardinal feature of systemic sclerosis. Rheumatology (2009) 48(Suppl 3):4–8. doi: 10.1093/rheumatology/kep108
153. Forbes A, Marie I. Gastrointestinal complications: the most frequent internal complications of systemic sclerosis. Rheumatology (2009) 48(Suppl. 3):iii36–9. doi: 10.1093/rheumatology/ken485
154. Distler O, Distler JH, Scheid A, Acker T, Hirth A, Rethage J, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circul Res. (2004) 95:109–16. doi: 10.1161/01.RES.0000134644.89917.96
155. Staton CA, Valluru M, Hoh L, Reed MW, Brown NJ. Angiopoietin-1, angiopoietin-2 and Tie-2 receptor expression in human dermal wound repair and scarring. Br J Dermatol. (2010) 163:920–7. doi: 10.1111/j.1365-2133.2010.09940.x
156. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. (2014) 5:491. doi: 10.3389/fimmu.2014.00491
157. Claman HN. On scleroderma. mast cells, endothelial cells, and fibroblasts. JAMA (1989) 262:1206–9. doi: 10.1001/jama.1989.03430090068035
158. Overed-Sayer C, Rapley L, Mustelin T, Clarke DL. Are mast cells instrumental for fibrotic diseases? Front Pharmacol. (2013) 4:174. doi: 10.3389/fphar.2013.00174
159. Macdonald RA, Robbins SL, Mallory GK. Dermal fibrosis following subcutaneous injections of serotonin creatinine sulphate. Proc Soc Exp Biol Med. (1958) 97:334–7. doi: 10.3181/00379727-97-23734
160. Ruoss SJ, Hartmann T, Caughey GH. Mast cell tryptase is a mitogen for cultured fibroblasts. J Clin Invest. (1991) 88:493–9. doi: 10.1172/JCI115330
161. Cairns JA, Walls AF. Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J Clin Invest. (1997) 99:1313–21. doi: 10.1172/JCI119290
162. Pincha N, Hajam EY, Badarinath KS, Batta PR, Masudi T, Dey R, et al. PAI1 mediates fibroblast-mast cell interactions in skin fibrosis. J Clin Invest. (2018) 128:1807–19. doi: 10.1172/JCI99088
163. Tecchio C, Micheletti A, Cassatella MA. Neutrophil-derived cytokines: facts beyond expression. Front Immunol. (2014) 5:508. doi: 10.3389/fimmu.2014.00508
164. Barnes TC, Anderson ME, Edwards SW, Moots RJ. Neutrophil-derived reactive oxygen species in SSc. Rheumatology (2012) 51:1166–9. doi: 10.1093/rheumatology/ker520
165. Richter K, Kietzmann T. Reactive oxygen species and fibrosis: further evidence of a significant liaison. Cell Tissue Res. (2016) 365:591–605. doi: 10.1007/s00441-016-2445-3
166. Gregory AD, Kliment CR, Metz HE, Kim KH, Kargl J, Agostini BA, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukocyte Biol. (2015) 98:143–52. doi: 10.1189/jlb.3HI1014-493R
167. Takemasa A, Ishii Y, Fukuda T. A neutrophil elastase inhibitor prevents bleomycin-induced pulmonary fibrosis in mice. Eur Respir J. (2012) 40:1475–82. doi: 10.1183/09031936.00127011
168. Khalil N, Bereznay O, Sporn M, Greenberg AH. Macrophage production of transforming growth factor beta and fibroblast collagen synthesis in chronic pulmonary inflammation. J Exp Med. (1989) 170:727–37. doi: 10.1084/jem.170.3.727
169. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am J Resp Critic Care Med. (2011) 184:569–81. doi: 10.1164/rccm.201010-1719OC
170. Fasbender F, Widera A, Hengstler JG, Watzl C. Natural killer cells and liver fibrosis. Front Immunol. (2016) 7:19. doi: 10.3389/fimmu.2016.00019
171. Horikawa M, Hasegawa M, Komura K, Hayakawa I, Yanaba K, Matsushita T, et al. Abnormal natural killer cell function in systemic sclerosis: altered cytokine production and defective killing activity. J Invest Dermatol. (2005) 125:731–7. doi: 10.1111/j.0022-202X.2005.23767.x
172. O'Reilly S, Hugle T, van Laar JM. T cells in systemic sclerosis: a reappraisal. Rheumatology (2012) 51:1540–9. doi: 10.1093/rheumatology/kes090
173. Barron L, Wynn TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J physiol. (2011) 300:G723–8. doi: 10.1152/ajpgi.00414.2010
174. Kolahian S, Fernandez IE, Eickelberg O, Hartl D. Immune mechanisms in pulmonary fibrosis. Am J Respir Cell Mol Biol. (2016) 55:309–22. doi: 10.1165/rcmb.2016-0121TR
175. Francois A, Chatelus E, Wachsmann D, Sibilia J, Bahram S, Alsaleh G, et al. B lymphocytes and B-cell activating factor promote collagen and profibrotic markers expression by dermal fibroblasts in systemic sclerosis. Arthrit Res Ther. (2013) 15:R168. doi: 10.1186/ar4352
176. Chakravarty EF, Martyanov V, Fiorentino D, Wood TA, Haddon DJ, Jarrell JA, et al. Gene expression changes reflect clinical response in a placebo-controlled randomized trial of abatacept in patients with diffuse cutaneous systemic sclerosis. Arthrit Res Ther. (2015) 17:159. doi: 10.1186/s13075-015-0669-3
177. Postlethwaite AE, Wong WK, Clements P, Chatterjee S, Fessler BJ, Kang AH, et al. A multicenter, randomized, double-blind, placebo-controlled trial of oral type I collagen treatment in patients with diffuse cutaneous systemic sclerosis: I. oral type I collagen does not improve skin in all patients, but may improve skin in late-phase disease. Arthritis Rheum. (2008) 58:1810–22. doi: 10.1002/art.23501
178. Lafyatis R, Mantero JC, Gordon J, Kishore N, Carns M, Dittrich H, et al. Inhibition of beta-catenin signaling in the skin rescues cutaneous adipogenesis in systemic sclerosis: a randomized, double-blind, placebo-controlled trial of C-82. J Invest Dermatol. (2017) 137:2473–83. doi: 10.1016/j.jid.2017.06.032
179. Martyanov V, Kim GJ, Hayes W, Du S, Ganguly BJ, Sy O, et al. Novel lung imaging biomarkers and skin gene expression subsetting in dasatinib treatment of systemic sclerosis-associated interstitial lung disease. PLoS ONE (2017) 12:e0187580. doi: 10.1371/journal.pone.0187580
180. Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J Clin Invest. (2015) 125:2795–807. doi: 10.1172/JCI77958
181. Spiera RF, Gordon JK, Mersten JN, Magro CM, Mehta M, Wildman HF, et al. Imatinib mesylate (Gleevec) in the treatment of diffuse cutaneous systemic sclerosis: results of a 1-year, phase IIa, single-arm, open-label clinical trial. Ann Rheum Dis. (2011) 70:1003–9. doi: 10.1136/ard.2010.143974
182. Prey S, Ezzedine K, Doussau A, Grandoulier AS, Barcat D, Chatelus E, et al. Imatinib mesylate in scleroderma-associated diffuse skin fibrosis: a phase II multicentre randomized double-blinded controlled trial. Br J Dermatol. (2012) 167:1138–44. doi: 10.1111/j.1365-2133.2012.11186.x
183. Pope J, McBain D, Petrlich L, Watson S, Vanderhoek L, de Leon F, et al. Imatinib in active diffuse cutaneous systemic sclerosis: Results of a six-month, randomized, double-blind, placebo-controlled, proof-of-concept pilot study at a single center. Arthritis Rheum. (2011) 63:3547–51. doi: 10.1002/art.30549
184. Fraticelli P, Gabrielli B, Pomponio G, Valentini G, Bosello S, Riboldi P, et al. Imatinib in Scleroderma Italian Study, Low-dose oral imatinib in the treatment of systemic sclerosis interstitial lung disease unresponsive to cyclophosphamide: a phase II pilot study. Arthrit Res Ther. (2014) 16:R144. doi: 10.1186/ar4606
185. Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, et al. Cat-192 Study, and C. Scleroderma Clinical Trials, Recombinant human anti-transforming growth factor beta1 antibody therapy in systemic sclerosis: a multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. (2007) 56:323–33. doi: 10.1002/art.22289
186. Gordon JK, Martyanov V, Magro C, Wildman HF, Wood TA, Huang WT, et al. Nilotinib (Tasigna) in the treatment of early diffuse systemic sclerosis: an open-label, pilot clinical trial. Arthrit Res Ther. (2015) 17:213. doi: 10.1186/s13075-015-0721-3
187. Khanna D, Albera C, Fischer A, Khalidi N, Raghu G, Chung L, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: the LOTUSS trial. J Rheumatol. (2016) 43:1672–9. doi: 10.3899/jrheum.151322
188. Khanna D, Clements PJ, Furst DE, Korn JH, Ellman M, Rothfield N, et al. The Scleroderma Clinical Trials, Recombinant human relaxin in the treatment of systemic sclerosis with diffuse cutaneous involvement: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2009) 60:1102–11. doi: 10.1002/art.24380
189. Khanna D, Denton CP, Jahreis A, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet (2016) 387:2630–40. doi: 10.1016/S0140-6736(16)00232-4
190. Khanna D, Denton CPC, Lin JF, van Laar JM, Frech TM, Anderson ME, et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann Rheum Dis. (2018) 77:212–20. doi: 10.1136/annrheumdis-2017-211682
191. Iyer SN, Wild JS, Schiedt MJ, Hyde DM, Margolin SB, Giri SN. Dietary intake of pirfenidone ameliorates bleomycin-induced lung fibrosis in hamsters. J Lab Clin Med. (1995) 125:779–85.
192. Conte E, Gili E, Fagone E, Fruciano M, Iemmolo M, Vancheri C. Effect of pirfenidone on proliferation, TGF-beta-induced myofibroblast differentiation and fibrogenic activity of primary human lung fibroblasts. Eur J Pharm Sci. (2014) 58:13–9. doi: 10.1016/j.ejps.2014.02.014
193. Lehtonen ST, Veijola A, Karvonen H, Lappi-Blanco E, Sormunen R, Korpela S, et al. Pirfenidone and nintedanib modulate properties of fibroblasts and myofibroblasts in idiopathic pulmonary fibrosis. Resp Res. (2016) 17:14. doi: 10.1186/s12931-016-0328-5
194. Xiao H, Zhang GF, Liao XP, Li XJ, Zhang J, Lin H, et al. Anti-fibrotic effects of pirfenidone by interference with the hedgehog signalling pathway in patients with systemic sclerosis-associated interstitial lung disease. Int J Rheum Dis. (2018) 21:477–86. doi: 10.1111/1756-185X.13247
195. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1434–45. doi: 10.1183/09031936.00174914
196. Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther. (2014) 349:209–20. doi: 10.1124/jpet.113.208223
197. Huang J, Beyer C, Palumbo-Zerr K, Zhang Y, Ramming A, Distler A, et al. Nintedanib inhibits fibroblast activation and ameliorates fibrosis in preclinical models of systemic sclerosis. Ann Rheum Dis. (2016) 75:883–90. doi: 10.1136/annrheumdis-2014-207109
198. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
199. Akhmetshina A, Dees C, Pileckyte M, Maurer B, Axmann R, Jungel A, et al. Dual inhibition of c-abl and PDGF receptor signaling by dasatinib and nilotinib for the treatment of dermal fibrosis. FASEB J. (2008) 22:2214–22. doi: 10.1096/fj.07-105627
200. Distler JH, Jungel A, Huber LC, Schulze-Horsel U, Zwerina J, Gay RE, et al. Imatinib mesylate reduces production of extracellular matrix and prevents development of experimental dermal fibrosis. Arthritis Rheum. (2007) 56:311–22. doi: 10.1002/art.22314
201. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Design Dev Ther. (2015) 9:4479–99. doi: 10.2147/DDDT.S86621
202. Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, et al. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature (2013) 503:126–30. doi: 10.1038/nature12614
203. Puthawala K, Hadjiangelis N, Jacoby SC, Bayongan E, Zhao Z, Yang Z, et al. Inhibition of integrin alpha(v)beta6, an activator of latent transforming growth factor-beta, prevents radiation-induced lung fibrosis. Am J Resp Critic Care Med. (2008) 177:82–90. doi: 10.1164/rccm.200706-806OC
204. Garcia-Gonzalez E, Selvi E, Balistreri E, Lorenzini S, Maggio R, Natale MR, et al. Cannabinoids inhibit fibrogenesis in diffuse systemic sclerosis fibroblasts. Rheumatology (2009) 48:1050–6. doi: 10.1093/rheumatology/kep189
205. Balistreri E, Garcia-Gonzalez E, Selvi E, Akhmetshina A, Palumbo K, Lorenzini S, et al. The cannabinoid WIN55, 212-2 abrogates dermal fibrosis in scleroderma bleomycin model. Ann Rheum Dis. (2011) 70:695–9. doi: 10.1136/ard.2010.137539
206. Fu Q, Zheng Y, Dong X, Wang L, Jiang CG. Activation of cannabinoid receptor type 2 by JWH133 alleviates bleomycin-induced pulmonary fibrosis in mice. Oncotarget (2017) 8:103486–98. doi: 10.18632/oncotarget.21975
Keywords: myofibroblast, systemic scleroderma, immune system, treatment, cytokine
Citation: van Caam A, Vonk M, van den Hoogen F, van Lent P and van der Kraan P (2018) Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 9:2452. doi: 10.3389/fimmu.2018.02452
Received: 17 June 2018; Accepted: 04 October 2018;
Published: 13 November 2018.
Edited by:
Urs Christen, Goethe-Universität Frankfurt am Main, GermanyReviewed by:
Dimitrios Petrou Bogdanos, University of Thessaly, GreeceGrazyna Kwapiszewska, Ludwig Boltzmann Institute for Lung Vascular Research, Austria
Copyright © 2018 van Caam, Vonk, van den Hoogen, van Lent and van der Kraan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arjan van Caam, YXJqYW4udmFuY2FhbUByYWRib3VkdW1jLm5s