 Milena Vukelic†
Milena Vukelic† Yi Li
Yi Li Vasileios C. Kyttaris
Vasileios C. Kyttaris- Division of Rheumatology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, United States
Purpose of Review: The standard treatment options for systemic lupus erythematosus (SLE) are focused on non-specific immunosuppression. Over the past few years, scientific studies and ongoing clinical trials have shifted the paradigm with rapid advances in developing biologics and small molecules. A number of monoclonal antibodies and small molecule inhibitors have been developed to target specific pathways involved in SLE. Many of these novel therapeutic agents are already being tested in clinical trials and they may 1 day reshape the landscape of SLE treatment. Herein we review potential future therapeutic options for SLE.
Introduction
In the past few years, greater understanding of the pathogenesis of SLE has translated into the development of more targeted therapeutic agents in various stages of clinical trials. Current treatment regiments for SLE typically comprise some combination of glucocorticoids, antimalarials, immune suppressive drugs, and cytotoxic agents in severe cases. The first biologic agent approved for SLE, Belimumab, has been in clinical practice for more than 5 years with overall positive albeit modest results (1). Therefore, developing more effective treatment for lupus remains a priority in the field.
Recent studies have identified numerous immunological checkpoints that are dysregulated in SLE and contribute to the loss of self-tolerance. A pipeline of novel agents are being developed to specifically target intracellular signaling pathways, inflammatory cytokines, chemokines, cell surface costimulation molecules, and the proteasome (Figure 1). Herein we will review the potential novel treatment options that are currently being tested in clinical trials for SLE.
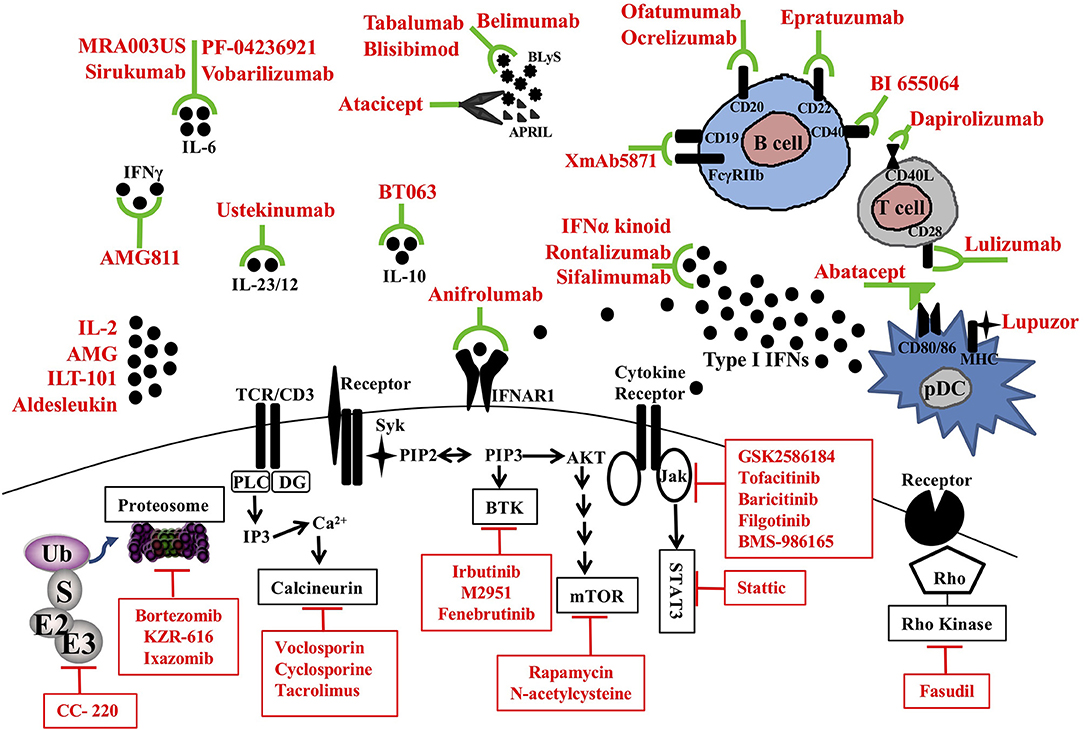
Figure 1. Therapeutic targets and novel treatments in SLE. Target molecules (black) and corresponding therapeutic agents (red) in clinical trials are illustrated. pDC, plasmacytoid dendritic cell; MHC, major histocompatibility complex; CD, cluster of differentiation; APRIL, a proliferating-induced ligand; BLys, B-lymphocyte stimulator; IL, interleukin; TCR, T cell receptor; IFNAR1, Interferon alpha receptor 1; BTK, Bruton's tyrosine kinase; mTOR, mammalian target of rapamycin; Jak, Janus kinases; STAT, signal transducer and activator of transcription proteins; Ub, ubiquitin; E3, Ubiquitin protein ligases.
B Cell Inhibition
Systemic lupus erythematosus is a multisystem autoimmune disease characterized by the production of autoantibodies that primarily target a variety of nuclear antigens, deposit in tissues and activate complement. Plasma cells and their precursors, B cells, are fundamental to the development of these antibodies, and therefore are a prime therapeutic target for intervention in the disease.
B Lymphocyte Stimulator (BLyS)/A Proliferation-Inducing Ligand (APRIL) Belimumab was the first FDA approved fully humanized monoclonal anti-BLyS antibody for use in SLE more than 5 years ago. Administration of Belimumab was found to benefit SLE patients who had positive anti-double-stranded (ds) DNA and low complement (C3 or C4) levels. Moreover, the pivotal trials that lead to the approval of belimumab used a novel at the time composite outcome measure, the SLEDAI response index (SRI). Patients would be regarded SRI responders if they have (1) at least 4 points decrease in their SLEDAI scores over the period of the study, (2) No worsening of the physician global assessment (PGA), and (3). No new BILAG A or more than one BILAG B scores. The original SRI can be modified to require higher decrease in SLEDAI score e.g., by 5 or 6 points (SRI-5, SRI-6, respectively).
Compared to the placebo group's 44% SLEDAI response index (SRI)-4, the belimumab group had SRI-4 of 58%, indicating a statistically significant but yet modest effect. This modest effectiveness was later confirmed by an extensive post- marketing surveillance, as well as, its overall safety profile. Subcutaneously administered belimumab had similar efficacy at week 52 with SRI-4 response of 61.4% vs. placebo 48.4% (p = 0.0006) (2). It is worth mentioning that the effectiveness of belimumab remains unclear in severe renal and CNS disease as patients with these manifestations were excluded from the initial studies (1, 3). A study investigating the usefulness of belimumab in patients with lupus nephritis is currently ongoing (NCT01639339).
Tabalumab, a monoclonal antibody against BLyS that neutralizes membrane-bound and soluble BLyS, was assessed for its effectiveness in moderately active lupus in two large phase III clinical trials (ILLUMINATE-1 & 2). Although tabalumab treatment resulted in favorable changes in disease biomarkers (anti-dsDNA abs and complement levels), efficacy was marginal with SRI-5 of 38.4% in the tabalumab-treated vs. 27.7% in the placebo group (p = 0.002) in one trial. There was no statistical difference between the two groups in the second trial. Again, the treatment with tabalumab was found to be relatively safe as was the case with belimumab (4–6).
Blisibimod is a BLyS -neutralizing agent composed of a tetrameric BLyS binding domain fused to a human IgG1 Fc region. It binds both soluble and membrane-bound BLyS. In the recently completed phase III trial (CHABLISSC1) in patients with severe disease (SELENA-SLEDAI score ≥10), blisibimod showed a statistically significant steroid-sparing effect, reduction in SLE autoantibodies, B cell count, and proteinuria while increasing complement levels (7). SRI-6 as a primary end point was not met since response rate in the control subjects in this study was very high compared to prior SLE trials; 46.9% in the Blisibimod vs. 42.3% jn the control group. Higher steroid dosing in the placebo arm may have contributed to the relatively high response rates, confounding the primary efficacy outcome. Blisibimod was well-tolerated and the most common adverse events were upper respiratory or urinary tract infection and diarrhea (7).
Atacicept is a fully human recombinant fusion protein made of the extracellular portion of the TACI receptor and the Fc portion of human IgG. As atacicept blocks both BLyS and APRIL (8), it was predicted that atacicept may have a more potent effect on immunoglobulin production. Indeed, a significant high risk of severe infection and a decreased in immunoglobulin levels lead to a terminated phase II/III APRIL-SLE trial in nephritis (9). Similar effect on immunoglobulin levels was seen in the ADDRESS II trial (10) where effectiveness of atacicept to improve serologic markers and prevent lupus flares was superior to placebo only with 150 mg twice weekly dosing. The safety profile was acceptable with no reportedly increase in the overall frequency of serious adverse effects as compared to placebo. However, further assessment of the long-term safety of atacicept is warranted as this study only evaluated the safety and efficacy at 24-weeks (10). Given these results, the initial enthusiasm with this molecule has largely dissipated.
Overall, anti-BLyS but probably not anti-APRIL therapies, represent a moderately effective and safe approach in the management of patients with moderately active SLE with musculoskeletal and skin manifestations, especially if they remain corticosteroid dependent.
Anti-CD 20
Unlike BLyS inhibition with the capacity of altering B cell maturation, CD20 targeting therapy depletes mature B cells without affecting plasma cells. Rituximab (RTX) is the most widely used anti-CD20 antibody; due to its chimeric nature, it was found to cause allergic reactions in approximately 10% of patients. Therefore, in the past few years several fully humanized anti-CD20 antibodies have been developed, such as ocrelizumab, ofatumumab, and obinutuzumab. Small uncontrolled trials showed that rituximab, already known to be effective in rheumatoid arthritis (11), can also ameliorate lupus (12). The non-randomized “Rituxilup” trial (n = 50) used rituximab and methyl prednisolone followed by mycophenolate mofetil in newly diagnosed lupus nephritis. Ninety percent of patients achieving a partial or complete remission by 37 weeks of treatement. A randomized multicenter clinical trial conducted by Rovin et al., was recently terminated prematurely due to slow recruitment (CTN84054592). But other pivotal trials in lupus nephritis (LUNAR) (13) and non-renal SLE (EXPLORER) were largely negative (14). Currently, the European League Against Rheumatism (EULAR) recommends rituximab as a treatment of last resort in severe lupus (15).
More recently, a few case reports (16, 17) and a phase 2A open-label proof-of-concept suggested that the combination of RTX and anti-BLyS (belimumab, BLM) could be effective. 11/16 patients with refractory SLE achieved renal responses (defined as proteinuria decreased to ≤0.7 g/24 h, normal serum albumin, stable creatinine and a normal urinary sediment). Importantly, RTX + BLM reduced nuclear autoantibody titers, and prevented the spike of circulating BLyS that is common after B-cell depletion. Similar results were observed in another small case series of patients with refractory SLE, who entered long term remission and discontinued corticosteroids (18, 19).
Similarly, the humanized anti-CD20 ocrelizumab failed to show significant efficacy in two early terminated phase III trials (BELONG and BEGIN). Patients with class III and IV nephritis were enrolled in BELONG (20) that compared ocrelizumab to placebo. Patients also received mycophenolate mofetil or cyclophosphamide (euro-lupus nephritis treatment protocol). Non-renal SLE patients were enrolled in the BEGIN trial. Both trials were terminated after significant increases of severe infections were noted in the ocrelizumab group. It has to be noted that efficacy analysis showed a trend favoring ocrelizumab over placebo.
Ofatumumab was administered to 16 SLE patients who could not tolerate rituximab; 87% (14/16) of these patients tolerated the infusion. About 85% patient achieved B cell-depletion with associated improvements in serological markers of disease activity (ANA, anti-dsDNA and complement levels). Half of the patients with lupus nephritis achieved renal remission by 6 months. Overall safety profile seems acceptable with 5/16 patients developing grade III infections; no malignancies or deaths were reported during the 28 months follow up (21). One SLE patient with autoimmune hemolytic anemia who failed rituximab, achieved clinical remission after ofatumumab (22). Four patients with nephritis achieved reduction of proteinuria and anti-dsDNA levels (23). There are no active formal clinical trials in SLE. A 52-week, phase II trial studying safety and efficacy of obinutuzumab, a different anti-CD20 antibody, in lupus neprhritis is currently active with estimated completion date in December 2019 (NCT02550652).
As per EULAR recommendations, anti-CD20 treatment can be tried in refractory SLE patients. Severe infections remain a concern as many of these patients are already receiving other immunosuppressive medications.
Anti-CD22
CD22 is a surface molecule that modulates B cell activation and migration. Epratuzumab is a humanized anti-CD22 antibody, initially showed positive results reaching its primary endpoint (BICLA response) in the EMBLEM phase II trials (24). However, the beneficial effect was not replicated in the larger and more stringently performed phase III EMBODY trial (25). The reason of failure was thought due to sub-optimal dosing, high placebo response rates, and inadequate optimization of standard of care. Interestingly, some promising response was observed in subgroups of patient with features of Sjogren's syndrome and positive anti-SSA antibodies. Further research is needed to explore this and other potential sub-groups that might respond (26).
Anti-CD19
CD19 is a surface receptor found exclusively on B cells. XmAb5871 is an antibody that co-engages CD19 and the inhibitory FcγRIIb receptor, resulting in B cell inhibition but not ablation (27). There is an ongoing randomized, double-blinded, placebo-controlled study of XmAb5871 to determine its ability to maintain SLE remission achieved by a brief course of steroid therapy (NCT02725515). This clinical trial has an interesting design: Moderately active SLE patients are taken off traditional therapies and are given high dose parenteral steroids. In theory most patients will become clinically inactive. Then the patients of both XmAb5871 and placebo group are observed for development of flare. The outcome measure would be time to relapse after the initial induction of remission. This unconventional study design is likely to reduce the placebo response by eliminating the effect of background therapy.
Proteasome Inhibitors
The lack of expression of CD20 on plasma cells, especially long-lived plasma cells may be one of the reasons for the poor performance of anti-CD20 therapies in SLE (28). This can be addressed by targeting specifically the plasma cell ability to produce immune globulins by inhibiting the proteasome. This organelle handles misfolded proteins, produced at high levels during immune-globulin assembly, and has proven critical for plasma cell function.
Bortezomib is a proteasome inhibitor, that is efficacious in plasma cell cancers. In lupus prone MPL/lpr mice, administration of bortezomib was found effective in preventing and more importantly treating established disease (29). Similar effects were observed with two other proteasome inhibitors, carfilzomib (30), and delanzomib (31) when used in preclinical models of lupus nephritis (NZBW F1 and MRL/lpr mice). Following these successful studies, bortezomib was infused in 12 patients with refractory SLE. The patients had significant improvement in several clinical parameters such as dermatitis, proteinuria, arthritis, and serositis. Neuropathy developed in 2/12 patients as a major side effect. Currently, bortezomib is assessed in a phase II clinical trial in SLE patients (NCT02102594) with estimated completion date in December 2018.
Although proteasome inhibition is attractive, the main concern for adding these seemingly potent medications in the SLE therapeutic armamentarium remains the severe toxicity associated with their chronic use.
Intracellular Signaling
Bruton's Tyrosine Kinase (Btk)
Btk is a B-cell receptor (BCR) associated kinase that activates the NFkB pathway (32). Importantly though it also associates with the Fc receptor in monocytes (33) and can bridge BCR and TLR9 signaling (34). Mutations in the Btk gene result in agammaglobulinemia (35). Ibrutinib is already in use for B cell malignancies and has good safety profile (36). In mouse models of lupus nephritis treatment with Btk inhibitors PF-06250112 (37) and ibrutinib (38) resulted in less severe nephritis. This served as the base for the Btk inhibitor MSC2364447C (M2951) to be evaluated in a phase Ib trial in SLE patients with mild to moderate disease (NCT02537028). Fenebrutinib (GDC-0853), an orally available inhibitor of Btk, is evaluated in a phase II clinical study in patients with moderate to severe active SLE (NCT03407482). Participants will receive GDC-0853 twice daily for 48 weeks and will be followed for additional 8 weeks to evaluate the long-term safety and efficacy.
Cereblon Modulator (CC-220)
CC-220 binds to cereblon (CRBN), a substrate receptor of CUL4CRBN E3 ubiquitin ligase complex. As an immunomodulatory compound, CC-220 can lead a substrate specific ubiquitination of transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) both essential for antibody production (38–40). A Pilot phase II randomized, placebo-controlled, double-blind study is underway to evaluate efficacy, safety, tolerability, pharmacokinetics of CC-220 in patients with SLE (NCT02185040). CC-220 showed some efficacy but there were important safety issues in a 12-week, phase II, dose-escalation study of 42 patients and 14% of patients stopped treatment because of adverse effects. Higher doses of CC-220 were associated with neutropenia, pneumonia, and dermatitis (41).
Calcineurin Inhibitors
Activated lupus T cells show an exaggerated calcium response, which leads to early and sustained activation of the phosphatase calcineurin and its substrate, the transcription factor nuclear factor of activated T cells (NFAT). NFAT upregulates a number of genes, including CD154 (also called CD40L) (42), a critical molecule for T:B cell interaction. Calcineurin inhibitors cyclosporine and tacrolimus, have been successfully used in preventing transplant rejection through blocking this important pathway. Moreover, calcineurin inhibitors may have an antiproteinuric effect, rendering them an important treatment alternative or adjuvant therapy for lupus nephritis. Compared to mycophenolate mofetil, tacrolimus was found to be non-inferior in induction of remission (62 vs. 59%). However, there was a trend for more flares in the tacrolimus group (43).
Voclosporin, a novel calcineurin inhibitor, was investigated in a phase II trial for lupus nephritis as a combination therapy with mycophenolate mofetil (AURA trial). Patients received mycophenolate alone or Voclosporin at 39.5 or 27.5 mg combined with mycophenolate. Remission rates at 6 months favored the combination therapy over mycophenolate alone (OR = 2.03). Unfortunately, the voclosporin-mycophenolate combination resulted in severe side effects including 12 deaths vs. 1 death in the mycophenolate group alone (44) despite the use of rather low corticosteroid doses. Double-blind, placebo-controlled AURORA (NCT03021499) phase 3 clinical trial has started with a plan to include 320 patients with nephritis. It will determine if a combination of voclosporin and a standard of care therapy with mycophenolate mofetil increases kidney function, compared with to standard of therapy alone.
Overall, calcineurin inhibitors alone or combined with mycophenolate represent acceptable alternatives for lupus nephritis treatment; the combination though may carry a significant risk for serious infections.
Mammalian Target of Rapamycin (mTOR) Signaling Inhibitor
mTOR is a highly conserved serine/threonine kinase and is well known to be essential for the regulation of cell metabolism, growth, and proliferation (45, 46). Activated mTOR in lupus T cells is associated with several abnormalities including expansion of both TH17 and CD3+CD4−CD8− double negative T, as well as, contraction of Tregs (47, 48).
Administration of the mTOR inhibitor rapamycin, results in immediate inhibition of mTORC1 signaling and delayed inhibition of mTORC2 signaling. It has a marked effect on the immune system, partly by interrupting metabolic demands associated with lymphocyte proliferation and effector function. In the context of SLE, rapamycin ameliorated nephritis and improved IL-2 production in MRL/lpr mice (49). In an open-label clinical trial, rapamycin improved the clinical and laboratory parameters in patients with recalcitrant SLE (50). A larger clinical trial in SLE (NCT00779194) is under way.
The N-acetylcysteine (NAC), a potent anti-oxidant and glutathione precursor, also inhibits mTOR. NAC administration in SLE patients resulted in improvement of disease activity (51). NAC also led to the expansion of CD4+CD25+FoxP3+ Tregs and depletion of phospho-S6RPhi DN T cells (52). NAC is well tolerated with its major side-effect being nausea at high doses (over 4.8 g per day).
JAK/STAT Inhibitors
The Janus kinase (JAK)/signal transducer and activator of transcription (STAT) system fine-tunes immune cell activation (53), defining their differentiation. In SLE, there is mounting evidence of the critical involvement of this system in disease pathogenesis. In a recent study, it was shown that increased STAT5 signaling in lupus T cells is related to changes in circulating CD4 T cell subsets and correlated with more aggressive disease (54).
Following the success in treating RA with tofacitinib, the first oral JAK inhibitor (55), several JAK inhibitors are currently under investigation. As type I interferons, known to be upregulated in SLE, transduce their signal through JAK, the JAK1 inhibitor GSK2586184 was used in a small trial to block the expression of interferon-related genes in SLE. The trial failed to show a difference (56) without being powered to address the broader effect JAK inhibition may have on disease activity.
Tofacitinib was studied in phase Ib trial in patients with mild to moderate lupus, stratified based on the presence or absence of STAT4 risk alleles. Although data are not available to date, this study is one of the first to address the link between genetic susceptibility and response to treatment in SLE. Baricitinib, a more selective JAK 1/2 inhibitor was evaluated in a phase II trial in SLE patients that was completed with positive yet modest results (57). A third JAK1 inhibitor, Filgotinib, is currently being evaluated in patients with moderate to severe active cutaneous lupus (NCT03134222) and with membranous lupus nephropathy (NCT03285711).
BMS-986165 is broad inhibitor against a panel of 265 kinases and pseudokinases. BMS-986165 protected NZB/W lupus-prone mice from nephritis possibly through its effect on interferon signaling (58). BMS-986165 also suppresses IL-23/IL-17 and IL-12. An ongoing phase 2 randomized, double-blind, placebo-controlled trial is exploring the efficacy and safety in patients with SLE (59). Finally, our group identified STAT3 that associates with JAK and mediates IL-6 and IL-23 signaling, as a potential therapeutic target in SLE (60, 61). STAT3 influences SLE T cell cytokine production, cell migration and B cell activity in lupus prone mice (62). In a preclinical study, Stattic, a small molecular STAT3 blocker, alleviated nephritis in lupus prone MRL/lpr mice (58, 63).
The Jak-STAT pathway therefore represents a very promising therapeutic target in both non-renal and renal lupus. Moreover, the use of small molecular oral agents to inhibit this pathway as opposed to biologic inhibitors of cytokines, makes this approach even more appealing.
Rho Kinase (ROCK) Inhibitors
ROCKs are a family of serine-threonine kinases (59) that function as downstream effectors for the GTPase Rho. The main isoforms ROCK1 and ROCK2 regulate multiple biological functions, including proliferation, differentiation, and migration by cytoskeletal reorganization. The potential of this pathway as a treatment target in SLE, was first shown by the ROCK inhibitor Y27632 that blocked the ability of SLE T cells to migrate in vitro (64, 65). Subsequently, ROCK2 was found to be selectively activated in murine lupus T cells. In these lupus models, ROCK2 regulates IRF4 and increases IL-17 and IL-21 production (66).
A wide array of available ROCK inhibitors has been investigated in SLE including Fasudil and Y27632. Fasudil is non-isoform selective ROCK inhibitor that attenuates disease activity in MRL/lpr mice and NZBWF1 mice (64). Fasudil can also cause vasodilation and hence was evaluated for the treatment of systemic sclerosis patients with Raynaud's phenomenon. A single oral dose of 40 or 80 mg fasudil though, did not improve skin temperature recovery or increase digital blood flow (67).
Although, there are no current clinical trials investigating ROCK inhibitors in patient with SLE, they are evaluated in patients with angina pectoris, pulmonary hypertension (68), idiopathic pulmonary fibrosis (NCT02688647) and psoriasis vulgaris (NCT02317627).
Co-stimulation
T cell activation is a tightly controlled process that consists of several steps to allow proper T cell differentiation. Each step in this process has the potential to serve as therapeutic target for autoimmune diseases. Following antigenic binding to the T-cell receptor, the strength of the immune response depends on the expression and interaction of costimulatory surface molecules on antigen presenting cells with those on T cells (68, 69). Here we review the role of disrupting the two most important co-stimulatory pairs, CD28/B7 and CD40/CD154 as a therapeutic strategy in SLE.
CD28/B7
Inhibition of co-stimulatory pathway has already been utilized in in the treatment of rheumatoid arthritis with abatacept. This is a fusion protein made of CTLA4 (cytotoxic T-lymphocyte-associated protein 4) and an immunoglobulin chain (CTLA4-Ig). It binds CD80/86 with a higher affinity than CD28. This interaction leads to both inhibition of T cell proliferation and B cell antibody production (70, 71). Efficacy and safety of abatacept added to standard of care with mycophenolate mofetil and steroids was evaluated in a phase III double-blind placebo-controlled trial that randomized over 400 lupus patients with class III or IV nephritis. The study end point was complete renal remission and corticosteroid dose assessed at 52 weeks. The trial did not reach its primary endpoint with 35% of patients treated with abatacept achieving remission vs. 33% in the placebo group (p = 0.73). Abatacept treatment was associated with improvement of immunologic markers (anti-ds DNA, C3, and C4 levels) as compared to placebo. In patients with nephrotic-range proteinuria, treatment with abatacept led to more rapid and greater reduction of proteinuria compared with placebo. Infection rates were similar as previously reported for RA patients. Thus, far, there have been several other trials of abatacept in active lupus nephritis but none achieved its primary endpoint (72, 73).
Targeting the CD28 instead of B7 has been challenging by the lack of inhibitory antibodies that would not crosslink CD28 and trigger a cytokine storm (74). One solution was to develop pegylated monovalent anti-CD28 antibodies such as lulizumab (75). Lulizumab was evaluated in phase II trial in non-renal lupus following acceptable safety profile in phase I trial. The trial was terminated early as it failed to meet protocol objectives (NCT02265744).
CD154/CD40
Another pair of co-stimulatory receptor-ligand system whose engagement has profound effects on B, dendritic and endothelial cells is the CD40-CD40 ligand (CD154). Following T cell activation, CD154 is expressed on the surface of the cell, allowing binding to B cells through CD40; that in turn leads to IgG class switching (76). Two drugs are currently explored as therapeutic agents in phase II trials for patients with moderately to severely active SLE. The first one, Dapirolizumab, a polyethylene glycol conjugated anti-CD40L Fab' fragment, was well tolerated in phase I study (77). At 12 weeks of treatment 46% of high disease activity patients showed reduction in disease activity measured by BILAG and 41% had improved SRI-4. The second one, BI 655064 is a humanized monoclonal anti-CD40 antibody. Its efficacy will be assessed in a double-blind, randomized, placebo-controlled trial for patients with active class III and IV lupus nephritis (NCT03385564). The study is actively enrolling and the primary endpoint is defined as complete renal response at 1 year.
Cytokines
SLE is characterized by skewed cytokine production that can directly cause local tissue damage and contribute to systemic symptomatology. Besides the interferons, other inflammatory and immunomodulatory cytokines have been investigated as therapeutic targets for SLE.
Interleukin-2 (IL-2)
There has been resurgent interest in interleukin-2 since the discovery of its homeostatic potential on CD4 T cells and its ability to redirect immune responses toward tolerance. IL-2 promotes the expansion and survival of regulatory T cells, and can be used in low doses to promote tolerance averting graft vs. host disease in bone marrow recipients (78–80). In SLE, low-dose IL-2 therapy is of particular interest, as these patients have low levels of IL-2, defective regulatory T cell function, and overactive T effector cells (81, 82). This imbalance can potentially be reversed with the addition of IL-2 (83). In preclinical studies, low dose IL-2 abrogated the development of nephritis in lupus-prone mice and mediated selective expansion of regulatory T cells in SLE patients (84, 85). This approach was tested in an open label phase I/II trial of subcutaneous low-dose IL-2 injection on alternate days for 3 cycles in 38 SLE patients. More than 80% of patients showed significant SRI-4 response by week 12, in addition to increased numbers of regulatory T cells, decreased Th17, follicular helper and double negative T cells (86, 87). This paved the way for larger placebo-controlled trials of low dose IL-2, using different IL-2 preparations and dosing schedules: AMG 592 (NCT03451422); LUPIL-2 trial with ILT-101 (NCT02955615); and Charact-IL-2 with Aldesleukin (NCT03312335).
Interleukin (IL)-12/23
Elevated IL-23 levels have been found in patients with lupus nephritis (83, 88, 89). Activation of IL-23/IL-17A axis induces expansion of highly pathogenic TH17 cells, ultimately contributing to pathogenesis of lupus nephritis by enhancing immunoglobulin and complement deposition (90, 91). Sole targeting of IL-17 in murine lupus nephritis models either with genetic deletion or utilizing a blocking antibody against IL-17A had no impact on the disease (92, 93). However, upstream targeting of this axis with ustekinumab, a monoclonal anti-IL-12/23 antibody that is already approved and well tolerated in patients with a variety of autoimmune diseases, showed promising data. Ustekinumab was evaluated in placebo-controlled phase II trial that recruited 102 SLE patients with active disease despite ongoing standard of care therapy (steroid, antimalarial and/or immunosuppressive therapies) (94). The protocol allowed intravenous loading, followed by subcutaneous administration every 8 weeks. At week 24, ustekinumab arm showed a significant improvement of the SLEDAI-2K score compared to placebo (SRI-4: 60 vs. 31%, respectively, p = 0.0046), which was the predetermined primary endpoint. A number of other metrics also improved, including anti-dsDNA, C3 levels, musculoskeletal and mucocutaneous manifestations. Moreover, a significant lower risk of a new BILAG flare was found in the ustekinumab group (p = 0.0078) but there was no difference in BILAG or BICLA scores at week 24 among the groups. Safety and adverse events of ustekinumab were similar to safety profile reported for other indications. Overall, this is very promising therapeutic option with an ongoing phase III trial that will address ultimately its usefulness in SLE.
IL-10
IL-10 has been shown to be increased in the serum of SLE patients and levels do correlate with disease activity (95). Its exact role in the propagation of the disease is unclear as IL-10 has both pro and anti-inflammatory effects. The anti-IL-10 antibody, BT063, is currently undergoing a phase II trial (NCT02554019); the trial aims at recruiting 36 patients with SLE who are to receive 50 mg of BT063. Safety and efficacy will be compared to standard of care. The drug will be administered over 8 cycles of intravenous infusion in 12 weeks period. Although no data are available, the clinical development program for this molecule is active.
IL-6
IL-6 is a proinflammatory cytokine found to be elevated in patients with active SLE (96). Rationale for its therapeutic blockade comes from data that showed diverse biologic function, spanning from promoting terminal differentiation of B and TH17 cells to locally driving tissue damage (97–99). Additionally, in animal models of SLE, disrupting IL-6 signaling either by utilizing an anti-IL-6 monoclonal antibody or anti-IL-6 receptor antibody led to improved survival, decreased levels of ds-DNA and proteinuria (100, 101). The opposite was found when mice were injected with recombinant human IL-6 (101, 102). Unfortunately, the clinical trials though have not been as encouraging.
PF-04236921, a monoclonal IL-6 antibody failed to meet its primary efficacy endpoint (SRI-4) a phase II trial that enrolled 183 patients assigned to receive subcutaneous 10 mg, 50 mg or 200 mg drug or placebo (103). In the 200 mg dose group, there were four deaths secondary to infections and thrombosis. A subgroup analysis showed that benefit can be seen in patients with high disease activity at baseline who received the 10 mg dose. They had significantly improved SRI-4 and BICLA response rates compared to placebo (49 vs. 25.1%, p < 0.05) and decreased incidence of severe lupus flares. Two other monoclonal antibodies failed to demonstrate efficacy in phase II trials, sirukumab (104), and vobarilizumab. Finally, the monoclonal antibody MRA003US is currently in a phase I trial (NCT00046774) and no results have been released to date.
The Interferons (IFN)
The hypothesis that IFNs have an important role in SLE pathogenesis is supported by plethora of findings both in humans and animals. Patients with active lupus have elevated levels of type I IFN. Moreover, patients with active and quiescent disease have evidence of continuous exposure to type I interferons based on multiple gene expression studies that show upregulated interferon responsive genes, collectively known as “IFN signature” (105–108). Given modulatory potential of IFNs to initiate or amplify immune responses leading to organ damage in lupus, this cytokine system became an excellent therapeutic target.
Sifalimumab, an anti-IFNα monoclonal antibody was evaluated in a phase II clinical trial in patients with moderate to severe SLE (109). Compared to placebo, patients receiving monthly IV infusions of sifalimumab (1,200, 600, and 200 mg groups) had statistically superior SRI-4 response index compared to placebo that received standard of care treatment at 52 weeks (sifalimumab: 59.8, 56.5, and 58.3 vs. 45.4% placebo, respectfully). At baseline, approximately 80% of patients had the IFN signature of gene expression at baseline, and tended to have better responses. The most common infectious complication was herpes zoster in patients receiving high dose (9.3 vs. 0.9% in the placebo group) that responded to treatment. There was one recurrence among patients who continued receiving sifalimumab. Overall, this was a positive study but given the modest effect size, there was no further development of this drug.
Rontalizumab is another humanized IgG1 anti-IFNα antibody that can neutralize all 12 subtypes of interferon-alpha. It was evaluated in the placebo control phase II trial, ROSE (110). At baseline, 76% of patients had high IFN regulated gene expression. At 24 weeks of treatment, the drug failed to meet the primary endpoint. Unexpectedly, treatment with rontalizumab showed consistent benefits with higher SRI-4 responses compared to controls in the subgroup of patients who had low interferon signature detected (72.7 vs. 41.7% placebo) and this group achieved meaningful rates of prednisone dose reduction to ≤10 mg daily. Rontalizumab was well tolerated and serious adverse events were 14.6 vs. 8.3% in the placebo group, all classified as unrelated to the study drug. Therefore, higher doses of rontalizumab would be well tolerated and possibly more effective but no additional trials are planned to answer this question. There are currently two phase I trials with other monoclonal antibodies against IFN-α: IAGS-009 completed phase I (NCT00960362) and JNJ-55920839 is in recruiting phase (NCT02609789).
Another strategy to inhibit the IFN-I pathway is to block its receptor, the interferon alpha receptor 1 (IFNAR1) that binds all type I IFNs including IFNα and IFNβ. Anifrolumab, a monoclonal antibody against IFNAR1, was granted fast- track status by the FDA and was successful in phase II open-label trial (108, 109). In this study, 75% of patients had high IFN signature at baseline. The primary endpoint consisting of composite SRI-4 combined with a measure of steroid sparing to <10 mg/day was achieved in both anifrolumab dose groups at higher rates than placebo (28.8% in the 1,000 mg IV monthly, 34.3% in 300 mg IV monthly vs. 17.6% in the placebo group). At 1 year, 56.4% of the patients taking a 300 mg dose met the SRI end-point, as compared to 31.7% receiving 1000 mg dose (p = 0.595) and 26.6% on placebo. Regarding the infections rate, patients receiving anifrolumab had dose-dependent increase in herpes zoster cases (placebo: 2.0%; 300 mg: 5.1%; 1,000 mg: 9.5%) and a greater number of influenza infections (placebo: 2.0%; 300 mg: 6.1%; 1,000 mg: 7.6%). However, most cases of influenza were unconfirmed. With this positive data, two phase III studies are currently underway via the TULIP (NCT02547922) program. TULIP-LN1 on the other hand is phase IIb study designed to assess efficacy and safety of two intravenous doses of anifrolumab vs. placebo while taking standard of care treatment with mycophenolate mofetil and corticosteroids in adults with active proliferative lupus nephritis. On August 31, 2018, it was reported that in one of the phase III trials (TULIP I), anifrolumab failed to meet its primary endpoint.
In conclusion, IFNAR blocking has been a promising therapeutic approach for SLE patients who fail to respond to available therapies. The recently released result though from the phase III trial dampen the enthusiasm for the usefulness of this approach.
Interferon-γ (IFNγ)
The pathogenic role of IFNγ has been better characterized in mice, as opposed to humans (111), where elevated levels and correlation with disease activity is found in both NZB/W and MRL/lpr mice. Administration of IFN-γ accelerates murine lupus while early treatment with anti-IFNγ antibody rescues mice from disease (112). Unfortunately two phase I studies of the anti-IFNγ antibody AMG811 in the treatment of mild to moderate systemic and cutaneous lupus (113) showed safety with favorable immunogenicity profile but did not show significant therapeutic effect despite decreasing IFNγ-related gene expression (114). The negative results from these small studies led to discontinuation of the development program for AMG811.
IFNα Kinoid (IFNα-K)
This is another interesting approach for neutralizing I IFN that received fast track status by the FDA. It is currently in phase IIb trial aiming at recruiting 185 patients (NCT02665364). Patients are assigned to receive IFNα-K immunotherapy or placebo in addition to standard treatment with immunosuppressives, antimalarials, and/or steroids. The drug is composed of inactivated IFN-α coupled to the keyhole limpet haemocyanin protein and when injected, leads to induction of polyclonal anti-IFNα responses with transient immunity against all 13 subtypes of interferon alpha. In preclinical stage, IFNα-K was able to slow disease progression in NZB/W mice (115). From an infectious standpoint, this would be an advantageous approach as cellular tolerance and host defense against viral infections would remain intact. The primary end point in the phase II is the BILAG-based Composite Lupus Assessment (BICLA) response at week 36.
Other
Lupuzor/P140 peptide or regiremod, is a 21-mer linear peptide derived from nuclear ribonucleoprotein U1-70K that needs to be phosphorylated at the Ser140 position in order to exert its immunomodulatory properties via binding to MHC class II. This allows recognition in the context of T cell receptor, both in lupus patients and mice and alters autoreactive T cell phenotype. In a phase IIb data, patients receiving Lupuzor 200 μg subcutaneously every 4 weeks achieved the SRI response at week 12 at higher rates than standard of care therapy that included stroids, antimalarials, azathioprine or methotrexate (53.1 vs. 36.2%). These positive results in IIb trial were more pronounced among patients with SLEDAI-2K ≥6 at baseline, showing that 61.9% achieved SRI response at week 12 vs. 38.6% in the placebo group. Nevertheless, in the phase III clinical trial, Lupuzor failed to meet the primary endpoint (p = 0.2631 vs. placebo). The investigational treatment in this trial though holds promise for patients with anti-dsDNA autoantibodies as 7.6% of these patients in the Lupuzor group went into full remission, compared with none in the placebo treated group. The company launched 6 months open label extension study for all participants of phase III trial, allowing continuation of Lupuzor treatment in combination with standard therapy for additional 48 weeks. Finally, this study confirmed the already known Lupuzor's good safety profile with zero adverse effects reported.
Conclusions
To date, belimumab (anti-BLyS) is the only FDA approved biologic for treating SLE. Over the last decade and despite the setbacks including the recent failure of the highly promising anti-IFNαR therapy, our understanding of the mechanisms of SLE contributed to expansion of the drug pipeline for SLE (Table 1). Currently, drugs representing a variety of therapeutic strategies are moving to phase III trials. These include: cytokine infusions (low dose IL-2); antibodies against cytokines (ustekinumab); and finally, small molecule inhibitors against kinases (Jak inhibitors) and phosphatases (calcineurin inhibitors). It is highly likely that these targeted therapies in conjunction with biomarker development and more rigorous outcome measures will finally result in a fundamental change of the stagnant therapeutic paradigm in SLE.
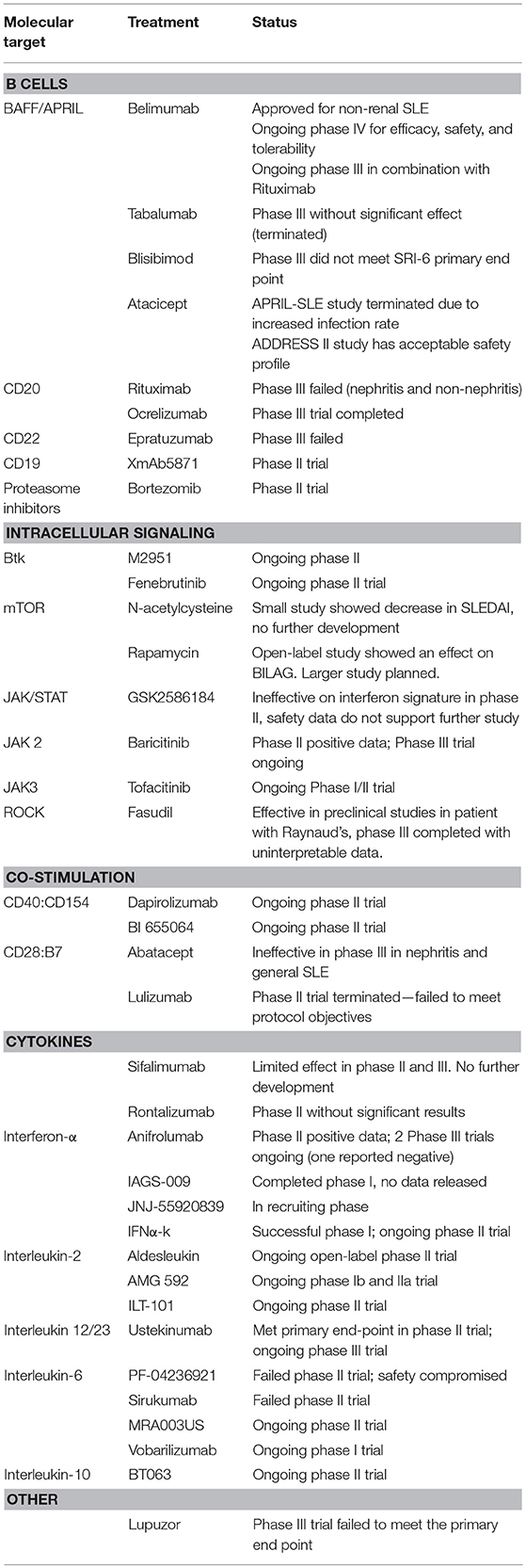
Table 1. New and emerging therapies in SLE.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
VK is a site PI for the EMD Serono sponsored phase II, study to evaluate the safety and efficacy of M2951 in subjects with SLE.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzova D, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. (2011) 63:3918–30. doi: 10.1002/art.30613
2. Stohl W, Schwarting A, Okada M, Scheinberg M, Doria A, Hammer AE, et al. Efficacy and safety of subcutaneous belimumab in systemic lupus erythematosus: a fifty-two-week randomized, double-blind, placebo-controlled study. Arthritis Rheumatol. (2017) 69:1016–27. doi: 10.1002/art.40049
3. Doria A, Stohl W, Schwarting A, Okada M, Scheinberg M, van Vollenhoven R, et al. Efficacy and safety of subcutaneous belimumab in anti-double-stranded DNA-positive, hypocomplementemic patients with systemic lupus erythematosus. Arthritis Rheumatol. (2018) 70:1256–64. doi: 10.1002/art.40511
4. Isenberg DA, Petri M, Kalunian K, Tanaka Y, Urowitz MB, Hoffman RW, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. (2016) 75:323–31. doi: 10.1136/annrheumdis-2015-207653
5. Isenberg D. Further thoughts about the ILLUMINATE studies of tabalumab in SLE. Ann Rheum Dis. (2016) 75:e11. doi: 10.1136/annrheumdis-2015-208709
6. Merrill JT, van Vollenhoven RF, Buyon JP, Furie RA, Stohl W, Morgan-Cox M, et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann Rheum Dis. (2016) 75:332–40. doi: 10.1136/annrheumdis-2015-207654
7. Merrill JT, Shanahan WR, Scheinberg M, Kalunian KC, Wofsy D, Martin RS. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2018) 77:883–9. doi: 10.1136/annrheumdis-2018-213032
8. Dall'Era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. (2007) 56:4142–50. doi: 10.1002/art.23047
9. Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann Rheum Dis. (2015) 74:2006–15. doi: 10.1136/annrheumdis-2013-205067
10. Merrill JT, Wallace DJ, Wax S, Kao A, Fraser PA, Chang P, et al. Efficacy and safety of atacicept in patients with systemic lupus erythematosus: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled, parallel-arm, phase IIb study. Arthritis Rheumatol. (2018) 70:266–76. doi: 10.1002/art.40360
11. Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, et al. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. (2004) 350:2572–81. doi: 10.1056/NEJMoa032534
12. Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: a systematic review of off-label use in 188 cases. Lupus (2009) 18:767–76. doi: 10.1177/0961203309106174
13. Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. (2012) 64:1215–26. doi: 10.1002/art.34359
14. Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. (2010) 62:222–33. doi: 10.1002/art.27233
15. Bertsias GK, Tektonidou M, Amoura Z, Aringer M, Bajema I, Berden JH, et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann Rheum Dis. (2012) 71:1771–82. doi: 10.1136/annrheumdis-2012-201940
16. Simonetta F, Allali D, Roux-Lombard P, Chizzolini C. Successful treatment of refractory lupus nephritis by the sequential use of rituximab and belimumab. Joint Bone Spine (2017) 84:235–6. doi: 10.1016/j.jbspin.2016.01.008
17. Gonzalez-Echavarri C, Ugarte A, Ruiz-Irastorza G. Rituximab-refractory lupus nephritis successfully treated with belimumab. Clin Exp Rheumatol. (2016) 34:355–6.
18. Kraaij T, Kamerling SWA, de Rooij ENM, van Daele PLA, Bredewold OW, Bakker JA, et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J Autoimmun. (2018) 91:45–54. doi: 10.1016/j.jaut.2018.03.003
19. Gualtierotti R, Borghi MO, Gerosa M, Schioppo T, Larghi P, Geginat J, et al. Successful sequential therapy with rituximab and belimumab in patients with active systemic lupus erythematosus: a case series. Clin Exp Rheumatol. (2018) 36:643–7.
20. Mysler EF, Spindler AJ, Guzman R, Bijl M, Jayne D, Furie RA, et al. Efficacy and safety of ocrelizumab in active proliferative lupus nephritis: results from a randomized, double-blind, phase III study. Arthritis Rheum. (2013) 65:2368–79. doi: 10.1002/art.38037
21. Masoud S, McAdoo SP, Bedi R, Cairns TD, Lightstone L. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology (2018) doi: 10.1093/rheumatology/key042. [Epub ahead of print].
22. Karageorgas T, Zomas A, Kazakou P, Katsimbri P, Mantzourani M, Boumpas D. Successful treatment of life-threatening autoimmune haemolytic anaemia with ofatumumab in a patient with systemic lupus erythematosus. Rheumatology (2016) 55:2085–7. doi: 10.1093/rheumatology/kew267
23. Haarhaus ML, Svenungsson E, Gunnarsson I. Ofatumumab treatment in lupus nephritis patients. Clin Kidney J. (2016) 9:552–5. doi: 10.1093/ckj/sfw022
24. Wallace DJ, Gordon C, Strand V, Hobbs K, Petri M, Kalunian K, et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic lupus erythematosus: results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology (2013) 52:1313–22. doi: 10.1093/rheumatology/ket129
25. Clowse ME, Wallace DJ, Furie RA, Petri MA, Pike MC, Leszczynski P, et al. Efficacy and safety of epratuzumab in moderately to severely active systemic lupus erythematosus: results from two phase III randomized, double-blind, placebo-controlled trials. Arthritis Rheumatol. (2017) 69:362–75. doi: 10.1002/art.39856
26. Gottenberg JE, Dorner T, Bootsma H, Devauchelle-Pensec V, Bowman SJ, Mariette X, et al. Efficacy of epratuzumab, an anti-CD22 monoclonal IgG antibody, in systemic lupus erythematosus patients with associated sjogren's syndrome: post hoc analyses from the EMBODY trials. Arthritis Rheumatol. (2018) 70:763–73. doi: 10.1002/art.40425
27. Chu SY, Yeter K, Kotha R, Pong E, Miranda Y, Phung S, et al. Suppression of rheumatoid arthritis B cells by XmAb5871, an anti-CD19 antibody that coengages B cell antigen receptor complex and Fcgamma receptor IIb inhibitory receptor. Arthritis Rheumatol. (2014) 66:1153–64. doi: 10.1002/art.38334
28. Slifka MK, Ahmed R. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr Opin Immunol. (1998) 10:252–8. doi: 10.1016/S0952-7915(98)80162-3
29. Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. (2008) 14:748–55. doi: 10.1038/nm1763
30. Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, et al. Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. (2012) 64:493–503. doi: 10.1002/art.33333
31. Seavey MM, Lu LD, Stump KL, Wallace NH, Ruggeri BA. Novel, orally active, proteasome inhibitor, delanzomib (CEP-18770), ameliorates disease symptoms and glomerulonephritis in two preclinical mouse models of SLE. Int Immunopharmacol. (2012) 12:257–70. doi: 10.1016/j.intimp.2011.11.019
32. Kurosaki T. Regulation of BCR signaling. Mol Immunol. (2011) 48:1287–91. doi: 10.1016/j.molimm.2010.12.007
33. Jongstra-Bilen J, Puig Cano A, Hasija M, Xiao H, Smith CI, Cybulsky MI. Dual functions of Bruton's tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J Immunol. (2008) 181:288–98. doi: 10.4049/jimmunol.181.1.288
34. Kenny EF, Quinn SR, Doyle SL, Vink PM, van Eenennaam H, O'Neill LA. Bruton's tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS ONE (2013) 8:e74103. doi: 10.1371/journal.pone.0074103
35. Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science (1993) 261:355–8. doi: 10.1126/science.8332900
36. Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. (2015) 373:2425–37. doi: 10.1056/NEJMoa1509388
37. Rankin AL, Seth N, Keegan S, Andreyeva T, Cook TA, Edmonds J, et al. Selective inhibition of BTK prevents murine lupus and antibody-mediated glomerulonephritis. J Immunol. (2013) 191:4540–50. doi: 10.4049/jimmunol.1301553
38. Hutcheson J, Vanarsa K, Bashmakov A, Grewal S, Sajitharan D, Chang BY, et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res Ther. (2012) 14:R243. doi: 10.1186/ar4086
39. Schafer PH, Ye Y, Wu L, Kosek J, Ringheim G, Yang Z, et al. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis. (2018) 77:1516–23. doi: 10.1136/annrheumdis-2017-212916
40. Lessard CJ, Adrianto I, Ice JA, Wiley GB, Kelly JA, Glenn SB, et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am J Hum Genet. (2012) 90:648–60. doi: 10.1016/j.ajhg.2012.02.023
41. Gaudy AYY, Korish S, Houg D, Weiswasser S, Choi S, Furie R, Werth V, Schafer P. Cereblon Modulator CC-220 Decreases Naïve and Memory B cells and Plasmacytoid Dendritic Cells in Systemic Lupus Erythematosus (SLE) Patients: Exposure-Response Results From a Phase 2A Proof of Concept Study. EULAR Abstract, Madrid (2017).
42. Crist SA, Sprague DL, Ratliff TL. Nuclear factor of activated T cells (NFAT) mediates CD154 expression in megakaryocytes. Blood (2008) 111:3553–61. doi: 10.1182/blood-2007-05-088161
43. Mok CC, Ying KY, Yim CW, Siu YP, Tong KH, To CH, et al. Tacrolimus versus mycophenolate mofetil for induction therapy of lupus nephritis: a randomised controlled trial and long-term follow-up. Ann Rheum Dis. (2016) 75:30–6. doi: 10.1136/annrheumdis-2014-206456
44. Dooley MA, Pendergraft I, Ginzler EM, Olsen NJ, Tumlin J, Rovin BH, et al., editors. Speed of remission with the use of voclosporin, MMF and low dose steroids: results of a global lupus nephritis study [abstract]. Arthritis Rheumatol. (2016).
45. Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. (2005) 280:39505–9. doi: 10.1074/jbc.M506096200
46. Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science (2011) 334:678–83. doi: 10.1126/science.1207056
47. Fernandez DR, Telarico T, Bonilla E, Li Q, Banerjee S, Middleton FA, et al. Activation of mammalian target of rapamycin controls the loss of TCRzeta in lupus T cells through HRES-1/Rab4-regulated lysosomal degradation. J Immunol. (2009) 182:2063–73. doi: 10.4049/jimmunol.0803600
48. Kato H, Perl A. Mechanistic target of rapamycin complex 1 expands Th17 and IL-4+ CD4-CD8- double-negative T cells and contracts regulatory T cells in systemic lupus erythematosus. J Immunol. (2014) 192:4134–44. doi: 10.4049/jimmunol.1301859
49. Warner LM, Adams LM, Sehgal SN. Rapamycin prolongs survival and arrests pathophysiologic changes in murine systemic lupus erythematosus. Arthritis Rheum. (1994) 37:289–97. doi: 10.1002/art.1780370219
50. Fernandez D, Bonilla E, Mirza N, Niland B, Perl A. Rapamycin reduces disease activity and normalizes T cell activation-induced calcium fluxing in patients with systemic lupus erythematosus. Arthritis Rheum. (2006) 54:2983–8. doi: 10.1002/art.22085
51. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. (2012) 64:2937–46. doi: 10.1002/art.34502
52. Perl A, Hanczko R, Lai ZW, Oaks Z, Kelly R, Borsuk R, et al. Comprehensive metabolome analyses reveal N-acetylcysteine-responsive accumulation of kynurenine in systemic lupus erythematosus: implications for activation of the mechanistic target of rapamycin. Metabolomics (2015) 11:1157–74. doi: 10.1007/s11306-015-0772-0
53. Schwartz DM, Bonelli M, Gadina M, O'Shea JJ. Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nat Rev Rheumatol. (2016) 12:25–36. doi: 10.1038/nrrheum.2015.167
54. Goropevsek A, Gorenjak M, Gradisnik S, Dai K, Holc I, Hojs R, et al. STAT5 phosphorylation in CD4 T cells from patients with SLE is related to changes in their subsets and follow-up disease severity. J Leukoc Biol. (2017) 101:1405–18. doi: 10.1189/jlb.5A0416-194R
55. Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. (2012) 367:495–507. doi: 10.1056/NEJMoa1109071
56. Kahl L, Patel J, Layton M, Binks M, Hicks K, Leon G, et al. Safety, tolerability, efficacy and pharmacodynamics of the selective JAK1 inhibitor GSK2586184 in patients with systemic lupus erythematosus. Lupus (2016) 25:1420–30. doi: 10.1177/0961203316640910
57. Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet (2018) 392:222–31. doi: 10.1016/S0140-6736(18)31363-1
58. Gillooly K, Yifan Y, Yang X, Zupa-Fernandez A, Cheng L, Strnad J, Cunningham M editors. BMS-986165 is a highly potent and selective allosteric inhibitor of Tyk2, blocks IL-12, IL-23 and type I interferon signaling and provides for robust efficacy in preclinical models of systemic lupus erythematosus and inflammatory bowel disease [abstract]. Arthritis Rheumatol. (2016).
59. Isgro J, Gupta S, Jacek E, Pavri T, Duculan R, Kim M, et al. Enhanced rho-associated protein kinase activation in patients with systemic lupus erythematosus. Arthritis Rheum. (2013) 65:1592–602. doi: 10.1002/art.37934
60. Harada T, Kyttaris V, Li Y, Juang YT, Wang Y, Tsokos GC. Increased expression of STAT3 in SLE T cells contributes to enhanced chemokine-mediated cell migration. Autoimmunity (2007) 40:1–8. doi: 10.1080/08916930601095148
61. Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, et al. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. (2007) 178:1938–47. doi: 10.4049/jimmunol.178.3.1938
62. Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. (1996) 392:189–93. doi: 10.1016/0014-5793(96)00811-3
63. Edwards LJ, Mizui M, Kyttaris V. Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin Immunol. (2015) 158:221–30. doi: 10.1016/j.clim.2015.04.004
64. Stirzaker RA, Biswas PS, Gupta S, Song L, Bhagat G, Pernis AB. Administration of fasudil, a ROCK inhibitor, attenuates disease in lupus-prone NZB/W F1 female mice. Lupus (2012) 21:656–61. doi: 10.1177/0961203312436862
65. Vicari RM, Chaitman B, Keefe D, Smith WB, Chrysant SG, Tonkon MJ, et al. Efficacy and safety of fasudil in patients with stable angina: a double-blind, placebo-controlled, phase 2 trial. J Am Coll Cardiol. (2005) 46:1803–11. doi: 10.1016/j.jacc.2005.07.047
66. Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, et al. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J Clin Invest. (2010) 120:3280–95. doi: 10.1172/JCI42856
67. Fava A, Wung PK, Wigley FM, Hummers LK, Daya NR, Ghazarian SR, et al. Efficacy of Rho kinase inhibitor fasudil in secondary Raynaud's phenomenon. Arthritis Care Res. (2012) 64:925–9. doi: 10.1002/acr.21622
68. Fukumoto Y, Yamada N, Matsubara H, Mizoguchi M, Uchino K, Yao A, et al. Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ J. (2013) 77:2619–25. doi: 10.1253/circj.CJ-13-0443
69. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. (2013) 13:227–42. doi: 10.1038/nri3405
70. Genovese MC, Pacheco-Tena C, Covarrubias A, Leon G, Mysler E, Keiserman M, et al. Longterm safety and efficacy of subcutaneous abatacept in patients with rheumatoid arthritis: 5-year results from a Phase IIIb trial. J Rheumatol. (2018) 45:1085–92. doi: 10.3899/jrheum.170344
71. Picchianti Diamanti A, Rosado MM, Scarsella M, Germano V, Giorda E, Cascioli S, et al. Abatacept (cytotoxic T lymphocyte antigen 4-immunoglobulin) improves B cell function and regulatory T cell inhibitory capacity in rheumatoid arthritis patients non-responding to anti-tumour necrosis factor-alpha agents. Clin Exp Immunol. (2014) 177:630–40. doi: 10.1111/cei.12367
72. Furie R, Nicholls K, Cheng TT, Houssiau F, Burgos-Vargas R, Chen SL, et al. Efficacy and safety of abatacept in lupus nephritis: a twelve-month, randomized, double-blind study. Arthritis Rheumatol. (2014) 66:379–89. doi: 10.1002/art.38260
73. Group AT. Treatment of lupus nephritis with abatacept: the Abatacept and Cyclophosphamide Combination Efficacy and Safety Study. Arthritis Rheumatol. (2014) 66:3096–104. doi: 10.1002/art.38790
74. Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. (2006) 355:1018–28. doi: 10.1056/NEJMoa063842
75. Shi R, Honczarenko M, Zhang S, Fleener C, Mora J, Lee SK, et al. Pharmacokinetic, pharmacodynamic, and safety profile of a novel anti-CD28 domain antibody antagonist in healthy subjects. J Clin Pharmacol. (2017) 57:161–72. doi: 10.1002/jcph.791
76. Crow MK, Kirou KA. Regulation of CD40 ligand expression in systemic lupus erythematosus. Curr Opin Rheumatol. (2001) 13:361–9. doi: 10.1097/00002281-200109000-00004
77. Chamberlain C, Colman PJ, Ranger AM, Burkly LC, Johnston GI, Otoul C, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis. (2017) 76:1837–44. doi: 10.1136/annrheumdis-2017-211388
78. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. (2012) 12:180–90. doi: 10.1038/nri3156
79. Yu A, Zhu L, Altman NH, Malek TR. A low interleukin-2 receptor signaling threshold supports the development and homeostasis of T regulatory cells. Immunity (2009) 30:204–17. doi: 10.1016/j.immuni.2008.11.014
80. Mizui M, Tsokos GC. Low-Dose IL-2 in the treatment of lupus. Curr Rheumatol Rep. (2016) 18:68. doi: 10.1007/s11926-016-0617-5
81. Lieberman LA, Tsokos GC. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J Biomed Biotechnol. (2010) 2010:740619. doi: 10.1155/2010/740619
82. Comte D, Karampetsou MP, Tsokos GC. T cells as a therapeutic target in SLE. Lupus (2015) 24:351–63. doi: 10.1177/0961203314556139
83. Dai H, He F, Tsokos GC, Kyttaris VC. IL-23 Limits the production of IL-2 and promotes autoimmunity in lupus. J Immunol. (2017) 199:903–10. doi: 10.4049/jimmunol.1700418
84. Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP III et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med. (2011) 365:2055–66. doi: 10.1056/NEJMoa1108188
85. Mizui M, Koga T, Lieberman LA, Beltran J, Yoshida N, Johnson MC, et al. IL-2 protects lupus-prone mice from multiple end-organ damage by limiting CD4-CD8- IL-17-producing T cells. J Immunol. (2014) 193:2168–77. doi: 10.4049/jimmunol.1400977
86. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. (2008) 181:8761–6. doi: 10.4049/jimmunol.181.12.8761
87. He J, Zhang X, Wei Y, Sun X, Chen Y, Deng J, et al. Low-dose interleukin-2 treatment selectively modulates CD4(+) T cell subsets in patients with systemic lupus erythematosus. Nat Med. (2016) 22:991–3. doi: 10.1038/nm.4148
88. Zhang Z, Kyttaris VC, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. (2009) 183:3160–9. doi: 10.4049/jimmunol.0900385
89. Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol. (2008) 127:385–93. doi: 10.1016/j.clim.2008.01.019
90. Zickert A, Amoudruz P, Sundstrom Y, Ronnelid J, Malmstrom V, Gunnarsson I. IL-17 and IL-23 in lupus nephritis - association to histopathology and response to treatment. BMC Immunol. (2015) 16:7. doi: 10.1186/s12865-015-0070-7
91. Schmidt T, Paust HJ, Krebs CF, Turner JE, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol. (2015) 67:475–87. doi: 10.1002/art.38955
92. Kyttaris VC, Zhang Z, Kuchroo VK, Oukka M, Tsokos GC. Cutting edge: IL-23 receptor deficiency prevents the development of lupus nephritis in C57BL/6-lpr/lpr mice. J Immunol. (2010) 184:4605–9. doi: 10.4049/jimmunol.0903595
93. Kyttaris VC, Kampagianni O, Tsokos GC. Treatment with anti-interleukin 23 antibody ameliorates disease in lupus-prone mice. Biomed Res Int. (2013) 2013:861028. doi: 10.1155/2013/861028
94. van Vollenhoven R, Hahn BH, Tsokos GC, Wagner C, Lipsky P, Hsu B, et al., editors. Efficacy and safety of ustekinumab, an interleukin 12/23 inhibitor, in patients with active systemic lupus erythematosus: results of a phase 2, Randomized Placebo-Controlled Study [abstract]. Arthritis Rheumatol. (2017).
95. Godsell J, Rudloff I, Kandane-Rathnayake R, Hoi A, Nold MF, Morand EF, et al. Clinical associations of IL-10 and IL-37 in systemic lupus erythematosus. Sci Rep. (2016) 6:34604. doi: 10.1038/srep34604
96. Chun HY, Chung JW, Kim HA, Yun JM, Jeon JY, Ye YM, et al. Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J Clin Immunol. (2007) 27:461–6. doi: 10.1007/s10875-007-9104-0
97. Ripley BJ, Goncalves B, Isenberg DA, Latchman DS, Rahman A. Raised levels of interleukin 6 in systemic lupus erythematosus correlate with anaemia. Ann Rheum Dis. (2005) 64:849–53. doi: 10.1136/ard.2004.022681
98. Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood (2010) 115:4699–706. doi: 10.1182/blood-2009-07-230631
99. Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
100. Ryffel B, Car BD, Gunn H, Roman D, Hiestand P, Mihatsch MJ. Interleukin-6 exacerbates glomerulonephritis in (NZB x NZW)F1 mice. Am J Pathol. (1994) 144:927–37.
101. Finck BK, Chan B, Wofsy D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J Clin Invest. (1994) 94:585–91. doi: 10.1172/JCI117373
102. Mihara M, Takagi N, Takeda Y, Ohsugi Y. IL-6 receptor blockage inhibits the onset of autoimmune kidney disease in NZB/W F1 mice. Clin Exp Immunol. (1998) 112:397–402. doi: 10.1046/j.1365-2249.1998.00612.x
103. Wallace DJ, Strand V, Merrill JT, Popa S, Spindler AJ, Eimon A, et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: a phase II dose-ranging randomised controlled trial. Ann Rheum Dis. (2017) 76:534–42. doi: 10.1136/annrheumdis-2016-209668
104. Szepietowski JC, Nilganuwong S, Wozniacka A, Kuhn A, Nyberg F, van Vollenhoven RF, et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum. (2013) 65:2661–71. doi: 10.1002/art.38091
105. Kirou KA, Lee C, George S, Louca K, Papagiannis IG, Peterson MG, et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheum. (2004) 50:3958–67. doi: 10.1002/art.20798
106. Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. (2007) 8:492–502. doi: 10.1038/sj.gene.6364408
107. Coit P, Jeffries M, Altorok N, Dozmorov MG, Koelsch KA, Wren JD, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun. (2013) 43:78–84. doi: 10.1016/j.jaut.2013.04.003
108. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
109. Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-alpha monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. (2016) 75:1909–16. doi: 10.1136/annrheumdis-2015-208562
110. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. (2016) 75:196–202. doi: 10.1136/annrheumdis-2014-206090
111. Welcher AA, Boedigheimer M, Kivitz AJ, Amoura Z, Buyon J, Rudinskaya A, et al. Blockade of interferon-gamma normalizes interferon-regulated gene expression and serum CXCL10 levels in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2015) 67:2713–22. doi: 10.1002/art.39248
112. Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest. (1998) 101:364–71. doi: 10.1172/JCI750
113. Werth VP, Fiorentino D, Cohen SB, Fivenson D, Hansen C, Zoog S, et al. A Phase I single-dose crossover study to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and clinical efficacy of AMG 811 (anti-IFN-gamma). In: Subjects With Discoid Lupus Erythematosus. Boston, MA: American College of Rheumatology Annual Meeting 2014 (2014).
114. Martin D, Amoura Z, Romero-Diaz J, Chong Y, Sanchez-Guerrero J, Chan T, et al. A multiple dose study of AMG 811 (anti-IFNgamma) in subjects with systemic lupus erythematosus and active nephritis [abstract]. Ann Rheum Dis. (2015) 74:337. doi: 10.1136/annrheumdis-2015-eular.2916
115. Ducreux J, Houssiau FA, Vandepapeliere P, Jorgensen C, Lazaro E, Spertini F, et al. Interferon alpha kinoid induces neutralizing anti-interferon alpha antibodies that decrease the expression of interferon-induced and B cell activation associated transcripts: analysis of extended follow-up data from the interferon alpha kinoid phase I/II study. Rheumatology (2016) 55:1901–5. doi: 10.1093/rheumatology/kew262
Keywords: lupus, biologics, small molecules, treatment, clinical trails
Citation: Vukelic M, Li Y and Kyttaris VC (2018) Novel Treatments in Lupus. Front. Immunol. 9:2658. doi: 10.3389/fimmu.2018.02658
Received: 03 September 2018; Accepted: 29 October 2018;
Published: 16 November 2018.
Edited by:
José Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoReviewed by:
Pier Luigi Meroni, Istituto Auxologico Italiano (IRCCS), ItalyCheng-De Yang, Shanghai Jiao Tong University, China
Copyright © 2018 Vukelic, Li and Kyttaris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasileios C. Kyttaris, dmt5dHRhcmlAYmlkbWMuaGFydmFyZC5lZHU=
†These authors have contributed equally to this work