 Kelly A. Pike1,2*
Kelly A. Pike1,2* Michel L. Tremblay1,3,4,5*
Michel L. Tremblay1,3,4,5*- 1Department of Microbiology and Immunology, McGill University, Montréal, QC, Canada
- 2Inception Sciences Canada, Montréal, QC, Canada
- 3Rosalind and Morris Goodman Cancer Centre, McGill University, Montréal, QC, Canada
- 4Division of Experimental Medicine, Department of Medicine, McGill University, Montréal, QC, Canada
- 5Department of Biochemistry, McGill University, Montréal, QC, Canada
Protein tyrosine phosphatases (PTPs) play a critical role in co-ordinating the signaling networks that maintain lymphocyte homeostasis and direct lymphocyte activation. By dephosphorylating tyrosine residues, PTPs have been shown to modulate enzyme activity and both mediate and disrupt protein-protein interactions. Through these molecular mechanisms, PTPs ultimately impact lymphocyte responses to environmental cues such as inflammatory cytokines and chemokines, as well as antigenic stimulation. Mouse models of acute and chronic intestinal inflammation have been shown to be exacerbated in the absence of PTPs such as PTPN2 and PTPN22. This increase in disease severity is due in part to hyper-activation of lymphocytes in the absence of PTP activity. In accordance, human PTPs have been linked to intestinal inflammation. Genome wide association studies (GWAS) identified several PTPs within risk loci for inflammatory bowel disease (IBD). Therapeutically targeting PTP substrates and their associated signaling pathways, such as those implicated in CD4+ T cell responses, has demonstrated clinical efficacy. The current review focuses on the role of PTPs in controlling CD4+ T cell activity in the intestinal mucosa and how disruption of PTP activity in CD4+ T cells can contribute to intestinal inflammation.
Introduction
The gastrointestinal track is a large mucosal surface at which the host's immune system is juxtaposed with a dense microbial population and a diverse array of dietary antigens. Immune recognition of enteric antigens however, is minimized by physical compartmentalization. Bacteria and dietary products are retained within the gut lumen, while the host immune system is localized in the mucosal tissue. This physical separation is preserved by the intestinal mucosal barrier, which includes a mucin layer, a single epithelial cell lining sealed by tight and adherens junctions and a continuous secretion of anti-inflammatory soluble mediators (Figure 1) (1–5).
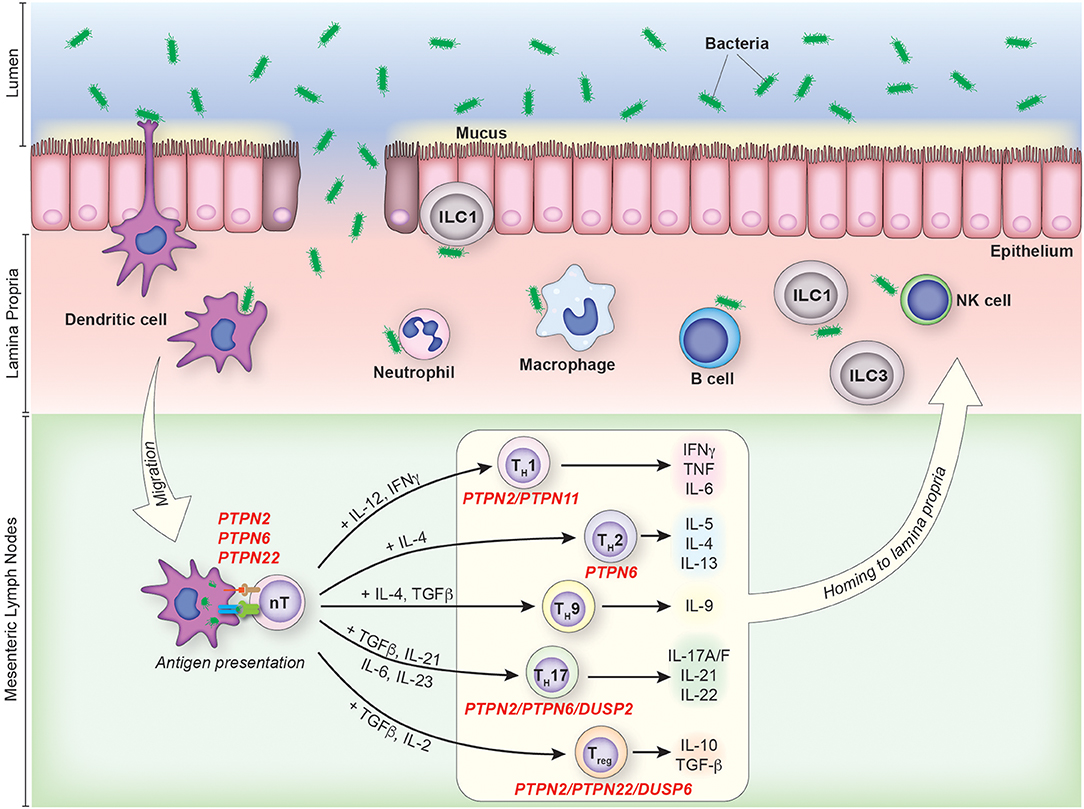
Figure 1. The contribution of PTPs to CD4+ T cell activation and differentiation in colonic inflammation. IBD pathophysiology is associated with the disruption of the intestinal mucosal barrier due to genetic, environmental and/or immunological factors. Under such circumstances, an increase in the uptake and processing of luminal antigens by innate immune cells initiates and maintains the chronic inflammatory response characteristic of IBD. CD4+ T cells are activated in the mesenteric lymph nodes following recognition of their cognate antigen presented in the context of MHC on the surface of antigen presenting cells. Activated CD4+ T cells then enter the lamina propria from circulation and perpetuate inflammation, secreting pathogenic pro-inflammatory mediators and chemokines which recruit additional leukocytes. PTPs involved in the activation and/or differentiation of specific T cell subsets are indicated.
The mucosal barrier is not absolute however, and a small number of bacteria do translocate from the lumen to the underlying lamina propria. In such instances, a complex network of innate and adaptive immune cells impedes the spread of intestinal bacteria in a manner that limits tissue damage (6, 7). This cellular network includes dendritic cells, resident macrophages and the largest population of T cells in the body.
Physical compartmentalization and mucosal immunity therefore establish and maintain microbiome-host mutualism (8). The loss of mutualism and a hyper-activation of the innate and adaptive immune system can result in intestinal inflammatory diseases such as Crohn's disease (CD) and ulcerative colitis (UC), the two main forms of inflammatory bowel disease (IBD). UC and CD are polygenic diseases characterized by chronic relapsing inflammation that results in intestinal pain, intestinal bleeding and diarrhea (9–11). While UC is restricted to the large intestine where it manifests as a uniform continuous pattern of inflammation, CD can occur anywhere throughout the gastrointestinal track in patches. The thickness of the inflammation also distinguishes the two diseases, UC being confined to the mucosa and CD presenting in both the mucosa and underlying muscle tissue (transmural).
The mechanisms that initiate and sustain IBD are incompletely understood. Current evidence supports a model in which genetic alterations and environmental factors, increase IBD susceptibility by deregulating the interplay between the microbiome, the intestinal epithelial barrier and the immune system. While the nature of pre-disposing environmental factors remains under debate, functional annotation of IBD-associated genes has identified gene variants that impact processes such as intestinal barrier function, anti-microbial activity, and autophagy (12–14). In addition, the disruption of the adaptive immune system has been implicated, with multiple IBD susceptibility genes being shown to contribute to CD4+ T cell development and function. Examples include IL23R, JAK2, and STAT3 (12, 15–21).
CD4+ T Cells and IBD
CD4+ T cells direct suitable immune responses, maintain immune tolerance and support the differentiation of endurable immunological memory. However, CD4+ T cell subsets have also been shown to contribute to chronic intestinal inflammation, accumulating in the mucosa of both UC and CD patients (22). Additional evidence supporting a role for CD4+ T cells in IBD, is based on HIV+ IBD patients who, with a reduced total CD4 T cell count, have a higher incidence of remission as compared to non-HIV IBD patients (23, 24). Therapeutically, CD4+ T cell-depleting and blocking antibodies (cM-T412, MAX.16H5, and B-F5) have been shown to induce remission in both CD and UC patients (25, 26), while alternate therapies that inhibit the differentiation of CD4+ T cell subsets and the cytokines they secrete, have proven to be efficacious in IBD patients, These would include Tofacitinib (oral JAK inhibitor), Ustekinumab (human monoclonal antibody directed against IL-12 and Il-23) and Infliximab (chimeric hiamn/mouse monoclonal antibody directed against TNFα) (27–33). It should be noted, that such therapies also target other immune cell lineages and as such, efficacy may not be solely driven through a CD4+ T cell specific mechanism.
CD4+ T cells are classified into distinct subsets based on their inducing cytokines, transcription factor expression, and effector cytokine secretion. The initial classification of CD4+ T cells as TH1 IFNγ producers vs. TH2 IL-4 producers, has been broadened to include multiple additional subsets (34, 35). These subsets, and the cytokines they secrete, include TH9 (IL-9), TH17 (IL-17A, IL-17F, and IL-22), TH22 (IL-22), T follicular helper TFH (IL-21) cells, as well as thymic-derived and peripherally-induced T regulatory cells (IL-10, TGFβ) (36–40) (Figure 1).
The contribution of the various CD4+ T cell subsets to CD and UC remains an area of ongoing research. Originally, CD was thought to be driven by TH1 T cells and UC by TH2 T cells. The use of such a TH1/TH2 paradigm to describe the different T cell responses involved in CD and UC has proven over simplistic however. It did not account for the role of more recently identified subsets such as TH17 T cells and Tregs. Moreover, the recent discovery of ongoing T cell plasticity in the intestinal mucosa of both CD and UC patients, has added further complexity to the CD4+ T cell response in these diseases (41, 42).
Protein Phosphorylation and CD4+ T Cell Differentiation
Protein tyrosine phosphorylation is required for CD4+ T cell differentiation and activation. Cascades of reversible protein phosphorylation events downstream of cytokine receptors (CytR), co-stimulatory molecules, and the T cell receptor (TCR), converge to induce gene expression profiles that drive CD4+ T cell activation and differentiation into distinct subsets (40).
Naive T cells in peripheral circulation are activated upon TCR recognition of its cognate antigen in the context of major histocompatibility complex (MHC) expressed on antigen presenting cells. Upon TCR engagement, Src-family kinases (Lck, Fyn) are activated and phosphorylate tyrosine residues within the immune-receptor tyrosine-based activation motifs (ITAMs) in the TCR-associated CD3 and zeta chains (43–46). Phosphorylated ITAMs then provide docking sites for the recruitment and activation of the zeta-associated protein kinase (ZAP-70) (47). Cooperatively, Src-family kinases and Zap70 phosphorylate downstream signaling pathways which dictate the cellular response (Figure 2).
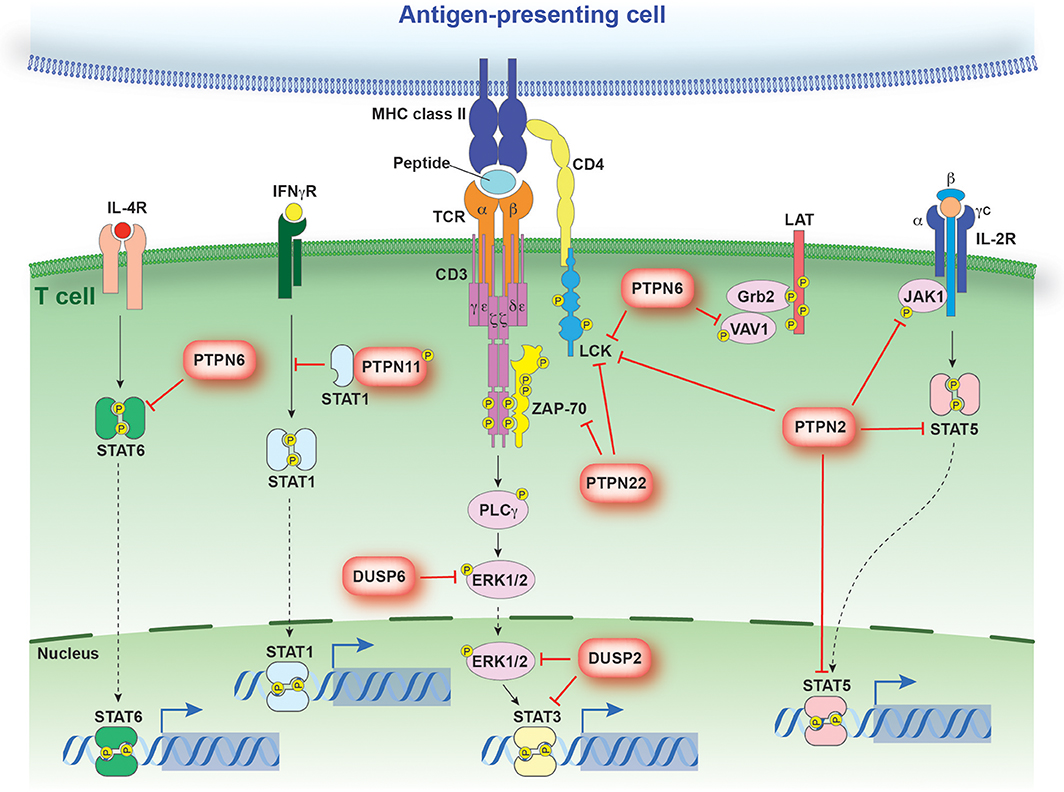
Figure 2. PTP regulation of antigen and cytokine receptor signaling. Schematic representation of signaling events regulated by PTPs discussed in the text. PTPs are linked to their respective substrates by a red bar-headed line. Dotted arrows depict translocation while solid black lines identify molecules linked in a signaling cascade. The direct interaction between STAT1 and PTPN11 models the sequestration of STAT1 from the IFNγR.
The strength of TCR signaling has a direct impact on CD4+ T cell differentiation (48). For example, Foxp3+ peripheral T regulatory (Treg) cells are generated primarily from CD4+ Foxp3− T cells exposed to antigen under tolerogenic conditions or during homeostatic proliferation (49–52). In the presence of TGFβ, IL-2 and co-stimulatory signaling, intermediate TCR signaling induces Foxp3 expression and Treg differentiation (iTreg). By comparison, weak and strong TCR signaling are less potent at inducing Foxp3 expression (53, 54). Regulatory mechanisms are therefore required to establish and maintain the strength of TCR signaling within a given range. Such regulatory mechanisms include the modulation of protein tyrosine phosphorylation.
Critically, engagement of the TCR and co-stimulatory molecules is not sufficient to drive the polarization of CD4+ T cells (40). Rather, the presence of inductive cytokines and the activation of the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway is also required. Binding of cytokines to their corresponding receptor results in receptor dimerization, allowing for the juxtaposition and subsequent cross-phosphorylation of associated JAK molecules (55). Activated JAK molecules are then responsible for the phosphorylation of STAT family members. Tyrosine phosphorylated STAT molecules dimerize and translocate to the nucleus, where they promote specific gene expression profiles (Figure 2) (56–62).
Thus a network of signaling pathways, heavily dependent on tyrosine phosphorylation, directs CD4+ T cell activation and differentiation. The current review will examine the role of protein tyrosine phosphatases (PTP) in safeguarding this network, and how PTP deletion can perturb CD4+ T cell function and consequently contribute to intestinal inflammation.
The Protein Tyrosine Phosphatase Family
The PTP family comprises a heterogeneous set of enzymes that were first defined by Tonks and colleagues by their capacity to dephosphorylate phospho-tyrosine residues and by their structurally related phosphatase catalytic domain (63, 64). PTP1B was the first phosphatase identified. Its sequence homology with a segment of the CD45 receptor protein (65), pointed to the existence of a conserved catalytic domain that became the main feature of the PTP gene family. CD45 also brought an interesting first link between PTPs and the immune system.
The human PTP family members are divided into distinct classes based on their structural and biochemical properties (Figure 3). The majority of PTPs have a conserved catalytic domain that contains a cysteine which executes a nucleophilic attack on substrate residues. There are 3 classes of such Cys-based PTPs. Class I is comprised of both classical PTPs that target phosphorylated tyrosine residues, and dual-specific phosphatases (DUSP) that target phosphorylated tyrosine, serine and threonine residues. Class II includes two PTPs, namely low molecular weight PTP and SSU72, whereas Class III comprises the three cell cycle Cdc25 regulatory proteins.
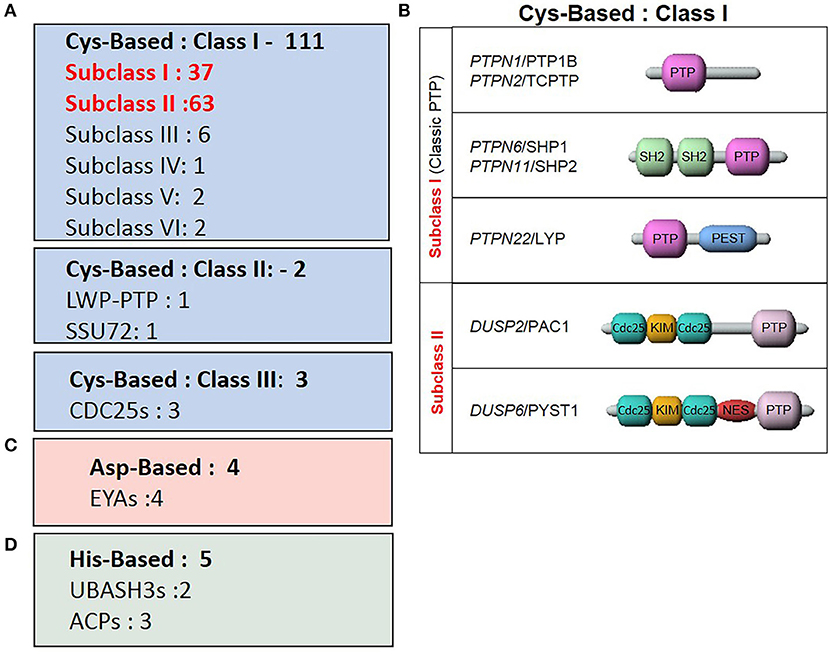
Figure 3. Classes of the tyrosine phosphatase family. (A) Sub-classes of Cys-based PTPs and the number of associated genes. (B) Representative PTPs from Class I Cys-based enzymes described in the review. (C) ASP-based PTPs. (D) His-based PTPs.
More recent studies characterized enzymes using alternate nucleophilic catalytic residues which were then added to the list of PTP family members. These two additional classes are the aspartic-based and histidine-based acid phosphatase enzymes. Thus, within this classification frame, Alonso and Pulido expanded the PTP gene superfamily to 125 members (66).
Tyrosine phosphorylation is a reversible mechanism that promotes protein–protein interactions and propagates signaling cascades in all cell types. It therefore stands to reason that the PTPs targeting these phosphorylated amino acids play important roles in modulating the strength and duration with which a signaling pathway is activated. Moreover, given that PTPs are a heterogeneous gene family (expression, function, and regulation) it is not surprising that PTPs have been associated with many distinct pathologies in both animal models and in human disease (67). Of importance herein, these include multiple immune-related disorders.
The immune system relies heavily on the use of protein phosphorylation to recognize and transmit intra- and extracellular signals. Signaling cascades initiated at the cell surface by receptor kinases, or from the recruitment of cytoplasmic kinases such as the JAK and Src-like kinase families, translate into multiple immune cell responses like cellular adhesion, division, migration and others. Hence, from the list of over 100 classical Cys-based PTPs, Arimura and Yagi demonstrated that between 58 and 76 of them are expressed in various cell lineages of the immune system (68) and that the vast majority of those (40 to 50) are present in T-cells (69). In T cells, examples of PTP activity mediating both positive and negative regulation of intracellular signaling events have been reported (69).
Multiple genome wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) linked with IBD, in or near PTP coding units (70, 71). Examples include the PTPN22, PTPN2, PTPRC, PTPRK, and MTMR3 loci (72). Murine pre-clinical colitis models and tissue-specific PTP knockout mouse strains have been used to discern the role of such PTPs in T cells in the inflamed intestine. Such studies, demonstrated that the perturbation of PTPs such as PTP22, PTPN2, PTPN11, DUSP2, and DUSP6 impacts IBD relevant T cell subsets and/or deregulates T-cell function (73–77). Yet only PTPN22 and PTPN2 have been clearly validated in both patients and animal models for their involvement in IBD.
PTP Regulation of CD4+ T Cell Activity in IBD
PTPN22
PTPN22 is a non-receptor protein tyrosine phosphatase that is expressed primarily in hematopoietic cells (78). It has been linked with multiple inflammatory disorders and in fact, PTPN22 genetic variation is among the strongest genetic risk factors for autoimmunity diseases such as type I diabetes (T1D), rheumatoid arthritis and systemic lupus erythematosus (79–83). One of the most extensively studied SNPs within the PTPN22 locus is SNP rs33996649 (G788>C) which causes a substitution of arginine 263 with a glutamine residue (263Q variant). While this variant increases susceptibility to multiple autoimmune diseases, it is in fact protective against CD (12, 79). The mechanism mediating this differential effect on disease susceptibility remains unclear. It has been hypothesized that it is due to the different role innate vs. adaptive immune cells play in the onset of these distinct inflammatory diseases (77). Further studies examining the cell type specific effects of the G788>C variant will provide clarity, although the continued debate regarding whether the variant encodes a gain-of-function or altered-function protein will need to be resolved (84–87).
PTPN22 deficient mice exhibit a lymphoproliferative disease and accumulate memory-phenotype T cells with age, but do not display signs of spontaneous autoimmunity (88). It is important to note that a PTPN22 deficiency can cooperate with the E613R CD45 mutant to induce autoimmune diseases. This suggests that the loss of PTPN22 can play a role in increasing susceptibility to certain inflammatory disorders (89).
A comprehensive examination of the different T cell compartments in PTPN22−/− mice provided an example of how cellular context can change the importance of the regulatory function of a given PTP. During the first 2 days following TCR activation, PTPN22+/+ and PTPN22−/− T cells display comparable proliferation, cytokine production and expression of activation markers. Beyond 2 days however, PTPN22−/− T cells exhibit an increased rate of cell cycling. In addition, reactivated knockout effector T cells undergo a more rapid and robust proliferation and secrete higher levels of cytokines compared to controls. Likewise ex vivo stimulation of CD44HI CD62LLO PTPN22−/− effector/memory cells display an increased proliferative response upon stimulation. This has been attributed to enhanced and prolonged phosphorylation of the Lck auto-regulatory (Tyr394) tyrosine following TCR ligation and not due to changes to IL-2 or IL-15 receptor signaling (88).
PTP localization also contributes to the regulation of PTP activity. In addition to Lck, Zap70 has been shown to be a substrate of PTPN22 and both Csk and Vav have been shown to be interacting partners. Csk plays a critical role in sequestering PTPN22 from the TCR complex, thereby limiting the capacity of PTPN22 to inhibit TCR signaling (90, 91–93). The kinetics and localization of PTPN22 activity, and its binding partners in antigen experienced vs. inexperienced T cells, remains unclear. Such an understanding would provide insight into why PTPN22 is particularly important beyond the initial stage of T cell activation.
Murine colitis models indicate that PTPN22 does not solely play a role in the function of conventional T cells. Using the T cell transfer colitis model, PTPN22 was shown to contribute to both effector and regulatory T cells in colitis. The transfer of PTPN22−/− naïve CD4+CD45RBHI T cells into an immune-deficient recipient resulted in a more severe colitis phenotype, characterized by rapid weight loss and decreased survival. Strikingly, while co-transfer of WT Tregs with PTPN22−/− naïve CD4+CD45RBHI T cells did not suppress disease, co-transfer of PTPN22−/− Tregs did. Protection was attributed to the increased secretion of anti-inflammatory cytokines by PTPN22−/− Tregs (76). It then follows that the lack of spontaneous autoimmunity in the PTPN22−/− mice may be due to a coincident increase in both effector and regulatory T cell activity.
Further investigation is required to determine if human PTPN22 variants contribute to IBD pathology through their effects on T cell homeostasis in the gut. Although two independent studies confirmed the role of PTPN22 in acute colitis using the dextran-sodium sulfate (DSS) induced model, these studies attributed the increase in disease severity to changes in TLR4 signaling, type I interferon production and macrophage polarization (94, 95).
PTPN2
PTPN2 is a classical cytoplasmic phosphatase expressed ubiquitously, with the highest levels of expression being detected in lymphoid cells. Its critical role in the homeostasis of the immune system is evident by the progressive systemic inflammatory disease that develops in PTPN2 deficient mice within 1–2 weeks of birth (96). Pro-inflammatory mediators IFNγ, TNFα, and inducible nitric oxide synthase (iNOS) are readily detectable as early as 3 days after birth, followed by the infiltration of mononuclear cells into lymphoid and non-lymphoid organs (97). In comparison, T-cell specific PTPN2 deficient mice do not develop systemic inflammation but rather develop spontaneous autoimmunity at older ages. A reduction in the TCR threshold in both PTPN2−/− CD4 and CD8 was identified as causing increased T cell activation due to hyper-phosphorylation of the activating tyrosine residue of Lck (98). It was then proposed that through its negative regulation of TCR signaling, PTPN2 contributes to the maintenance of immune homeostasis.
The loss of PTPN2 has been shown to alter CD4+ T cell differentiation, impacting disease severity in multiple murine colitis models. In the T cell transfer colitis model, the transfer of naïve PTPN2−/− CD4+ T cells resulted in an earlier onset of disease, as well as an increased in weight loss, spleen weight and macroscopic signs of colitis. These clinical signs correlated with more pronounced intestinal inflammation as detected by histological scoring of immune infiltration and epithelial damage. The more pronounced disease severity was confirmed in both acute and chronic DSS colitis models, which both demonstrated a more dramatic weight loss and colonic shortening (99).
Mechanistically, an increase in STAT1 and STAT3 phosphorylation was detected in whole colon lysates. An increase in STAT1/3 phosphorylation was also detected in PTPN2−/− CD4+ T cells cultured under polarization conditions. The effects of PTPN2 loss on TH differentiation in vivo were also examined. In the T cell transfer colitis model, the introduction of PTPN2 deficient CD4+ T cells resulted in an almost 3-fold increase in the frequency of IFNγ producers and a 2-fold increase in the frequency of IFNγ+ IL17+ double producers. In contrast, the frequency of Tregs was reduced over 3-fold. These findings were also recapitulated in the DSS model, although with a less pronounced effect that most likely reflected the fact that pathogenic T cells are not induced robustly in this model (99). One point for further investigation is whether the observed increase in STAT1/3 phosphorylation alters the propensity to commit to a given subset, or if it heightens proliferation rates following commitment.
Importantly, the consequences of PTPN2 loss in murine CD4+ T cell differentiation are in line with the sequencing of human PTPN2 variants that have been to be associated with CD. Specifically, transcriptional profiling of CD patients expressing the PTPN2 loss-of-function variant (rs1893217) presented higher expression of TH1 and TH17-related transcription factors and cytokines (serum and intestinal biopsies) in comparison to PTPN2 expressing CD patients. Such human studies solidify the important role of PTPN2 CD4+ T cell responses in the context of human IBD (99).
PTPN6
The Src homology region 2 domain-containing tyrosine phosphatase-1 (SHP-1, PTPN6) is a cytoplasmic phosphatase expressed in all hematopoietic cell lineages throughout development and activation (100–103). Functionally, PTPN6 has been shown to negatively regulate signaling downstream of multiple receptors which co-ordinate immune cell homeostasis and function. These include antigen, cytokine, chemokine and integrin receptors (104–115). As such, it is not surprising that systemic inflammation is the dominant phenotype of the motheaten (me) and motheaten viable (mev) mice. These mice harbor mutations that result in undetectable and reduced PTPN6 expression respectively. Similar to PTPN2 deficient mice, me/me mice succumb to disease 2–3 weeks following birth (116, 117).
Due to the complex inflammatory pathology of the me/me mice, it has proven difficult to dissect the T cell intrinsic effects of a PTPN6 deficiency. Extensive characterization of T cells derived from me/me mice suggests PTPN6 plays a role in regulating the threshold for TCR activation in thymic and peripheral T cells. Ex vivo stimulation of me/me-derived T cells, as well as T cells expressing a PTPN6 dominant-negative allele, results in a hyper-proliferative response at low antigen concentrations (104, 118–121). Mechanistically, PTPN6 can dephosphorylate multiple proteins downstream of the TCR complex (104, 122–125). Whether one substrate is the physiological substrate, critical to setting the threshold of TCR activation remains a matter of debate.
In vivo, the generation of T cell specific PTPN6 deficient mice by different groups produced conflicting findings regarding the role of PTPN6 in T cell polarization. Johnson et al. reported that the loss of PTPN6 does not alter thymocyte or peripheral T cell sensitivity to TCR activation. Rather, the authors note a higher frequency of memory phenotype T cells in the peripheral T cell pool. Given that memory-phenotype T cells respond more robustly upon TCR activation, a hyper-proliferative response is observed when a heterogenous pool of peripheral T cells is stimulated ex vivo. The same study demonstrated that a PTPN6 deficiency causes a skewing toward the TH2 lineage, associated with sustained IL-4-STAT6 signaling (126). In direct contrast, Martinez et al., also using a T cell specific conditional knockout mouse, showed that PTPN6 depletion lowers the threshold of TCR activation and causes an increase in thymic negative selection and impairs the T cell repertoire (127). Most recently, it has been reported that in an alternate conditional knockout model, in which PTPN6 is deleted in post-selection thymocytes, CD4+ T cells are hyper-responsive to TCR stimulation and are intrinsically more resistant to Treg suppression (128).
An understanding of the link between PTPN6 and IBD still needs to be established. For example, a characterization of colitis induction and progression in a PTPN6 T cell conditional knockout mouse has not been published. It remains unknown therefore, whether PTPN6 plays a role in CD4+ T cell biology in the inflamed gut. Although limited in scope, two published reports suggest a link between PTPN6 in human colitis though. The levels of PTPN6 were quantified in 98 colonic biopsies, and found to be significantly reduced in active UC, quiescent UC and active CD when compared to healthy controls (129). In a separate study, 2 PTPN6 SNPs (rs7310161 and rs759052) were genotyped in 107 IBD patients and 162 healthy controls from Southern Tunisia (130). A weak association with UC was identified which requires confirmation in a larger cohort. Noteworthy, such studies do not demonstrate that it is the deficiency of PTPN6 in CD4+ T cells that is implicated in the disease pathology.
PTPN11
PTPN11 encodes the Src homology 2-containing protein tyrosine phosphatase 2 (SHP-2). Similar to PTPN6, PTPN11 regulates signaling downstream of multiple surface receptors including growth factor receptors, cytokine receptors and integrins. Its broad tissue expression is in line with the embryonic lethality of PTPN11 knockout mouse at mid-gestation in (131–133).
Extensive human genetic data indicates that PTPN11 has an important role in human disease. Specifically, somatic PTPN11 mutations in patients with multiple cancer types including leukemias, breast cancer and gastric cancer (134) have been genotyped. As well, in a cohort of 114 Japanese patients, a genetic association has been made between a SNP within the PTPN11 locus and UC (135).
Surprisingly, characterization of T cells from multiple PTPN11 dominant negative knock-in mice and PTPN11 knockout mice, has not lead to a conclusive understanding of this PTP's role in T cell biology. Examples of both deficient- and hyper-T cell activation can be found in these studies. This has been in part attributed to the scaffolding properties of PTPN11 that are retained in the catalytically dead knock-in mouse (73, 136–141). As an example of the scaffolding properties of PTPN11, it has been shown to directly interact with STAT1 and retain STAT1 in the cytoplasm thereby impeding its recruitment to the IFNγ receptor. This disruption of IFNγ signaling, was sufficient to inhibit the production of TH1 cytokines and improve 2, 4, 6-trinitrobenzene sulfonic acid induced colitis (142).
By comparison, a PTPN11 conditional T-cell specific knockout mouse has been reported to exhibit increased susceptibility to DSS induced acute colitis. Phenotypic analysis of the T cell compartments in the PTPN11 T cell deficient mice, did not identify any change in the frequency of peripheral T cells as compared to control mice. However, the severity of DSS-induced colitis was found to be much more pronounced in the deficient mice. This increase in severity manifested as a higher body weight loss, disease activity index and colon shortening. It was also reported that an increase in the infiltration of immune cells was observed. Cytokine profiling identified an increase in pro-inflammatory cytokines including IFN-γ, IL-17A, TNF-α, and IL-6 in the mucosa, which correlated with an increased number of TH1 and TH17 cells in the spleen and lamina propria of T cell specific PTPN11 deficient mice (73).The increases risk of development cancer in UC and CD patients in consistent with the role chronic inflammation plays in tumor initiation and progression. It was therefore surprising that despite the increased inflammation observed in mice harboring a PTPN11 deficiency in CD4+ T cells, mice were protected against colitis associated cancer. Specifically, PTPN11 CD4+ T cell deficient mice display enhanced TH1 immunity and aggravated colitis, but the deficiency inhibited the development of AOM-DSS colitis-associated carcinoma. Mice contained fewer and smaller tumors which expressed reduced levels of proliferation markers. In direct contrast, the progression and metastasis of melanoma was accelerated in the same mouse model. This apparent contradiction suggests that the effects of PTP loss in T cells on tumor progression are highly dependent on the location and stage of the tumor. It must be noted however, that an understanding of how the modulation of PTPs such as SHP-2, impacts the interface between CD4+ T cells and tumors, remains largely unexplored (73).
It is important to note that PTPN11's role in maintaining intestinal homeostasis is not restricted to CD4+ T cells. In fact, an intestinal epithelial specific PTPN11 knockout mouse has been generated and develops severe colitis. Such mice have a severe defect in the intestinal barrier which is associated with impaired tight junction formation, goblet cell differentiation and IEC migration (143, 144).
DUSP2
DUSP2 is a mitogen- and stress-inducible nuclear DUSP that is enriched in lymphoid tissue and differentially expressed in CD4+ T cell subsets. Possibly due to compensation by other DUSP family members, DUSP2 deficient mice are viable and exhibit no gross defects and no alterations in the frequency or absolute number of lymphoid populations in the thymus, spleen or lymph nodes. However, using in vitro models of T cell polarization, it was demonstrated that in the absence of DUSP2, TH17 cell differentiation is promoted while inducible Treg (iTreg) differentiation is suppressed (74).
To question whether such modulation of in vitro TH differentiation relates to effects observed in vivo, two mouse models of intestinal inflammation were used. First, in acute and chronic DSS colitis models, Dusp2−/− mice exhibited a more severe disease as assessed by weight loss, clinical scoring, survival and histopathology scoring. Higher colonic inflammation was attributed to an increased proportion of colonic Il-17A+ CD4+ T cells in Dusp2−/− mice as compared to control mice, whereas TH1 and Treg populations appeared to be unaltered in DSS treated mice. Second, a T cell transfer colitis model has confirmed the T cell intrinsic effects of a DUSP2 deficiency. To do so, severe combined immunodeficiency (SCID) mice were reconstituted with either WT or DUSP2−/− CD4+CD45RBhi T cells. In this model, an increase in body weight loss, severity of histopathology scoring, and colon thickening were observed following the transfer of DUSP2−/− T cells compared to controls. Such clinical signs were corroborated by a rise in the number of colon infiltrating CD4+ T cells expressing a TH17 gene signature. Moreover, such TH17 T cells in the inflamed tissue expressed elevated levels of pathogenic TH17-associated genes but not non-pathogenic genes (74).
Given evidence that Treg differentiation is suppressed in in vitro differentiation assays, the ability of DUSP2−/− Tregs to suppress colitis in the T cell transfer model was evaluated. Surprisingly, DUSP2 was found to be dispensable, as DUSP2−/− Tregs were as efficient as WT Tregs in suppressing inflammation when co-transferred with WT CD4+CD45RBhi cells into SCID mice (74).
To identify the molecular mechanism by which DUSP2 limits TH17 differentiation, phosphoproteomic analysis was performed on colon homogenates from DSS treated WT and DUSP2−/− mice. Along with follow-up biochemical studies, STAT3 was identified as a direct substrate of DUSP2. DUSP2 was shown to dephosphorylate residues Tyr705 and Ser727. Although comparable levels of basal colonic STAT3 phosphorylation were reported between WT and DUSP2−/− mice, the levels were higher in DUSP2−/− DSS-challenged mice compared to WT DSS-challenged mice (74). STAT3 is critical in driving disease in T-cell transfer models of colitis (145), the authors propose that the DUSP2−/− phenotype is due to elevated STAT3 activity. It should be noted however, that the authors have not formally proven that the phenotype is dependent on STAT3 activity.
DUSP2 has also been implicated in human IBD. The expression of DUPS2 mRNA is reduced in the peripheral blood of UC patients (n = 24) and is only increased minimally following PMA stimulation. The low levels of DUPS2 expression were found to be attributed to CpG methylation. Unfortunately, the levels of DUSP2 in colonic biopsies, or colonic T cells, were not quantified. As such, further evidence is required to definitely demonstrate that a reduction in T cell DUSP2 occurs in IBD patients and that this reduction impacts human T cell polarization in the disease state (74).
Based on these findings it has been proposed that under healthy conditions, T cell activation results in a transient increase in DUSP2 which reduces STAT3 activation below levels required to promote TH17 differentiation. In the absence of DUSP2, elevated levels of STAT3 phosphorylation result in the differentiation of pathogenic TH17 cells which promote inflammation (74).
DUSP6
Extracellular signal-regulated kinase (ERK) signaling regulates multiple cellular responses to activation including T cell proliferation, cytokine production, survival and adhesion. CD4+ T cell polarization in vivo has also been shown to be modulated by ERK signaling, numerous reports suggesting ERK activity impacts TH1 and TH2 differentiation and the TH17:Treg balance. (146–153).
DUSP6 is a cytoplasmic ERK-specific DUSP (154, 155). Surprisingly, genetic ablation of DUSP6 does not cause developmental defects despite an increase in the basal levels of ERK phosphorylation in the heart, spleen, kidney, and brain. Rather, DUSP6 deficient mice are viable with no reported phenotype in the steady state (156). Initial cellular in vitro assays suggested a role for DUSP6 in the regulation of CD4+ T cell activation. Specifically, DUSP6 was shown to be upregulated following TLR4 stimulation and then restrained ERK activation and suppressed IFNγ production by TCR stimulation (157).
More recently, a more in depth characterization of DUSP6 in CD4+ T cells has been reported (75). DUSP6−/− mice exhibited no change in the frequency or absolute number of peripheral CD4+ or CD8+ T cells. Nevertheless, a higher percentage of memory-effector CD4+ T cells and a lower percentage of naïve CD4+ T cells in DUSP6−/− mice, indicates an increase in CD4+ T cell activation. Ex vivo TCR-stimulated of DUSP6−/− CD4+ T cells display increased ERK activation and proliferation but also an elevated rate of activation induced cell death. Polarization studies demonstrated that DUSP6 depletion promotes TH1 differentiation and increases IFNγ production, whereas expression levels of IL-2, IL-4, IL-6, or IL-10 are not altered. In contrast TH17 CD4+ T cells displayed decreased survival and IL-17A secretion, leading to the conclusion that DUSP6 suppresses the TH1 lineage and promotes TH17 differentiation (75).
The role of DUSP6 in intestinal inflammation was addressed using an IL-10−/− colitis model. IL-10−/−DUSP6−/− double knockout mice were generated and phenotyped. Such mice had accelerated and exacerbated colitis. Histological examination identified elevated epithelial crypt hyperplasia, goblet-cell depletion, and infiltration of mononuclear cells. Moreover, colonic explants were found to secrete higher levels of TNFα and IFNγ, while IL-17A levels were reduced. Strikingly, ERK inhibition was shown to significantly reduce colitis in IL-10−/−DUSP6−/− mice, both prophylactically and therapeutically. The severity of both crypt hyperplasia and immune cell infiltration was reduced. Despite, these intriguing findings, future studies are needed to demonstrate that the protective role of DUSP6 in colitis is intrinsic to CD4+ T cells (75).
PTPs as Therapeutic Targets for IBD?
In the late 1990s, multiple research programs sought to identify the PTP(s) that recognize and dephosphorylate the insulin receptor (IR). The regulation of IR activation by a PTP was hypothesized to decrease IR mediated signaling events, and subsequent entry of glucose into insulin receptive tissues. It was believed that a rise in the expression or activity of such a regulatory PTP would result in high levels of blood glucose in the circulation, which would then instigate type II diabetes. This hypothesis was confirmed by two seminal publications which described the PTP1B knockout mouse that presented with an increase in IR phosphorylation. These studies validated PTP1B as an outstanding candidate to be targeted for the treatment of type II diabetes (158, 159) and led to major efforts to identify small inhibitors of PTP1B. Indeed, since their publications these reports were cited over 2500 times, primarily in the context of depicting the isolation of novel PTP1B inhibitors.
In spite of all these reports, there are no small molecule inhibitors against PTP1B (or any PTP) that have been successfully developed beyond initial clinical trials. Multiple reviews have already examined the difficulties associated with developing PTP competitive inhibitors and this subject is beyond the scope of this review (160–163). In brief, the main challenges in developing inhibitors are the high polarity and homology of their catalytic pockets. Hence, chemical screens for competitive small molecule PTP inhibitors, have only isolated inhibitors with poor cell permeability and low specificity. Excitement has been garnered by the recent development of allosteric PTP inhibitors such as the PTP1B carboxyl domain compound (164) and a PTPN11 inhibitor from Novartis (165) which may lead to the development of novel pharmaceuticals.
It is worthy to note that novel therapeutic strategies are also being developed to target PTPs. These include the forced dimerization and inhibition of receptors PTPs (166, 167), intrabodies inhibitors (168), small molecular caged compound inhibitors (169) and even RNA aptamers (170) that modulate the enzymatic activity of PTPs. As well, genetic modulation through CRISPR technologies (171), as well as protein degradation technologies (PDTs) such as PROTACs (PROteolysis TArgeting Chimeras) (172) and SNIPERS (Specific and Non-genetic IAP-dependent Protein Erasers) (173) are now exciting avenues for tackling the difficult PTP gene family.
Significant effort is also being made in the development of cell based therapies. Potential adverse effects that could be associated with systemically inhibiting PTPs are mitigated by using PTP inhibitors in ex-vivo cell cultures. This allows for the modulation of PTP activity in a cell-type specific and temporary fashion. For example, we reported a protocol for dendritic cell (DC) vaccination that employs inhibitors of PTPN1-PTPN2 (174). This work demonstrated that in the proper context, PTP inhibitors may have broad application in cancer and infectious diseases. Beyond DCs, other cell types such as macrophages, natural killer cells, and of course T-cells as described above, may also be malleable by PTP inhibition and useful in various cell therapies.
From an IBD perspective, it is clear that inhibiting the PTPs presented above would most likely exacerbate disease by potentiating the effects of pro-inflammatory cytokines and promoting the differentiation of pathogenic T cells. Indeed this contention is supported by the fact that, biologics and small molecules that suppress cytokine receptor signaling have been clinically successful in IBD disease management. One example would be tofacitinib, a JAK inhibitor found to be effective in phase 2 and 3 trials in moderate to severe ulcerative colitis (175).
Our understanding of PTP function in immune cells is expanding beyond cytokine receptor signaling, and our capacity to modulate cellular responses by titrating PTP expression is also evolving at a rapid pace. The cell-dependent context of positive or/and negative modulation bestowed by the nearly 80 PTPs expressed in immune cells, remains an exciting ground for study and clinical improvement. It remains to be seen if this renewed interest in PTP inhibitors would be applicable to inflammatory diseases such as UC and CD.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by an operating grant from the Canadian Institutes of Health Research (CIHR MOP 142497) and by support to MT from the Strauss -Aclon Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful for Dr. Noriko Uetani for the art work. MT is a Jeanne and Jean-Louis Lévesque chair in Cancer Research.
References
1. Farquhar MG, Palade GE. Junctional complexes in various epithelia. J Cell Biol. (1963) 17:375–412. doi: 10.1083/jcb.17.2.375
2. Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. (2008) 105:15064–9. doi: 10.1073/pnas.0803124105
3. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med. 5:e54. doi: 10.1371/journal.pmed.0050054
4. Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. (2000) 1:113–8. doi: 10.1038/77783
5. Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science (2011) 334:255–8. doi: 10.1126/science.1209791
6. Slack E, Hapfelmeier S, Stecher B, Velykoredko Y, Stoel M, Lawson M, et al. Innate and adaptive immunity cooperate flexibly to maintain host-microbiota mutualism. Science (2009) 325:617–20. doi: 10.1126/science.1172747
7. Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science (2004) 303:1662–5. doi: 10.1126/science.1091334
8. Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science (2001) 292:1115–8. doi: 10.1126/science.1058709
9. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. (2009) 361:2066–78. doi: 10.1056/NEJMra0804647
10. Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet (2007) 369:1641–57. doi: 10.1016/S0140-6736(07)60751-X
11. Bernstein CN, Fried M, Krabshuis JH, Cohen H, Eliakim R, Fedail S, et al. World Gastroenterology Organization Practice Guidelines for the diagnosis and management of IBD in 2010. Inflamm Bowel Dis. (2010) 16:112–24. doi: 10.1002/ibd.21048
12. Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. (2008) 40:955–62. doi: 10.1038/ng.175
13. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui K, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature (2012) 491:119–24. doi: 10.1038/nature11582
14. Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. (2015) 47:979–86. doi: 10.1038/ng.3359
15. Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly M, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science (2006) 314:1461–3. doi: 10.1126/science.1135245
16. Franke A, Balschun T, Karlsen TH, Hedderich J, May S, Lu T, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. (2008) 40:713–5. doi: 10.1038/ng.148
17. Sato K, Shiota M, Fukuda S, Iwamoto E, Machida H, Inamine T, et al. Strong evidence of a combination polymorphism of the tyrosine kinase 2 gene and the signal transducer and activator of transcription 3 gene as a DNA-based biomarker for susceptibility to Crohn's disease in the Japanese population. J Clin Immunol. (2009) 29:815–25. doi: 10.1007/s10875-009-9320-x
18. Cenit MC, Alcina A, Marquez A, Mendoza JL, Diaz-Rubio MV, de las Heras V, et al. STAT3 locus in inflammatory bowel disease and multiple sclerosis susceptibility. Genes Immun. (2010) 11:264–8. doi: 10.1038/gene.2010.10
19. Peter I, Mitchell AA, Ozelius L, Erazo M, Hu J, Doheny D, et al. Evaluation of 22 genetic variants with Crohn's disease risk in the Ashkenazi Jewish population: a case-control study. BMC Med Genet. (2011) 12:63. doi: 10.1186/1471-2350-12-63
20. Polgar N, Csongei V, Szabo M, Zambo V, Melegh BI, Sumegi K, et al. Investigation of JAK2, STAT3 and CCR6 polymorphisms and their gene-gene interactions in inflammatory bowel disease. Int J Immunogenet. (2012) 39:247–52. doi: 10.1111/j.1744-313X.2012.01084.x
21. Prager M, Buttner J, Haas V, Baumgart DC, Sturm A, Zeitz M, et al. The JAK2 variant rs10758669 in Crohn's disease: altering the intestinal barrier as one mechanism of action. Int J Colorectal Dis. (2012) 27:565–73. doi: 10.1007/s00384-011-1345-y
22. Globig AM, Hennecke N, Martin B, Seidl M, Ruf G, Hasselblatt P, et al. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-gamma+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm Bowel Dis. (2014) 20:2321–9. doi: 10.1097/MIB.0000000000000210
23. Greenwald B, James SP. Long-term HIV infection with Crohn's disease. Am J Gastroenterol. (1995) 90:167–8.
24. Skamnelos A, Tatsioni A, Katsanos KH, Tsianos V, Christodoulou D, Tsianos EV. CD4 count remission hypothesis in patients with inflammatory bowel disease and human immunodeficiency virus infection: a systematic review of the literature. Ann Gastroenterol. (2015) 28:337–46.
25. Stronkhorst A, Radema S, Yong SL, Bijl H, ten Berge IJ, Tytgat GN, et al. CD4 antibody treatment in patients with active Crohn's disease: a phase 1 dose finding study. Gut (1997) 40:320–7. doi: 10.1136/gut.40.3.320
26. Emmrich J, Seyfarth M, Fleig WE, Emmrich F. Treatment of inflammatory bowel disease with anti-CD4 monoclonal antibody. Lancet (1991) 338:570–1. doi: 10.1016/0140-6736(91)91133-F
27. Sandborn WJ, Su C, Panes Tofacitinib as induction and maintenance therapy for ulcerative colitis. J. N Engl J Med. (2017) 377:496–7. doi: 10.1056/NEJMc1707500
28. Feagan BG, Sandborn WJ, Gasink C, Jacobstein D, Lang Y, Friedman JR, et al. Ustekinumab as induction and maintenance therapy for Crohn's Disease. N Engl J Med. (2016) 375:1946–60. doi: 10.1056/NEJMoa1602773
29. Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, et al. (2014). Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology 146:85–95. doi: 10.1053/j.gastro.2013.05.048
30. Colombel JF, Sandborn WJ, Rutgeerts P, Enns R, Hanauer SB, Panaccione R, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology (2007) 132:52–65. doi: 10.1053/j.gastro.2006.11.041
31. Schreiber S, Khaliq-Kareemi M, Lawrance IC, Thomsen OO, Hanauer SB, McColm J, et al. Maintenance therapy with certolizumab pegol for Crohn's disease. N Engl J Med. (2007) 357:239–50. doi: 10.1056/NEJMoa062897
32. Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet (2002) 359:1541–9. doi: 10.1016/S0140-6736(02)08512-4
33. Duijvestein M, Battat R, Vande Casteele N, D'Haens GR, Sandborn WJ, Khanna R, et al. Novel therapies and treatment strategies for patients with inflammatory bowel disease. Curr Treat Options Gastroenterol. (2018) 16:129–46. doi: 10.1007/s11938-018-0175-1
34. Mosmann TR Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone I Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (1986) 136:2348–57.
35. Del Prete GF, De Carli M, Mastromauro C, Biagiotti R, Macchia D, Falagiani P, et al. Purified protein derivative of Mycobacterium tuberculosis and excretory-secretory antigen(s) of Toxocara canis expand in vitro human T cells with stable and opposite (type 1 T helper or type 2 T helper) profile of cytokine production. J Clin Invest. (1991) 88:346–50. doi: 10.1172/JCI115300
36. Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest. (2015) 125:2211–9. doi: 10.1172/JCI78085
37. Schmitt E, Klein M, Bopp T. Th9 cells, new players in adaptive immunity. Trends Immunol. (2014) 35:61–8. doi: 10.1016/j.it.2013.10.004
38. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41:529–42. doi: 10.1016/j.immuni.2014.10.004
39. Wing JB, Sakaguchi S. Multiple treg suppressive modules and their adaptability. Front Immunol. (2012) 3:178. doi: 10.3389/fimmu.2012.00178
40. DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol. (2016) 16:149–63. doi: 10.1038/nri.2015.18
41. Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology (2011) 140:957–65. doi: 10.1053/j.gastro.2010.12.002
42. Ueno A, Jijon H, Chan R, Ford K, Hirota C, Kaplan G, et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm Bowel Dis. (2013) 19:2522–34. doi: 10.1097/MIB.0b013e3182a85709
43. Iwashima M, Irving BA, van Oers NS, Chan AC, Weissm A. Sequential interactions of the TCR with two distinct cytoplasmic tyrosine kinases. Science (1994) 263:1136–9. doi: 10.1126/science.7509083
44. Samelson LE, Davidson WF, Morse HC III, Klausner RD. Abnormal tyrosine phosphorylation on T-cell receptor in lymphoproliferative disorders. Nature (1986) 324:674–6. doi: 10.1038/324674a0
45. Straus DB, Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell (1992) 70:585–93. doi: 10.1016/0092-8674(92)90428-F
46. van Oers NS, Killeen N, Weiss A. Lck regulates the tyrosine phosphorylation of the T cell receptor subunits and ZAP-70 in murine thymocytes. J Exp Med. (1996) 183:1053–62. doi: 10.1084/jem.183.3.1053
47. Chan AC, Iwashima M, Turck CW, Weiss A. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell (1992) 71:649–62. doi: 10.1016/0092-8674(92)90598-7
48. van Panhuys N, Klauschen F, Germain RN. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization in vivo. Immunity (2014) 41:63–74. doi: 10.1016/j.immuni.2014.06.003
49. Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. (2004) 199:1401–8. doi: 10.1084/jem.20040249
50. Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. (2004) 173:7259–68. doi: 10.4049/jimmunol.173.12.7259
51. Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, et al. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med. (2007) 204:1775–85. doi: 10.1084/jem.20070602
52. Fousteri G, Jasinski J, Dave A, Nakayama M, Pagni P, Lambolez F, et al. Following the fate of one insulin-reactive CD4 T cell: conversion into Teffs and Tregs in the periphery controls diabetes in NOD mice. Diabetes (2012) 61:1169–79. doi: 10.2337/db11-0671
53. Turner MS, Kane LP, Morel PA. Dominant role of antigen dose in CD4+Foxp3+ regulatory T cell induction and expansion. J Immunol. (2009) 183:4895–903. doi: 10.4049/jimmunol.0901459
54. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. (2010) 207:1701–11. doi: 10.1084/jem.20091999
55. Brooks AJ, Dai W, O'Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 344:1249783. doi: 10.1126/science.1249783
56. Fu XY, Zhang JJ. Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos gene promoter. Cell (1993) 74:1135–45. doi: 10.1016/0092-8674(93)90734-8
57. Shuai K, Horvath CM, Huang LH, Qureshi SA, Cowburn D, Darnell JE Jr. et al. Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell (1994) 76:821–8. doi: 10.1016/0092-8674(94)90357-3
58. Horvath CM, Wen Z, Darnell JE Jr. A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. (1995) 9:984–94. doi: 10.1101/gad.9.8.984
59. Shuai K, Stark GR, Kerr IM, Darnell JE Jr. A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science (1993) 261:1744–6. doi: 10.1126/science.7690989
60. Muller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell J, et al. Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. (1993) 12:4221–8. doi: 10.1002/j.1460-2075.1993.tb06106.x
61. Xu X, Sun YL, Hoey T. Cooperative DNA binding and sequence-selective recognition conferred by the STAT amino-terminal domain. Science (1996) 273:794–7. doi: 10.1126/science.273.5276.794
62. Villarino AV, Kanno Y, O'Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol. (2017) 18:374–84. doi: 10.1038/ni.3691
63. Tonks NK, Diltz CD, Fischer EH. Purification of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. (1988) 263:6722–30. 64. Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. (1988) 263:6731–7. 65. Thomas ML, Barclay AN, Gagnon J, Williams AF. Evidence from cDNA clones that the rat leukocyte-common antigen (T200) spans the lipid bilayer and contains a cytoplasmic domain of 80,000 Mr. Cell (1985) 41:83–93. doi: 10.1016/0092-8674(85)90063-7
64. Tonks NK, Diltz CD, Fischer EH. Characterization of the major protein-tyrosine-phosphatases of human placenta. J Biol Chem. (1988) 263:6731–7.
65. Thomas ML, Barclay AN, Gagnon J, Williams AF. Evidence from cDNA clones that the rat leukocyte-common antigen (T200) spans the lipid bilayer and contains a cytoplasmic domain of 80,000 Mr. Cell (1985) 41:83–93. doi: 10.1016/0092-8674(85)90063-7
66. Alonso A, Pulido R. The extended human PTPome: a growing tyrosine phosphatase family. FEBS J. (2016) 283:2197–201. doi: 10.1111/febs.13748
67. Hale AJ, Ter Steege E, den Hertog J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev Biol. (2017) 428:283–92. doi: 10.1016/j.ydbio.2017.03.023
68. Arimura Y, Yagi J. Comprehensive expression profiles of genes for protein tyrosine phosphatases in immune cells. Sci Signal. 3:rs1. doi: 10.1126/scisignal.2000966
69. Mustelin T, Vang T, Bottini N. Protein tyrosine phosphatases and the immune response. Nat Rev Immunol. (2005) 5:43–57. doi: 10.1038/nri1530
70. Spalinger MR, McCole DF, Rogler G, Scharl M. Protein tyrosine phosphatase non-receptor type 2 and inflammatory bowel disease. World J Gastroenterol. (2016) 22:1034–44. doi: 10.3748/wjg.v22.i3.1034
71. Sharp RC, Abdulrahim M, Naser ES, Naser SA. Genetic variations of PTPN2 and PTPN22: role in the pathogenesis of Type 1 Diabetes and Crohn's Disease. Front Cell Infect Microbiol. 5:95. doi: 10.3389/fcimb.2015.00095
72. Peloquin JM, Goel G, Kong L, Huang H, Haritunians T, Sartor R, et al. Characterization of candidate genes in inflammatory bowel disease-associated risk loci. JCI Insight (2016) 1:e87899. doi: 10.1172/jci.insight.87899
73. Liu W, Guo W, Shen L, Chen Z, Luo Q, Luo X, et al. T lymphocyte SHP2-deficiency triggers anti-tumor immunity to inhibit colitis-associated cancer in mice. Oncotarget (2017) 8:7586–97. doi: 10.18632/oncotarget.13812
74. Lu D, Liu L, Ji X, Gao Y, Chen X, Liu Y, et al. The phosphatase DUSP2 controls the activity of the transcription activator STAT3 and regulates TH17 differentiation. Nat Immunol. (2015) 16:1263–73. doi: 10.1038/ni.3278
75. Bertin S, Lozano-Ruiz B, Bachiller V, Garcia-Martinez I, Herdman S, Zapater P, et al. Dual-specificity phosphatase 6 regulates CD4+ T-cell functions and restrains spontaneous colitis in IL-10-deficient mice. Mucosal Immunol. (2015) 8:505–15. doi: 10.1038/mi.2014.84
76. Brownlie RJ, Miosge LA, Vassilakos D, Svensson LM, Cope A, Zamoyska R Lack of the phosphatase PTPN22 increases adhesion of murine regulatory T cells to improve their immunosuppressive function. Sci Signal. (2012) 5:ra87. doi: 10.1126/scisignal.2003365
77. Spalinger MR, McCole DF, Rogler G, Scharl M. Role of protein tyrosine phosphatases in regulating the immune system: implications for chronic intestinal inflammation. Inflamm Bowel Dis. (2015) 21:645–55. doi: 10.1097/MIB.0000000000000297
78. Cloutier JF, Veillette A. Association of inhibitory tyrosine protein kinase p50csk with protein tyrosine phosphatase PEP in T cells and other hemopoietic cells. EMBO J. (1996) 15:4909–18. doi: 10.1002/j.1460-2075.1996.tb00871.x
79. Diaz-Gallo LM, Espino-Paisan L, Fransen K, Gomez-Garcia M, van Sommeren S, Cardena C, et al. Differential association of two PTPN22 coding variants with Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. (2011) 17:2287–94. doi: 10.1002/ibd.21630
80. Kyogoku C, Langefeld CD, Ortmann WA, Lee A, Selby S, Carlton VE, et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet. (2004) 75:504–7. doi: 10.1086/423790
81. Michou L, Lasbleiz S, Rat AC, Migliorini P, Balsa A, Westhovens R, et al. Linkage proof for PTPN22, a rheumatoid arthritis susceptibility gene and a human autoimmunity gene. Proc Natl Acad Sci USA. (2007) 104:1649–54. doi: 10.1073/pnas.0610250104
82. Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. (2004) 75:330–7. doi: 10.1086/422827
83. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. (2004) 36:337–8. doi: 10.1038/ng1323
84. Vang T, Congia M, Macis MD, Musumeci L, Orru V, Zavattari P, et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. (2005) 37:1317–9. doi: 10.1038/ng1673
85. Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, et al. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci USA. (2007) 104:19767–72. doi: 10.1073/pnas.0706233104
86. Fiorillo E, Orru V, Stanford SM, Liu Y, Salek M, Rapini N, et al. Autoimmune-associated PTPN22 R620W variation reduces phosphorylation of lymphoid phosphatase on an inhibitory tyrosine residue. J Biol Chem. (2010) 285:26506–18. doi: 10.1074/jbc.M110.111104
87. Zhang J, Zahir N, Jiang Q, Miliotis H, Heyraud S, Meng X, et al. The autoimmune disease-associated PTPN22 variant promotes calpain-mediated Lyp/Pep degradation associated with lymphocyte and dendritic cell hyperresponsiveness. Nat Genet. (2011) 43:902–7. doi: 10.1038/ng.904
88. Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science (2004) 303:685–9. doi: 10.1126/science.1092138
89. Zikherman J, Hermiston M, Steiner D, Hasegawa K, Chan A, Weiss A. PTPN22 deficiency cooperates with the CD45 E613R allele to break tolerance on a non-autoimmune background. J Immunol. (2009) 182:4093–106. doi: 10.4049/jimmunol.0803317
90. Wu J, Katrekar A, Honigberg LA, Smith AM, Conn MT, et al. (2006). Identification of substrates of human protein-tyrosine phosphatase PTPN22. J Biol Chem. 281:11002–10. doi: 10.1074/jbc.M600498200
91. Gjorloff-Wingren A, Saxena M, Williams S, Hammi D, Mustelin T. Characterization of TCR-induced receptor-proximal signaling events negatively regulated by the protein tyrosine phosphatase PEP. Eur J Immunol. (1999) 29:3845–54. doi: 10.1002/(SICI)1521-4141(199912)29:12 < 3845::AID-IMMU3845>3.0.CO;2-U
92. Hill RJ, Zozulya S, Lu YL, Ward K, Gishizky M, Jallal B. The lymphoid protein tyrosine phosphatase Lyp interacts with the adaptor molecule Grb2 and functions as a negative regulator of T-cell activation. Exp Hematol. (2002) 30:237–44. doi: 10.1016/S0301-472X(01)00794-9
93. Ghose R, Shekhtman A, Goger MJ, Ji H, Cowburn D. A novel, specific interaction involving the Csk SH3 domain and its natural ligand. Nat Struct Biol. (2001) 8:998–1004. doi: 10.1038/nsb1101-998
94. Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity (2013) 39:111–22. doi: 10.1016/j.immuni.2013.06.013
95. Chang HH, Miaw SC, Tseng W, Sun YW, Liu CC, Tsao HW, et al. PTPN22 modulates macrophage polarization and susceptibility to dextran sulfate sodium-induced colitis. J Immunol. (2013) 191:2134–43. doi: 10.4049/jimmunol.1203363
96. You-Ten KE, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, et al. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice J Exp Med. (1997) 186:683–93. doi: 10.1084/jem.186.5.683
97. Heinonen KM, Nestel FP, Newell EW, Charette G, Seemayer TA, Tremblay ML, et al. T-cell protein tyrosine phosphatase deletion results in progressive systemic inflammatory disease. Blood (2004) 103:3457–64. doi: 10.1182/blood-2003-09-3153
98. Wiede F, Shields BJ, Chew SH, Kyparissoudis K, van Vliet C, Galic S, et al. T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J Clin Invest. (2011) 121:4758–74. doi: 10.1172/JCI59492
99. Spalinger MR, Kasper S, Chassard C, Raselli T, Frey-Wagner I, Gottier C, et al. (2015). Scharl PTPN2 controls differentiation of CD4(+) T cells and limits intestinal inflammation and intestinal dysbiosis. Mucosal Immunol. 8:918–29. doi: 10.1038/mi.2014.122
100. Matthews RJ, Bowne DB, Flores E, Thomas ML. Characterization of hematopoietic intracellular protein tyrosine phosphatases: description of a phosphatase containing an SH2 domain and another enriched in proline-, glutamic acid-, serine-, and threonine-rich sequences. Mol Cell Biol. (1992) 12:2396–405. doi: 10.1128/MCB.12.5.2396
101. Plutzky J, Neel BG, Rosenberg RD. Isolation of a src homology 2-containing tyrosine phosphatase. Proc Natl Acad Sci USA. (1992) 89:1123–7. doi: 10.1073/pnas.89.3.1123
102. Yi TL, Cleveland JL, Ihle JN. Protein tyrosine phosphatase containing SH2 domains: characterization, preferential expression in hematopoietic cells, and localization to human chromosome 12p12-p13. Mol Cell Biol. (1992) 12:836–46. doi: 10.1128/MCB.12.2.836
103. Shen SH, Bastien L, Posner BI, Chretien P. A protein-tyrosine phosphatase with sequence similarity to the SH2 domain of the protein-tyrosine kinases. Nature (1991) 352:736–9. doi: 10.1038/352736a0
104. Plas DR, Johnson R, Pingel JT, Matthews RJ, Dalton M, Roy G, et al. Direct regulation of ZAP-70 by SHP-1 in T cell antigen receptor signaling. Science (1996) 272:1173–6. doi: 10.1126/science.272.5265.1173
105. Burshtyn DN, Scharenberg AM, Wagtmann N, Rajagopalan S, Berrada K, Yi T, et al. Recruitment of tyrosine phosphatase HCP by the killer cell inhibitor receptor. Immunity (1996) 4:77–85. doi: 10.1016/S1074-7613(00)80300-3
106. Burshtyn DN, Shin J, Stebbins C, Long EO. Adhesion to target cells is disrupted by the killer cell inhibitory receptor. Curr Biol. (2000) 10:777–80. doi: 10.1016/S0960-9822(00)00568-6
107. Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. (1996) 16:6985–92. doi: 10.1128/MCB.16.12.6985
108. Kashiwada M, Giallourakis CC, Pan PY, Rothman PB. Immunoreceptor tyrosine-based inhibitory motif of the IL-4 receptor associates with SH2-containing phosphatases and regulates IL-4-induced proliferation. J Immunol. (2001) 167:6382–7. doi: 10.4049/jimmunol.167.11.6382
109. Kim CH, Qu CK, Hangoc G, Cooper S, Anzai N, et al. Abnormal chemokine-induced responses of immature and mature hematopoietic cells from motheaten mice implicate the protein tyrosine phosphatase SHP-1 in chemokine responses. J Exp Med. (1999) 190:681–90. doi: 10.1084/jem.190.5.681
110. Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell (1995) 80:729–38. doi: 10.1016/0092-8674(95)90351-8
111. Minoo P, Zadeh MM, Rottapel R, Lebrun JJ, Ali S. A novel SHP-1/Grb2-dependent mechanism of negative regulation of cytokine-receptor signaling: contribution of SHP-1 C-terminal tyrosines in cytokine signaling. Blood (2004) 103:1398–407. doi: 10.1182/blood-2003-07-2617
112. Nakamura MC, Niemi EC, Fisher MJ, Shultz LD, Seaman WE, Ryan JC. Mouse Ly-49A interrupts early signaling events in natural killer cell cytotoxicity and functionally associates with the SHP-1 tyrosine phosphatase. J Exp Med. (1997) 185:673–84. doi: 10.1084/jem.185.4.673
113. Pani G, Kozlowski M, Cambier JC, Mills GB, Siminovitch KA. Identification of the tyrosine phosphatase PTP1C as a B cell antigen receptor-associated protein involved in the regulation of B cell signaling. J Exp Med. (1995) 181:2077–84. doi: 10.1084/jem.181.6.2077
114. Roach TI, Slater SE, White LS, Zhang X, Majerus PW, Brown EJ, et al. The protein tyrosine phosphatase SHP-1 regulates integrin-mediated adhesion of macrophages. Curr Biol. (1998) 8:1035–8. doi: 10.1016/S0960-9822(07)00426-5
115. Xiao W, Hong H, Kawakami Y, Kato Y, Wu D, Yasudo H, et al. Tumor suppression by phospholipase C-beta3 via SHP-1-mediated dephosphorylation of Stat5. Cancer Cell (2009) 16:161–71. doi: 10.1016/j.ccr.2009.05.018
116. Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, et al. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell (1993) 73:1445–54. doi: 10.1016/0092-8674(93)90369-2
117. Tsui HW, Siminovitch KA, de Souza L, Tsui FW. Motheaten and viable motheaten mice have mutations in the haematopoietic cell phosphatase gene. Nat Genet. (1993) 4:124–9. doi: 10.1038/ng0693-124
118. Carter JD, Neel BG, Lorenz U. The tyrosine phosphatase SHP-1 influences thymocyte selection by setting TCR signaling thresholds. Int Immunol. (1999) 11:1999–2014. doi: 10.1093/intimm/11.12.1999
119. Zhang J, Somani AK, Yuen D, Yang Y, Love PE, Siminovitch KA. Involvement of the SHP-1 tyrosine phosphatase in regulation of T cell selection. J Immunol. (1999) 163:3012–21. 120. Sathish JG, Johnson KG, LeRoy FG, Fuller KJ, Hallett MB, Brennan P, et al. Requirement for CD28 co-stimulation is lower in SHP-1-deficient T cells. Eur J Immunol. (2001) 31:3649–58. doi: 10.1002/1521-4141(200112)31:12 < 3649::AID-IMMU3649>3.0.CO;2-8
120. Sathish JG, Johnson KG, LeRoy FG, Fuller KJ, Hallett MB, Brennan P, et al. Requirement for CD28 co-stimulation is lower in SHP-1-deficient T cells. Eur J Immunol. (2001) 31:3649–58. doi: 10.1002/1521-4141(200112)31:12 < 3649::AID-IMMU3649>3.0.CO;2-8
121. Su MW, Yu CL, Burakoff SJ, Jin YJ. Targeting Src homology 2 domain-containing tyrosine phosphatase (SHP-1) into lipid rafts inhibits CD3-induced T cell activation. J Immunol. (2001) 166:3975–82. doi: 10.4049/jimmunol.166.6.3975
122. Chen Z, Chen L, Qiao SW, Nagaishi T, Blumberg RS. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J Immunol. (2008) 180:6085–93. doi: 10.4049/jimmunol.180.9.6085
123. Lorenz U, Ravichandran KS, Burakoff SJ, Neel BG. Lack of SHPTP1 results in src-family kinase hyperactivation and thymocyte hyperresponsiveness. Proc Natl Acad Sci USA. (1996) 93:9624–9. doi: 10.1073/pnas.93.18.9624
124. Mizuno K, Tagawa Y, Watanabe N, Ogimoto M, Yakura H. SLP-76 is recruited to CD22 and dephosphorylated by SHP-1, thereby regulating B cell receptor-induced c-Jun N-terminal kinase activation. Eur J Immunol. (2005) 35:644–54. doi: 10.1002/eji.200425465
125. Stefanova I, Hemmer B, Vergelli M, Martin R, Biddison WE, Germain RN. TCR ligand discrimination is enforced by competing ERK positive and SHP-1 negative feedback pathways. Nat Immunol. (2003) 4:248–54. doi: 10.1038/ni895
126. Johnson DJ, Pao LI, Dhanji S, Murakami K, Ohashi PS, Neel BG. Shp1 regulates T cell homeostasis by limiting IL-4 signals. J Exp Med. (2013) 210:1419–31. doi: 10.1084/jem.20122239
127. Martinez RJ, Morris AB, Neeld DK, Evavold BD. Targeted loss of SHP1 in murine thymocytes dampens TCR signaling late in selection. Eur J Immunol. (2016) 46:2103–10. doi: 10.1002/eji.201646475
128. Mercadante ER, Lorenz UM. T cells deficient in the tyrosine phosphatase SHP-1 resist suppression by regulatory T cells. J Immunol. (2017) 199:129–37. doi: 10.4049/jimmunol.1602171
129. Christophi GP, Rong R, Holtzapple PG, Massa PT, Landas SK. Immune markers and differential signaling networks in ulcerative colitis and Crohn's disease. Inflamm Bowel Dis. (2012) 18:2342–56. doi: 10.1002/ibd.22957
130. Bouzid D, Fourati H, Amouri A, Marques I, Abida O, Haddouk S, et al. Association of ZAP70 and PTPN6, but Not BANK1 or CLEC2D, with inflammatory bowel disease in the Tunisian population. Genet Test Mol Biomarkers (2013) 17:321–6. doi: 10.1089/gtmb.2012.0372
131. Feng GS, Hui CC, Pawson T. SH2-containing phosphotyrosine phosphatase as a target of protein-tyrosine kinases. Science (1993) 259:1607–11. doi: 10.1126/science.8096088
132. Freeman RMJr, Plutzky J, Neel BG. Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proc Natl Acad Sci USA. (1992) 89:11239–43. doi: 10.1073/pnas.89.23.11239
133. Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, et al. Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J. (1997) 16:2352–64. doi: 10.1093/emboj/16.9.2352
134. Zhang J, Zhang F, Niu R. Functions of Shp2 in cancer. J Cell Mol Med. (2015) 19:2075–83. doi: 10.1111/jcmm.12618
135. Narumi Y, Isomoto H, Shiota M, Sato K, Kondo S, Machida H, et al. Polymorphisms of PTPN11 coding SHP-2 as biomarkers for ulcerative colitis susceptibility in the Japanese population. J Clin Immunol. (2009) 29:303–10. doi: 10.1007/s10875-008-9272-6
136. Dong B, Gao Y, Zheng X, Gao G, Gu H, Chen X, et al. T cell activation is reduced by the catalytically inactive form of protein tyrosine phosphatase SHP-2. Int J Clin Exp Med. (2015) 8:6568–77.
137. Miah SMS, Jayasuriya CT, Salter AI, Reilly EC, Fugere C, Yang W, et al. Ptpn11 Deletion in CD4(+) cells does not affect t cell development and functions but causes cartilage tumors in a T cell-independent manner. Front Immunol. 8:1326. doi: 10.3389/fimmu.2017.01326
138. Nguyen TV, Ke Y, Zhang EE, Feng GS. Conditional deletion of Shp2 tyrosine phosphatase in thymocytes suppresses both pre-TCR and TCR signals. J Immunol. (2006) 177:5990–6. doi: 10.4049/jimmunol.177.9.5990
139. Salmond RJ, Huyer G, Kotsoni A, Clements L, Alexander DR. The src homology 2 domain-containing tyrosine phosphatase 2 regulates primary T-dependent immune responses and Th cell differentiation. J Immunol. (2005) 175:6498–508. doi: 10.4049/jimmunol.175.10.6498
140. Zhang T, Guo W, Yang Y, Liu W, Guo L, Gu Y, et al. Loss of SHP-2 activity in CD4+ T cells promotes melanoma progression and metastasis. Sci Rep. 3:2845. doi: 10.1038/srep02845
141. Kwon J, Qu CK, Maeng JS, Falahati R, Lee C, Williams MS. Receptor-stimulated oxidation of SHP-2 promotes T-cell adhesion through SLP-76-ADAP. EMBO J. (2005) 24:2331–41. doi: 10.1038/sj.emboj.7600706
142. Wu X, Guo W, Wu L, Gu Y, Gu L, Xu S, et al. Selective sequestration of STAT1 in the cytoplasm via phosphorylated SHP-2 ameliorates murine experimental colitis. J Immunol. (2012) 189:3497–507. doi: 10.4049/jimmunol.1201006
143. Coulombe G, Leblanc C, Cagnol S, Maloum F, Lemieux E, Perreault N, et al. Epithelial tyrosine phosphatase SHP-2 protects against intestinal inflammation in mice. Mol Cell Biol. (2013) 33:2275–84. doi: 10.1128/MCB.00043-13
144. Yamashita H, Kotani T, Park JH, Murata Y, Okazawa H, et al. Role of the protein tyrosine phosphatase Shp2 in homeostasis of the intestinal epithelium. PLoS ONE 9:e92904. doi: 10.1371/journal.pone.0092904
145. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
146. Chang CF, D'Souza WN, Ch'en IL, Pages G, Pouyssegur J, Hedrick SM. Polar opposites: Erk direction of CD4 T cell subsets. J Immunol. (2012) 189:721–31. doi: 10.4049/jimmunol.1103015
147. Luo X, Zhang Q, Liu V, Xia Z, Pothoven KL, Lee C. Cutting edge: TGF-beta-induced expression of Foxp3 in T cells is mediated through inactivation of ERK. J Immunol. (2008) 180:2757–61. doi: 10.4049/jimmunol.180.5.2757
148. Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. PLoS Biol. 3:e356. doi: 10.1371/journal.pbio.0030356
149. Liu H, Yao S, Dann SM, Qin H, Elson CO, Cong Y. ERK differentially regulates Th17- and Treg-cell development and contributes to the pathogenesis of colitis. Eur J Immunol. (2013) 43:1716–26. doi: 10.1002/eji.201242889
150. Tan AH, Lam KP. Pharmacologic inhibition of MEK-ERK signaling enhances Th17 differentiation. J Immunol. (2010) 184:1849–57. doi: 10.4049/jimmunol.0901509
151. Li G, Yu M, Lee WW, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med. (2012) 18:1518–24. doi: 10.1038/nm.2963
152. Agrawal S, Agrawal A, Doughty B, Gerwitz A, Blenis J, Van Dyke T, et al. Cutting edge: different Toll-like receptor agonists instruct dendritic cells to induce distinct Th responses via differential modulation of extracellular signal-regulated kinase-mitogen-activated protein kinase and c-Fos J Immunol. (2003) 171:4984–9. doi: 10.4049/jimmunol.171.10.4984
153. Dillon S, Agrawal A, Van Dyke T, Landreth G, McCauley L, Koh A, et al. A Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via induction of extracellular signal-regulated kinase mitogen-activated protein kinase and c-Fos in dendritic cells. J Immunol. (2004) 172:4733–43. doi: 10.4049/jimmunol.172.8.4733
154. Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, et al. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J Biol Chem. (1996) 271:27205–8. doi: 10.1074/jbc.271.44.27205
155. Stewart AE, Dowd S, Keyse SM, McDonald NQ. Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation. Nat Struct Biol. (1999) 6:174–81. doi: 10.1038/5861
156. Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. (2008) 283:31246–55. doi: 10.1074/jbc.M806085200
157. Gonzalez-Navajas JM, Fine S, Law J, Datta SK, Nguyen KP, Yu M, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. (2010) 120:570–81. doi: 10.1172/JCI40055
158. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy A, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science (1999) 283:1544–8. doi: 10.1126/science.283.5407.1544
159. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, et al. (2000). Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 20:5479–5489. doi: 10.1128/MCB.20.15.5479-5489.2000
160. Lazo JS, McQueeney KE, Burnett JC, Wipf P, Sharlow ER. Small molecule targeting of PTPs in cancer. Int J Biochem Cell Biol. (2018) 96:171–81. doi: 10.1016/j.biocel.2017.09.011
161. He R. J., Yu ZH, Zhang RY, Zhang ZY. Protein tyrosine phosphatases as potential therapeutic targets. Acta Pharmacol Sin. (2014) 35:1227–46. doi: 10.1038/aps.2014.80
162. Hendriks W, Bourgonje A, Leenders W, Pulido R. Proteinaceous regulators and inhibitors of protein tyrosine phosphatases. Molecules (2018) 23. doi: 10.3390/molecules23020395
163. Stanford SM, Bottini N. Targeting tyrosine phosphatases: time to end the stigma. Trends Pharmacol Sci (2017) 38:524–40. doi: 10.1016/j.tips.2017.03.004
164. Krishnan N, Koveal D, Miller DH, Xue B, Akshinthala SD, Kragelj J, et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat Chem Biol. (2014) 10:558–66. doi: 10.1038/nchembio.1528
165. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker M, et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature (2016) 535:148–52. doi: 10.1038/nature18621
166. Perron MD, Chowdhury S, Aubry I, Purisima E, Tremblay ML, Saragovi HU. Allosteric noncompetitive small molecule selective inhibitors of CD45 tyrosine phosphatase suppress T-cell receptor signals and inflammation in vivo. Mol Pharmacol. (2014) 85:553–63. doi: 10.1124/mol.113.089847
167. Wu CL, Hardy S, Aubry I, Landry M, Haggarty A, Saragovi H, et al. Identification of function-regulating antibodies targeting the receptor protein tyrosine phosphatase sigma ectodomain. PLoS ONE 12:e0178489. doi: 10.1371/journal.pone.0178489
168. Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK. Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell (2011) 147:185–98. doi: 10.1016/j.cell.2011.08.036
169. Tan XF, Uddin Z, Park C, Song YH, Son M, et al. Competitive protein tyrosine phosphatase 1B (PTP1B) inhibitors, prenylated caged xanthones from Garcinia hanburyi and their inhibitory mechanism. Bioorg Med Chem. (2017) 25:2498–506. doi: 10.1016/j.bmc.2017.03.010
170. Townshend B, Aubry I, Marcellus RC, Gehring K, Tremblay ML. An RNA aptamer that selectively inhibits the enzymatic activity of protein tyrosine phosphatase 1B in vitro. Chembiochem (2010) 11:1583–93. doi: 10.1002/cbic.201000208
171. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller B, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547:413–8. doi: 10.1038/nature23270
172. Crew AP, Raina K, Dong H, Qian Y, Wang J, Vigil D, et al. Identification and characterization of von hippel-lindau-recruiting proteolysis targeting chimeras (PROTACs) of TANK-Binding Kinase 1. J Med Chem. (2018) 61:583–98. doi: 10.1021/acs.jmedchem.7b00635
173. Ohoka N, Okuhira K, Ito M, Nagai K, Shibata N, Hattori T, et al. In Vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) J Biol Chem. (2017) 292:4556–70. doi: 10.1074/jbc.M116.768853
174. Penafuerte C, Feldhammer M, Mills JR, Vinette V, Pike KA, Hall A, et al. Downregulation of PTP1B and TC-PTP phosphatases potentiate dendritic cell-based immunotherapy through IL-12/IFNgamma signaling. Oncoimmunology (2017) 6:e1321185. doi: 10.1080/2162402X.2017.1321185
Keywords: protein tyrosine phosphatase, CD4 T cells, cytokine, JAK-STAT, inflammatory bowel disease
Citation: Pike KA and Tremblay ML (2018) Protein Tyrosine Phosphatases: Regulators of CD4 T Cells in Inflammatory Bowel Disease. Front. Immunol. 9:2504. doi: 10.3389/fimmu.2018.02504
Received: 25 July 2018; Accepted: 10 October 2018;
Published: 31 October 2018.
Edited by:
Paul E. Love, National Institutes of Health (NIH), United StatesReviewed by:
Thomas Ciucci, National Cancer Institute (NCI), United StatesUlrike Lorenz, University of Virginia, United States
Copyright © 2018 Pike and Tremblay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kelly A. Pike, a2VsbHktYW5uZS5waWtlQG1haWwubWNnaWxsLmNh
Michel L. Tremblay, bWljaGVsLnRyZW1ibGF5QG1jZ2lsbC5jYQ==