 Paulina García-González
Paulina García-González Felipe Cabral-Miranda
Felipe Cabral-Miranda Claudio Hetz
Claudio Hetz Fabiola Osorio
Fabiola Osorio- 1Laboratory of Immunology and Cellular Stress, Program of Immunology, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, Santiago, Chile
- 2Faculty of Medicine, Biomedical Neuroscience Institute, University of Chile, Santiago, Chile
- 3Brain Health and Metabolism, FONDAP Center for Geroscience, Santiago, Chile
- 4Program of Cellular and Molecular Biology, Institute of Biomedical Sciences, University of Chile, Santiago, Chile
- 5Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil
- 6Buck Institute for Research on Aging, Novato, CA, United States
- 7Department of Immunology and Infectious Diseases, Harvard School of Public Health, Boston, MA, United States
Emerging evidence suggests that the immune and nervous systems are in close interaction in health and disease conditions. Protein aggregation and proteostasis dysfunction at the level of the endoplasmic reticulum (ER) are central contributors to neurodegenerative diseases. The unfolded protein response (UPR) is the main transduction pathway that maintains protein homeostasis under conditions of protein misfolding and aggregation. Brain inflammation often coexists with the degenerative process in different brain diseases. Interestingly, besides its well-described role in neuronal fitness, the UPR has also emerged as a key regulator of ontogeny and function of several immune cell types. Nevertheless, the contribution of the UPR to brain inflammation initiated by immune cells remains largely unexplored. In this review, we provide a perspective on the potential role of ER stress signaling in brain-associated immune cells and the possible implications to neuroinflammation and development of neurodegenerative diseases.
Introduction
The Unfolded Protein Response (UPR)
Proteostasis encompasses the dynamic interrelation of processes governing generation and localization of functional proteins (1). Physiological and pathological factors can impair the balance between protein load and protein processing, resulting into accumulation of improperly folded proteins (2, 3). Abnormal protein aggregation is a key feature of several neurodegenerative diseases, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease (HD) and prion-related disorders amongst others, collectively classified as protein misfolding diseases (PMDs) (4, 5).
Protein misfolding is sensed by dedicated stress-response pathways that include the cytoplasmic heat shock response (HSR) and the unfolded protein response originated in the mitochondria and in the endoplasmic reticulum (ER) (3). Activation of these intracellular mechanisms by the presence of misfolded proteins leads to ameliorating the protein folding load and resolving proteotoxic stress (1, 3). In this context, the ER is a central node of the proteostasis network controlling folding, processing and trafficking of up to a third of the protein load in the cell (6). The UPR originated in the ER (for now referred as “UPR”) is a main intracellular mechanism responsible to safeguard the fidelity of the cellular proteome and for this reason, it will be the main focus of the current review (6, 7). The UPR is an adaptive reaction controlled by three ER-located signal transducers: inositol requiring enzyme 1 (IRE1) α and β, protein kinase R-like ER kinase (PERK) and activating transcription factor 6 (ATF6) alpha and beta (6) (Figure 1). Upon activation, these signal transducers activate gene expression programs through specific downstream transcription factors, restoring proteostasis and increasing ER and Golgi biogenesis (6, 8). IRE1α cleaves the mRNA encoding for the X-box binding protein (XBP1), removing a 26 nucleotide intron, which followed by RTCB (RNA 2′,3′-Cyclic Phosphate and 5′-OH ligase) ligation changes the coding reading frame, prompting the translation of a protein with transcription factor activity termed XBP1s (XBP1 spliced) (7). XBP1s controls the expression of genes involved in ER-associated degradation (ERAD), lipid biosynthesis, folding and quality control (9, 10). IRE1α RNase also directly degrades diverse mRNAs and microRNAs through a process termed “Regulated IRE1-Dependent Decay” (RIDD) (11), originally proposed to contribute to alleviating the detrimental effects of ER stress by reducing the protein folding load (12), in addition to regulating inflammation and apoptosis (13). Activation of PERK mediates protein translation shutdown via phosphorylation of eukaryotic initiation factor 2α (P-eIF2α), which also favors selective translation of certain mRNAs encoding proteins involved in cell survival, ER homeostasis and anti-oxidant responses, such as ATF4 and nuclear erythroid related factor 2 (NRF2) (6, 14). ATF6, translocates to the Golgi apparatus where it is cleaved by site-1 and site-2 proteases, releasing a transcription factor that directs the expression of genes encoding ERAD components, ER chaperones and molecules involved in lipid biogenesis (15, 16). XBP1s and ATF6 can also heterodimerize to control selective gene expression patterns (9). Moreover, the activity (signaling amplitude and kinetics) of the three UPR stress sensors is controlled by several cofactors through the assembling of distinct platforms termed the UPRosome (17). Binding of adapter proteins to the IRE1α UPRosome also mediates the crosstalk with other stress pathways including MAP kinases and NF-κB (6). Thus, the UPR integrates information regarding intensity and duration of the stress stimuli toward cell fate control in cells suffering from ER stress.
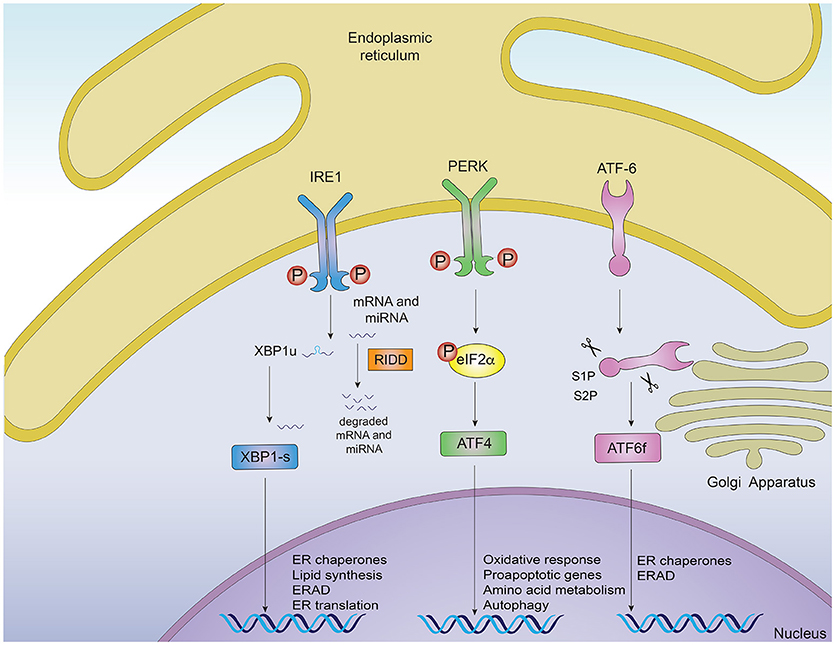
Figure 1. Signaling pathways of the unfolded protein response. Noxious stimuli in cells may induce endoplasmic reticulum (ER) stress and trigger an adaptive response known as the unfolded protein response (UPR), which is controlled by three main ER-resident sensors: IRE1α, PERK and ATF6. Upon ER stress, IRE1α autophosphorylates, leading to the activation of its RNase domain and the processing of the mRNA encoding for XBP1s, a transcriptional factor that upregulates genes involved in protein folding and quality control, in addition to regulating ER/Golgi biogenesis and ER-mediated degradation (ERAD). Additionally, IRE1α RNase also degrades a subset of specific RNAs and microRNAs, a process termed Regulated IRE1α-Dependent Decay (RIDD). The second ER sensor, PERK, phosphorylates the translation of the eukaryotic initiation factor eIF2α, decreasing the synthesis of proteins and the overload of misfolded proteins at the ER. PERK phosphorylation also leads to the specific translation of ATF4, a transcription factor that promotes the expression of genes related to amino acid metabolism, anti-oxidant response, autophagy and apoptosis. The third UPR sensor, ATF6, is a type II ER transmembrane protein that encodes a bZIP transcriptional factor in its cytosolic domain. Following ER stress, ATF6 translocates to the Golgi apparatus where it is processed, releasing a transcription factor which directs the expression of genes encoding ER chaperones, ERAD components and molecules involved in lipid biogenesis.
UPR in Brain Homeostasis and Protein Misfolding Diseases
ER stress signaling has a physiological as well as pathological role in brain function and development (18–20). In neurodegeneration, the UPR influences several aspects including cell survival, synaptic plasticity, axonal regeneration, protein aggregation and control of the secretory pathway (21–23). By mediating synthesis and secretion of the brain-derived neurotrophic factor (BDNF), XBP1s regulates neuronal plasticity at a structural, molecular and behavioral level (18, 24–27). Moreover, postmortem tissue analyses revealed that ER stress markers often co-localize with cells containing protein aggregates in brain of patients affected with PMDs (4, 5, 22, 28). In AD, the expression of Grp78/BiP, PDI and HRD1 is increased in the hippocampus and temporal cortex; and the phosphorylated forms of PERK, IRE1α and eIF2α are found in AD neurons and substantia nigra of PD patients (22, 29, 30). Phosphorylated IRE1α levels directly correlate with the degree of histopathological changes, where most cells showing neurofibrillary tangles exhibit signs of ER stress (31). Furthermore, ER stress signs are also observed in different brain areas in PD patients, a phenomenon also observed in incidental cases of subjects who died without PD symptoms but presented α-synuclein inclusions in the brain (32). Moreover, components of all UPR signaling branches are overexpressed in spinal cord samples of patients with familial and sporadic forms of ALS (33), as well as in striatum, parietal cortex and caudate putamen of HD and Prion disease patients (22, 34–39).
In support of a dual role of UPR in controlling cell fate in neurodegenerative diseases, genetic disruption and pharmacological intervention modulating ER stress signaling revealed that depending on disease type and the UPR component targeted, distinct and even opposite effects are observed [reviewed in (21, 40)]. Conditional deletion of XBP1 in the central nervous system (CNS) provides protective effects through upregulation of autophagy levels, improving motor performance in ALS, PD and Huntington's disease models (35, 37, 41), whereas XBP1 deficiency does not affect Prion pathogenesis in vivo (42). Ablation of IRE1α signaling in neurons decreases astrogliosis and amyloid β accumulation in an animal model of AD, correlating with improved neuronal function (31). Conversely, therapeutic gene delivery of active UPR components or ER chaperones to specific brain areas has shown outstanding effects in different animal models of PMDs (43). Different studies have shown that ectopic delivery of XBP1s into the hippocampus restored synaptic plasticity in an AD model (27), promoted axonal regeneration (44), reduced mutant huntingtin aggregation (45) and protected dopaminergic neurons against PD-inducing neurotoxins (41, 46).
Targeting the PERK pathway also provides contradicting results. PERK signaling supports oligodendrocyte survival in animal models of multiple sclerosis (MS) (47) and enhancement of eIF2α phosphorylation is protective in ALS and other models (32, 48), whilst ATF4 deficiency has a detrimental effect in spinal cord injury models, diminishing locomotor recovery following lesion, also impacting oligodendrocyte survival (49). Conditional deletion of PERK in the brain however, improved cognition in an AD model, correlating with decreased amyloidogenesis and restoration of normal expression of plasticity-related proteins (50, 51). Similarly, genetic targeting of CHOP has neuroprotective effects in a PD model, and ATF4 ablation protects against ALS (52, 53). Consistent with this, sustained PERK signaling has been shown to enhance neurodegeneration due to acute repression of synaptic proteins, resulting in abnormal neuronal function, as demonstrated through PERK inhibitors in Prion disease (54), frontotemporal dementia (48) and PD models (32). ATF6, on the other hand, protected dopaminergic neurons in another PD model, by upregulating ER chaperones and ERAD components (55, 56). Overall, UPR mediators have a pivotal role in the progression of various PMDs, nurturing the hypothesis that UPR components could be used as therapeutic targets in neurodegeneration (21, 22, 43).
UPR in Neuroinflammation
Immune surveillance is an active process in the brain. The mammalian CNS harbors several subtypes of leukocytes, which display physiological roles related to tissue homeostasis and regulation of the inflammatory response (57, 58). However, if unrestrained, inflammation can have detrimental effects in the CNS, contributing to the type of tissue malfunction that precedes pathological processes (59). During neuroinflammation, the immune response in the CNS is drastically altered, and it is typified by activation of resident microglia and invasion of peripheral immune cells into the parenchyma, including granulocytes, monocytes and, in pathologies like multiple sclerosis, lymphocytes (60–63). Interestingly, the UPR has shown to regulate inflammation in peripheral tissues, emerging as an interesting candidate for targeting CNS-associated inflammation in a field that remains largely unexplored. Thus, in addition to the well-described role of the UPR in neuronal fitness, it is also plausible that UPR activation in CNS-associated immune cells could contribute to modulating PMD development.
One hallmark of neuroinflammation is the presence of tumor necrosis factor (TNF), interleukin (IL)-1β, and IL-6 in brain, cerebrospinal fluid (CSF) and serum of patients with AD, PD and HD (63–65). Production of pro-inflammatory cytokines across tissues depends on activation of innate immune sensors (known as pattern recognition receptors, PRRs) specialized in the recognition of microbes and stress signals (63). In the brain, PRRs can promote pro-inflammatory cytokine production upon recognition of “neurodegeneration associated molecular patterns” (NAMPs) that consists in CNS-specific danger signals such as extracellular protein aggregates, molecules exposed by dying neurons, lipid degradation byproducts and myelin debris, among others (66). The most relevant PRRs associated to the development of PMDs are TLRs (Toll-like Receptors) and NLR (Nucleotide-binding domain, leucine-rich repeat containing) inflammasomes (63). These receptors are broadly expressed in CNS-myeloid cells including microglia, macrophages and infiltrating cells such as monocytes and dendritic cells (DCs) (63, 67). Interestingly, PRR-signaling and the UPR converge on several levels for amplification of inflammatory responses via activation of NF-kB, IRF-3, JNK and JAK/STAT modules (68–71). Signaling via TLR2 and TLR4 induces ER stress in peripheral macrophages and activates IRE1α and XBP1s, which in turn is required to increase production of IL-6 and TNF, thus connecting activation of the IRE1α-XBP1s branch of the UPR with TLR-dependent pro-inflammatory programs (68). In the CNS, misfolded α-synuclein and Fibrillar Aβ, characteristic in patients with PD and AD, can be sensed by TLR1/2 and TLR4, further promoting inflammation (63) (Figure 2). Moreover, injection of lipopolysaccharide (LPS), an agonist of TLR4, into the substantia nigra induces dopaminergic neuronal death resembling animal PD models (73). LPS-induced neurotoxicity and LPS-derived inducible nitric oxide synthase (iNOS) expression was shown to be mediated by the UPR related chaperone BiP/Grp78 and NF-kB (74, 75). Correspondingly, Tlr4 null mice are protected from PD in a mouse model induced with neurotoxins (63, 76). Overall, TLR pathways activating the IRE1α-XBP1s axis are relevant drivers of PMDs, although the precise contribution of this UPR branch to TLR-induced neuroinflammation remains to be formally demonstrated.
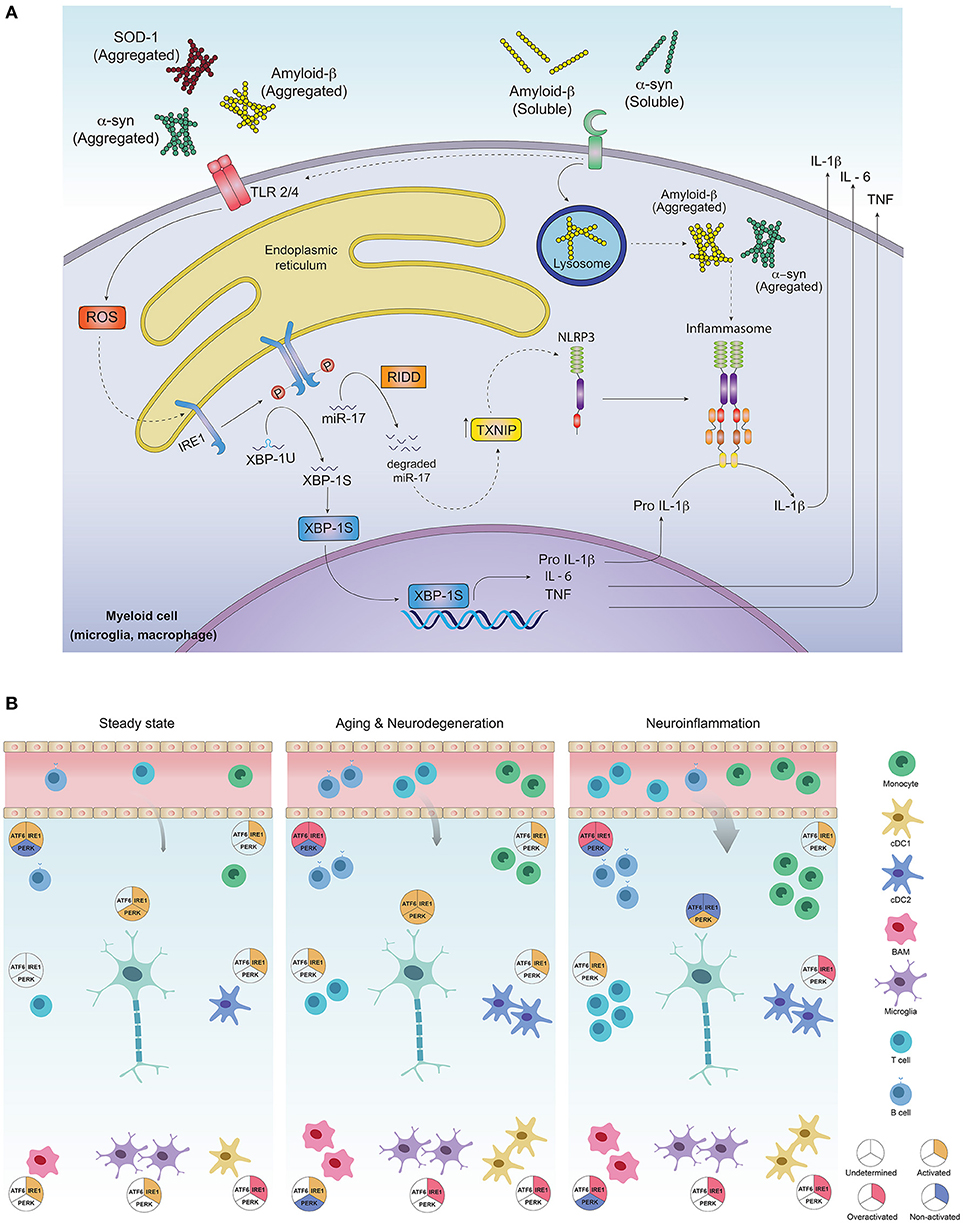
Figure 2. Activation of the unfolded protein response in CNS-residing immune cells may contribute to neuroinflammation and PMDs development. (A) Protein aggregates can promote inflammation via triggering of innate receptors and activation of the UPR. Neurodegeneration associated molecular patterns (NAMPS) such as protein aggregates are recognized by pattern recognition receptors (TLRs and PRRs) present on immune cells and signal through ROS production, which in turn could activate the IRE1α/XBP1s axis for co-transcriptional activation of IL-6, TNF and IL-1β. On the other hand, through RIDD, IRE1α induces degradation of the TXNIP-destabilizing microRNA mir-17, allowing activation of the NLRP3 inflammasome and processing of IL-1β into its active form. (B) Most of the immune cell lineages residing in the healthy and pathogenic brain are known targets of the UPR in peripheral tissues. In steady state, the most abundant immune cells in the brain are microglia, which along with border associated macrophages (“BAMs”) and dendritic cells act as sentinels, sampling the environment and clearing cell debris, maintaining CNS homeostasis. Except for dendritic cells and macrophages, which exhibit IRE1α/XBP1s activation, little is known about UPR activation in additional myeloid subsets, although microglia, macrophages and monocytes could potentially activate this axis downstream of PRR signaling. While very rare, B and T cells have been identified in the steady state brain, and activation of IRE1α/XBP1s has been proposed to be critical for their differentiation and activation. ATF6 axis is also necessary for B cell development and activation whilst absence of PERK contributes to plasma cell differentiation and immunoglobulin synthesis. Basal activation of UPR in neurons is still a matter of debate in literature as the function of IRE1α and PERK pathways has just begun to be understood in the context of normal neuronal physiology (72). In aging and neurodegeneration, the number of immune cells within the brain increases, due to higher cell activation as well as blood brain barrier infiltrates. Extracellular protein aggregation promotes activation of immune cells via PRRs, in addition to inducing ER stress and activation of the UPR, mainly the IRE1α/XBP1s axis. Microglia and dendritic cells become more activated, with higher production of pro-inflammatory and oxidative mediators and loss of their protein clearance function. This is further aggravated by antibodies against CNS-derived antigens by B cells accumulated in the CSF, mediated by the activation of IRE1α and ATF6 signaling. Activation of infiltrating T cells reactive to α-synuclein, amyloid-β and myelin constituents further amplify inflammation, resulting in more protein aggregation and neuronal loss. In neurons, UPR triggering may elicit both, adaptive or neurodegenerative responses, since all three UPR pathways are engaged in brain diseases and have been found to be altered during the normal aging process. Different inducers of neuroinflammation, have shown to engage the UPR in neurons and promote a greater inflammatory response due to immune cell infiltration, mainly B and T cells. The cDC1 subset of dendritic cells could activate IRE1α for cross presentation of antigens to infiltrating CD8+ T cells, and cDC2 as well as monocyte-derived DCs may set an inflammatory environment through cytokine secretion and activation of infiltrating CD4+ T cells. Macrophages and microglia also become highly activated and could tune IRE1α/XBP1s upon recognition of NAMPs. Inflammatory mediators such as cytokines prime axonal destruction and neuronal loss. It remains to be addressed weather UPR triggering in these cells corresponds to a homeostatic (adaptive) response, or a terminal (neurodegenerative) response due to sustained unresolved ER stress.
Another PRR relevant in neurodegeneration modulated by the UPR, is the NLRP3 (NLR Family Pyrin Domain-Containing-3) inflammasome, a multimeric protein complex composed of the NLRP3 sensor, the adaptor ASC and activated caspase 1, which mediates the proteolytic activation of IL-1β and IL-18 and promotes a type of inflammatory cell death referred to as pyroptosis (63). In the brain, the NLRP3 inflammasome is activated by amyloid β and α-synuclein aggregates (63). The relevance of this protein complex is underscored by studies with Nlrp3 deficient mice carrying mutations associated with familiar AD, which are protected from the disease (77). On a mechanistic level, the interplay between the UPR and inflammasome activation has been connected to IRE1α signaling (78), where the RNase domain of IRE1α increases the expression of TXNIP, an activator of the NLRP3 inflammasome, through degradation of the TXNIP-destabilizing microRNA miR-17 (78) (Figure 2). Considering the relevance of the NLRP3 inflammasome in AD progression and its dependence on IRE1α endonuclease, it is tempting to speculate that IRE1α activation in CNS-resident myeloid cells may contribute to the development of AD (79–84). Additionally, the B-class scavenger receptor CD36, upon recognition of amyloid β fibrils, forms a complex with TLR4/6, which triggers activation of the NLRP3 inflammasome, promoting cytokine and ROS production (67, 85).
On the other hand, in models of peripheral nerve damage, XBP1 expression has been shown to enhance nerve regeneration after injury, involving increased expression of the chemokine MCP-1 and macrophage infiltration, essential to remove myelin debris and allow axonal regeneration (44). PERK expression correlates with astroglial activation and production of IL-6 and the chemokines CCL2 and CCL20, which promotes microglial activation (71, 86). In spinal cord injury, ATF4 deficiency reduced microglial activation, which is associated with altered levels of IL-6, TNFα, and IL-1β (44–49). Similarly, ATF6 deficiency in the context of PD induced by neurotoxins leads to suppression of astroglial activation and decreased production of BDNF and anti-oxidative genes, such as heme oxygenase-1 (HO-1) and xCT (56). To sum up, ER stress and inflammation are both prevalent in many neurodegenerative diseases and NAMPs can alter neuronal function as well as promote inflammation through the activation of innate defense mechanisms of immune cells in the CNS, which can be modulated by UPR activity and vice versa.
Immune Targets of the UPR in the Central Nervous System
Although it is clear that inflammation contributes to neurodegeneration (61), there has been limited knowledge about the homeostasis of immune cells residing in the CNS. Recent technological advances in single cell analysis have provided insights into the identification and characterization of the vast diversity of immune cell lineages present in the healthy and pathogenic brain (61, 62). The potential role of the UPR in immune cell lineages in the CNS is illustrated in Figure 2.
Microglia
Microglia is the CNS-resident macrophage and most prominent myeloid cell in the brain (87). Microglia fine-tunes the development of neuronal circuits, neurogenesis and synaptic plasticity through the production of neurotrophic factors (88, 89). Given that several PRRs that signal via IRE1α and XBP1s such as TLR1/2 and TLR4, the NLRP3 inflammasome and nucleic acid sensors are expressed in this cell lineage, it is plausible that microglial XBP1s activation may contribute to the initiation of neuroinflammation. The ATF6 branch has also been associated with microglial activation and production of inflammatory mediators via NF-kB (90). Furthermore, although long conceived as a homogeneous cell type that becomes destructive in neurodegeneration (62), comprehensive single cell RNA analysis has demonstrated that a subset known as “disease-associated microglia” plays an important role in several CNS diseases including AD, ALS, MS and also in aging (62, 91–93). Thus, it is vital to elucidate whether protective microglial populations engage the UPR upon innate recognition of NAMPs, and whether microglial UPR is an intrinsic mechanism of sensing danger in the CNS.
Border Associated Macrophages
Border associated macrophages (BAMs) are a recently characterized population distinct from microglia and from infiltrating monocyte-derived macrophages, which display high heterogeneity and are classified per phenotype, development and location in the CNS (62, 94). Single cell analysis, fate mapping and parabiosis experiments revealed that these cells express distinct surface markers and differentially populate the pia mater, perivascular space, choroid plexus and dura mater (62, 94). Most of these subsets sample the environment, clear apoptotic cells and amyloid β plaques, and help maintaining CNS homeostasis in steady state. Up to date, there is no evidence available on the extent of UPR activation in BAMs. However, it has been described that splenic F4/80 macrophages display basal levels of IRE1α RNase activity and upon bacterial infection, peripheral macrophages induce XBP1s for enhancing cytokine production in a mechanism mediated by TLRs and reactive oxygen species (68, 95). However, whether CNS macrophages show a functional analogy to peripheral macrophages and also engage the IRE1α-XBP1s branch upon recognition of NAMPs (68) remains undetermined.
Dendritic Cells
DCs are major APCs in the CNS, acting as sentinels between brain and periphery (87, 96–99). Steady-state CNS is populated by most DC subtypes, including plasmacytoid DCs (pDCs), and conventional DC type 1 (cDC1) and type 2 (cDC2) (62). These cells locate in the choroid plexus, pia mater and dura mater, but not in the perivascular space, suggesting that these compartments may serve as entry sites for MHC-dependent T cells (62, 96, 97). Importantly, DCs are key targets of the UPR. XBP1s is constitutively expressed by DCs and high XBP1s is a hallmark of cDC1s across tissues, although the CNS remains to be examined (95, 100, 101). Furthermore, cDC1s activate the IRE1α -XBP1 axis for development, survival in mucosal tissues and cross-presentation of antigens to CD8+ T cells, which may be of relevance in infections with neurotropic viruses (2, 102). In addition, cDC1s are highly sensitive to perturbations in XBP1 signaling and counter activate RIDD upon XBP1 loss (95, 101). The implication of RIDD and XBP1s signaling in DC subtypes in the CNS has not been explored so far but relevant aspects downstream of XBP1s and RIDD may encompass cytokine production upon recognition of protein aggregates, cell survival and cross-presentation of antigens to CD8+ T cells.
Lymphocytes
T and B cells survey the steady-state CNS exerting a neuroprotective role, but can become pathogenic under unresolved inflammation (57, 103–106). T cell numbers have been found to be increased in AD, PD, ALS and MS, and to contribute both to inflammation and neuronal dysfunction as well as to deferring inflammatory responses leading to nerodegeneration (107, 108). The immune response elicited by these cells in the CNS depends on their functional phenotype, although observations regarding cell number and T cell subset involved varies between different disease types and model of study (108–113). UPR activation in T cells is not completely elucidated, however the IRE1α-XBP1s branch has shown to regulate cell differentiation and cytokine production in CD8+ and CD4+ T cells under infection and chronic ER stress (114–118). During neuroinflammation and aging, B cells play a pathogenic role by producing pro-inflammatory cytokines, promoting effector T cells and activating macrophages via Fc receptors (62, 119–123). B cell development, activation and differentiation is critically regulated by IRE1α-XBP1s and ATF6, whilst absence of PERK favors plasma cell differentiation and immunoglobulin synthesis (124–128).
Overall, as proposed on Figure 2, activation of UPR components could occur in CNS-residing and infiltrating immune cells upon PRR recognition of protein aggregates, or due to noxious threats. The IRE1α-XBP1s axis has a key role in immune cell development from hematopoietic progenitors, cell survival and effector function, and it could be activated by NAMPs through PRR signaling in microglia, macrophages or dendritic cells, inducing cell maturation and activation (66, 68, 88, 97). The PERK pathway in contrast, is mostly deactivated to allow immune cells to fulfill their function under different inflammatory settings without going through apoptosis. In AD or PD however, sustained stimulation triggered by amyloid β or α-synuclein aggregates could lead to a dysfunctional activated phenotype associated to defective clearance and increased production of inflammatory mediators. This process could, in turn, attract more immune cells that exert a neurotoxic effect, promoting the accumulation of more protein aggregates, axonal destruction and neuronal malfunction (129, 130). Under this chronic ER stress, UPR signaling would be expected to be highly activated in CNS-related immune cells, in line with observations in brain samples of patients. Nevertheless, it remains to be addressed whether the UPR output in CNS-associated immune cells proves to be beneficial or detrimental for the development of PMDs, as is the case of neurons and astrocytes (131, 132).
Concluding Remarks
The interplay between the UPR, the immune system and the CNS in neurodegenerative diseases remains in its early stages. Intensive research will be required to accurately understand the role of ER stress in the immune-related aspects of CNS pathology and to determine whether UPR signaling in immune cells answers to a homeostatic or a terminal fate. It is also important to keep in mind the potential differences between human and mice immune cell types, since most knowledge gained in this matter emerges from studies in murine models. Through our knowledge on the UPR role in peripheral immunity and neurodegeneration models, better access to human samples and the advent of novel analytic tools for identification of the diversity of cell lineages, the cell-specific contribution of the UPR to neural and CNS-associated immune cells will begin to be elucidated, generating valuable knowledge that may provide therapeutic opportunities.
Author Contributions
All authors read and approved the final version of the manuscript. PG-G and FC-M contributed equally to the work. PG-G, FO, FC-M, and CH participated in manuscript conception and design.
Funding
This work was primary funded by FONDECYT 1161212 and an International Research Scholar grant from the Howard Hughes Medical Institute # 55008744 (FO); and by FONDECYT 1140549, FONDAP program 15150012, Millennium Institute P09-015-F, European Commission R&D MSCA-RISE 734749 (CH). We thank the support from Michael J Fox Foundation for Parkinson's Research–Target Validation grant 9277, FONDEF ID16I10223, FONDEF D11E1007, US Office of Naval Research-Global N62909-16-1-2003, U.S. Air Force Office of Scientific Research FA9550-16-1-0384, ALSRP Therapeutic Idea Award AL150111, Muscular Dystrophy Association 382453, and CONICYT-Brazil 441921/2016-7 (CH), We also thank FONDECYT for postdoctoral fellowships 3180195 and from FONDAP program 15150012 (FC-M).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the laboratory of immunology and cellular stress for helpful discussions.
References
1. Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science (2008) 319:916–9. doi: 10.1126/science.1141448
2. Tavernier SJ, Lambrecht BN, Janssens S. The unfolded protein response in the immune cell development: putting the caretaker in the driving seat. Curr Top Microbiol Immunol. (2018) 414:45–72. doi: 10.1007/82_2017_1
3. Taylor RC, Berendzen KM, Dillin A. Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat Rev Mol Cell Biol.(2014) 15:211. doi: 10.1038/nrm3752
4. Cabral-Miranda F, Hetz C. ER Stress and neurodegenerative disease: a cause or effect relationship? Curr Top Microbiol Immunol. (2018) 414:131–57. doi: 10.1007/82_2017_52
5. Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. (2003) 4:49–60. doi: 10.1038/nrn1007
6. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol. Cell (2018) 69:169–81. doi: 10.1016/j.molcel.2017.06.017
7. Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. (2015) 17:829–38. doi: 10.1038/ncb3221
8. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (2011) 334:1081–6. doi: 10.1126/science.1209038
9. Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, et al. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. (2013) 3:1279–92. doi: 10.1016/j.celrep.2013.03.024
10. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. (2003) 23:7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003
11. Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. (2014) 39:245–54. doi: 10.1016/j.tibs.2014.02.008
12. Coelho DS, Domingos PM. Physiological roles of regulated Ire1 dependent decay. Front Genet. (2014) 5:76. doi: 10.3389/fgene.2014.00076
13. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. (2015) 10:173–94. doi: 10.1146/annurev-pathol-012513-104649
14. Kranz P, Neumann F, Wolf A, Classen F, Pompsch M, Ocklenburg T, et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. (2017) 8:e2986. doi: 10.1038/cddis.2017.369
15. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell (2007) 13:365–76. doi: 10.1016/j.devcel.2007.07.018
16. Hillary RF, FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. J Biomed Sci. (2018) 25:48. doi: 10.1186/s12929-018-0453-1
17. Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell (2009) 35:551–61. doi: 10.1016/j.molcel.2009.08.021
18. Martínez G, Vidal RL, Mardones P, Serrano FG, Ardiles AO, Wirth C, et al. Regulation of memory formation by the transcription factor XBP1. Cell Rep. (2016) 14:1382–94. doi: 10.1016/j.celrep.2016.01.028
19. Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, et al. eIF2α phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell (2007) 129:195–206. doi: 10.1016/j.cell.2007.01.050
20. Martínez G, Khatiwada S, Costa-Mattioli M, Hetz C. ER proteostasis control of neuronal physiology and synaptic function. Trends Neurosci. (2018) 41:610–24. doi: 10.1016/j.tins.2018.05.009
21. Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. (2017) 13:477–91. doi: 10.1038/nrneurol.2017.99
22. Scheper W, Hoozemans JJ. The unfolded protein response in neurodegenerative diseases: a neuropathological perspective. Acta Neuropathol. (2015) 130:315–31. doi: 10.1007/s00401-015-1462-8
23. Smith HL, Mallucci GR. The unfolded protein response: mechanisms and therapy of neurodegeneration. Brain (2016) 139(Pt 8):2113–21. doi: 10.1093/brain/aww101
24. Hayashi A, Kasahara T, Iwamoto K, Ishiwata M, Kametani M, Kakiuchi C, et al. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem. (2007) 282:34525–34. doi: 10.1074/jbc.M704300200
25. Hayashi A, Kasahara T, Kametani M, Kato T. Attenuated BDNF-induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem Biophys Res Commun. (2008) 376:758–63. doi: 10.1016/j.bbrc.2008.09.059
26. Saito A, Cai L, Matsuhisa K, Ohtake Y, Kaneko M, Kanemoto S, et al. Neuronal activity-dependent local activation of dendritic unfolded protein response promotes expression of brain-derived neurotrophic factor in cell soma. J Neurochem. (2018) 144:35–49. doi: 10.1111/jnc.14221
27. Cissé M, Duplan E, Lorivel T, Dunys J, Bauer C, Meckler X, et al. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol Psychiatry (2017) 22:1562–75. doi: 10.1038/mp.2016.152
28. Hetz C, Mollereau B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci. (2014) 15:233–49. doi: 10.1038/nrn3689
29. Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, et al. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. (2005) 110:165–72. doi: 10.1007/s00401-005-1038-0
30. Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W, et al. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. (2007) 354:707–11. doi: 10.1016/j.bbrc.2007.01.043
31. Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles ÁO, Medinas DB, Salazar C, et al. IRE1 signaling exacerbates Alzheimer's disease pathogenesis. Acta Neuropathol. (2017) 134:489–506. doi: 10.1007/s00401-017-1694-x
32. Mercado G, Castillo V, Soto P, López N, Axten JM, Sardi SP, et al. Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson's disease. Neurobiol Dis. (2018) 112:136–48. doi: 10.1016/j.nbd.2018.01.004
33. Medinas DB, Valenzuela V, Hetz C. Proteostasis disturbance in amyotrophic lateral sclerosis. Hum Mol Genet. (2017) 26:R91–104. doi: 10.1093/hmg/ddx274
34. Ilieva EV, Ayala V, Jové M, Dalfó E, Cacabelos D, Povedano M, et al. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain (2007) 130(Pt 12):3111–23. doi: 10.1093/brain/awm190
35. Hetz C, Thielen P, Matus S, Nassif M, Court F, Kiffin R, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. (2009) 23:2294–306. doi: 10.1101/gad.1830709
36. Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. (2008) 30:400–7. doi: 10.1016/j.nbd.2008.02.009
37. Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Targeting the UPR transcription factor XBP1 protects against Huntington's disease through the regulation of FoxO1 and autophagy. Hum Mol Genet. (2012) 21:2245–62. doi: 10.1093/hmg/dds040
38. Fernandez-Fernandez MR, Ferrer I, Lucas JJ. Impaired ATF6alpha processing, decreased Rheb and neuronal cell cycle re-entry in Huntington's disease. Neurobiol Dis. (2011) 41:23–32. doi: 10.1016/j.nbd.2010.08.014
39. Medinas DB, Rozas P, Martínez Traub F, Woehlbier U, Brown RH, Bosco DA, et al. Endoplasmic reticulum stress leads to accumulation of wild-type SOD1 aggregates associated with sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. (2018) 115:8209–14 doi: 10.1073/pnas.1801109115
40. Remondelli P, Renna M. The endoplasmic reticulum unfolded protein response in neurodegenerative disorders and its potential therapeutic significance. Front Mol Neurosci. (2017) 10:187. doi: 10.3389/fnmol.2017.00187
41. Valdés P, Mercado G, Vidal RL, Molina C, Parsons G, Court FA, et al. Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci USA. (2014) 111:6804–9. doi: 10.1073/pnas.1321845111
42. Hetz C, Lee AH, Gonzalez-Romero D, Thielen P, Castilla J, Soto C et al. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc Natl Acad Sci USA. (2008) 105:757–62. doi: 10.1073/pnas.0711094105
43. Valenzuela V, Jackson KL, Sardi SP, Hetz C. Gene therapy strategies to restore ER proteostasis in disease. Mol Ther.(2018) 26:1404–13 doi: 10.1016/j.ymthe.2018.04.004
44. Oñate M, Catenaccio A, Martínez G, Armentano D, Parsons G, Kerr B, et al. Activation of the unfolded protein response promotes axonal regeneration after peripheral nerve injury. Sci Rep. (2016) 6:21709. doi: 10.1038/srep21709
45. Zuleta A, Vidal RL, Armentano D, Parsons G, Hetz C. AAV-mediated delivery of the transcription factor XBP1s into the striatum reduces mutant Huntingtin aggregation in a mouse model of Huntington's disease. Biochem Biophys Res Commun. (2012) 420:558–63. doi: 10.1016/j.bbrc.2012.03.033
46. Sado M, Yamasaki Y, Iwanaga T, Onaka Y, Ibuki T, Nishihara S, et al. Protective effect against Parkinson's disease-related insults through the activation of XBP1. Brain Res. (2009) 1257:16–24. doi: 10.1016/j.brainres.2008.11.104
47. Clayton BLL, Popko B. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res. (2016) 1648(Pt B):594–602. doi: 10.1016/j.brainres.2016.03.046.
48. Hughes D, Mallucci GR. The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. Febs J. (2018) doi: 10.1111/febs.14422. [Epub ahead of print].
49. Valenzuela V, Collyer E, Armentano D, Parsons GB, Court FA, Hetz C. Activation of the unfolded protein response enhances motor recovery after spinal cord injury. Cell Death Dis. (2012) 3:e272. doi: 10.1038/cddis.2012.8
50. Ma T, Trinh MA, Wexler AJ, Bourbon C, Gatti E, Pierre P, et al. Suppression of eIF2α kinases alleviates AD-related synaptic plasticity and spatial memory deficits. Nat Neurosci. (2013) 16:1299–305. doi: 10.1038/nn.3486
51. Halliday M, Hughes D, Mallucci GR. Fine-tuning PERK signaling for neuroprotection. J Neurochem. (2017) 142:812–26. doi: 10.1111/jnc.14112
52. Matus S, Lopez E, Valenzuela V, Nassif M, Hetz C. Functional contribution of the transcription factor ATF4 to the pathogenesis of amyotrophic lateral sclerosis. PLoS ONE (2013) 8:e66672. doi: 10.1371/journal.pone.0066672
53. Silva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, et al. CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem. (2005) 95:974–86. doi: 10.1111/j.1471-4159.2005.03428.x
54. Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med (2013) 5:206ra138. doi: 10.1126/scitranslmed.3006767
55. Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, et al. The endoplasmic reticulum stress sensor, ATF6alpha, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem. (2011) 286:7947–57. doi: 10.1074/jbc.M110.156430
56. Hashida K, Kitao Y, Sudo H, Awa Y, Maeda S, Mori K, et al. ATF6alpha promotes astroglial activation and neuronal survival in a chronic mouse model of Parkinson's Disease. PLOS ONE (2012) 7:e47950. doi: 10.1371/journal.pone.0047950
57. Tanabe S, Yamashita T. The role of immune cells in brain development and neurodevelopmental diseases. Int Immunol. (2018) 30:437–44 doi: 10.1093/intimm/dxy041
58. Schwartz M, Kipnis J, Rivest S, Prat A. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci. (2013) 33:17587–96. doi: 10.1523/JNEUROSCI.3241-13.2013
59. Medzhitov R. Origin and physiological roles of inflammation. Nature (2008) 454:428–35. doi: 10.1038/nature07201
60. Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. (2015) 15:545–58. doi: 10.1038/nri3871
61. Kempuraj D, Thangavel R, Selvakumar GP, Zaheer S, Ahmed ME, Raikwar SP, et al. Brain and peripheral atypical inflammatory mediators potentiate neuroinflammation and neurodegeneration. Front Cell Neurosci. (2017) 11:216. doi: 10.3389/fncel.2017.00216
62. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity (2018) 48:599. doi: 10.1016/j.immuni.2018.02.014
63. Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. (2018) 69:437–49. doi: 10.1146/annurev-med-050715-104343
64. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. (2014) 14:463–77. doi: 10.1038/nri3705
65. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol. (2017) 17:49–59. doi: 10.1038/nri.2016.123
66. Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell (2018) 173:1073–81. doi: 10.1016/j.cell.2018.05.003
67. El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to β-Amyloid. J Exp Med. (2003) 197:1657–66. doi: 10.1084/jem.20021546
68. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. (2010) 11:411–8. doi: 10.1038/ni.1857
69. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. STING Senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell (2017) 171:809–23.e13. doi: 10.1016/j.cell.2017.09.034
70. Beisel C, Ziegler S, Martrus Zapater G, Chapel A, Griesbeck M, Hildebrandt H, et al. TLR7-mediated activation of XBP1 correlates with the IFNalpha production in humans. Cytokine (2017) 94:55–8. doi: 10.1016/j.cyto.2017.04.006
71. Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, et al. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol Cell Biol. (2014) 34:3911–25. doi: 10.1128/MCB.00980-14
72. Godin JD, Creppe C, Laguesse S, Nguyen L. emerging roles for the unfolded protein response in the developing nervous system. Trends Neurosci. (2016) 39:394–404. doi: 10.1016/j.tins.2016.04.002
73. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55:453–62. doi: 10.1002/glia.20467
74. Bellezza I, Grottelli S, Mierla AL, Cacciatore I, Fornasari E, Roscini L, et al. Neuroinflammation and endoplasmic reticulum stress are coregulated by cyclo(His-Pro) to prevent LPS neurotoxicity. Int J Biochem Cell Biol. (2014) 51:159–69. doi: 10.1016/j.biocel.2014.03.023
75. Ho HJ, Huang DY, Ho FM, Lee LT, Lin WW. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase expression by endoplasmic reticulum stress. Cell Signal. (2012) 24:2166–78. doi: 10.1016/j.cellsig.2012.07.018
76. Noelker C, Morel L, Lescot T, Osterloh A, Alvarez-Fischer D, Breloer M, et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci Rep. (2013) 3:1393. doi: 10.1038/srep01393
77. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A., et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature (2013) 493:674–8. doi: 10.1038/nature11729
78. Lerner AG, Upton JP, Praveen PV, Ghosh R, Nakagawa Y, Igbaria A, et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. (2012) 16:250–64. doi: 10.1016/j.cmet.2012.07.007
79. Kim S, Joe Y, Kim HJ, Kim YS, Jeong SO, Pae HO, et al. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J Immunol. (2015) 194:4498–506. doi: 10.4049/jimmunol.1401399
80. Tam AB, Mercado EL, Hoffmann A, Niwa M. ER stress activates NF-kappaB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE (2012) 7:e45078. doi: 10.1371/journal.pone.0045078
81. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (2000) 287:664–6. doi: 10.1126/science.287.5453.664
82. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell (2006) 124:587–99. doi: 10.1016/j.cell.2005.11.040
83. Hosoi T, Honda M, Oba T, Ozawa K. ER stress upregulated PGE/IFNgamma-induced IL-6 expression and down-regulated iNOS expression in glial cells. Sci Rep. (2013) 3:3388. doi: 10.1038/srep03388
84. Lin W, Harding HP, Ron D, Popko B. Endoplasmic reticulum stress modulates the response of myelinating oligodendrocytes to the immune cytokine interferon-gamma. J Cell Biol. (2005) 169:603–12. doi: 10.1083/jcb.200502086
85. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
86. Guthrie LN, Abiraman K, Plyler ES, Sprenkle NT, Gibson SA, McFarland BC, et al. Attenuation of PKR-like ER Kinase (PERK) signaling selectively controls endoplasmic reticulum stress-induced inflammation without compromising immunological responses. J Biol Chem. (2016) 291:15830–40. doi: 10.1074/jbc.M116.738021
87. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J., et al. Myeloid cells in the central nervous system. Immunity, (2017) 46:943–56. doi: 10.1016/j.immuni.2017.06.007
88. Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. (2017) 35:441–68. doi: 10.1146/annurev-immunol-051116-052358
89. Vezzani A, Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology (2015) 96(Pt A):70–82. doi: 10.1016/j.neuropharm.2014.10.027
90. Ta HM, Le TM, Ishii H, Takarada-Iemata M, Hattori T, Hashida K, et al. Atf6alpha deficiency suppresses microglial activation and ameliorates pathology of experimental autoimmune encephalomyelitis. J Neurochem. (2016) 139:1124–37. doi: 10.1111/jnc.13714
91. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's Disease. Cell (2017) 169:1276–90 e17. doi: 10.1016/j.cell.2017.05.018
92. Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's Disease. Cell (2017) 170:649–63.e13. doi: 10.1016/j.cell.2017.07.023
93. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity (2017) 47:566–81.e9. doi: 10.1016/j.immuni.2017.08.008
94. Goldmann T, Wieghofer P, Jordão MJ, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol. (2016) 17:797–805. doi: 10.1038/ni.3423
95. Osorio F, Tavernier SJ, Hoffmann E, Saeys Y, Martens L, Vetters J, et al. The unfolded-protein-response sensor IRE-1alpha regulates the function of CD8alpha+ dendritic cells. Nat Immunol. (2014) 15:248–57. doi: 10.1038/ni.2808
96. Anandasabapathy N, Victora GD, Meredith M, Feder R, Dong B, Kluger C, et al. Flt3L controls the development of radiosensitive dendritic cells in the meninges and choroid plexus of the steady-state mouse brain. J Exp Med. (2011) 208:1695–705. doi: 10.1084/jem.20102657
97. Bossù P, Spalletta G, Caltagirone C, Ciaramella A. Myeloid dendritic cells are potential players in human neurodegenerative diseases. Front Immunol. (2015) 6:632. doi: 10.3389/fimmu.2015.00632
98. Fischer HG, Reichmann G. Brain dendritic cells and macrophages/microglia in central nervous system inflammation. J Immunol. (2001) 166:2717–26. doi: 10.4049/jimmunol.166.4.2717
99. Pashenkov M, Huang YM, Kostulas V, Haglund M, Söderström M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain (2001) 124(Pt 3):480–92. doi: 10.1093/brain/124.3.480
100. Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J Exp Med. (2007) 204:2267–75. doi: 10.1084/jem.20070525
101. Tavernier SJ, Osorio F, Vandersarren L, Vetters J, Vanlangenakker N, Van Isterdael G, et al. Regulated IRE1-dependent mRNA decay sets the threshold for dendritic cell survival. Nat Cell Biol. (2017) 19:698–710. doi: 10.1038/ncb3518
102. Osorio F, Lambrecht BN, Janssens S. Antigen presentation unfolded: identifying convergence points between the UPR and antigen presentation pathways. Curr Opin Immunol. (2018) 52:100–7. doi: 10.1016/j.coi.2018.04.020
103. Ellwardt E, Walsh JT, Kipnis J, Zipp F. Understanding the role of T cells in CNS homeostasis. Trends Immunol. (2016) 37:154–65. doi: 10.1016/j.it.2015.12.008
104. Kurnellas MP, Ghosn EE, Schartner JM, Baker J, Rothbard JJ, Negrin RS, et al. Amyloid fibrils activate B-1a lymphocytes to ameliorate inflammatory brain disease. Proc Natl Acad Sci USA. (2015) 112:15016–23. doi: 10.1073/pnas.1521206112
105. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity (2014) 41:1040–51. doi: 10.1016/j.immuni.2014.10.016
106. Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. (2017) 17:179–94. doi: 10.1038/nri.2016.144
107. Bryson KJ, Lynch MA. Linking T cells to Alzheimer's disease: from neurodegeneration to neurorepair. Curr Opin Pharmacol. (2016) 26:67–73. doi: 10.1016/j.coph.2015.10.003
108. González H, Pacheco R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J Neuroinflamm. (2014) 11:201. doi: 10.1186/s12974-014-0201-8
109. Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H, et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity (2013) 39:949–62. doi: 10.1016/j.immuni.2013.10.016
110. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4(+) lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest. (2009) 119:182–92. doi: 10.1172/JCI36470
111. Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, et al. Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest. (2003) 112:415–22. doi: 10.1172/JCI200318104
112. Saresella M, Piancone F, Tortorella P, Marventano I, Gatti A, Caputo D, et al. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin Immunol. (2013) 148:79–88. doi: 10.1016/j.clim.2013.04.010
113. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson's disease recognize alpha-synuclein peptides. Nature (2017) 546:656–61. doi: 10.1038/nature22815
114. Kamimura D, Bevan MJ. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J Immunol. (2008) 181:5433–41. doi: 10.4049/jimmunol.181.8.5433
115. Kemp KL, Lin Z, Zhao F, Gao B, Song J, Zhang K, et al. The serine-threonine kinase inositol-requiring enzyme 1alpha (IRE1alpha) promotes IL-4 production in T helper cells. J Biol Chem. (2013) 288:33272–82. doi: 10.1074/jbc.M113.493171
116. Brunsing R, Omori SA, Weber F, Bicknell A, Friend L, Rickert R, et al. B- and T-cell development both involve activity of the unfolded protein response pathway. J Biol Chem. (2008) 283:17954–61. doi: 10.1074/jbc.M801395200
117. Omar I, Lapenna A, Cohen-Daniel L, Tirosh B, Berger M. Schlafen2 mutation unravels a role for chronic ER stress in the loss of T cell quiescence. Oncotarget (2016) 7:39396–407. doi: 10.18632/oncotarget.9818
118. Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. (2006) 7:644–51. doi: 10.1038/ni1338
119. Cepok S, von Geldern G, Grummel V, Hochgesand S, Celik H, Hartung H, et al. Accumulation of class switched IgD-IgM- memory B cells in the cerebrospinal fluid during neuroinflammation. J Neuroimmunol. (2006) 180:33–9. doi: 10.1016/j.jneuroim.2006.06.031
120. Sudduth TL, Greenstein A, Wilcock DM. Intracranial injection of gammagard, a human IVIg, modulates the inflammatory response of the brain and lowers Aβ in APP/PS1 mice along a different time course than anti-Aβ antibodies. J Neurosci. (2013) 33:9684–92. doi: 10.1523/JNEUROSCI.1220-13.2013
121. Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer's Disease. Front Neurosci. (2014) 8:235. doi: 10.3389/fnins.2014.00235
122. Peress NS, Fleit HB, Perillo E, Kuljis R, Pezzullo C. Identification of Fc gamma RI, II and III on normal human brain ramified microglia and on microglia in senile plaques in Alzheimer's disease. J Neuroimmunol. (1993) 48:71–9. doi: 10.1016/0165-5728(93)90060-C
123. Biragyn A, Aliseychik M, Rogaev E. Potential importance of B cells in aging and aging-associated neurodegenerative diseases. Semin Immunopathol. (2017) 39:283–94. doi: 10.1007/s00281-016-0615-8
124. Kharabi Masouleh B, Geng H, Hurtz C, Chan LN, Logan AC, Chang MS, et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc Natl Acad Sci USA. (2014) 111:E2219–28. doi: 10.1073/pnas.1400958111
125. Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis. J Clin Invest. (2005) 115:268–81. doi: 10.1172/JCI200521848
126. Gass JN, Jiang HY, Wek RC, Brewer JW. The unfolded protein response of B-lymphocytes: PERK-independent development of antibody-secreting cells. Mol Immunol. (2008) 45:1035–43. doi: 10.1016/j.molimm.2007.07.029
127. Hu CC, Dougan SK, McGehee AM, Love JC, Ploegh HL. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. (2009) 28:1624–36. doi: 10.1038/emboj.2009.117
128. Gass JN, Gifford NM, Brewer JW. Activation of an unfolded protein response during differentiation of antibody-secreting B cells. J Biol Chem. (2002) 277:49047–54. doi: 10.1074/jbc.M205011200
129. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science (2013) 339:156–61. doi: 10.1126/science.1227901
130. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. (2015) 16:358–72. doi: 10.1038/nrn3880
131. Jiang P, Gan M, Ebrahim AS, Lin WL, Melrose HL, Yen SH. ER stress response plays an important role in aggregation of alpha-synuclein. Mol Neurodegener. (2010) 5:56. doi: 10.1186/1750-1326-5-56
Keywords: UPR, neurodegeneration, immune system, inflammation, protein protein misfolding diseases, ER stress, immune cells, misfolded proteins
Citation: García-González P, Cabral-Miranda F, Hetz C and Osorio F (2018) Interplay Between the Unfolded Protein Response and Immune Function in the Development of Neurodegenerative Diseases. Front. Immunol. 9:2541. doi: 10.3389/fimmu.2018.02541
Received: 05 September 2018; Accepted: 15 October 2018;
Published: 02 November 2018.
Edited by:
Marco Cosentino, Università degli Studi dell'Insubria, ItalyCopyright © 2018 García-González, Cabral-Miranda, Hetz and Osorio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudio Hetz, Y2hldHpAbWVkLnVjaGlsZS5jbA==
Fabiola Osorio, ZmFiaW9sYW9zb3Jpb0BtZWQudWNoaWxlLmNs
†These authors have contributed equally to this work