 Wolfgang Schwinger1Christian Urban1
Wolfgang Schwinger1Christian Urban1 Raphael Ulreich2Daniela Sperl1Anna Karastaneva1
Raphael Ulreich2Daniela Sperl1Anna Karastaneva1 Volker Strenger1Herwig Lackner1
Volker Strenger1Herwig Lackner1 Kaan Boztug3,4,5,6Michael H. Albert7Martin Benesch1
Kaan Boztug3,4,5,6Michael H. Albert7Martin Benesch1 Markus G. Seidel1,8*
Markus G. Seidel1,8*- 1Division of Pediatric Hematology-Oncology, Department of Pediatrics and Adolescent Medicine, Medical University Graz, Graz, Austria
- 2Pediatric Intensive Care Unit, Division of General Pediatrics, Department of Pediatrics and Adolescent Medicine, Medical University Graz, Graz, Austria
- 3Ludwig Boltzmann Institute for Rare and Undiagnosed Diseases, Vienna, Austria
- 4CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria
- 5Department of Pediatrics and Adolescent Medicine, Medical University of Vienna, Vienna, Austria
- 6Department of Pediatrics, St. Anna Kinderspital and Children's Cancer Research Institute, Medical University of Vienna, Vienna, Austria
- 7Dr. von Hauner University Children's Hospital, Ludwig Maximilian Universität, Munich, Germany
- 8Research Unit for Pediatric Hematology and Immunology, Medical University Graz, Graz, Austria
Early diagnosis of primary immunodeficiency disorders (PID) is vital and allows directed treatment, especially in syndromes with severe or profound combined immunodeficiency. In PID patients with perinatal CMV or other opportunistic, invasive infections (e.g., Pneumocystis or Aspergillus), multi-organ morbidity may already arise within the first months of life, before hematopoietic stem cell transplantation (HSCT) or gene therapy can be undertaken, compromising the definitive treatment and outcome. Deficiency of Wiskott-Aldrich syndrome (WAS) protein-interacting protein (WIP deficiency) causes an autosomal recessive, WAS-like syndrome with early-onset combined immunodeficiency that has been described in three pedigrees to date. While WAS typically includes combined immunodeficiency, microthrombocytopenia, and eczema, the clinical and laboratory phenotypes of WIP-deficient patients–including lymphocyte subsets, platelets, lymphocyte proliferation in vitro, and IgE—varied widely and did not entirely recapitulate WAS, impeding early diagnosis in the reported patients. To elucidate the phenotype of WIP deficiency, we provide a comprehensive synopsis of clinical and laboratory features of all hitherto-described patients (n = 6) and WIP negative mice. Furthermore, we summarize the treatment modalities and outcomes of these patients and review in detail the course of one of them who was successfully treated with serial, unconditioned, maternal, HLA-identical donor lymphocyte infusions (DLI) against life-threatening, invasive CMV infection, followed by a TCRαβ/CD19-depleted, treosulfan/melphalan-conditioned, peripheral blood HSCT and repetitive, secondary-prophylactic, CMV-specific DLI with 1-year post-HSCT follow-up. This strategy could be useful in other patients with substantial premorbidity, considered “too bad to transplant,” who have an HLA-identical family donor, to eliminate infections and bridge until definitive treatment.
Introduction
Wiskott-Aldrich syndrome (WAS) is an X-linked, classical combined immunodeficiency with syndromic features, defined by a clinical trias of immunodeficiency, eczema, and microthrombocytopenia that bears a risk of autoimmunity and lymphoma (1–5). Allogeneic hematopoietic stem cell transplantation (HSCT) is the treatment of choice, if a suitable donor is available. Deficiency of WAS protein-interacting protein (WIP) is a recessive disorder that leads to premature degradation of WAS protein (WASP) and thus highly resembles WAS. However, the clinical presentation and courses of previously reported patients with WIP deficiency were more heterogeneous than those of WAS patients (6–8), especially with regards to very early onset of severe, life-threatening immunodeficiency. The conditioning pretreatment before HSCT of patients with WAS and other profound combined immunodeficiencies includes strongly immunosuppressive chemotherapy, and the outcome depends on the degree of preexisting and acquired invasive infections until engraftment enables the generation of an intact immune system.
Functional Aspects of WIP or WASP Deficiency
WASP is present in the cytoplasm of hematopoietic cell lineages in an auto-inhibited, inactive form that is stabilized in a chaperone-like fashion by WIP binding (9). Upon activation by cell division cycle 2 (CDC42), a Rho family GTPase, activated by a variety of transmembrane receptors, or by proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1), and phosphatidylinositol-4,5-biphosphate (PIP2), the inhibitory, intramolecular, interaction is released, and actin polymerization is initiated [reviewed in Cotta-de-Almeida et al. (10)]. Besides the WASP-stabilizing function of WIP, it was shown to facilitate WASP activation by recruiting WASP to the immunological synapse upon activation of the T cell receptor and by enhancing CDC42-dependent activation (11). Actin polymerization is necessary for the immunological synapse formation, its stability, intracellular signaling, cytokine secretion, cellular migration, and lytic granule polarization, but there are also other actin-independent effects of WASP such as activity as transcription factor for T-bet and as anti-apoptotic survival factor (10). In addition to the T cell pathology notoriously associated with WAS, many of these functional defects also affect B cells [such as migration toward a chemokine gradient (8)] and NK cells (12, 13), and there is evidence from murine studies, that WIP has WASP-independent effects on B cell function, being a regulator of CD19 activation and PI3K/Akt signaling in B cells (14). Active WASP is stabilized by tyrosine phosphorylation, and its activity is counter-balanced by ubiquitin-dependent proteasomal degradation [reviewed in Cotta-de-Almeida et al. (10)].
Synopsis of Six WIP-Deficient Patients From Three Pedigrees
The first description of a WIP-deficient patient was from a girl from Morocco in 2012, who had been diagnosed and successfully transplanted with cord-blood-derived stem cells in Brescia, Italy, at 4.5 months and followed-up at that time for 16 months (6) (Table 1, pedigree A). Her appearance largely resembled WAS, but at that time, except for rare cases of imbalanced X chromosomal inactivation (15, 16), WAS in females or a recessive form was unknown or genetically unresolved (17). Results of immunological analyses, including the T and NK cell functions and an absence of WAS-protein despite normal WAS DNA and mRNA sequences, lead physicians to suspect degradation of WAS-protein due to WIP deficiency (6). Four years later, a large, branched family in Saudi Arabia was reported in which four (of 12 affected) individuals with WIP deficiency were described (7) (Table 1, pedigree B). Of these four patients, two underwent HSCT from a matched sibling and two from unrelated cord blood; one patient died shortly after HSCT from CMV-mediated, respiratory distress syndrome (7). The platelet phenotype varied inter-individually between normal-sized or microplatelets and moderate to severe thrombocytopenia (Table 1). Similarly, skin lesions varied, but an increased risk of infections, chronic diarrhea, and respiratory symptoms were constantly observed. The diagnosis of these patients was established by panel sequencing. The sixth patient was described in a report on the effect of WIP deficiency on the actin cytoskeleton and functional lymphocyte architecture recently (8); and a detailed clinical description with longer follow-up of this patient is provided below and in Table 1 (pedigree C). All hitherto-described patients displayed an extremely early onset of symptoms of primary immunodeficiency, but varying syndromic and laboratory features (Table 1).
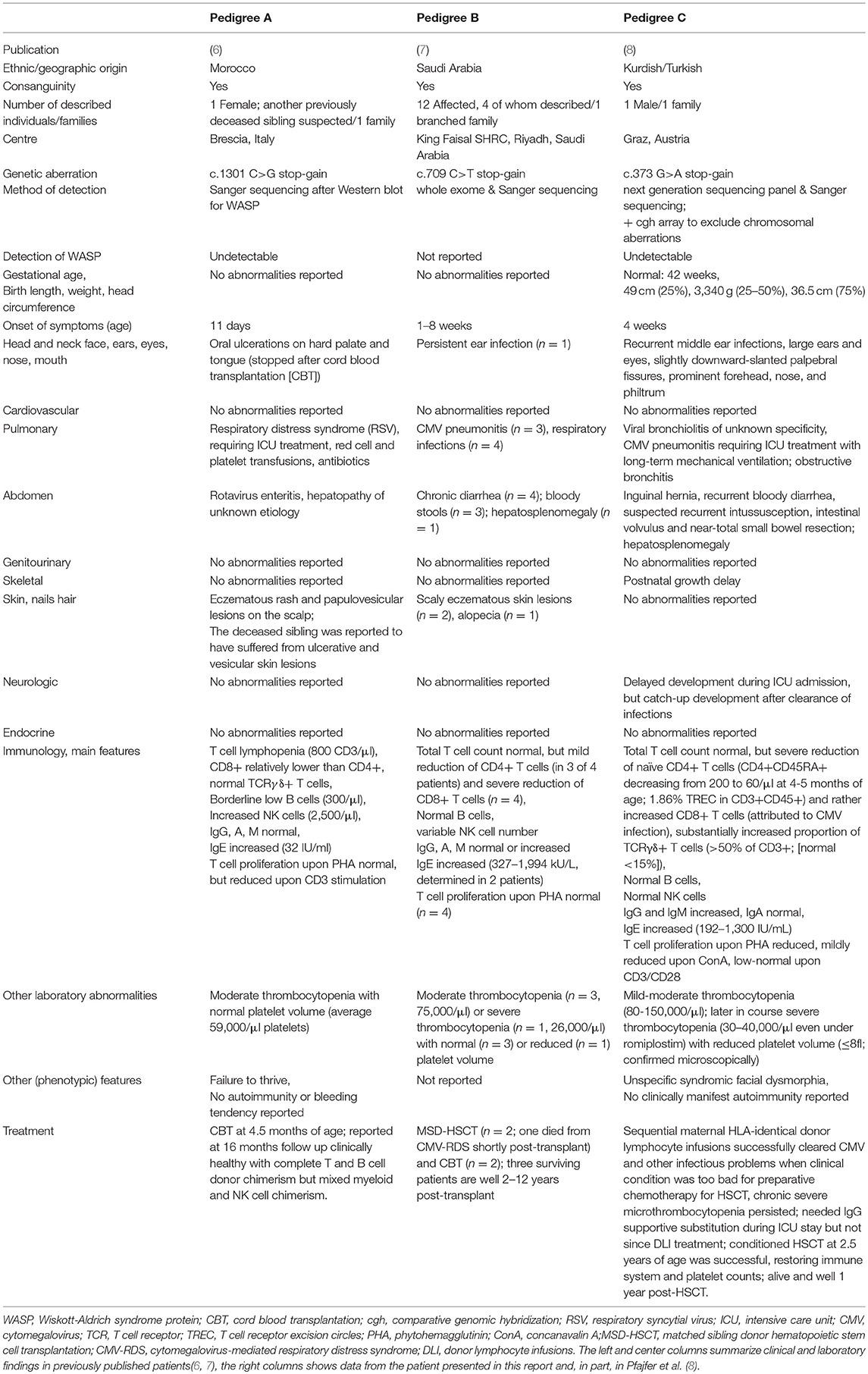
Table 1. Clinical synopsis of WIP deficiency.
As described briefly previously, the sixth WIP-deficient patient in this cohort (pedigree C, Table 1) became symptomatic with viral bronchiolitis 4 weeks after birth, required hospital admission and mechanical ventilation for pneumonitis at 3 months, due to a systemic CMV infection (detectable in plasma, urine, stool, saliva, bronchial secretion, cerebrospinal fluid) (8). Immunoglobulin-G was needed to be substituted intravenously. Platelet concentrates were given sporadically for intermittent bloody diarrhea with progressive thrombocytopenia (Table 1; and Figure 1), suggesting the presence of a relapsing intussusception, inflammatory bowel disease, or both. Endoscopy was performed at 6 months of age and biopsies showed signs of chronic erosive focal duodenitis (similar to infection but without detection of CMV or Cryptosporidium), further, a chronic, active gastritis and duodenitis (resembling reflux-association) were found. At that time, the colon was normal, and CMV, HSV, or other microorganisms could not be detected in situ. At 9 months of age, he had another endoscopy which showed active duodenitis with CMV-positive stroma cells and mild colitis with lymphoplasmacellular and patchy neutrophil infiltrations, but no microorganisms or viruses detectable despite CMV nucleic acid positivity in many body fluids at earlier time points. An acute clinical deterioration was observed at 11 months of age accompanied by clinical signs of intussusception. Emergency surgery was performed and necrotic bowel was resected, showing perforations, erosions, and ulcerations, as well as peritonitis. The mucosa could not be evaluated due to necrosis, the surgery yielded a severe short bowel syndrome. His psychomotor development was found to be impaired, despite displaying no signs of encephalitis (imaging; EEG; clinical) or meningitis (CSF; imaging). However, after 25 days of invasive and 94 days of nasal CPAP ventilation, his neurological status was difficult to assess.
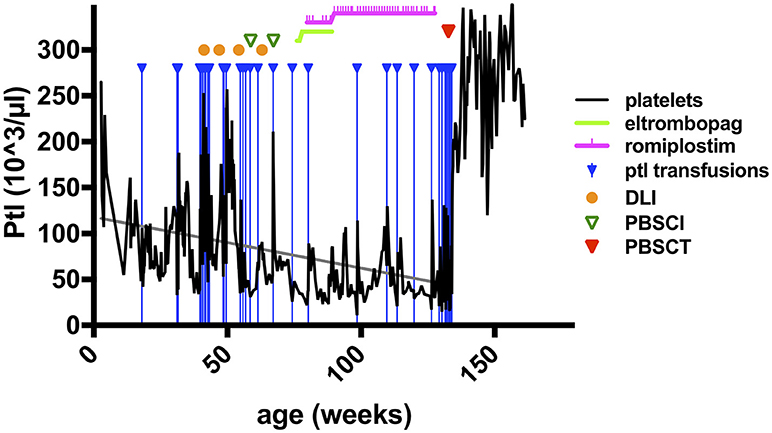
Figure 1. Platelet counts and therapeutic interventions in a patient with WIP deficiency. The course of platelet counts of the patient of pedigree C (Table 1, right column) is shown as black solid line (x1,000/μl) in weeks of life (x-axis). Treatment interventions are shown in the figure legend as indicated. Doses of cell transfer and stem cell transplantation are stated in the main text. Ptl, platelets; DLI, donor lymphocyte infusions; PBSCI, peripheral stem cell infusion (without prior conditioning treatment); PSCT, peripheral stem cell transplantation (HSCT after conditioning pretreatment as described in the main text).
The results of immunological analyses performed at 4 and 5 months of age showed clear signs of combined immunodeficiency with reduced naïve T cells, reduced T cell proliferation in vitro, and an increased proportion of T cell receptor gamma-delta (TCRγδ)-positive T cells (Table 1). Because ganciclovir treatment did not efficiently eliminate CMV, the clinical situation was considered too unstable for a conditioned HSCT. Since the mother was HLA-identical and CMV-seropositive, an attempt of maternal DLI (1 × 106/kg CD3+ T cells) was performed in his 10th month of life. Previous reports had shown the long-term efficacy of unconditioned DLI in the setting of a patient series with 22q11 syndrome, ZAP70 deficiency, and other syndromes (18, 19). The first DLI was given under sirolimus to prevent the onset of graft-vs. host disease (GvHD), the following DLIs were administered without immunosuppression, and all were tolerated well. This led to clinical improvement and a gradual reduction in the CMV plasma load, with complete clearance after six doses (four DLI at 1 × 106-1x107/kg CD3+ cells and two peripheral stem cell concentrates at 2 × 107/kg CD34+ plus 2 × 107/kg CD3+ cells), and a stable mixed T cell donor chimerism over >12 months (>50–70% donor T cells), but expectedly did not lead to the engraftment of myeloid cells (chimerism of CD33+ cells <2–3% in peripheral blood) (8). Meanwhile, findings from NGS panel diagnostics led to the diagnosis of WIP deficiency due to the identification of a novel, stop-gain mutation in WIPF1 [c.C373T, p.R125X; Table 1 and Pfajfer et al. (8)].
There were three indications to proceed to full conditioning and HSCT after the elimination of CMV in this patient: (i), an increasing frequency of ear-nose-throat (ENT) and respiratory infections despite the predominant donor T cell chimerism and IgG substitution; (ii), the uncertainty about the durability of the DLI-mediated T cell engraftment; and, (iii), most urgently, the continuous decline in thrombocyte counts (Figure 1). The latter did not sustainably respond to thrombopoietin receptor agonists, as treatment with eltrombopag had been attempted without effect due to suspected malresorption in short bowel syndrome, and even a switch to romiplostim was ineffective, although both substances were given at standard and increased dose intensities (Figure 1). Consequently, the patient was dependent on platelet transfusions. Furthermore, the psychoneurological situation had improved substantially, indicating that severe brain damage may not have occurred. The transplantation was carried out from his mother after conditioning with ATG-Grafalon (3 × 10mg/kg; days −8, −7, −6), treosulfan (3 × 12g/m2; days −5, −4, −3), and melphalan (140 mg/m2; day −2) at the age of 2.5 years, 15 months after receiving the last DLI and peripheral stem-cell infusion. The graft contained peripheral TCRαβ/CD19-negatively selected cells (5.7 × 107/kg CD34+ and 1.9 × 107/kg TCRγδ+CD3+ cells), and a second fraction of CD34+ positively selected stem cells (7.2 × 106/kg CD34+ and 2.2 × 104/kg CD3+ cells), providing high numbers of stem cells to facilitate rapid engraftment. GvHD prophylaxis consisted of mycophenolate mofetil (1,200 mg/m2), administered until day +46. Engraftment was excellent with 35,000/μl leukocytes on day +8; platelet transfusion was last required on day +9, with persisting full donor chimerism. Ganciclovir was administered from day −10 until day +53 as a secondary prophylaxis. In addition, five doses of CMV-specific DLI were given until day+84 in 2–4-weekly intervals to prevent CMV reactivation. CMV nucleic acid was detectable despite these measures in low-copy numbers (1 × 102-1.5 × 103/mL blood) without clinical correlates until day +90, at which point it was again negative. Apart from experiencing challenging alimentation problems due to the short bowel syndrome and a transient, acute, toxic pancreatitis, the patient did not experience any severe complications. He has been progressing wonderfully, and parenteral nutrition was gradually reduced until it was provisionally terminated at 9 months post-HSCT. He is currently well with >12 months follow-up after conditioned HSCT.
Because WASP undergoes proteasomal degradation, proteasome inhibition in a condition with unimpaired WASP synthesis could hypothetically result in intracellular stabilization of WASP and functional restoration even in WIP deficiency. Therefore, we performed an extensive in vitro screen with various proteasome inhibitors, including bortezomib, with inconclusive results. Finally, proteasome inhibition in vivo was not attempted in any WIP deficient patient reported so far.
Comparison of the Human Phenotype With Mice Deficient in WIP
Newborn and young WIP null mice did not display a pronounced phenotype, neither anatomically, developmentally, nor hematologically, or immunologically (20, 21). However, after 8–10 weeks of life, granulocytosis and T- and B cell lymphopenia developed, splenomegaly was noted, while platelet numbers and volume were normal (21). A progressive lymphopenia was noted in lymph nodes and Peyer's plaques. Most prominently in the clinical phenotype, mice suffered from autoimmune inflammatory disease in many organs such as the gut, lungs, kidneys, joints, but not skin or other organs; and antinuclear and anti-cytoplasmatic antibodies were detected (21). The reported differentiation block, signaling and functional alterations of lymphocytes in WIP null mice including synapse formation and CD19 malfunction (14, 20) associated with autoimmunity and inflammation is similar to what was observed in humans with WIP deficiency. However, the degree of immunodeficiency in humans was not reflected by mice under laboratory conditions of animal facilities, and the platelet and megakaryocyte phenotype, which is variable in humans with WIP deficiency but rather constantly seen in WASP deficient humans and mice (1, 22), was not reported in WIP-deficient mice at all (21).
Discussion
We provide a comprehensive synopsis of clinical and laboratory/biological features of all hitherto-published patients with WIP deficiency. Highly resembling WAS, subtle differences in presentation of WIP deficiency can be observed in addition to the obvious difference that stems from the autosomal recessive pattern of inheritance. The thrombocytopenia in WIP deficiency seems to be less severe and inconstant, and microthrombocytopenia is less frequent than in WAS, raising the question whether other molecules could substitute for WIP as chaperones for WASP specifically in platelets and megakaryocytes. This might be supported by the absence of thrombocytopenia or microplatelets in WIP deficient mice, as reported so far. It is striking that the T-cell immunodeficiency presents quite early in WIP deficient patients, while even classic WAS patients often don't present with severe opportunistic infections before the age of one. The patient reviewed in more detail here (the sixth of all previously published patients; from pedigree C) displayed nearly absent naïve CD4 T-cells and TRECs, which was not tested in the other patients, and had reduced T cell proliferation upon stimulation with PHA, a pathology not detected in previously tested individuals. This and the fact that maternal T-cells were able to engraft without conditioning may implicate a more severe T-cell deficiency than is commonly found in WAS. On the contrary, we cannot explain why in that patient T cell proliferation was tested low-normal upon stimulation with CD3/CD28 in contrast to a previously tested individual (from pedigree A). For these reasons WIP deficiency will be more difficult to diagnose. It does suggest that all infants with clinically severe T-cell deficiency, especially if accompanied by thrombocytopenia, should be tested for WIP deficiency.
Given the severe T-cell deficiency, early diagnosis of WIP deficiency seems prudent. Certainly, newborn screening (NBS) for TRECs would have accelerated the diagnosis in our patient [reviewed in Kwan and Puck (23)]. However, patients with combined immunodeficiency might not always be detected by TREC-based NBS with cutoffs designed to detect SCID, and NBS is not globally available. At the time of PID diagnosis, the patient from pedigree C showed many contraindications for conditioned HSCT, such as a systemic, invasive CMV infection, neurological impairment, and others. Similar to previous observations (18, 19), the patient had an HLA-identical, and, in his case, CMV-competent and WIP-proficient, family member. Despite the risk of GvHD, the potential anti-infectious activity of peripheral blood lymphocytes from such a donor, even with long-term “repopulating” capacity (18, 19), was an attempt to stabilize the clinical condition and buy time. Together, this clinical observation (8) indicated that, also for other patients with SCID or profound CID, an HLA-identical lymphocyte donor might be identified, especially, in highly consanguineous pedigrees, and a patient in a condition that is “too bad to transplant” might be rescued by taking a DLI approach and bridging to a subsequent HSCT.
Ethics Statement
The study was performed with informed consent and according to an IRB approval (24-334 ex 11/12; IRB00002556) in accordance with the Declaration of Helsinki.
Author Contributions
WS, CU, MB, RU, AK, DS, HL, VS, and MGS cared for the patient and collected the data. MGS wrote the manuscript, and designed table and figure. MB, MA, and KB analyzed and interpreted clinical and laboratory/genetic data, respectively, and corrected the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Manfred Hönig, Ulm, for constructive discussions on the pre-HSCT DLI approach and critical review of the manuscript, and the team at CCI Freiburg, especially Drs S. Ehl and C. Klemann, for their cooperation in challenging diagnostic situations of profound combined immunodeficiency. Further, We thank Dr. S. Crockett for scientific editing, and the patient family for cooperativity. MGS was in part funded by Steirische Kinderkrebshilfe (Styrian Children's Cancer Aid).
Abbreviations
WASP, Wiskott-Aldrich syndrome protein; WIP, WASP interacting protein; PID, primary immunodeficiency; NGS, next generation sequencing; DLI, donor lymphocyte infusions; CMV, cytomegalovirus.
References
1. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. (2009) 15:84–90. doi: 10.1016/j.bbmt.2008.10.007
2. Thrasher AJ. New insights into the biology of Wiskott-Aldrich syndrome (WAS). Hematology Am Soc Hematol Educ Program (2009) 132–8. doi: 10.1182/asheducation-2009.1.132
3. Bousfiha A, Jeddane L, Picard C, Ailal F, Bobby Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
4. Candotti F. Clinical manifestations and pathophysiological mechanisms of the wiskott-aldrich syndrome. J Clin Immunol. (2018) 38:13–27. doi: 10.1007/s10875-017-0453-z
5. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International union of immunological societies: 2017 primary immunodeficiency diseases committee report on inborn errors of immunity. J Clin Immunol. (2018) 38:96–128. doi: 10.1007/s10875-017-0464-9
6. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. (2012) 209:29–34. doi: 10.1084/jem.20110896
7. Al-Mousa H, Hawwari A, Al-Ghonaium A, Al-Saud B, Al-Dhekri H, Al-Muhsen S, et al. Hematopoietic stem cell transplantation corrects WIP deficiency. J Allergy Clin Immunol. (2017) 139:1039–40 e1034. doi: 10.1016/j.jaci.2016.08.036
8. Pfajfer L, Seidel MG, Houmadi R, Rey-Barroso J, Hirschmugl T, Salzer E, et al. WIP deficiency severely affects human lymphocyte architecture during migration and synapse assembly. Blood (2017) 130:1949–53. doi: 10.1182/blood-2017-04-777383
9. de la Fuente MA, Sasahara Y, Calamito M, Anton IM, Elkhal A, Gallego MD, et al. WIP is a chaperone for Wiskott-Aldrich syndrome protein (WASP). Proc Natl Acad Sci USA. (2007) 104:926–31. doi: 10.1073/pnas.0610275104
10. Cotta-de-Almeida V, Dupre L, Guipouy D, Vasconcelos Z. Signal integration during T lymphocyte activation and function: lessons from the Wiskott-Aldrich syndrome. Front Immunol. (2015) 6:47. doi: 10.3389/fimmu.2015.00047
11. Sasahara Y, Rachid R, Byrne MJ, de la Fuente MA, Abraham RT, Ramesh N, et al. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol Cell (2002) 10:1269–81. doi: 10.1016/S1097-2765(02)00728-1
12. Orange JS, Ramesh N, Remold-O'Donnell E, Sasahara Y, Koopman L, Byrne M, et al. Wiskott-Aldrich syndrome protein is required for NK cell cytotoxicity and colocalizes with actin to NK cell-activating immunologic synapses. Proc Natl Acad Sci USA. (2002) 99:11351–6. doi: 10.1073/pnas.162376099
13. Krzewski K, Chen X, Strominger JL. WIP is essential for lytic granule polarization and NK cell cytotoxicity. Proc Natl Acad Sci USA. (2008) 105:2568–73. doi: 10.1073/pnas.0711593105
14. Keppler SJ, Gasparrini F, Burbage M, Aggarwal S, Frederico B, Geha RS, et al. Wiskott-Aldrich syndrome interacting protein deficiency uncovers the role of the co-receptor CD19 as a generic hub for PI3 kinase signaling in B cells. Immunity (2015) 43:660–73. doi: 10.1016/j.immuni.2015.09.004
15. Parolini O, Ressmann G, Haas OA, Pawlowsky J, Gadner H, Knapp W, et al. X-linked Wiskott-Aldrich syndrome in a girl. N Engl J Med. (1998) 338:291–5. doi: 10.1056/NEJM199801293380504
16. Boonyawat B, Dhanraj S, Al Abbas F, Zlateska B, Grunenbaum E, Roifman CM, et al. Combined de-novo mutation and non-random X-chromosome inactivation causing Wiskott-Aldrich syndrome in a female with thrombocytopenia. J Clin Immunol. (2013) 33:1150–5. doi: 10.1007/s10875-013-9927-9
17. Conley ME, Wang WC, Parolini O, Shapiro DN, Campana D, Siminovitch KA. Atypical Wiskott-Aldrich syndrome in a girl. Blood (1992) 80:1264–9.
18. Hoenig M, Schulz A, Schuetz C, Debatin KM, Friedrich W. Treatment of complete digeorge syndrome by repeat transfusions or Blood lymphocytes from an HLA-identical sibling donor. Blood (2004) 104:1332.
19. Honig M, Schuetz C, Schwarz K, Rojewski M, Jacobsen E, Lahr G, et al. Immunological reconstitution in a patient with ZAP-70 deficiency following transfusion of blood lymphocytes from a previously transplanted sibling without conditioning. Bone Marrow Transplant (2012) 47:305–7. doi: 10.1038/bmt.2011.71
20. Anton IM, de la Fuente MA, Sims TN, Freeman S, Ramesh N, Hartwig JH, et al. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity (2002) 16:193–204. doi: 10.1016/S1074-7613(02)00268-6
21. Curcio C, Pannellini T, Lanzardo S, Forni G, Musiani P, Anton IM. WIP null mice display a progressive immunological disorder that resembles Wiskott-Aldrich syndrome. J Pathol. (2007) 211:67–75. doi: 10.1002/path.2088
22. Snapper SB, Rosen FS, Mizoguchi E, Cohen P, Khan W, Liu CH, et al. Wiskott-Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity (1998) 9:81–91. doi: 10.1016/S1074-7613(00)80590-7
Keywords: Wiskott-Aldrich syndrome protein-interacting protein (WIP), primary immunodeficiency (PID), Wiskott-Aldrich syndrome (WAS), hematopoietic stem cell transplantation (HSCT), donor lymphocyte infusions (DLI), combined immunodeficiency (CID) with syndromic features, inborn errors of immunity
Citation: Schwinger W, Urban C, Ulreich R, Sperl D, Karastaneva A, Strenger V, Lackner H, Boztug K, Albert MH, Benesch M and Seidel MG (2018) The Phenotype and Treatment of WIP Deficiency: Literature Synopsis and Review of a Patient With Pre-transplant Serial Donor Lymphocyte Infusions to Eliminate CMV. Front. Immunol. 9:2554. doi: 10.3389/fimmu.2018.02554
Received: 26 July 2018; Accepted: 17 October 2018;
Published: 02 November 2018.
Edited by:
Satoshi Okada, Hiroshima University, JapanReviewed by:
Saul Oswaldo Lugo Reyes, Instituto Nacional de Pediatria, MexicoYoji Sasahara, Tohoku University School of Medicine, Japan
Copyright © 2018 Schwinger, Urban, Ulreich, Sperl, Karastaneva, Strenger, Lackner, Boztug, Albert, Benesch and Seidel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus G. Seidel, bWFya3VzLnNlaWRlbEBtZWR1bmlncmF6LmF0