 Peter Bannas1
Peter Bannas1 Friedrich Koch-Nolte2*
Friedrich Koch-Nolte2*- 1Deptartment of Radiology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
- 2Institute of Immunology University, Medical Center Hamburg-Eppendorf, Hamburg, Germany
The NAD+-metabolizing ectoenzyme CD38 is an established therapeutic target in multiple myeloma. The CD38-specific monoclonal antibodies daratumumab and isatuximab show promising results in the clinic. Nanobodies correspond to the single variable domains (VHH) derived from heavy chain antibodies that naturally occur in camelids. VHHs display high solubility and excellent tissue penetration in vivo. We recently generated a panel of CD38-specific nanobodies, some of which block or enhance the enzymatic activity of CD38. Fusion of such a nanobody to the hinge, CH2, and CH3 domains of human IgG1 generates a chimeric llama/human hcAb of about half the size of a conventional moAb (75 vs. 150 kDa). Similarly, a fully human CD38-specific hcAb can be generated using a CD38-specific human VH3 instead of a CD38-specific camelid nanobody. Here we discuss the advantages and disadvantages of CD38-specific hcAbs vs. conventional moAbs and provide an outlook for the potential use of CD38-specific hcAbs as novel therapeutics for multiple myeloma.
Introduction
CD38 is a cell surface ectoenzyme that metabolizes NAD+ released from damaged cells in inflammation (1). In concert with the ecto-enzymes CD203 and CD73, CD38 contributes to the conversion of NAD+ to immunosuppressive extracellular adenosine. In the tumor microenvironment, CD38 may promote tumor growth by suppressing effector T cell responses (1, 2). Since CD38 is overexpressed by multiple myeloma cells and other hematological tumors, it has attracted interest as a target for therapeutic antibodies (3–5).
Nanobodies are single domain antibody fragments derived from the heavy chain IgG antibodies naturally occurring in llamas and other camelids (6–8). In these animals, the IgG2 and IgG3 isotypes lack the CH1 domain and do not bind to light chains. Nanobodies correspond to the variable domain (VHH) of these heavy chain antibodies. VHHs carry characteristic residues in the framework region 2 (FR2) that render them highly soluble in the absence of a paired light chain (8–10). VHHs often have a long complementarity determining region 3 (CDR3) that can mediate binding to the catalytic cavity of an enzyme and other hidden epitopes that are not accessible for conventional antibodies (11–13). Their robust, soluble single domain format renders nanobodies amenable for genetic fusion to the hinge and Fc domains of other antibody isotypes (14, 15). Owing to their high solubility, it is much easier to link two or more VHHs into bi- or multivalent formats than the corresponding VH+VL domains of conventional antibodies.
CD38-Specific Therapeutic Chimeric Mouse/Human and Fully Human Conventional Antibodies
The conventional CD38-specific moAbs daratumumab and isatuximab have proven therapeutic efficacy in multiple myeloma (5, 16). Both antibodies were derived from mice immunized with human CD38. While daratumumab was generated from CD38-immunized transgenic mice that carry genomic loci encoding human IgH and IgL (17), isatuximab was generated from CD38-immunized wild type mice (18). The VH and VL domains of the murine moAb were genetically fused to the CH1-hinge-CH2-CH3 domains of human IgG1 and to the constant domain of the kappa light chain (Cκ), respectively, generating a classic mouse/human chimeric antibody (Figure 1A). The crystal structure of isatuximab in complex with CD38 indicates that its capacity to inhibit the enzymatic activity of CD38 is by an allosteric mechanism (18). Recently, the VH and VL domains of daratumumab were used to construct a single-chain human anti-CD38 cytokine-antibody fusion protein termed IL2-αCD38-αCD38-scTRAIL (19). The bivalent tandem scFv of daratumumab mediated specific binding to CD38 expressing myeloma cells, while the engineered homotrimeric format of TRAIL induced apoptosis of these cells, presumably by binding to cognate death receptors.
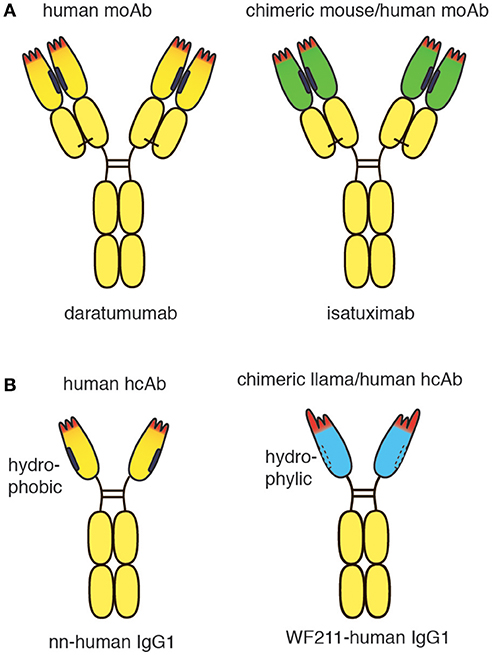
Figure 1. Schematic diagram of conventional and heavy chain CD38-specific antibodies. (A) Schematic diagrams of therapeutic CD38-specific conventional antibodies. Daratumumab is derived from a CD38-immunized human-antibody transgenic mouse. Isatuximab is derived from a CD38-immunized wildtype mouse. The chimeric antibody was generated by genetic fusion of the VH and VL domains of the mouse monoclonal antibody to the constant domains of human IgG1 and kappa, respectively. (B) Schematic diagrams of CD38-specific heavy chain antibodies. A fully human CD38-specific heavy chain antibody has been derived from a CD38-immunized human heavy chain transgenic rat. The chimeric WF211-human IgG1 heavy chain antibody was generated by genetic fusion of the VHH domain of a llama heavy chain antibody to the hinge, CH2 and CH3 domains of human IgG1. Human heavy chain antibodies display a natural “stickiness,” i.e., tendency to bind light chains via the hydrophobic interface that helps to stabilize the orientation of the VH-VL pair in conventional antibodies. In contrast, chimeric llama-human heavy chain antibodies do not show any natural “stickiness” or tendency to bind light chains. VHH domains have been shaped by 50 Mio years of evolution for high solubility in the absence of a light chain.
CD38-Specific Chimeric Llama/Human and Fully Human Heavy Chain Antibodies
Recently, CD38-specific nanobodies were generated from CD38-immunized llamas (20, 21). Some of these nanobodies inhibited or enhanced CD38 enzymatic activity in a dose dependent manner and effectively targeted CD38 on human tumor cells in a mouse Xenograft model (20). Several nanobodies bind independently of daratumumab. Such nanobodies have proven useful for detecting cell surface CD38 in patients treated with daratumumab (22). Owing to their high solubility, the nanobodies can readily be fused to other protein domains, including the hinge and Fc domains of human IgG1 (Figure 1B). Such chimeric llama/human heavy chain antibodies acquire the capacity to induce classic Fc-mediated effector functions, including ADCC and CDC (23).
In contrast to the hydrophylic nature of camelid VHH domains, human VH domains display a natural “stickiness” and tendency to aggregate in the absence of a light chain (24–26). This “stickiness” is attributed to the hydrophobic interface that helps to properly orient VH and VL domains for joint interaction with the target antigen (27). “Camelization” of human VH domains by substituting hydrophobic amino acid residues in FR2 with hydrophilic residues can greatly enhance the solubility of human VH domains (28–30). In order to express human heavy chain antibodies in transgenic mice or rats it is therefore advisable to inactivate not only the endogenous rodent heavy chain locus, but also the kappa and lambda light chain loci. Interestingly, during the ensuing immune response, somatic hypermutation and selection drive the expansion of VH variants that increase the solubility of heavy chain antibodies (31, 32). A similar mechanism was observed when human VH domains were affinity matured in vitro (33). Recently, CD38-specific human heavy chain antibodies were generated successfully from CD38-immunized human heavy chain-only transgenic rats (32).
Advantages and Disadvantages of CD38-Specific Heavy Chain Antibodies vs. Conventional moAbs
Tissue Penetration and in vivo Half Life
Heavy chain antibodies are only half the size of conventional moAbs (75 vs. 150 kDa). HcAbs may therefore penetrate more effectively into CD38-expressing tumors than full size moAbs, particularly when the tumors promote increased interstitial pressure. Better tissue penetration has indeed been demonstrated for nanobodies vs. conventional antibodies in solid tumors and subcutaneous tumors (34, 35). Tissue penetration is a highly relevant issue, in particular when considering that multiple myeloma resides in the bone marrow and is surrounded by a dense immune suppressive microenvironment (1). It will be important to determine whether nanobody-based hcAbs do reach myeloma cells in the bone marrow more efficiently than conventional antibodies.
The half life of therapeutic antibodies is influenced by several factors, including size, glycosylation, and affinity to the neonatal Fc receptor. While a smaller size may facilitate tissue penetration, a small size may also facilitate renal filtration and thereby shorten the persistence of the therapeutic in vivo. Several strategies have been employed successfully to prolong the in vivo half life of nanobodies, including conjugation to polyethylene glycol polymers (36), genetic fusion to an albumin-specific nanobody (34, 37). In case of nanobody based hcAbs, Fc engineering could be used to introduce mutations that enhance binding to the neonatal Fc receptor and thereby prolong persistence in vivo (38, 39).
Developability of Bispecific Therapeutics
The soluble nature of the nanobody VHH domain, facilitates the construction and production of bispecific antibodies. For example, a bispecific nanobody-based heavy chain antibody can readily be generated simply by fusing a second nanobody to the N-terminus of a nanobody-based hcAb. Importantly, nanobody-based bispecific hcAbs are composed of two identical polypeptide chains, i.e., their production does not require any “knob in hole” technology or adjusting the of expression levels of two or more vectors (40, 41). This simplifies the production and developability of bispecific hcAbs, although the moderate increase in size of a bispecific vs. a mono-specific hcAb (from ~75 to ~100 kDa) may compromise tissue penetration. By tandem fusion of two nanobodies that recognize independent epitopes of CD38 to the Fc domain of human IgG, we recently generated tetravalent biparatopic hcAbs that exhibit a markedly enhanced capacity to induce CDC of CD38-expressing myeloma cells.
Modulation of Enzyme Activity
Owing to the inherent capacity of nanobodies to extend into and block active site crevices (11, 12), a heavy chain antibody containing a CD38-antagonistic nanobody may provide an additional therapeutic benefit by inhibiting the production of immunosuppressive adenosine (1, 2). Conceivably, the potency of enzyme inhibition may be enhanced by fusion of an enzyme-inhibiting nanobody to a nanobody recognizing a distinct epitope of CD38, e.g., in a biparatopic activity blocking hcAb.
Immunogenicity
The potential immunogenicity of antibody therapeutics is a relevant concern (5, 42, 43). The development of neutralizing antibodies against the therapeutic antibody by the patient usually renders the patient resistant to the therapeutic. This risk for developing such antibodies is larger for chimeric antibodies that contain murine VH and VL domains such as rituximab and isatuximab than for fully human antibodies such as dartumumab which is composed only of human domains. However, it is impossible to fully humanize the idiotype of an antibody without losing specificity of effectivity since the unique CDR loops of the VH and VL domains are required for specificity. Hence, the potential development of antibodies directed against the unique CDR loops remains a concern for any therapeutic antibody. Drug antibodies have not yet been detected in any daratumumab-treated patients (5). However, it is uncertain to what extent this is due to the lack of a sensitive assay for such antibodies.
The human germline encodes ~50 distinct VH domains and 4 distinct IgG isotypes (Figure 2A) (44, 45). V-D-J recombination during B-cell development generates millions of distinct idiotypes (antigen binding paratopes). Subsequent to antigen encounter, somatic hypermutation generates many more variant VH domains. During pregnancy, maternal IgG is translocated from the maternal blood through placental trophoblasts into the blood stream of the fetus, leading to tolerization of the new born immune system against millions of VH variants, but only 4 distinct IgG isotypes. In germline configuration, llama VHH domains show ~80–90% amino acid sequence identity to human VH3 domains, i.e., the predominant VH subset found in human immunoglobulins (46). As a result of somatic hypermutation, two matured human VH domains often differ more from one another than a germline human VH3 domain from a llama VHH3 domain. A few hydrophilic amino acid residues in framework region 2 and the long CDR3 that can partially fold back onto the former interface to the VL domain largely account for the dramatically improved solubility of camelid VHH domains vs. human VH3 domains. These residues cannot be fully humanized without compromising solubility. Notwithstanding, the idiotype (CDR regions 1, 2, and 3) cover a much larger space (both, in the literal sense and in terms of potential immunogenicity) than these hydrophilic amino acids in the former VL interface.
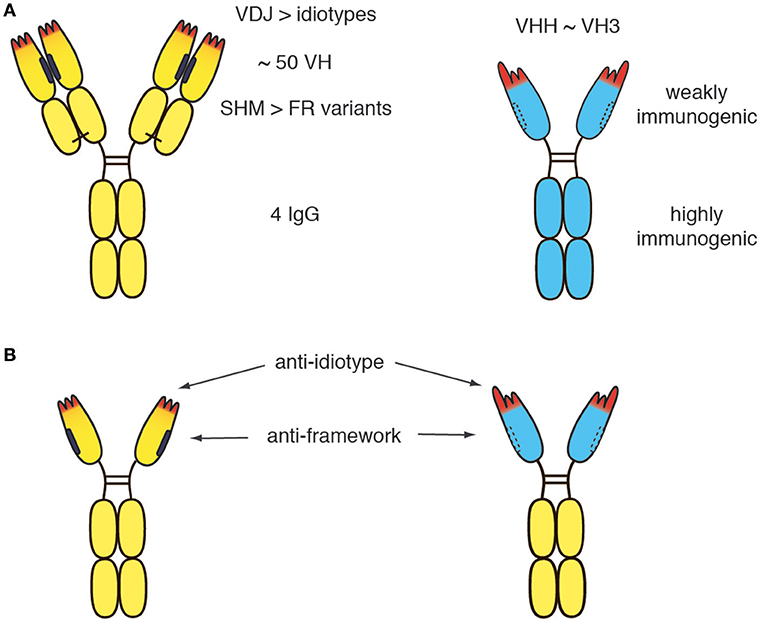
Figure 2. Potential immunogenicity of heavy chain antibodies. (A) The human germline encodes ~50 distinct VH domains and 4 distinct IgG isotpyes. V-D-J recombination during B-cell development generates millions of distinct idiotypes (antigen binding paratopes, CDR regions 1, 2 indicated in red). Subsequent to antigen encounter, somatic hypermutation generates many more variant VH domains. During pregnancy, maternal IgGs are translocated through the placental trophoblasts to the fetus, leading to tolerization of the new born human immune system against millions of VH variants, but only 4 distinct IgG isotypes. (B) In germline configuration, llama VHH domains show ~80-90% amino acid sequence identitiy to human VH3 domains. A few amino acid substitutions in the VL face (mainly framework region 2, indicated by dashed lines) and a long CDR3 that can partially fold back onto this face largely account for the dramatically improved solubility of camelid VHH domains vs. human VH3 domains. The solubility of human VH can be improved by “camelization,” i.e., by replacing hydrophobic residues at the interface of the VL domain (indicated in black) with hydrophylic residues resembling those found in VHH domains. Conversely, camelid VHH domains can be “humanized,” i.e., by replacing amino acid residues in the framework with residues corresponding to germlin human VH domains. However, the idiotype of a therapeutic moAb or hcAb cannot be fully humanized without compromising binding to the target antigen. Similarly, the VL face cannot be fully humanized without compromising solubility. Therefore, small risks remain, that the patient will develop antibodies against the idiotype and/or against the (much smaller) hydrophilic VL face.
Although the immunogenicity of a therapeutic antibody can be reduced by humanization, the residual risk remains for any therapeutic antibody that the patient develops antibodies directed against the idiotype (Figure 2B). Such anti-drug antibodies usually render the therapeutic useless for the patient. If more than one therapeutic antibody is available for a particular target, an option in such cases is to switch to a different biologic targeting the same molecule (e.g., from daratumumab to isatuximab or vice versa). It is conceivable that in the future, the risk of developing anti-drug antibodies can be reduced further by tolerization strategies that will become available and permit tolerization of the patient to the therapeutic antibody before treatment is initiated.
Conclusions and Outlook
Later this year, Caplacizumab, a dimeric nanobody directed against the van Willebrand factor, is expected to receive FDA approval as the first nanobody in the clinic (47, 48). Nanobodies that antagonize CD38 provide proof of concept for the notion that these small biologics represent attractive alternatives to small molecule inhibitors for inhibiting the production of immunosuppressive adenosine. Nanobody-based heavy chain antibodies retain all effector functions of full sized moAbs, at half the size. This size advantage will likely facilitate targeting of tumor cells in vivo, even under conditions of increased interstitial pressure within tumors. Owing to their excellent solubility, it is much easier to link different nanobodies in a single therapeutic than the combined VH+VL domains of conventional moAbs. The high solubility of CD38-specific heavy chain antibodies may come at the price of a slightly higher risk for inducing anti-drug antibodies compared to conventional human CD38-specific moAbs. In addition to the complementarity determining regions, the VL face of heavy chain antibodies may provide a second, albeit much smaller, vulnerability than the idiotype. It will be interesting to see whether “humanized” nanobody heavy chain antibodies or “camelized” human heavy chain antibodies will hold the leading nose in the race to the clinic.
Author Contributions
PB and FK-N conceived the project. FK-N wrote the manuscript. Both authors reviewed and approved the manuscript.
Funding
This work was supported by grants from the Sander Stiftung to FK-N and PB, and by grant SFB1328-Z2 from the DFG to FK-N.
Conflict of Interest Statement
FK-N receives a share of antibody sales via MediGate GmbH, a wholly owned subsidiary of the University Medical Center Hamburg-Eppendorf. PB and FK-N are co-inventors on a patent application on CD38-specific nanobodies.
The Handling Editor declared a past co-authorship with one of the authors, FK-N.
Abbreviations
ADCC, antibody-dependent cytotoxicity; CDC, complement-dependent cytotoxicity; CDR, compelementarity determining region; Fc, crystallizing fragment; FR, framework region; Ig, immunoglobulin; kD, kilodalton; NAD, nicotinamide adenine dinucleotide; moAb, monoclonal antibody; Nb, nanobody; Nb-hcAb, nanobody-based (human) heavy chain antibody; scFv, single chain variable fragment; VH, variable domain of a conventional heavy chain; VHH, variable domain of a camelid heavy chain antibody.
References
1. Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, et al. NAD(+)-Metabolizing ectoenzymes in remodeling tumor-host interactions: the human myeloma model. Cells (2015) 4:520–37. doi: 10.3390/cells4030520
2. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci. (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
3. Afifi S, Michael A, Lesokhin A. Immunotherapy: A new approach to treating multiple myeloma with daratumumab and elotuzumab. Ann Pharmacother. (2016) 50:555–68. doi: 10.1177/1060028016642786
4. Rajan AM, Kumar S. New investigational drugs with single-agent activity in multiple myeloma. Blood Cancer J. (2016) 6:e451. doi: 10.1038/bcj.2016.53
5. van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood (2017) 131:13–29. doi: 10.1182/blood-2017-06-740944
6. Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature (1993) 363:446–8. doi: 10.1038/363446a0
7. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. (2013) 82:775–97. doi: 10.1146/annurev-biochem-063011-092449
8. Ingram JR, Schmidt FI, Ploegh HL. Exploiting Nanobodies' Singular Traits. Annu Rev Immunol. (2018) 36:695–715. doi: 10.1146/annurev-immunol-042617-053327
9. Goldman ER, Liu JL, Zabetakis D, Anderson GP. Enhancing stability of camelid and shark single domain antibodies: an overview. Front Immunol. (2017) 8:865. doi: 10.3389/fimmu.2017.00865
10. Konning D, Zielonka S, Grzeschik J, Empting M, Valldorf B, Krah S, et al. Camelid and shark single domain antibodies: structural features and therapeutic potential. Curr Opin Struct Biol. (2017) 45:10–6. doi: 10.1016/j.sbi.2016.10.019
11. Lauwereys M, Arbabi Ghahroudi M, Desmyter A, Kinne J, Holzer W, De Genst E, et al. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. (1998) 17:3512–20. doi: 10.1093/emboj/17.13.3512
12. De Genst E, Silence K, Decanniere K, Conrath K, Loris R, Kinne J, et al. Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc Natl Acad Sci USA. (2006) 103:4586–91. doi: 10.1073/pnas.0505379103
13. Wesolowski J, Alzogaray V, Reyelt J, Unger M, Juarez K, Urrutia M, et al. Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med Microbiol Immunol. (2009) 198:157–74. doi: 10.1007/s00430-009-0116-7
14. Bannas P, Hambach J, Koch-Nolte F. Nanobodies and nanobody-based human heavy chain antibodies as antitumor therapeutics. Front Immunol. (2017) 8:1603. doi: 10.3389/fimmu.2017.01603
15. Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, et al. Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc Natl Acad Sci USA. (2018) 115:3912–7. doi: 10.1073/pnas.1801524115
16. Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood (2017) 129:3294–303. doi: 10.1182/blood-2016-09-740787
17. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
18. Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallee F, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. (2014) 20:4574–83. doi: 10.1158/1078-0432.CCR-14-0695
19. De Luca R, Kachel P, Kropivsek K, Snijder B, Manz MG, Neri D. A novel dual-cytokine-antibody fusion protein for the treatment of CD38-positive malignancies. Protein Eng Des Sel. (2018) 31:173–9. doi: 10.1093/protein/gzy015
20. Fumey W, Koenigsdorf J, Kunick V, Menzel S, Schutze K, Unger M, et al. Nanobodies effectively modulate the enzymatic activity of CD38 and allow specific imaging of CD38(+) tumors in mouse models in vivo. Sci Rep. (2017) 7:14289. doi: 10.1038/s41598-017-14112-6
21. Li T, Qi S, Unger M, Hou YN, Deng QW, Liu J, et al. Immuno-targeting the multifunctional CD38 using nanobody. Sci Rep. (2016) 6:27055. doi: 10.1038/srep27055
22. Oberle A, Brandt A, Alawi M, Langebrake C, Janjetovic S, Wolschke C, et al. Long-term CD38 saturation by daratumumab interferes with diagnostic myeloma cell detection. Haematologica (2017) 102:e368–370. doi: 10.3324/haematol.2017.169235
23. Nolte F, Bannas P, Schütze K, Fumey F, Schriewer L. Antigen-Binding Polypeptides directed against CD38. WO2017081211A2 (2017).
24. Barthelemy PA, Raab H, Appleton BA, Bond CJ, Wu P, Wiesmann C, et al. Comprehensive analysis of the factors contributing to the stability and solubility of autonomous human VH domains. J Biol Chem. (2008) 283:3639–54. doi: 10.1074/jbc.M708536200
25. Ewert S, Cambillau C, Conrath K, Pluckthun A. Biophysical properties of camelid V(HH) domains compared to those of human V(H)3 domains. Biochemistry (2002) 41:3628–36. doi: 10.1021/bi011239a
26. Feige MJ, Buchner J. Principles and engineering of antibody folding and assembly. Biochim Biophys Acta (2014) 1844:2024–31. doi: 10.1016/j.bbapap.2014.06.004
27. Teplyakov A, Obmolova G, Malia TJ, Luo J, Muzammil S, Sweet R, et al. Structural diversity in a human antibody germline library. MAbs (2016) 8:1045–63. doi: 10.1080/19420862.2016.1190060
28. Davies J, Riechmann L. ‘Camelising' human antibody fragments: NMR studies on VH domains. FEBS Lett. (1994) 339:285–90.
29. Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, et al. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci USA. (2012) 109:10879–84. doi: 10.1073/pnas.1202866109
30. Kim DY, Hussack G, Kandalaft H, Tanha J. Mutational approaches to improve the biophysical properties of human single-domain antibodies. Biochim Biophys Acta (2014) 1844:1983–2001. doi: 10.1016/j.bbapap.2014.07.008
31. Nilvebrant J, Tessier PM, Sidhu SS. Engineered Autonomous Human Variable Domains. Curr Pharm Des. (2016) 22:6527–37. doi: 10.2174/1381612822666160921143011
32. Aldred SF, Bourdreau A, Buelow B, Clarke S, Dank K, Davison L, et al. Multispecific antibodies targeting CD38 show potent tumor-specific cytotoxicity. (Abstract). J Clin Oncol. (2018) 36(5Suppl.):57. doi: 10.1200/JCO.2018.36.5_suppl.57
33. Julian MC, Li L, Garde S, Wilen R, Tessier PM. Efficient affinity maturation of antibody variable domains requires co-selection of compensatory mutations to maintain thermodynamic stability. Sci Rep. (2017) 7:45259. doi: 10.1038/srep45259
34. Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier, T, de Haard HJ, et al. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther. (2008) 7:2288–97. doi: 10.1158/1535-7163.MCT-07-2384
35. Bannas P, Well L, Lenz A, Rissiek B, Haag F, Schmid J, et al. In vivo near-infrared fluorescence targeting of T cells: comparison of nanobodies and conventional monoclonal antibodies. Contrast Media Mol Imaging (2014) 9:135–42. doi: 10.1002/cmmi.1548
36. Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, et al. Noninvasive imaging of immune responses. Proc Natl Acad Sci USA. (2015) 112:6146–51. doi: 10.1073/pnas.1502609112
37. Danquah W, Meyer-Schwesinger C, Rissiek B, Pinto C, Serracant-Prat A, Amadi M, et al. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Sci Transl Med. (2016) 8:366ra162. doi: 10.1126/scitranslmed.aaf8463
38. Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, et al. Increasing the serum persistence of an IgG fragment by random mutagenesis. Nat Biotechnol. (1997) 15:637–40. doi: 10.1038/nbt0797-637
39. Scheuplein F, Rissiek B, Driver JP, Chen YG, Koch-Nolte F, Serreze DV. A recombinant heavy chain antibody approach blocks ART2 mediated deletion of an iNKT cell population that upon activation inhibits autoimmune diabetes. J Autoimmun. (2010) 34:145–54. doi: 10.1016/j.jaut.2009.08.012
40. Ridgway JB, Presta LG, Carter P. ‘Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. (1996) 9:617–21.
41. De Nardis C, Hendriks LJA, Poirier E, Arvinte T, Gros P, Bakker ABH, et al. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem. (2017) 292:14706–17. doi: 10.1074/jbc.M117.793497
42. Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. (2009) 157:220–33. doi: 10.1111/j.1476-5381.2009.00190.x
43. van Schie KA, Wolbink GJ, Rispens T. Cross-reactive and pre-existing antibodies to therapeutic antibodies–Effects on treatment and immunogenicity. MAbs (2015) 7:662–71. doi: 10.1080/19420862.2015.1048411
44. Chothia C, Lesk AM, Gherardi E, Tomlinson IM, Walter G, Marks JD, et al. Structural repertoire of the human VH segments. J Mol Biol. (1992) 227:799–817.
45. de Bono B, Madera M, Chothia C. VH gene segments in the mouse and human genomes. J Mol Biol. (2004) 342:131–43. doi: 10.1016/j.jmb.2004.06.055
46. Klarenbeek A, El Mazouari K, Desmyter A, Blanchetot C, Hultberg A, de Jonge N, et al. Camelid Ig V genes reveal significant human homology not seen in therapeutic target genes, providing for a powerful therapeutic antibody platform. MAbs (2015) 7:693–706. doi: 10.1080/19420862.2015.1046648
47. Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs (2018) 10:183–203. doi: 10.1080/19420862.2018.1415671
Keywords: antibody engineering, CD38, heavy chain antibody, monoclonal antibody, multiple myeloma, nanobody
Citation: Bannas P and Koch-Nolte F (2018) Perspectives for the Development of CD38-Specific Heavy Chain Antibodies as Therapeutics for Multiple Myeloma. Front. Immunol. 9:2559. doi: 10.3389/fimmu.2018.02559
Received: 12 June 2018; Accepted: 17 October 2018;
Published: 06 November 2018.
Edited by:
Fabio Malavasi, Università degli Studi di Torino, ItalyReviewed by:
Zsolt Sebestyen, University Medical Center Utrecht, NetherlandsCox Terhorst, Harvard Medical School, United States
Copyright © 2018 Bannas and Koch-Nolte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Friedrich Koch-Nolte, bm9sdGVAdWtlLmRl