Abstract
Pregnancy is an immunological challenge to the mother. The fetal tissues including the placenta must be protected from activation of the maternal immune system. On the other hand, the placental tissue sheds into the maternal circulation and must be adequately identified and phagocytized by the maternal immune system. During a healthy pregnancy, numerous immunosuppressive processes take place that allow the allograft fetus to thrive under exposure to humoral and cellular components of the maternal immune system. Breakdown of immune tolerance may result in sterile inflammation and cause adverse pregnancy outcomes such as preeclampsia, a vascular disease of the pregnancy with unpredictable course and symptoms from several organs. Immunological incompatibility between mother and fetus is strongly indicated in preeclampsia. Recently, genetic factors linking immunological pathways to predisposition to preeclampsia have been identified. In this mini-review genetic variation in immunological factors are discussed in the context of preeclampsia. Specifically, we explore immunogenetic and immunomodulary mechanisms contributing to loss of tolerance, inflammation, and autoimmunity in preeclampsia.
Introduction
Preeclampsia is a heterogeneous vascular disease of the human pregnancy that presents in a previously normotensive woman during the second half of the pregnancy with hypertension and proteinuria, or preeclampsia-associated signs in the absence of proteinuria (1, 2). Preeclampsia occurs in 3% of pregnancies (3), and it is one of the most important causes of maternal and fetal morbidity and mortality worldwide. The etiology of preeclampsia is incompletely understood, but it has its origins in early pregnancy and delivery of the placenta is the only cure (4). Two distinct subtypes has been frequently used in the literature based on the timing of the disease onset/delivery: early-onset <34 + 0 and late-onset ≥34 weeks of gestation. However, better understanding of the etiology and different subtypes is needed for early recognition and preventive measures.
Preeclampsia is considered a two stage-disease in which poorly perfused placenta produces factor(s) leading to systemic vascular disease and the clinical manifestations of preeclampsia (5).
At 8 weeks of gestation, the trophoblast cells invade from the placenta into the maternal tissue and into the uterine arteries. These endovascular trophoblast cells facilitate the remodeling of spiral uterine arteries, which is essential for a healthy pregnancy. In order for the placentation process to be sufficient to support a healthy pregnancy, the extravillous trophoblast cells must avoid detection by the alternative pathway, subsequent complement activation, and immune response ((4, 6); Figure 1). Immunogenetic susceptibility to preeclampsia may have effect in the early stages of pregnancy whereby through loss of maternal tolerance toward the fetal components, the process of placentation is impaired. On the other hand, during the third trimester, underlying immunogenetic predisposition may aggravate sterile inflammation, which is exacerbated by systemic endothelial dysfunction in the mother's vasculature and result in progression of preeclampsia ((10) Figure 2A).
Figure 1
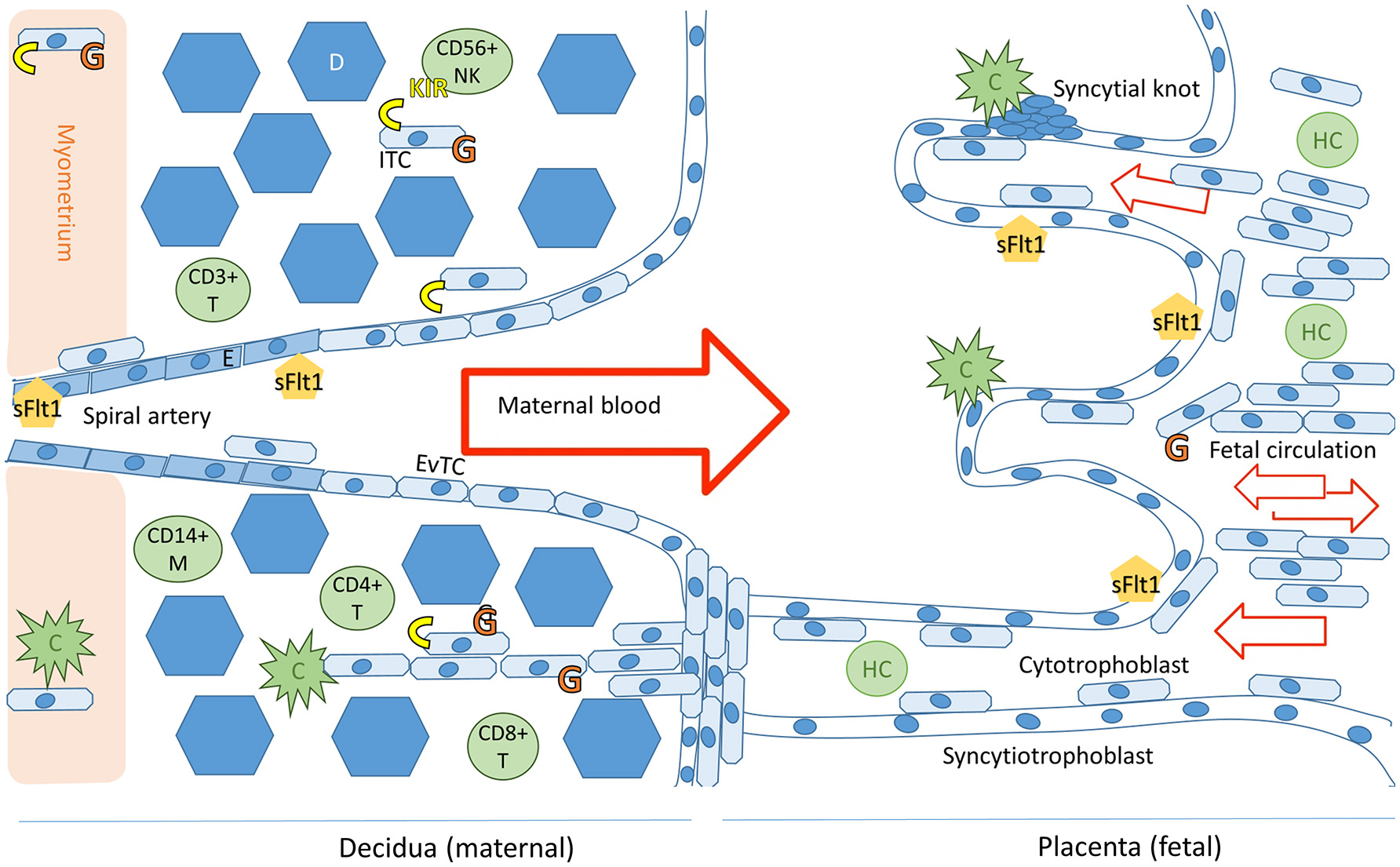
Schematic representation of the maternal-fetal interface and its immunologicalplayers. Trophoblast cells (fetal) and endothelial cells (maternal) express sFlt1. The placental villi are shown in the right side of the image and decidua on the left. Invading trophoblast cells will encounter maternal complement system (C) in the decidua and in the intervillous space. Invading extravillous trophoblasts express HLA-C and HLA-G receptors (in yellow and orange, respectively), expression of HLA-G by villous trophoblast cells in the placenta decreases during the course of the pregnancy (7). Successful trophoblast invasion will extend to the vascular layers of myometrium and invade the uterine spiral arteries, where endovascular trophoblast cells (EvTC) will replace endothelial cells (E) causing remodulation and relaxation of the spiral artery to allow for non-turbulent high volume low pressure circulation into the intervillous space. Interstitial trophoblast cells (ITC) will remain in the maternal tissue creating tolerance of the fetal tissue in the maternal immune system. Hofbauer cells (HC) are the predominant immune cell population in the villi throughout placental development. The decidual immune cell population consists of macrophages (M), natural killer cells (NK), and populations of T-cells (T). Tolerance inducing Treg and Breg cells in particular are essential for a healthy pregnancy (8). Figure adapted from Lokki 2017 PhD thesis (9).
Figure 2
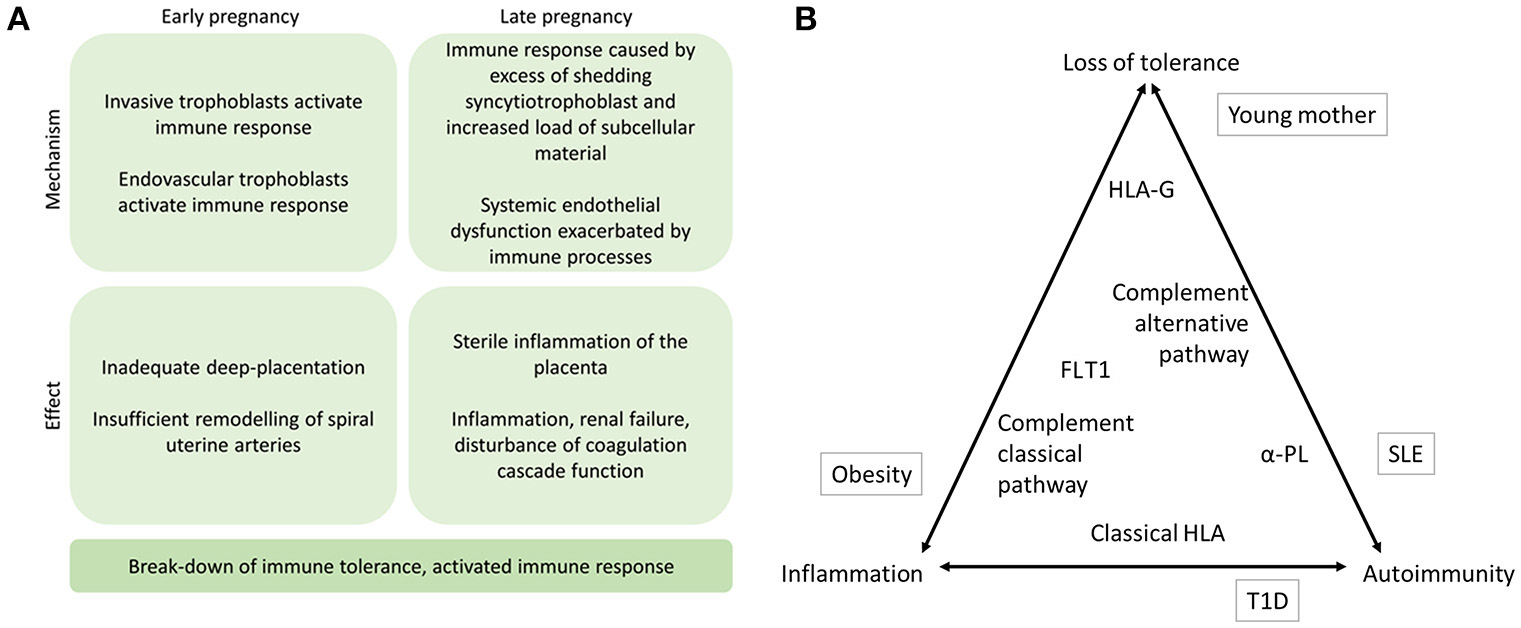
(A) The proposed mechanisms of immune response in the etiology of preeclampsia. Immune mechanisms contribute to preeclampsia in early and late stages of the pregnancy. Specifically, if alternative pathway (AP) of complement system fails to recognize the invading trophoblast cells at the placental bed or in the uterine arteries, placentation may remain superficial and maternal uterine spiral artery remodeling may be compromised. In later stages of the pregnancy, complement activation aiming to facilitate phagosytosis of excess syncytiotrophoblast debris may induce sterile inflammation of the placenta locally. Concurrently, systemic endothelial dysfunction including disturbed coagulation function may be aggravated by immune activation and result in inflammation and shifting of Th2 to Th1 helper cell as well as disturbance of Treg and Th17 cell balances thereby further contributing to activation of the maternal immune response including erroneous antigen presenting in patients with applicable HLA alleles (11). (B) The triad of immune mechanisms in preeclampsia. The placement of contributing factors represents their suggested role in contributing to loss of tolerance, inflammation, and autoimmunity. Environmental risk factors outside in boxes, and the immunogenetic factors discussed in this review are inside the triangle. α-PL, anti-phospholipid antibodies; HLA, human leukocyte antigen; FLT1, Fms-like tyrosine kinase 1; T1D, type 1 diabetes.
Data from epidemiological studies suggest that maternal and paternal genes through fetus affect the risk of preeclampsia, and its genetic basis is polygenic. Heritability of preeclampsia has estimated 55% with greater maternal (35%) than fetal (20%) contribution (12). Genes coding for components of the immune system are among the more important candidates in the quest to pinpoint clinically relevant genetic association. This is also evidenced by numerous non-genetic studies and observations involving components of the immune system (13–15). Activation of alternative pathway of complement activation has been shown to coincide with the critical weeks of placentation (16, 17). The role of decidual monocyte populations in healthy and pathological pregnancies have been reviewed elsewhere (18).
Genetic risk profile for preeclampsia is currently poorly characterized. The genetic studies have suffered from non-reproducibility and been lacking functional validation. Recently, in a large genome wide association study of preeclampsia a first robust association in the fetal genome was found in the common variant near Fms related tyrosine kinase gene (FLT1) encoding anti-angiogenic factor Fms-like tyrosine kinase 1 (FLT1) (19). Our group has published protective maternal low-frequency variants in the same gene (20).
In this mini-review we explore the immunogenetic role of FLT1 in preeclampsia and selected genetic studies implicating loss of immune tolerance in early pregnancy or late pregnancy inflammation in preeclampsia. Dysregulation of complement system and autoimmunity are discussed in detail as potential causes of loss of maternal tolerance, while obesity is considered a possible cause of inflammation.
Immunogenic FLT1? an evolutionary perspective
The anti-angiogenic factor, soluble FLT1 is also known to have an anti-inflammatory function (21). FLT1 is expressed on inflammatory cells in addition to endothelial and trophoblast cells (22). In areas of Africa, where Plasmodium falciparum malaria is endemic, first pregnancies share a particular risk of not only preeclampsia but also of placental malaria (23). In placental malaria, the fetal tissue will express an excess of sFLT1 apparently in an attempt to regulate the maternal inflammatory response thereby reducing the rate of spontaneous abortions (24). Consequently, positive selection on a genetic variant with capacity to resist placental malaria by increasing sFLT concentration may have influenced FLT1 allele frequencies within the general population enough to introduce a novel risk to preeclampsia (25).
Soluble FLT1 is conserved across vertebrates. The human FLT1 protein contains two tyrosine kinase catalytic (TyrKc) domains, three domains from the immunoglobulin (Ig) cell adhesion molecule (cam) subfamily (Igcam), one Ig-like domain, and one true Ig domain (26). In a detailed molecular evolutionary analysis, in contrast to other related proteins, in FLT1, only the TyrKc domains located at amino acids 819-933 and 991-1157 were found to be conserved across vertebrates (26). Large degree of variance between related proteins may be a reflection of recent evolutionary selection pressure on the FLT1. Malaria is known to be a potent source of immunological selection. Together this evidence is in support of possible thus far poorly understood immunological roles of the FLT1.
The major contributor to sFLT1 load in human pregnancy is the recently evolved isoform sFLT1-e15 (27). Overexpression of the primate specific isoform sFLT1-e15a is also associated with preeclampsia suggesting, that this novel isoform harbors thus far unexplained fitness advantages (27, 28). Assuming that sFLT1 is pathogenic, it is thereby possible that in non-primate mammals' conditions that lead to pregnancy-associated pathological rise in sFLT1 do not exist. On the other hand, it is also possible that the sFLT1 in humans has evolved specific functions, patterns of expression, or regulatory mechanisms that are essential for development of preeclampsia (25).
Further evidence of the immunological interactions of FLT1 is derived from a murine model, where increase in complement activation resulted in increased levels of FLT1 (29). Monocytes can be stimulated to express an excess of FLT1 when exposed to complement activation products C3a and C5a in vitro (29). Additionally, nuclear factor of activated T-cells (NFAT) transcription factors are involved in expression of mRNA of inflammatory cytokines, sFLT1-e15, and FLT1, as well as, and secretion of sFLT1 from primary human cytotrophoblasts (30). NFAT transcription factors may in further studies prove to be another link between FLT1 and immune response. Furthermore, angiogenic dysregulation may play a role in activation of the classical pathway in the kidney in a murine model of preeclampsia as evidenced by C4 deposition in the tissue in presence of excess sFLT1 (31).
FLT1 3'UTR dinucleotide repeat polymorphism have been shown to influence the expression of FLT1 and fetal outcome in the context of placental malaria with possible immunomodulatory effect (23). As far as we know, the distribution of these repeat polymorphisms in preeclampsia has not been explored.
Tolerating offspring: dual role of complement system in recognizing and clearing of fetal material
Complement system is an ancient part of innate immunity, which consists of cell surface-bound and freely circulating proteins that interact in a cascade of activation and regulation. Complement system has the capacity to discriminate between self- and non-self-cells and particles, and thereby maintain tolerance, or activate adaptive immunity. Complement activation can lead to inflammation, cell death, and tissue destruction. However, complement system also has a crucial role as a facilitator of phagocytosis thereby clearing debris and altered cells, in addition to removing pathogens. To protect own tissues from complement-mediated destruction and death, pathways of complement activation must be carefully regulated. Complement system has been studied extensively in preeclampsia, but genetic association studies linking components of the complement system to preeclampsia are not as plentiful.
Preeclampsia has previously been likened to thrombotic microangiopathies (TMA), which are caused by inadequate regulation of the complement system. In TMA, complement attacks against endogenous tissue structures such as endothelial cells and blood cells causing vascular damage and kidney failure. Pregnancy can act as a trigger of TMA syndromes. Atypical hemolytic uremic syndrome (aHUS) is a complement disease of the kidney with a TMA mechanism (32). Dysregulation of alternative pathway of complement system is indicated in aHUS (33). Similarly, most preeclampsia associations in complement system are found in the alternative pathway (34, 35).
The component C3 is in the core of the complement system. It can be activated by three different pathways. C3 can also become spontaneously activated in the human serum (32). Via the alternative pathway of complement activation, C3 is spontaneously activated and cleaved into activation products C3a and C3b in the absence of complement regulators. According to the sequence context, a haplotype spanning the active domains of C3 may predispose or protect from severe preeclampsia in a Finnish population (36, 37). We found a haplotype of 16 SNPs spanning the functionally critical sections in the middle of the gene. In this haplotype, three SNPs have most robust independent associations to severe preeclampsia further supporting its functional significance (36).
The results of this and other studies indicate that parallel to mice, C3 also plays a central role in the healthy human pregnancy (38, 39). The mechanism of haplotype association to severe preeclampsia is unclear but the effect may be due to functional or regulatory attributes of this region. For example, functional effect may affect the extravillous trophoblasts' capacity to evade complement activation by C3 binding, thereby compromising deep placentation and spiral artery remodeling in early pregnancy. Inflammation caused by excess complement activation may also be involved in a later stage of preeclampsia influencing severity of symptoms such as hypertension (40).
Immune complexes or opsonisation of the target surface by C1 complex triggers the activation of the classical pathway of complement system. Clearance of placenta-derived particles is crucial when preeclampsia symptoms develop in later pregnancy. Classical pathway activation results in cleavage of C4, which is a homolog of C3. C4 is present in two proteins, C4A and C4B, which are coded by usually two copies of each gene. While copy number variation of both C4 is common, zero copies of both resulting in complete C4 deficiency are very rare (41). Results of a pilot study conducted on mother-infant pairs with early-onset (delivery < 34 weeks of gestation) or late-onset (delivery ≥ 34 weeks of gestation) preeclampsia and non-preeclamptic controls suggest that deficiencies in C4 may predispose to preeclampsia (14). C4A or C4B deficiencies were found almost twice as often in women with early-onset preeclampsia than in healthy controls. C4A deficiencies are observed in 16% of general population in Finland (42). In preeclampsia, C4A deficiencies were found in 40% (2/5) of women with late-onset preeclampsia and in 43% (3/7) of women with early-onset preeclampsia. None were observed in controls (n = 7). The copy number of C4 seems to decrease with the severity of preeclamptic symptoms (14). C4A deficiencies have previously been linked to autoimmune diseases (43). The patients in the preeclampsia study did not suffer from autoimmune diseases. The high incidence of C4A deficiency in preeclampsia supports the importance of classical pathway of complement system in preeclampsia.
Membrane co-factor protein (MCP, CD46) has the capacity to regulate both alternative and classical pathways of complement activation by binding to C3b or C4b and acting as a co-factor to the inactivator enzyme Factor I. MCP is a widely expressed type 1 membrane bound protein. aHUS, can be caused by mutations in MCP (44–47). Proteinuria is one of the cornerstone symptoms of preeclampsia, but its degree varies between patients. Renal dysfunction due to uncontrolled complement activation has been suspected to be the underlying link between preeclampsia and kidney diseases. To investigate whether sequence variants in the CD46 might predispose to preeclampsia, we sequenced the MCP gene in preeclamptic women with severe proteinuria and in non-preeclamptic controls (37, 48). The results of this study do not corroborate the previously reported association of A304V to severe preeclampsia in a cohort of autoimmune pregnancies (49). We observed similar minor allele frequency (MAF) of ~6% in cases and controls. We also found one control woman, who was homozygous to 304*V allele. Heterozygosity for another functional single nucleotide polymorphism (SNP) K32N (rs150429980) was found in one preeclamptic woman and one control. Thereby results are inconclusive. It is possible, however, that MCP plays a part in a particular subtype of preeclampsia, due to the heterogenous nature of the disease.
The complement co-factor I (coded by CFI) is a serine protease that inactivates C3b and C4b in the presence of a co-factor protein, such as complement factor H (FH) and MCP. In the PROMISSE cohort consisting of patients with anti-phospholipid antibodies or systemic lupus erythematosus (SLE), two severe preeclamptic patients with a history of complicated pregnancies were found to carry the loss-of-function mutation I398L in CFI (49). Furthermore, a mutation in the CFH the gene coding for FH with unknown functional consequence was found in another patient (49). While FH is mainly effective in inhibition of the alternative pathway, MCP and factor I have the capacity to regulate both alternative and classical pathways of complement activation.
Immunogenetic predisposition for compromised tolerance
Major histocompatibility (MHC) in chromosome 6 (6p21.3), is the most polymorphic region of the human genome as a result of diverse and shifting immunological selection pressures. Many of the genes in the MHC code for proteins with immunological function. Genes coding for C4A and C4B are located in the MHC as are the genes coding for human leukocyte antigen (HLA) receptors.
At least two autoimmune diseases exist that harbor an increased susceptibility to preeclampsia. Among other immunological defects, aberrant NK cell biology has also been implicated in both, SLE and Type 1 Diabetes. Together these observations might shed light to the disease mechanisms in pregnancy complications.
The proportion of natural killer (NK) cells increases markedly in the uterus/endometrium during implantation and they likely have an important function during early stages of placentation (50, 51). Accordingly, genotypes of KIR receptors on the NK cells in combination with genotypes of their ligands, HLA-C on fetal trophoblast cells have been under investigation in preeclampsia (52–54).
SLE is characterized by a diminished number of NK cells with variety of functional abnormalities (55). SLE shares many symptoms with preeclampsia, including hypertension, proteinuria, and thrombocytopenia. SLE carries a 2- to 4-fold increase in risk of preeclampsia during pregnancy. In a Swedish population-based registry study, the risk for severe preeclampsia in SLE patients was 4.3% (56). Among the MHC loci, the HLA-DRB1*15:01, one of the strongest susceptibility loci for SLE in European-descent populations, is also associated to reproductive failure, i.e., recurrent pregnancy loss and secondary recurrent pregnancy loss (57, 58).
Among pregnant women with Type 1 diabetes (T1D), 15–20% develop preeclampsia (59), and nephropathy further increases the risk of preeclampsia to up to 42–52% (60), which raises questions of shared pathologies. NK cells have a crucial role in trophoblast invasion and spiral artery remodeling in the early stages of pregnancy, as well as in the recognition of the allograft fetal cells. Diverse aberrations of NK cell function are widely evidenced in T1D [reviewed in (61)]. It has been shown, that during the diabetic pregnancy, NK cells adhering to normal decidual endothelium are diminished in comparison to the non-diabetic control pregnancies suggested reduced number of NK cells homing to decidua in the diabetic pregnancy (62). Furthermore, the peripheral blood CD56bright NK cells from pregnant T1D patients expressed very low levels of selectin L (SELL) and alpha 4 integrin (ITGA4), which are important receptors for homing to the uterus (63). CXCL10 and CXCL12 chemokines are produced by the decidua. Their receptors CXCR3 and CXCR4, respectively, were expressed in lower levels on NK cells from T1D patients (63). Furthermore, the expression of activating receptor CD335 in the NK cell is increased during pregnancy in T1D patients. Aberrant NK cell function may result in the increased Th1/Th2 ratio and enhanced activation of intermediate and non-classical monocytes (64), both of which have been observed in preeclampsia, as well as T1D. This may contribute to the underlying mechanism of higher incidence of preeclampsia in T1D patients.
While the villous trophoblasts in the placenta are HLA null, the invasive extravillous trophoblasts (EVT) express genes belonging to the MHC, namely HLA-E, -F, -G, and -C genes (Figure 1). Trophoblast cells do not express HLA class II on the placental surface but syncytiotrophoblast, the outer most layer of placental villi, contains intracellular HLA class II antigens (65). Therefore, compatibility of maternal and fetal HLA genotypes may also influence the immune response in late pregnancy when fetal components are released from the disintegrating placenta. In this context, HLA-A, -B, -DR, and -DQ gene groups may also be relevant in preeclampsia, but thus far, this hypothesis has received little attention.
HLA-G is considered to be protective and tolerogenic during pregnancy. HLA-G is present in semen, which suggests that immune tolerance induction starts already before conception. There are several studies suggesting low or reduced levels of sHLA-G in preeclampsia patients and reduced levels of HLA-G mRNA has been observed in placentas of preeclamptic women (66–69). In a study of genetic polymorphisms, a 14-bp ins/del polymorphism in the 3'UTR of exon 8 of the HLA-G gene was associated with mRNA stability and overall HLA-G production (70). The role of HLA-G polymorphism in preeclampsia is still unresolved.
Antiangiogenic sFLT1 and increased inflammatory response in established preeclampsia
Inappropriate maternal immune responses to trophoblast in early pregnancy may lead to abnormal placentation and set the stage for clinical preeclampsia later in pregnancy. Established preeclampsia is characterized by endothelial dysfunction and systemic inflammatory response to placental oxidative stress [Figure 2A; (71)].
It has been suggested that antiangiogenic sFLT1 sensitizes endothelial cells to pro-inflammatory factors (72). The first genome wide association study of offspring from preeclamptic pregnancies reported that common variants near FLT1 on chromosome 13 were associated with preeclampsia (19). Incidence of preeclampsia is known to be increased in pregnancies with fetal Trisomy 13 (73) suggesting that increased placental production of sFLT1 has a role in preeclampsia susceptibility. Furthermore, the importance of the FLT1 in the disease is evidenced by the recently discovered protective variants in the gene (20).
Normal third-trimester pregnancy is characterized by activation of peripheral blood leukocytes, which is further increased in preeclampsia (74). Increase in the maternal circulating levels of proinflammatory cytokines tumor necrosis factor alpha (TNF α), interleukin (IL)-6, and also the anti-inflammatory cytokine IL-10 in the third trimester of pregnancy in women affected by preeclampsia have been demonstrated in a meta-analysis (75).
Obesity increases the risk of preeclampsia 2- to 3-fold (76–78), but the underlying mechanisms are not fully understood. Obesity is a state of uncontrolled inflammatory responses leading to systemic low-grade inflammation and increased insulin resistance (79). Even modestly overweight women have vascular endothelial dysfunction assessed by brachial artery ultrasound flow-mediated vasodilation (80). The secretion of pro-inflammatory cytokines including TNFα and IL-6 is increased in hypertrophic adipocytes (79). Herse and coworkers have also shown that TNFα decreased sFLT1 expression in mature adipocytes (81). We and others have found lower concentration of sFLT1 in obese compared to normal-weight preeclamptic women, but not in respective normotensive pregnant women (82, 83). Associations between maternal body mass index and proinflammatory cytokines TNFα and MCP-1 in maternal plasma have been demonstrated (84). Thus, the production of sFLT1 and proinflammatory cytokines by placenta and extraplacental sources may be different in obese and normal-weight women during pregnancy and in normal and complicated pregnancies.
Conclusion
The mechanisms regulating the immune response are central in normal pregnancy and the development of preeclampsia (Figure 2A). Mechanisms of autoimmunity, loss of tolerance, and inflammation in preeclampsia are evidenced in this mini-review (Figure 2B). Angiogenic proteins, continuous subclinical inflammation, and insulin resistance in preeclamptic women have been suggested to result in increased cardiovascular risk that with additional risk factors may result in cardiovascular disease (85–87). Thus far, investigation into the genetic background of the immunological pathogenesis of preeclampsia has mostly concentrated on the genes coding complement components and MHC. While both pathways are relevant to the early pregnancy and later clinical manifestations of preeclampsia, studies addressing other immunological mechanisms will be a welcome contribution to increase our understanding of the complex immunological interactions in the disease.
Statements
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Acknowledgments
Figure 1 is adapted from Ph.D. thesis by AL, University of Helsinki 2017, available at https://helda.helsinki.fi/handle/10138/228534 reprinted with permission of University of Helsinki.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1.
TranquilliALDekkerGMageeLRobertsJSibaiBMSteynWet al. The classification, diagnosis and management of the hypertensive disorders of pregnancy: a revised statement from the ISSHP. Pregnancy Hypertens (2014) 4:97–104. 10.1016/j.preghy.2014.02.001
2.
American College of Obstetricians and Gynecologists, Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists' Task Force on Hypertension in Pregnancy. Obstet Gynecol. (2013) 122:1122–31. 10.1097/01.AOG.0000437382.03963.88
3.
LisonkovaSJosephKS. Incidence of preeclampsia: risk factors and outcomes associated with early- versus late-onset disease. Am J Obstet Gynecol. (2013) 209:544.e1–12. 10.1016/j.ajog.2013.08.019
4.
RedmanCWSargentIL. Latest advances in understanding preeclampsia. Science (2005) 308:1592–4. 10.1126/science.1111726
5.
RobertsJMHubelCA. The two stage model of preeclampsia: variations on the theme. Placenta (2009) 30 (Suppl. A):S32–7. 10.1016/j.placenta.2008.11.009
6.
RegalJFBurwickRMFlemingSD. The complement system and preeclampsia. Curr Hypertens Rep. (2017) 19:87. 10.1007/s11906-017-0784-4
7.
KovatsSMainEKLibrachCStubblebineMFisherSJDeMarsR. A class I antigen, HLA-G, expressed in human trophoblasts. Science (1990) 248:220–3.
8.
GhaebiMNouriMGhasemzadehAFarzadiLJadidi-NiaraghFAhmadiMet al. Immune regulatory network in successful pregnancy and reproductive failures. Biomed Pharmacother. (2017) 88:61–73. 10.1016/j.biopha.2017.01.016
9.
LokkiAI. The Conundrum of Pre-eclampsia – From Candidate Genes to Complement System. PhD-thesis, University of Helsinki, Helsinki, Finland (2017).
10.
LynchAMSalmonJE. Dysregulated complement activation as a common pathway of injury in preeclampsia and other pregnancy complications. Placenta (2010) 31:561–7. 10.1016/j.placenta.2010.03.010
11.
Darmochwal-KolarzDKludka-SternikMTabarkiewiczJKolarzBRolinskiJLeszczynska-GorzelakBet al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol. (2012) 93:75–81. 10.1016/j.jri.2012.01.006
12.
CnattingiusSReillyMPawitanYLichtensteinP. Maternal and fetal genetic factors account for most of familial aggregation of preeclampsia: a population-based Swedish cohort study. Am J Med Genet A. (2004) 130A:365–71. 10.1002/ajmg.a.30257
13.
HeYXuBSongDYuFChenQZhaoM. Expression of the complement system's activation factors in plasma of patients with early/late-onset severe pre-eclampsia. Am J Reprod Immunol. (2016) 76:205–211. 10.1111/aji.12541
14.
LokkiAIHeikkinen-ElorantaJJarvaHSaistoTLokkiMLLaivuoriHet al. Complement activation and regulation in preeclamptic placenta. Front Immunol. (2014) 5:312. 10.3389/fimmu.2014.00312
15.
BanadakoppaMVidaeffACYallampalliURaminSMBelfortMAYallampalliC. Complement split products in amniotic fluid in pregnancies subsequently developing early-onset preeclampsia. Dis Markers (2015) 2015:263109. 10.1155/2015/263109
16.
LynchAMMurphyJRByersTGibbsRSNevilleMCGiclasPCet al. Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am J Obstet Gynecol. (2008) 198:385.e1–9. 10.1016/j.ajog.2007.10.793
17.
LynchAMWagnerBDGiclasPCWestNAGibbsRSHolersVM. The relationship of longitudinal levels of complement Bb during pregnancy with preeclampsia. Am J Reprod Immunol. (2016) 75:104–11. 10.1111/aji.12439
18.
NingFLiuHLashGE. The role of decidual macrophages during normal and pathological pregnancy. Am J Reprod Immunol. (2016) 75:298–309. 10.1111/aji.12477
19.
McGinnisRSteinthorsdottirVWilliamsNOThorleifssonGShooterSHjartardottirSet al. Variants in the fetal genome near FLT1 are associated with risk of preeclampsia. Nat Genet. (2017) 49:1255–60. 10.1038/ng.3895
20.
LokkiAIDalyETriebwasserMKurkiMIRobersonEDOHappolaPet al. Protective low-frequency variants for preeclampsia in the fms related tyrosine kinase 1 gene in the Finnish population. Hypertension (2017) 70:365–71. 10.1161/HYPERTENSIONAHA.117.09406
21.
YanoKLiawPCMullingtonJMShihSCOkadaHBodyakNet al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J Exp Med. (2006) 203:1447–58. 10.1084/jem.20060375
22.
LuttunATjwaMMoonsLWuYAngelillo-ScherrerALiaoFet al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. (2002) 8:831–40. 10.1038/nm731
23.
MuehlenbachsAFriedMLachowitzerJMutabingwaTKDuffyPE. Natural selection of FLT1 alleles and their association with malaria resistance in utero. Proc Natl Acad Sci USA. (2008) 105:14488–491. 10.1073/pnas.0803657105
24.
MuehlenbachsAMutabingwaTKEdmondsSFriedMDuffyPE. Hypertension and maternal-fetal conflict during placental malaria. PLoS Med. (2006) 3:e446. 10.1371/journal.pmed.0030446
25.
BrownEARuvoloMSabetiPC. Many ways to die, one way to arrive: how selection acts through pregnancy. Trends Genet. (2013) 29:585–92. 10.1016/j.tig.2013.03.001
26.
DormerABeckG. Evolutionary analysis of human vascular endothelial growth factor, angiopoietin, and tyrosine endothelial kinase involved in angiogenesis and immunity. In Silico Biol. (2005) 5:323–39.
27.
PalmerKRTongSKaitu'u-LinoTJ. Placental-specific sFLT-1: role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol Hum Reprod. (2017) 23:69–78. 10.1093/molehr/gaw077
28.
ThomasCPAndrewsJIRaikwarNSKelleyEAHerseFDechendRet al. A recently evolved novel trophoblast-enriched secreted form of fms-like tyrosine kinase-1 variant is up-regulated in hypoxia and preeclampsia. J Clin Endocrinol Metab. (2009) 94:2524–30. 10.1210/jc.2009-0017
29.
GirardiGYarilinDThurmanJMHolersVMSalmonJE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. (2006) 203:2165–75. 10.1084/jem.20061022
30.
YeLGrattonAHannanNJCannonPDeoMPalmerKRet al. Nuclear factor of activated T-cells (NFAT) regulates soluble fms-like tyrosine kinase-1 secretion (sFlt-1) from human placenta. Placenta (2016) 48:110–18. 10.1016/j.placenta.2016.10.013
31.
PenningMChuaJSvan KootenCZandbergenMBuurmaASchutteJet al. Classical complement pathway activation in the kidneys of women with preeclampsia. Hypertension (2015) 66:117–25. 10.1161/HYPERTENSIONAHA.115.05484
32.
MeriS. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med. (2013) 24:496–502. 10.1016/j.ejim.2013.05.009
33.
SridharanMGoRSWillrichMAV. Atypical hemolytic uremic syndrome: review of clinical presentation, diagnosis and management. J Immunol Methods (2018). 461:15–22. 10.1016/j.jim.2018.07.006
34.
AlrahmaniLWillrichMAV. The complement alternative pathway and preeclampsia. Curr Hypertens Rep. (2018) 20:40. 10.1007/s11906-018-0836-4
35.
LokkiAITriebwasserMDalyEAuroKPerolaMFINNPECet al. Rare mutations in factor H predispose to severe preeclampsia. Mol Immunol. (2018) 102:184–85. 10.1016/j.molimm.2018.06.147
36.
LokkiAIKaartokallioTHolmbergVOnkamoPKoskinenLLESaavalainenPet al. Analysis of complement C3 gene reveals susceptibility to severe preeclampsia. Front Immunol. (2017) 8:589. 10.3389/fimmu.2017.00589
37.
JääskeläinenTHeinonenSKajantieEKereJKivinenKPoutaAet al. Cohort profile: the finnish genetics of pre-eclampsia consortium (FINNPEC). BMJ Open (2016) 6:e01314810.1136/bmjopen-2016-013148
38.
XuCMaoDHolersVMPalancaBChengAMMolinaH. A critical role for murine complement regulator crry in fetomaternal tolerance. Science (2000) 287:498–501. 10.1126/science.287.5452.498
39.
MohlinFCGrosPMercierEGrisJRBlomAM. Analysis of C3 Gene variants in patients with idiopathic recurrent spontaneous pregnancy loss. Front Immunol. (2018) 9:1813. 10.3389/fimmu.2018.01813
40.
LillegardKEJohnsonACLojovichSJBauerAJMarshHCGilbertJSet al. Complement activation is critical for placental ischemia-induced hypertension in the rat. Mol Immunol. (2013) 56:91–7. 10.1016/j.molimm.2013.04.009
41.
LokkiMLColtenHR. Genetic deficiencies of complement. Ann Med. (1995) 27:451–9. 10.3109/07853899709002453
42.
WennerstromAVlachopoulouELahtelaLEPaakkanenREronenKTSeppanenMet al. Diversity of extended HLA-DRB1 haplotypes in the Finnish population. PLoS ONE (2013) 8:e79690. 10.1371/journal.pone.0079690
43.
SamanoESRibeiroLde MGorescuRGRochaKCGrumachAS. Involvement of C4 allotypes in the pathogenesis of human diseases. Rev Hosp Clin Fac Med Sao Paulo (2004) 59:138–44. 10.1590/S0041-87812004000300009
44.
ManchesterMValsamakisAKaufmanRLiszewskiMKAlvarezJAtkinsonJPet al. Measles virus and C3 binding sites are distinct on membrane cofactor protein (CD46). Proc Nat Acad Sci USA. (1995) 92:2303–7.
45.
RichardsAKempEJLiszewskiMKGoodshipJALampeAKDecorteRet al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Nat Acad Sci USA. (2003) 100:12966–71. 10.1073/pnas.2135497100
46.
Fremeaux-BacchiVMoultonEAKavanaghDDragon-DureyMBlouinJCaudyAet al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2006) 17:2017–25. 10.1681/ASN.2005101051
47.
FangCJFremeaux-BacchiVLiszewskiMKPianettiGNorisMGoodshipTHJet al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood (2008) 111:624–32. 10.1182/blood-2007-04-084533
48.
LokkiAIAalto-ViljakainenTMeriSLaivuoriH. Genetic analysis of membrane cofactor protein (CD46) of the complement system in women with and without preeclamptic pregnancies. PLoS ONE (2015) 10:e0117840. 10.1371/journal.pone.0117840
49.
SalmonJEHeuserCTriebwasserMLiszewskiMKKavanaghDRoumeninaLet al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med. (2011) 8:e1001013. 10.1371/journal.pmed.1001013
50.
AshkarAADiSanto JPCroyBA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. (2000) 192:259–70. 10.1084/jem.192.2.259
51.
BulmerJNWilliamsPJLashGE. Immune cells in the placental bed. Int J Dev Biol. (2010) 54:281–94. 10.1387/ijdb.082763jb
52.
HibySEWalkerJJO'ShaughnessyKMRedmanCWGCarringtonMTrowsdaleJet al. Combinations of maternal KIR and fetal HLA-C genes influence the risk of preeclampsia and reproductive success. J Exp Med. (2004) 200:957–65. 10.1084/jem.20041214
53.
SaitoSTakedaYSakaiMNakabayahiMHayakawaS. The incidence of pre-eclampsia among couples consisting of Japanese women and Caucasian men. J Reprod Immunol. (2006) 70:93–8. 10.1016/j.jri.2005.12.005
54.
HibySEAppsRChazaraOFarrellLEMagnusPTrogstadLet al. Maternal KIR in combination with paternal HLA-C2 regulate human birth weight. J Immunol. (2014) 192:5069–73. 10.4049/jimmunol.1400577
55.
SpadaRRojasJMBarberDF. Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J Leukoc Biol. (2015) 98:479–87. 10.1189/jlb.4RU0315-081RR
56.
SimardJFArkemaEVNguyenCSvenungssonEWikstromAKPalmstenKet al. Early-onset preeclampsia in lupus pregnancy. Paediatr Perinat Epidemiol. (2017) 31:29–36. 10.1111/ppe.12332
57.
KolteAMSteffensenRChristiansenOBNielsenHS. Maternal HY-restricting HLA class II alleles are associated with poor long-term outcome in recurrent pregnancy loss after a boy. Am J Reprod Immunol. (2016) 76:400–5. 10.1111/aji.12561
58.
GambinoCMDiBona DAielloACarruCDuroGGugginoGet al. HLA-C1 ligands are associated with increased susceptibility to systemic lupus erythematosus. Hum Immunol. (2018) 79:172–7. 10.1016/j.humimm.2018.01.005
59.
WeissgerberTLMuddLM. Preeclampsia and diabetes. Curr Diab Rep. (2015) 15:3–9. 10.1007/s11892-015-0579-4
60.
KlemettiMMLaivuoriHTikkanenMNuutilaMHiilesmaaVTeramoK. Obstetric and perinatal outcome in type 1 diabetes patients with diabetic nephropathy during 1988-2011. Diabetologia (2015) 58:678–86. 10.1007/s00125-014-3488-1
61.
MarcaVGianchecchiEFierabracciA. Type 1 diabetes and its multi-factorial pathogenesis: the putative role of NK cells. Int J Mol Sci. (2018) 19:E794. 10.3390/ijms19030794
62.
SeawardAVBurkeSDRamshawHSmithGNCroyBA. Circulating CD56+ cells of diabetic women show deviated homing potential for specific tissues during and following pregnancy. Hum Reprod. (2011) 26:1675–84. 10.1093/humrep/der114
63.
BurkeSDSeawardAVRamshawHSmithGNViraniSCroyBAet al. Homing receptor expression is deviated on CD56+ blood lymphocytes during pregnancy in Type 1 diabetic women. PLoS ONE (2015) 10:e0119526. 10.1371/journal.pone.0119526
64.
GroenBvan der WijkAEvan den BergPPLefrandtJDvan den BergGSollieKMet al. Immunological adaptations to pregnancy in women with type 1 diabetes. Sci Rep. (2015) 5:13618. 10.1038/srep13618
65.
RanellaAVassiliadisSMastoraCValentinaMDionyssopoulouEAthanassakisI. Constitutive intracellular expression of human leukocyte antigen (HLA)-DO and HLA-DR but not HLA-DM in trophoblast cells. Hum Immunol. (2005) 66:43–55. 10.1016/j.humimm.2004.10.002
66.
YieSMTaylorRNLibrachC. Low plasma HLA-G protein concentrations in early gestation indicate the development of preeclampsia later in pregnancy. Am J Obstet Gynecol. (2005) 193:204–08. 10.1016/j.ajog.2004.11.062
67.
HackmonRKoifmanAHyodoHGlickmanHSheinerEGeraghtyDE. Reduced third-trimester levels of soluble human leukocyte antigen G protein in severe preeclampsia. Am J Obstet Gynecol. (2007) 197:255.e1–255.e5. 10.1016/j.ajog.2007.06.033
68.
HeYChenSHuangHChenQ. Association between decreased plasma levels of soluble human leukocyte antigen-G and severe pre-eclampsia. J Perinat Med. (2016) 44:283–90. 10.1515/jpm-2015-0062
69.
MarozioLGarofaloABerchiallaPTavellaAMSaltonLCavalloFet al. Low expression of soluble human leukocyte antigen G in early gestation and subsequent placenta-mediated complications of pregnancy. J Obstet Gynaecol Res. (2017) 43:1391–6. 10.1111/jog.13377
70.
Lynge NilssonLDjurisicSHviidTV. Controlling the immunological crosstalk during conception and pregnancy: HLA-G in reproduction. Front Immunol. (2014) 5:198. 10.3389/fimmu.2014.00198
71.
RedmanCWSargentIL. Immunology of pre-eclampsia. Am J Reprod Immunol. (2010) 63:534–43. 10.1111/j.1600-0897.2010.00831.x
72.
Cindrova-DaviesTSandersDABurtonGJCharnock-JonesDS. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor-mediated signalling. Cardiovasc Res. (2011) 89:671–9. 10.1093/cvr/cvq346
73.
BdolahYPalomakiGEYaronYBdolah-AbramTGoldmanMLevineRJet al. Circulating angiogenic proteins in trisomy 13. Am J Obstet Gynecol. (2006) 194:239–45. 10.1016/j.ajog.2005.06.031
74.
SacksGPStudenaKSargentKRedmanCW. Normal pregnancy and preeclampsia both produce inflammatory changesin peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. (1998) 179:80–6. 10.1016/S0002-9378(98)70254-6
75.
LauSYGuildSJBarrettCJChenQMcCowanLJordanVet al. Tumor necrosis factor-alpha, interleukin-6, and interleukin-10 levels are altered in preeclampsia: a systematic review and meta-analysis. Am J Reprod Immunol. (2013) 70:412–27. 10.1111/aji.12138
76.
BodnarLMNessRBMarkovicNRobertsJM. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann Epidemiol. (2005) 15:475–82. 10.1016/j.annepidem.2004.12.008
77.
DuckittKHarringtonD. Risk factors for pre-eclampsia at antenatal booking: systematic review of controlled studies. BMJ (2005) 330:565. 10.1136/bmj.38380.674340.E0
78.
JeyabalanA. Epidemiology of preeclampsia: impact of obesity. Nutr Rev. (2013) 71 (Suppl. 1):S18–25. 10.1111/nure.12055
79.
ChoeSSHuhJYHwangIJKimJIKimJB. Adipose tissue remodeling: its role in energy metabolism and metabolic disorders. Front Endocrinol. (2016) 7:30. 10.3389/fendo.2016.00030
80.
PatelARHuiHKuvinJTPandianNGKarasRH. Modestly overweight women have vascular endothelial dysfunction. Clin Cardiol. (2009) 32:269–73. 10.1002/clc.20451
81.
HerseFFainJNJankeJEngeliSKuhnCFreyNet al. Adipose tissue-derived soluble fms-like tyrosine kinase 1 is an obesity-relevant endogenous paracrine adipokine. Hypertension (2011) 58:37–42. 10.1161/HYPERTENSIONAHA.111.171322
82.
SuwakiNMasuyamaHNakatsukasaHMasumotoASumidaYTakamotoNet al. Hypoadiponectinemia and circulating angiogenic factors in overweight patients complicated with pre-eclampsia. Am J Obstet Gynecol. (2006) 195:1687–92. 10.1016/j.ajog.2006.04.003
83.
JääskeläinenTHeinonenSHämäläinenEPulkkiKRomppanenJLaivuoriHFINNPEC. Impact of obesity on angiogenic and inflammatory markers in the Finnish Genetics of Pre-eclampsia Consortium (FINNPEC) cohort. Int J Obes. (2018). [Epub ahead of print]. 10.1038/s41366-018-0217-8
84.
AyeILLagerSRamirezVIGaccioliFDudleyDJJanssonTet al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. (2014) 90:129. 10.1095/biolreprod.113.116186
85.
SattarNGreerIA. Pregnancy complications and maternal cardiovascular risk: opportunities for intervention and screening?BMJ (2002) 325:157–60. 10.1136/bmj.325.7356.157
86.
KvehaugenASDechendRRamstadHBTroisiRFugelsethDStaffAC. Endothelial function and circulating biomarkers are disturbed in women and children after preeclampsia. Hypertension (2011) 58:63–9. 10.1161/HYPERTENSIONAHA.111.172387
87.
WolfMHubelCALamCSampsonMEckerJLNessRBet al. Preeclampsia and future cardiovascular disease: potential role of altered angiogenesis and insulin resistance. J Clin Endocrinol Metab. (2004) 89:6239–43. 10.1210/jc.2004-0548
Summary
Keywords
preeclampsia, genetics, complement, major histocompatibility complex, FLT1, autoimmunity, pregnancy
Citation
Lokki AI, Heikkinen-Eloranta JK and Laivuori H (2018) The Immunogenetic Conundrum of Preeclampsia. Front. Immunol. 9:2630. doi: 10.3389/fimmu.2018.02630
Received
28 June 2018
Accepted
25 October 2018
Published
13 November 2018
Volume
9 - 2018
Edited by
Nandor Gabor Than, Hungarian Academy of Sciences (MTA), Hungary
Reviewed by
Shigeru Saito, University of Toyama, Japan; Offer Erez, Soroka Medical Center, Israel
Updates
Copyright
© 2018 Lokki, Heikkinen-Eloranta and Laivuori.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: A. Inkeri Lokki inkeri.lokki@helsinki.fi
This article was submitted to Immunological Tolerance and Regulation, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.