 Muhammad Haseeb
Muhammad Haseeb Muhammad Ayaz Anwar
Muhammad Ayaz Anwar Sangdun Choi
Sangdun Choi- Department of Molecular Science and Technology, Ajou University, Suwon, South Korea
Innate immunity constitutes the first line of host defense against various anomalies in humans, and it also guides the adaptive immune response. The function of innate immune components and adaptive immune components are interlinked in hematological malignancies including chronic lymphocytic leukemia (CLL), and molecular interactions between innate and adaptive immune components are crucial for the development, progression and the therapeutic outcome of CLL. In this leukemia, genetic mutations in B cells and B cell receptors (BCR) are key driving factors along with evasion of cytotoxic T lymphocytes and promotion of regulatory T cells. Similarly, the release of various cytokines from CLL cells triggers the protumor phenotype in macrophages that further edges the CLL cells. Moreover, under the influence of various cytokines, dendritic cells are unable to mature and trigger T cell mediated antitumor response. The phenotypes of these cells are ultimately controlled by respective signaling pathways, the most notables are BCR, Wnt, Notch, and NF-κB, and their activation affects the cytokine profile that controls the pathogenesis of CLL, and challenge its treatment. There are several novel substances for CLL under clinical development, including kinase inhibitors, antibodies, and immune-modulators that offer new hopes. DC-based vaccines and CAR T cell therapy are promising tools; however, further studies are required to precisely dissect the molecular interactions among various molecular entities. In this review, we systematically discuss the involvement, common targets and therapeutic interventions of various cells for the better understanding and therapy of CLL.
Introduction
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western world, with ~4.5 cases per 100,000 individuals reported annually (1). The development and progression of CLL are accompanied by several genetic abnormalities and disorders, and CLL is characterized by the gradual accumulation of maturing-looking clonally expanded CD5+ B lymphocytes in peripheral lymphoid organs, secondary lymphoid organs, and bone marrow. There are various signals that induce proliferation and accumulation that lead to survival of malignant cells (2). Abnormalities in the development of B cells cause CLL, immune deficiencies, malignancies, and allergies. Various mutations in hematopoietic cells and immune deficiencies are recognized in CLL, and novel studies are being designed to gain a deeper understanding of these associations. Moreover, personalized forms of treatment are being developed to treat CLL (3).
CLL cells are largely derived from the continuum of maturation states observed in normal developmental stages when compared to normal B cells. Epigenetic maturation in CLL is associated with an indolent gene expression pattern and increasingly favorable clinical outcomes (4). In addition, previously reported tumor-specific methylation events are normally present in non-malignant B cells. Moreover, a potential pathogenic role has been identified for dysregulated transcription factors in CLL, including the induction of signaling by nuclear factor of activated T cells (NFAT) and early growth response (EGR) proteins, resulting in diminished early B cell factor (EBF) and AP-1 programming compared to that in the normal B cell epigenetic program.
The immune system response can be divided into two phases; (a) innate immunity that arise from myeloid lineage cells and mature into monocytes, macrophages, erythrocytes, platelets, and granulocytes, provide the first line of defense, (b) and the adaptive immune system arises from lymphoid progenitor cells and give rise to natural killer (NK) cells, B cells, and T cells, and provides the second line of defense against pathogens and other abnormalities (Figure 1) (5, 6). These myeloid and lymphoid cells affect the progression of CLL in an independent and collaborative manner. The CLL microenvironment is populated by macrophages, and the transfer of antigens is dependent on the contact between B cells and macrophages (7). Other than antigen transfer, the influence of various cell surface receptors, cytokine secretion, and immune suppression are frequently being interconnected among these cells. Therefore, in this review, we will address the recent advances in linking these blood cells and the therapeutic approaches to counter blood malignancies, particularly CLL.
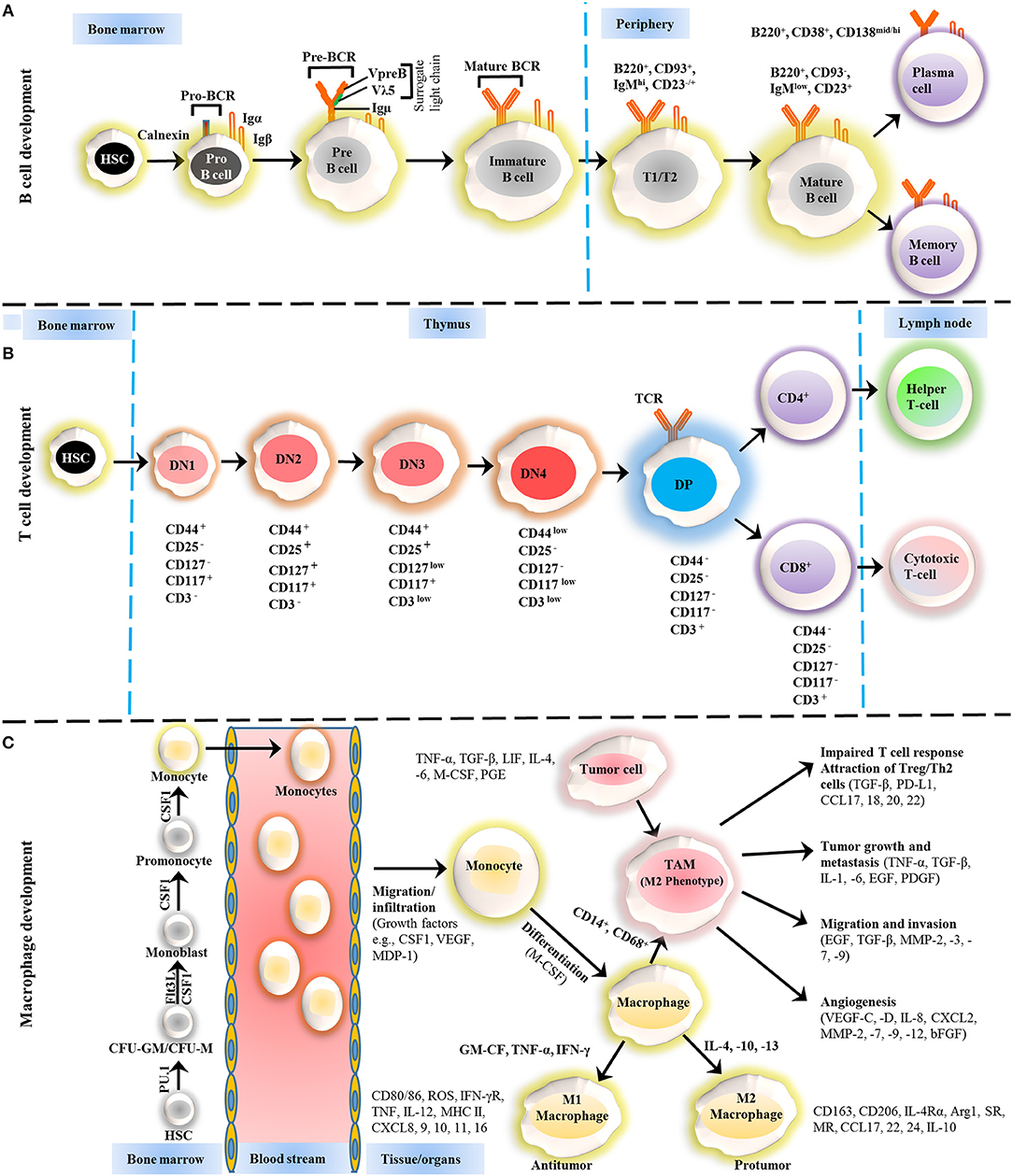
Figure 1. Development of B cells, T cells and macrophages. (A) The development of B cells occurs in the bone marrow and peripheral lymphoid tissues. Development progresses from hematopoietic precursor cells (HSCs) and proceeds through several stages, such as a pro-B cell, pre-B cell, and immature B cell. During differentiation, the pre-B-cell receptor (pre-BCR) is generated following immunoglobulin locus rearrangements and is expressed on the cell surface. This pre-BCR (consisting of the surrogate light chain [VpreB or Vλ5] and an Igμ heavy chain) undergoes further rearrangements of the light and heavy-chain genes to form a mature BCR that can bind to the antigen. A selection process occurs at this immature B cell stage that prevents self-reactive cells from developing further. This stage is accompanied by both clonal selection and receptor editing. Those cells that successfully pass through this checkpoint (named transitional B cells) leave the BM and acquire their mature form as mature follicular B cells. (B) T cells development starts from HSCs in bone marrow and progresses to the thymus, where it passes through a series of developmental stages that can be recognized based on the expression of different cell surface markers. In the beginning of development, the expression of co-receptors CD4 and CD8 are absent and called double negative (DN) cells. The DN cells (DN1, DN2, DN3, and DN4) are further sub-divided by the expression of CD117, CD44, CD25, CD127, and CD3 markers. Further differentiation takes place by the up-regulation of CD4 and CD8 expression, therefore, names as double positive (DP) cells. The negative selection against self-antigen occurs in the thymus (medulla), where antigens are presented to them by dendritic cells and macrophages. T cells with stronger affinity then eliminated and the remaining T cells downregulate either co-receptor CD4 or CD8 and give rise to naïve cells stay in thymus and periphery. (C) The macrophage development and maturation also take place in bone marrow and tissues. From HSC, myeloid colony forming units are derived in bone marrow, and further grow into monocytes under colony-stimulating factor 1 (CSF1) through various highly organized stages. These monocytes can give rise to common DC progenitor cells that can transform into blood monocytes and, upon homing to various tissues except brain and skin macrophages, tissue macrophages. During most of the developmental stages, various factors influence the macrophage lineage development, however, CSF1 is likely imparted the highest influence. Finally, to address the inflammation, monocytes are recruited to tissues and restricted to specific phenotypes, M1, M2, and tumor-associated macrophages (TAM) depending upon inflammatory milieu. The development of TAM could also be influenced by tumor cells and tissue-resident macrophages. On the other hand, some other factors released by TAMs suppress the local immune response by either directly suppressing T cell responses or recruiting Treg cells. Arg1, arginase 1; bFGF, basic fibroblast growth factor; CCL, chemokine (C-C motif) ligand; CD, cluster of differentiation; CSF1 colony-stimulating factor-1; CXCL, chemokine (C-X-C motif) ligand; FLT3, FMS–like tyrosine kinase receptor 3; IFN, interferon; Ig, Immunoglobulin; IL, interleukin; LIF, leukocyte inhibitory factor; MDP, macrophage-derived proteoglycan; MHC, major histocompatibility complex; MMP, matrix metalloproteases; MR, Mineralocorticoid receptor; PD-L, Programmed death-ligand; PGE, prostaglandin; SR, scavenger receptor; TAM, tumor-associated macrophage; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Adaptive immune system in CLL: lymphocytes involvement in CLL pathogenesis
The adaptive immune response is mainly carried out by B cells, T cells NK cells, where, B cells are the major player in various blood-related abnormalities. For the development and maturation of B cells, the B cell receptor (BCR) is the critical mediator of the proliferation and survival of mature B cells and other precursor tumor cells, as well as, BCR mutational status, is highly correlated with the pathology of disease (8). Moreover, mutations in the receptors are thought to play pivotal roles in CLL etiopathogenesis, as 20% of CLL in unrelated patients involves the display of extremely similar and sometimes even identical antigen receptors. BCR signaling is also critical for CLL cell trafficking, interaction with stromal microenvironment, impaired CLL response, and low expression of the BCR correlates with reduced induction of protein tyrosine kinase activity.
CD5+ B cells are the primary cells that give rise to CLL, although a few reports implicate T cells as well. BCR signaling is the most important feature and a diagnostic marker of CLL that can drive CLL progression in an antigen-independent manner (9). Along with BCR, several genetic modifications are also frequently reported to be causative agents of CLL, and such genetic abnormalities act in synergy with various cells types, such as stromal cells, T cells, and nurse-like cells (NLC) in the lymph nodes (10). Numerous defects affecting downstream signaling proteins in the BCR pathway and mutations in the interleukin 7 (IL-7) receptor are not direct causes of CLL; however, such factors predispose cells to develop into CLL cells, and they also influence the development of B and T cell malignancies and severe immunodeficiencies (11, 12). Moreover, B cell development in humans is heavily reliant on Bruton's tyrosine kinase (BTK) signaling activity, as this pathway regulates the activity of various transcription factors, and any errors in this pathway severely impact and even block B cell maturation (Table 1) (43).
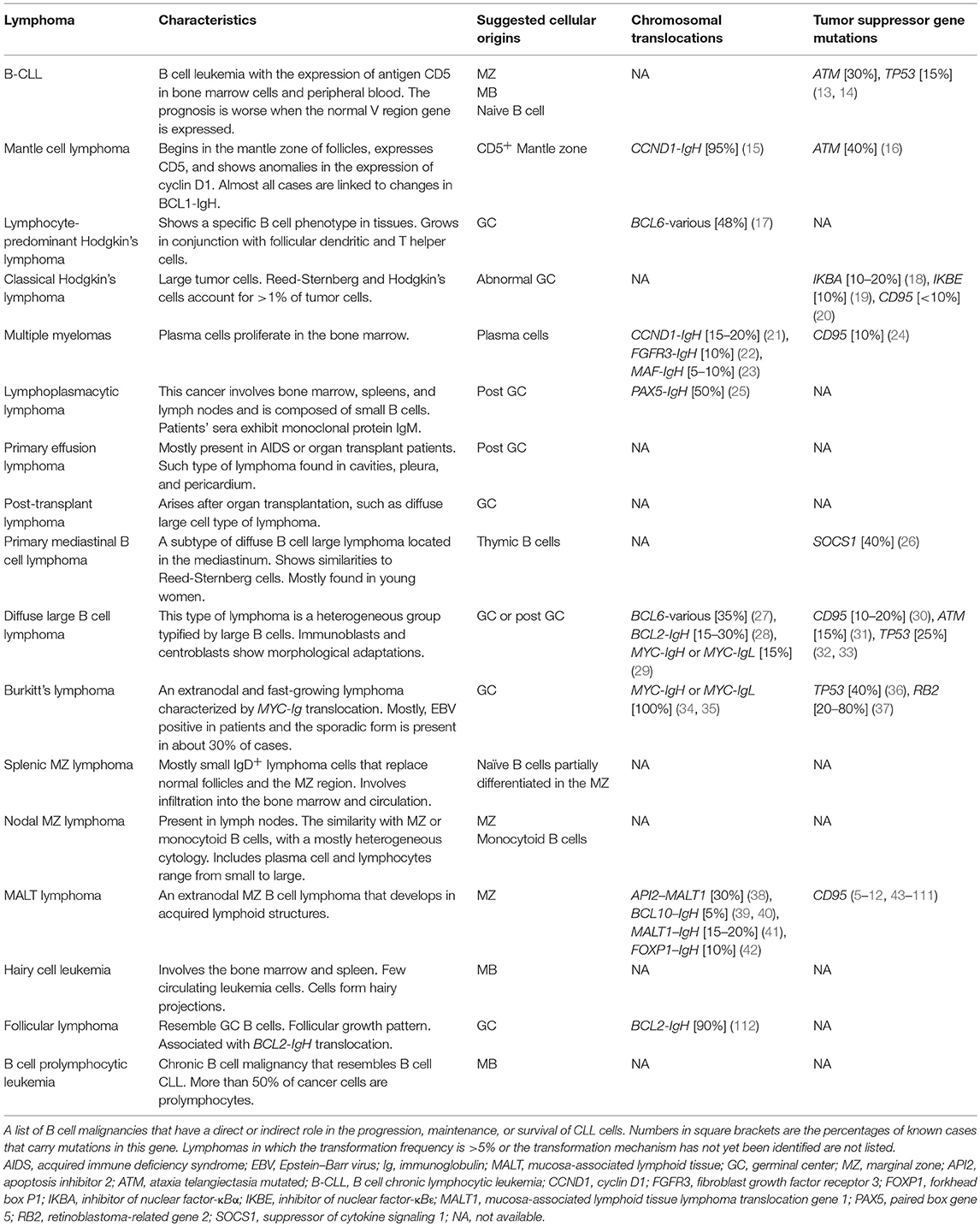
Table 1. B cell lymphomasm and mechanisms of pathogenesis.
CLL also manipulates T cells to gain a survival edge by turning off cytotoxic functionalities of T cells and increased expression of immune checkpoints with abnormal subset distributions. Prior treatment of CLL may also shape the T cells profile (44). CLL cells using their extracellular vesicles (EV) can also modulate T cells in favor of enhanced migration, interaction with tumor cells, and immunological synapse signaling, to avoid immune attack. When analyzed, these EVs contain miR-363 that suppresses the immune modulatory molecule, CD69, and the knockdown of miR-363 altered T cell phenotype (45).
Recently, it has been identified that the lymph nodes harbor the highest number of CLL cells (46), where, the CD4+ T cells can induce them to be adhesive toward hyaluronan through CD40L and CD44 interaction and antagonizing their motility (47). The higher number of CD8+ T cells as compared to CD4+ T cells predispose the CLL patients to a shorter lifespan, and this can be correlated to the immune checkpoint receptor PD-1 expression. CLL patients expressed higher amount of PD-1 due to hypomethylation of its promoter region (48). Further analysis revealed that CLL cell has CCR6 and KLRG1 as differentially methylated genes that have known immune regulatory functions. Moreover, a significant correlation was found between T cells and CLL in terms of PD1/PD-L1 interactions when studied in mice model, Eμ-Tcl1 CLL model, and T cells can express a higher level of PD-1 under leukemic cells influence (49).
CLL cells may also interfere with cytotoxic T cell (CTLs) activity and avoid immune surveillance. This can be attributed to the presence of defective linker for activation of T cells (LAT) that is manipulated by B cells. CLL forms a dysfunctional non-lytic immune synapse with CTLs and stimulates CTLs to release non-polarized lytic granules, thus escaping CTL mediated cytotoxicity (50). LAT involvement in clonal expansion and long-term memory was also reported via Ubiquitin Specific Peptidase 9 X-Linked (Usp9X). Ubiquitinated ZAP70 is unable to form functional signalosome with LAT, and Usp9X mediated deubiquitylation of ZAP70 improves signalosome formation in CD4+ T cells. Usp9X triggers deubiquitylation under TCR in T cells and similarly activates B cells under BCR for the induction of protein kinase C β (PKCβ) (51). In this way, Usp9X functions to sustain adaptive immune response. The restoration of CTL functions using a combination of GM-CSF and IL-4 (termed as GIFT-4) has been evaluated. GIFT-4 induces CTL to secrete IFN-γ and causes lysis of autologous CLL. GIFT-4 treatment also up-regulated CD40, CD80, and CD86, various interleukins and STAT5 phosphorylation that can convert CLL cells into immune helper-like cells (52).
The imbalances in T cells ratio are a critical proponent for CLL with supporting evidence that the expansion of CD8+ T cells in CLL possibly related to a CLL-specific adaptive immune response (53). These imbalances are perpetuated through CD4+ forkhead box P3 (FoxP3+) regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) (54, 55). Moreover, it has been reported in several studies that decrease in the number of IL-17+, T-helper 17 (Th17) and CTL numbers are associated with poor prognosis (56), while the expansion of Th17 accompanied by lenalidomide might have a defensive role (57). However, the induction of Th17 cells and related cytokines may have the possibility to increase the complication of autoimmune cytopenias (56, 58).
Innate immune system in CLL: macrophage-DC precursors in CLL pathogenesis
The prominent cells in a myeloid lineage that are crucial for tumor response include macrophages, monocytes, and DCs. Monocytes are acquired from ancestor cells and differentiate either into macrophages or DCs at marginal tissue sites (6). At the beginning of an adaptive immune response, stimulated DCs migrate to secondary lymphoid organs and present the antigen to other antigen presenting cells (59, 60), whereas, after activation, macrophages have tissue-specific functions and remain in the peripheral tissues (61).
Macrophages are usually considered phagocytes of cancer cells and disease-causing particles and they can acquire M1 (antitumor) or M2 (protumor) phenotypes depending on various stimuli and microenvironment (62). Normally, microbial components or IFNγ activates M1 macrophages, which activate T cells, while, the M2 phenotype is triggered by IL-4 and -13 and is involved in controlling the disease response (63), hence macrophage penetration in tumors can be either positive or negative, depending on the tumor type, which suggests the dual role of macrophages (64).
The development of a tumor niche occurs during neoplastic conversion when B cell factors attract circulating monocytes that differentiate into macrophages, which express CD163, CD206, CD14+, and CD68 and show an M2-like functionality (64). The differentiated macrophages are functionally indistinguishable from NLCs, rendering TAMs and NLCs functionally equivalent in CLL tissues (65–67). CD14+ monocytes from a healthy donor cultured in vitro can be transformed into NLCs with CD19+ in the presence of CLL B cells; however, normal B cells lack this ability to transform. Differentiation into NLCs promotes the survival of CLL cells via cytokine production. In contrast, healthy B cells are unable to induce an NLC phenotype in monocytes and do not support CLL cell survival (68). Thus, factors that increase the survival of CLL cells are released with the differentiation of NLCs. Moreover, NLCs are blood monocyte-derived cells that secret CXCL12 and 13, and that protect CLL cells from spontaneous apoptosis or drug-induced apoptosis in response to CXCL12, B cell-activating factor (BAFF), a proliferation-inducing ligand (APRIL), CD31, plexin-B1, and activation of the BCR signaling cascade (69–72). NLCs are known as TAMs and LAMs (lymphoma-associated macrophages) in other B cells malignancies; however, the signaling mechanism and roles are similar.
The essential role of chemokines and cytokines in stimulating the symbiotic relationship is supported by monocytes, TAMs/NLCs, and tumor cells. CLL cells secrete IL-4, -10, and -13 (73) that endorse M2-like properties in macrophages and stimulate pro-survival responses through the secretion of IL-8 (74), CCL2, CXCL2, 12 (75, 76), and insulin-like growth factor-1 (IGF-1) (77). Because of the sustained selection of circulating monocytes, the tissue niche is expanded and is rich in chemokines, such as CCL2, 3, and 4 and CXCL12, 13, 19, and 20 (72, 78). Consequently, TAMs/NLCs are promoted by leukemic cells and anti-tumor immunity is suppressed in CLL microenvironments. The properties of TAMs can be modulated by the phosphoinositol-3 kinase/mammalian target of rapamycin (PI3K/mTOR) pathway (79, 80), and they have been shown to suppress T cell-mediated antitumor responses through the PI3Kγ signaling pathway (81, 82). Moreover, gene expression profiling (GEP) of CLL-associated monocytes revealed aberrantly high PD-L1 expression and secretion of multiple inflammatory and immunosuppressive cytokines like IL-10, TNF-α, and CXCL9 that also contribute to worsening the situation.
The functional and clinical importance of DC is long been known, with the functional alteration of cytokine profile in CLL (83), these cells also showed phenotypically immature population with the absence of maturing antigen CD83 and CD80, reduced expression of IL-12 and unable to activate type 1 T cell response (84). Further, unable to induce T cell response can also be coupled to SOCS5 that negatively regulates STAT6, a downstream mediator of the IL-4Rα receptor. In CLL, overexpression of IL-4Rα is due to overactivation of STAT3 and to regulate IL-4Rα activation, SOCS5 comes into play. This decoupling reduced the pro-inflammatory cytokines from DCs and hinders its maturation (85). In another study, the involvement of CXCR5 has been studied in Eμ-Tcl1 CLL model and it has been concluded that CXCR5-deficient cells showed reduced leukemic transformation, and in this activity, follicular DCs play a critical role (86). The disruption of this link may offer a potential therapeutic window.
DC vaccines hold therapeutic potential, and when apo-DC based vaccine along with GM-CSF, lenalidomide, and cyclophosphamide has been evaluated, it triggers a T cell response against tumor cells, however, it showed toxicity that warrants careful administration of this combination (87). The lower induction of TH1 cytokine profile from DC vaccines is an impediment to their clinical efficacy. Recently, it has been reported that α-type-1 polarized DCs (αDC1s) have shown to produce superior TH1 cytokine profile, with an equivalent number of CD70 expression. These αDC1s can be an alternative to prostaglandin-mediated DC maturation for better CLL treatment (88).
Signaling pathways in CLL
In CLL, numerous pathways play essential roles in responding to external stimuli, making identification of the most significant pathway a challenge. Several pathways are involved in the proliferation and survival of CLL cells, including the mitogen-activated protein kinase/extracellular regulated kinase (MAPK/ERK), nuclear factor κB (NF-κB), Notch, Wnt, phosphatidylinositol-3-kinase/AKT (PI3K/AKT), and Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathways (89). The stimulation of signaling pathways, specifically MAPK/ERK, NF-κB, and PI3K/AKT, in a tissue microenvironment is dependent upon the composition of the microenvironment. This can differ among different tissues, triggering specific signaling pathways (Figure 2) (90).
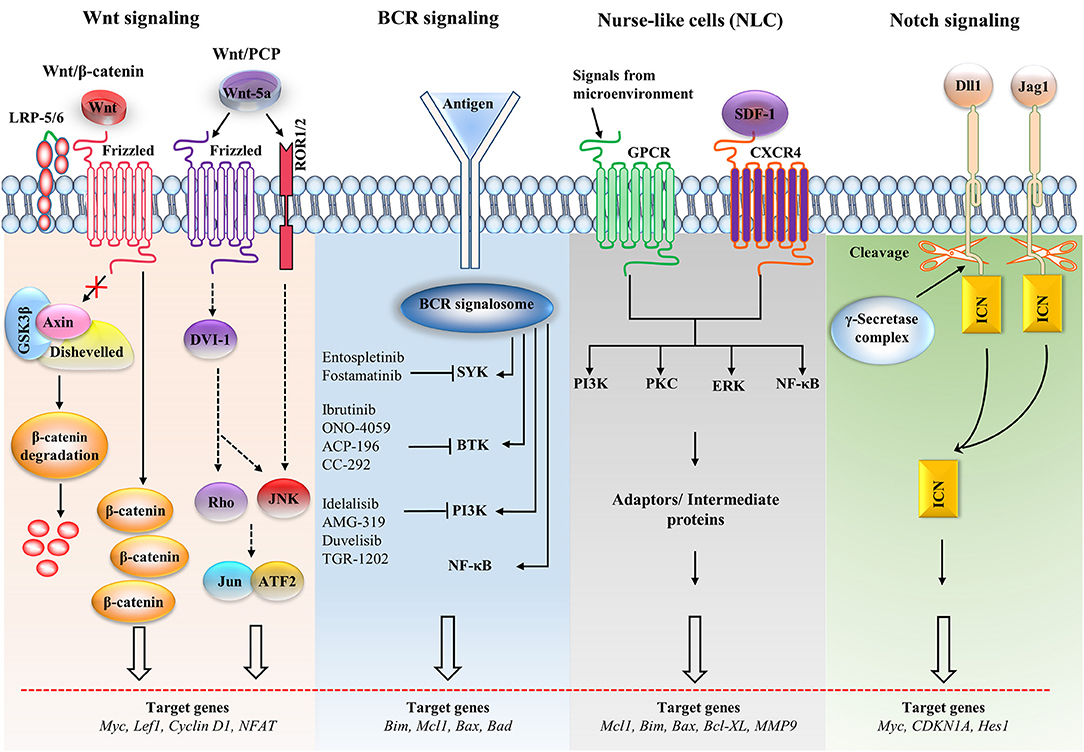
Figure 2. Major signaling pathways and therapeutic targets in CLL cells. The Wnt/β-catenin signaling pathway is activated by the Wnt ligand attached to Frizzled and LRP-5/6 receptors that activate disheveled (dsh), resulting in the inactivation of the destruction complex. This inactivation causes the accumulation of β-catenin, which enters the nucleus. The nuclear translocation of β-catenin allows it to interact with the transcription factor TCF/LEF and induces the transcription of the target genes. The non-canonical Wnt pathway [Wnt/planar cell polarity (PCP)] is triggered by Wnt-5a that enhances the heterodimerization of receptor tyrosine kinase-like orphan receptor 1 (ROR1) and ROR2. Receptor binding leads to the formation of the ligand-receptor complex and downstream activation of the dsh protein (DVI-l), Rho GTPases and c-Jun N-terminal kinase (JNK), which together regulate tissue polarity and cell motility, and has also been implicated in organogenesis and cancer metastasis. B cell receptor (BCR) signaling causes a signalosome containing spleen tyrosine kinase (SYK), Bruton's tyrosine kinase (BTK), phosphatidylinositol 3-kinase (PI3K), and adaptor proteins to form, which activates downstream pathways. Small molecule inhibitors are shown in the BCR pathway. In nurse-like cells (NLCs), the interaction of chemokines with receptors generates microenvironmental signaling. NLCs regulates several signaling pathways, including the protein kinase C (PKC), PI3K, extracellular regulated kinase (ERK), and nuclear factor-κB (NF-κB) pathways, which are similar to the BCR pathway. In the Notch signaling pathway, the ligands [Dll1, Jagged-1 (JAG1)] bind with the receptor and cause the intracellular Notch domain (ICN) to be cleaved by the g-secretase complex. The cleaved domain then translocates to the nucleus, forms complex with other proteins and activates target genes. BAD, Bcl2-associated agonist of cell death; Bcl-XL, B-cell lymphoma-extra-large; CDKN1A, cyclin-dependent kinase inhibitor 1A; GSK3β, glycogen synthase kinase-3β; GPCR, G-protein coupled receptor; LEF1, Lymphoid enhancer-binding factor 1; LRP, lipoprotein receptor-related protein; MMP, matrix metalloproteinase; SDF-1, stromal cell-derived factor 1.
The BCR signaling pathway is the most critical pathogenic factor, and it has long been considered a valuable target in CLL. In various studies, BTK (kinase factor of BCR) inhibitor has been evaluated with other chemotherapeutic agents to reduce the pathophysiology of CLL (72). Continuous activation of the BCR leads to the apoptosis-resistant CLL cells, and overexpression of the antiapoptotic proteins XIAP, MCL-1, and BCL-XL (91). Moreover, studies involving ibrutinib and CAL101 (PI3K inhibitor) have shown promising results in the treatment of CLL (92). Furthermore, evaluation of a highly selective oral AKT inhibitor, MK2206, indicated that this compound selectively inhibits BCR-induced cytokines, activates other lymphocytes, and in synergy with bendamustine, induces apoptosis in CLL cells (93).
The MAPK/ERK pathway conveys pro-survival signals in cancerous cells and is mainly activated by various growth factors, and CCL19, 21 and CXCL12, 13 stimulation (94, 95). The basic components of this pathway include one small G protein (Ras) accompanied by three other kinases (RAF, MEK, ERK) that, upon activation, leads to the translocation of the ERK to nucleus and activation of target genes (96). The entry of CLL cells into the S-phase of the cell cycle and expression of MYC are essential activities of MEK1/2, and MEK2 also upregulates antiapoptotic protein, XIAP (91, 95). MYC induces the cell cycle component cyclin D2 and downregulates p27, a cell cycle inhibitor (97, 98).
The NF-κB pathway generally promotes proliferation and is downstream of many cell surface receptors, including Toll-like receptors (TLRs). These receptors can be triggered through a multitude of signals, leading to the activation of NF-κB, which induces the transcription of CCL5, 9, 17, 20, and 22, IFNγ, IL-2, 6, 8, 9, and 10, MIPs, CCRs, CXCRs, TLR 2 and 9, and various other early response genes, transcription factors, and regulators (99). It also cross-talks with various other pathways; for instance, stimulation of NF-κB upregulates BFL-1 and B cell lymphoma-extra-large (BCL-XL) anti-apoptotic genes, which inhibit the apoptosis of CLL cells via PI3K/AKT signaling. Further, IL-4 and soluble CD40-ligand (sCD40L) were found to be most effective in preventing CLL apoptosis by triggering NF-κB (100). NF-κB also modulates the redox state of cells, and, when production of reactive oxygen species is blocked using IT901, tumor cells become susceptible to apoptosis (101).
Notch signaling pathway plays a role in embryonic development, cell fate determination, and neural differentiation. There are four different Notch receptors: Notch1, Notch2, Notch3, and Notch4. A mutation in the Notch1 receptor has been confirmed in 10–15% of CLL patients (102). The initial events in this pathway involve the interaction of Jagged and Delta-like ligand with the Notch receptor, induces its cleavage by γ-secretases, and the cleaved Notch1 receptor forms a complex with other factors that stimulate the transcription of target genes, including HES1 and Myc. In CLL, Notch1 and Notch2 and their ligands are constitutively expressed, which allow cells to resist apoptosis, upregulates NF-κB activity, and induces expression of XIAP and cIAP2 (cellular inhibitor of apoptosis protein 2). However, inhibition of Notch signaling by γ-secretase inhibitor and specific siRNAs promotes the death of CLL cells (103). Furthermore, Notch signaling is also involved in the induction of MCL-1 and the promotion of eukaryotic translation initiation factor 4E activity that leads to CLL survival (104). Inhibition of the Notch signaling pathway may also induce the expression of Kruppel-like factor 4, which was suppressed due to aberrant methylation (105).
The Wnt signaling pathway plays an important role in cell development, differentiation and oncogenesis. This pathway shows a high level of Wnt and Frizzled expression, and downregulation of antagonist genes, including WIF1 and secreted Frizzled-related members (SFRP). Wnt activation inhibits GSK3β mediated β-catenin phosphorylation, then the dephosphorylated β-catenin enters the nucleus and binds with lymphoid enhancing factor (LEF) to stimulate the transcription of Wnt target genes, such as LEF, CyclinD1, Myc, matrix metalloproteinases, and cyclooxygenase-2 (106–108). During CLL, overexpression of the Wnt pathway and enhanced translocation of β-catenin to the nucleus occurs in the absence of E-cadherin (106). In a recent report, ibrutinib, a classical BTK inhibitor, was used to target metadherin, that not only inhibited metadherin, but also LEF1 and β-catenin (109). Other than canonical Wnt, recently, Wnt5a was found to act as a regulator of ROR1, a receptor in the non-canonical Wnt/planar cell polarity (PCP) pathway, which may promote CLL cell survival and subvert apoptotic signals (110).
PI3K/AKT signaling pathway is a central mediator of cancerous cells and their microenvironment and transmits signals from CXCR4 and CD40 (91). The principal regulator of this pathway is phosphatase and tensin homolog (PTEN) (113), along with other transcription factors, such as FOXO. Upon activation of PTEN, FOXO proteins are translocated to the nucleus and induce p27, which arrests the cell cycle. Activation of the PI3K/AKT pathway by microRNA-22 induces the proliferation of CLL cells, which can be reversed by inhibiting the PI3K-Δγ signaling pathway using IPI-145 (114, 115). The role of the PI3K/AKT pathway in CLL proliferation can be attributed to a chemotactic response mediated by CXCL12 and 13 and CCL19 and 21 (69, 116) and assists in the survival of cancerous cells in response to various exterior stimuli conveyed through CD40L (117), BCR (91), CCL19 and 21, vascular cell adhesion molecule 1, and anti-apoptotic proteins, such as BCL-2 and MCL-1 (90). Moreover, synergy was observed in PI3K/AKT and Hedgehog (HH)/GLI pathway, when both these targeted simultaneously, a synergistic therapeutic effect is observed in CLL, which suggests a combinatorial therapy (118).
The JAK/STAT signaling pathway is another pathway crucial that mediates signals from cytokines, which are soluble messengers produced by various cells. The JAK/STAT pathway provides a direct link from the cell surface to the nucleus. The binding of the cytokine to its cognate receptor triggers this pathway that involves phosphorylation among JAK and STAT molecules. Later, STAT molecules translocate to the nucleus and bind with DNA as either hetero- or homodimers. There are multiple JAK and STAT molecules that combine in different ways to induce diverse transcriptional profiles (119). In CLL, inhibition of the JAK2/STAT3 pathway culminates in the apoptosis of cancerous cells. The dual inhibitor cerdulatinib, which targets both spleen tyrosine kinase (SYK, a BCR component) and JAK kinases, potently inhibits tumor growth and induces apoptosis in CLL cells at clinically feasible drug concentrations (120).
Innate vs. Adaptive Immune System: an Interplay of Myeloid and Lymphoid Cells in CLL
The interaction between lymphocytes and myeloid cells is crucial for immune response and any anomaly may prone the individual at risk of developing diseases. B cells present in the MZ, a region between red and white pulp of spleen and enriched with macrophages, are an essential link between innate and adaptive immunity (121, 122). Recently, acquisition of antigen through BCR and then transfer to macrophages through direct contact has been reported (7) that signifies B cells involvement in macrophage-mediated CD4+ T cells activation. Further, B cell development into memory or plasma cells needs to encounter an antigen, which can either be diffusion-controlled or be presented by macrophages, DCs, or FDC (123).
B cells and macrophages show bidirectional interactions via different soluble factors. Macrophages produce APRIL and BAFF in the presence of costimulatory signals, such as IL-6, TGFβ, IL-10, and TLR ligands (124, 125). In proliferating B cells, BAFF is significantly involved in signals exchanged between macrophages and B cells. Additionally, B lymphocyte stimulator (BLyS), a TNF family member, also induces the B cell growth. The B cell tropism of BLyS is consistent with its receptor expression on B-lineage cells. The biological profile of BLyS suggests that it is involved in monocyte-driven B cell activation (125, 126). IL-10 involved in the initial development of B1 cells, and IL-6 contributing to later B cell development (127, 128). Moreover, the accumulation of B cells in response to IL-10 can suppress murine macrophage functions in vitro (129) and transform macrophages into cells with a pro-tumor phenotype both in vivo and in vitro (Figure 3) (130).
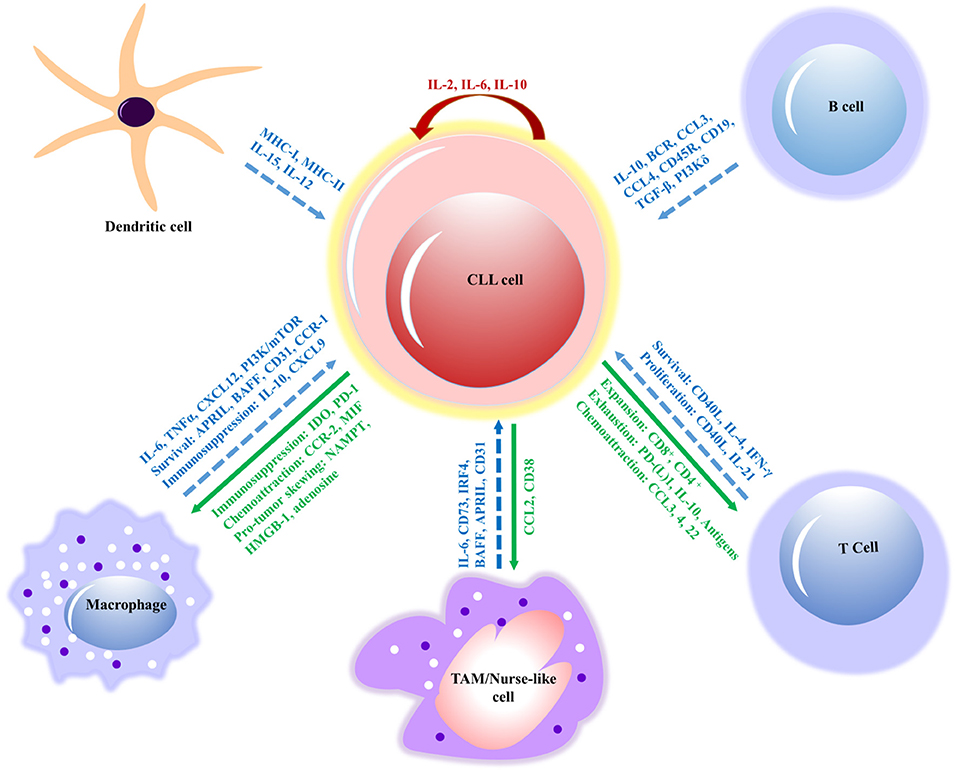
Figure 3. Interactions between CLL cells and bystander cells that subsidize the formation of a tumor-supportive microenvironment. The molecular interactions of innate immune cells (macrophages, dendritic and nurse-like cells) and adaptive immune cells (B and T cells) have relevant effects, such as survival, immunosuppression, proliferation and signaling molecules (in blue) involved in the interactions of CLL cell. The response of CLL cells includes exhaustion of T cells, expansion, chemoattraction, immunosuppression, and immune evasion (in green) on innate and adaptive immune cells. However, IL-2 and IL-10 (in red) contribute to the autocrine self-renewal and survival of CLL cells. APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; BCR, B cell receptor; CCL, chemokine (C-C motif) ligand; CD, cluster of differentiation; CLL, chronic lymphocytic leukemia; CSF1R, colony-stimulating factor-1 receptor; CSF1, colony-stimulating factor-1; CXCL, chemokine (C-X-C motif) ligand; (TGF-β), Transforming growth factor beta; NAMPT, extracellular nicotinamide phosphoribosyltransferase; HMGB1, High mobility group box 1; IL, interleukin; MIF, mini zinc finger; PI3K/mTOR, phosphatidylinositol-3-kinase/mammalian target of rapamycin; TAM, tumor-associated macrophage; MHC, major histocompatibility complex; TNF, tumor necrosis factor; CCR, chemokine receptor; IDO, indoleamine 2,3-dioxygenase; PD-1, program cell death; IRF4, interferon regulatory factor; IFN, interferon.
GEPs indicate complex interactions between macrophages and malignant B cells in the microenvironments that induce cellular changes. This analysis has shown that CLL cells are activated differently by bone marrow-derived stromal cells (BMSCs) and NLCs (72, 131). In particular, the expression pattern induced by BMSCs shows a characteristic upregulation of T cell leukemia/lymphoma 1 (TCL1), a lymphoid proto-oncogene, with a concomitant decrease in TCL1-interacting FOS proto-oncogene and Jun proto-oncogene (FOS/JUN) (131). However, an NLC-induced GEP pattern in leukemic cells showed the expression of genes in the NF-κB and BCR signaling pathways (72), which was astonishingly similar to the expression pattern of CLL cells that were extracted from a patient's lymph nodes (132). Additionally, BMSCs differentially induce many vital genes (e.g., TNF receptor superfamily member 17 (TNFRSF17), pre-B lymphocyte 3, TNFSF10), but their precise functions in the CLL microenvironment remain to be explored. MDSCs have also been widely studied in the context of immune and T cells suppression in malignant diseases. MDSC (CD14+/HLA-DR−/−) was increased in CLL that suppress T cell activation and caused suppressive Tregs activation and this can be attributed to increased indoleamine 2,3-dioxygenase activity in T cells (55).
The interaction of leukemic cells with bone marrow cells is perpetuated through adhesion molecules and chemokines (133), and cell-to-cell contact via bidirectional interplay as studied by Hacken and Burger (134). The CXCR5 expression on leukemic cells controls the approach to FDCs, which provide the proliferative stimuli and expression of myeloid cell leukemia-1 (MCL-1) antiapoptotic protein (86, 135). The stimulation or suppression of the development of CLL clones by T cells have been reported, suggesting that T cells are unable to form efficient immunological synapses alone (136). On the other hand, colocalization with CLL cells indicates that they may induce the development of leukemic clones (133). There are several T cell-attracting chemokines, such as CCL3 and 4, which are secreted at high levels by CLL cells in response to TNF-α, IFNα, and IL-2 that protect cancerous cells from apoptosis (72, 134).
Human signaling lymphocytic activation molecule family 1 (SLAMF1/CD150) receptor is expressed by T cells, B cells, macrophages, and DC, where it acts as a co-stimulatory through self-interactions on the hematopoietic cell surface, and plays critical roles for various T and B cells functions. In a subset of CLL patients, SLAMF1 was found to be downregulated that warranted the shorter life spans (137). Moreover, SLAMF1−/− positively affects chemotactic response to CXCL12 by overexpression of CD38, CD44, and CXCR4, and its activation through antibody can facilitate the autophagic flux. SLAMF1 along with other receptors, particularly, CD180, influences the leukemic cells pathobiology. This signaling crosstalk causes the inhibition of AKT and MAPK pathways and results in reduced phosphorylation of ERK1/2, AKT, ribosomal S6 kinase, c-Jun, and other vital intermediate kinases (138, 139). Thus, various studies revealed that SLAMF1 plays a critical role in CLL pathogenesis and restoring the expression of SLAMF1 would be a great therapeutic target for CLL.
Therapeutic Implications
CCL is either resistant to chemotherapy or the outcome is very poor. Consequently, various studies are underway to develop new therapies that either inhibit intracellular signaling or block extracellular signals using small molecules, antibodies, kinase inhibitors, immunomodulators, and antagonists (Table 2) (90).
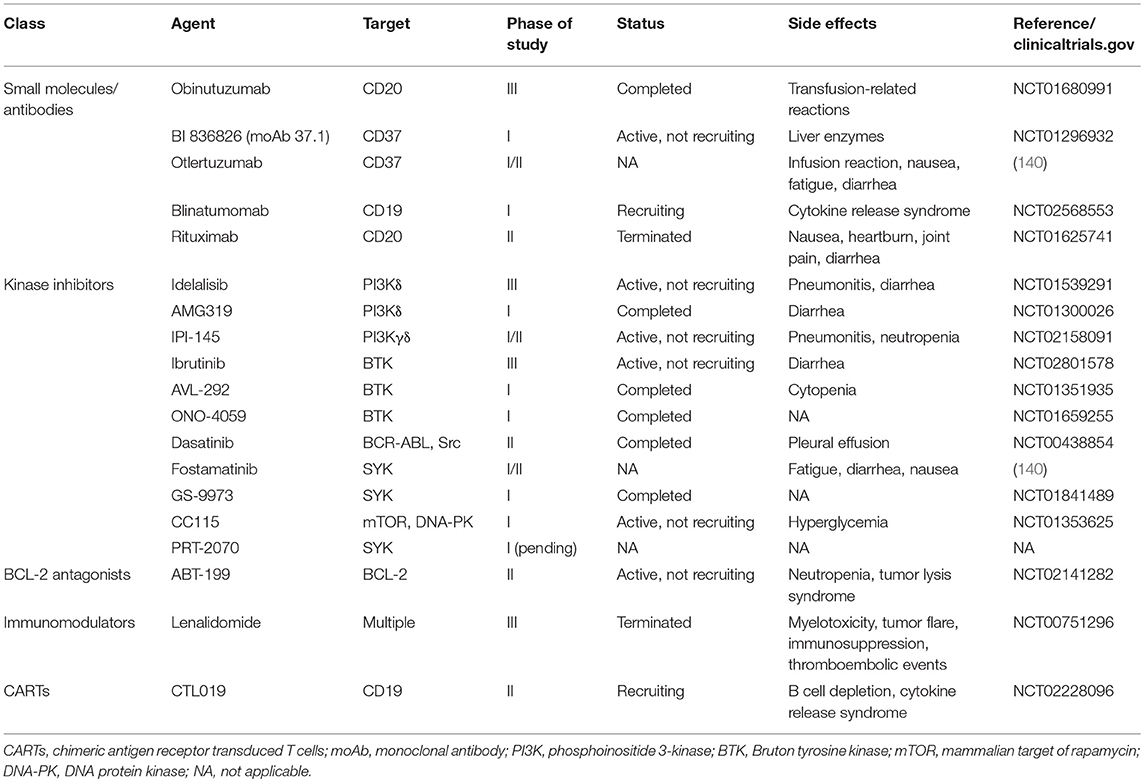
Table 2. Current therapies that are under investigation for use in chronic lymphocytic leukemia.
The role of colony stimulating factor 1 (CSF1/CSF1R) in regulating the survival and differentiation of macrophages is well-recognized, and blocking signaling through this pathway may have therapeutic significance (141, 142). The activity of CSF1R is blocked by the inhibitor pacritinib, which reduces NLCs and limits the progress of CLL (143). Importantly, a novel treatment, anti-CSF1R monoclonal antibody, is under clinical assessment in solid tumors (144). It has also been observed that CSF1R blockade targets TAMs and reprograms the microenvironment to an antileukemic phenotype. Targeting macrophages not only increases the death of CLL cells but also that of CD20+ leukemic cells (145). In a recent clinical trial, patients were given ibrutinib alone and in combination with anti-CD20 monoclonal antibodies (moAbs). The study revealed that ibrutinib had both positive and negative effects, as ibrutinib consistently downregulated CD20, rendering cells less susceptible to moAb. However, ibrutinib can impair trogocytosis, a major contributor to antigen loss and tumor escape during moAb therapy (146). Moreover, macrophage targeting through CSFR1 blockade can also increase CD20+ leukemic cells, and targeting of macrophages by either CSF1R signaling blockade or clodronate-induced macrophage killing can significantly inhibit established leukemia. The removal of macrophages facilitates TNF-mediated leukemic cell death and modifies the microenvironment to induce an antitumor response. By inhibiting CSF1R, it reduces the CLL cell population and increases CD20+ CLL cells that can be co-targeted along with TAM to achieve superior results (145).
Recently, a scavenger receptor, MARCO, was identified on TAMs that is immunosuppressive and is present in the tumor microenvironment. This receptor is specific for a subtype of TAMs and has been successfully targeted through moAbs in various solid tumors, where it has antitumor effects (80). The antitumor effects obtained by targeting this receptor is proposed to be mediated by the Fc-receptor FcγRIIB. Although it has not been evaluated in CLL, it presents a potential target for therapy. Trabectedin is used for the treatment of soft tissue sarcomas, where it reduces TAMs (147). It is cytotoxic for human monocytes and inhibits the production of IL-6 and CCL2; in addition, it may shape the microenvironment of chronic B lymphoid malignancies (147). CD47 is another target molecule that needs to be explored in CLL as a potential therapy (148). Lenalidomide is an immunomodulatory drug used for the treatment of CLL, (149) and showed rare cytotoxicity in malignant cells, but apparently, it acts on tumor-supporting microenvironments and affects the CLL myeloid compartment (150).
Valproic acid (VPA) is a histone deacetylase inhibitor (HDAC/HDI), which has long been used as an antiepileptic drug. VPA has also been evaluated in hematological malignancies for the apoptosis induction of cancerous cells, and has been clinically tested to cause differentiation in carcinoma cells, acute myeloid leukemia cells from patients, and transformed hematopoietic progenitor cells. It has also reduced the tumor growth in animal models (151, 152). VPA perpetuates its effects by up-regulating cyclin-dependent kinase inhibitor 1A, BCLA1, and p53, whereas proto-oncogene (c-Myc, BCL2, BCL-XL, Ataxia-Telangiectasia Mutated, protein kinase B) were downregulated, thereby augmenting mTOR inhibitor to induce autophagic cell death in several kinds of lymphomas including Burkitt leukemia/lymphoma, cutaneous T cell lymphoma and CLL (153, 154). Furthermore, VPA alone or in combination with other drugs, such as fludarabine, flavopiridol, cladribine, lenalidomide, and bortezomib is highly effective in inducing cell death in CLL cells and could be considered for therapeutic intervention (155, 156).
Immune checkpoint inhibitors (ICI) are gaining popularity for their therapeutic potential as these ICIs block the interaction of cytotoxic T lymphocyte-associated 4 (CTLA4) and PD-1 to their cognate ligands, CD80/CD86 and PD-1 ligand (PD-L1), respectively. These interactions are critical for cancer cell survival including CLL in the tumor microenvironment (157). Various ligands and antibodies have been developed to target this particular interaction. Recently, chimeric antigen receptor (CAR) T cell therapy is being studied that targets B cell-specific antigen in CLL, CD19 (anti-CD19 CART [CTL019]), and observed a promising response in clinical trials for CLL patients (158). In future, CAR-T cells combining with other therapeutic agents could be the treatment of choice for various malignancies.
Future Prospect and Conclusion
B cells and macrophages are crucial components of the immune system. These cells interact and influence each other at various levels, beginning in development and later inactivation and immune responses. There are several links between these cells that are either protumoral or antitumoral. In this review, we provided a sketch of B cells and macrophages and discussed their links. The molecular interactions between macrophages and B cells have been characterized in detail in CLL. There are various mutations that cause CLL, affecting different pathways as either causative agents or enhancers of CLL pathogenicity, as well as, a plethora of signals from the microenvironment and the interplay of various other cells that play roles in the progression of CLL. All these aspects complicate the mechanistic elucidative studies, hinder the formulating and devising therapeutic strategies, and complicate the outcome of already implemented therapeutic regimens.
Several pathways are involved in CLL. The real problem arises when these pathways interact with other pathways to increase the overall intensity or exaggerate the outcome. These interactions are surprisingly difficult to manage in cancer cases, and particularly in CLL. Moreover, CLL is a neoplasm of immune-related cells, adding an extra layer of complexity, because any immunotherapy approach that activates immune cells can also activate tumor cells.
Consequently, we assessed that single molecule therapy is often inadequate for CLL as these offer a wide range of interactions that simultaneously propagate the disease, suppress the immune system, and resist the therapeutic intervention. Therefore, we emphasized on the common targets such as, PD-1, BTK, CSF1R, and CD20 that were in one cell influence the phenotype of other cells rendering the possibility of co-targeting multiple cells simultaneously in order to achieve superior results.
Author Contributions
MH and MA conceptualized and drafted the manuscript. MH created the tables. MH and SC created the figures. SC critically analyzed and coordinated the project. All authors read and approved the final manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF-2015R1A2A2A09001059) and the Commercializations Promotion Agency for R&D Outcomes funded by the Ministry of Science and ICT (2018K000369).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ADP, antibody dependent phagocytosis; Ag, antigen; AIDS, acquired immune deficiency syndrome; APC, antigen-presenting cells; API2, apoptosis inhibitor 2; APRIL, A proliferation inducing ligand; ATM, ataxia telangiectasia mutated; BAFF, B cell activating factor; B-CLL, B cell chronic lymphocytic leukemia; BCR, B cell receptor; BM, bone marrow; BTK, bruton tyrosine kinase; CARTs, chimeric antigen receptor transduced T cells; CCL3, C-C motif chemokine ligand 3; CCL4, C-C motif chemokine ligand 4; CCND1, cyclin D1; CD, cluster of differentiation; CDR3, complementarity determining region 3; CEBPα, CCAAT/enhancer-binding protein α; CEBPβ, CCAAT/enhancer-binding protein β; CLL, chronic lymphocytic leukemia; COX-2, cyclooxygenase-2; CSF1R, colony-stimulating factor 1 receptor; CXCL13, chemokine (C-X-C motif) ligand 13; CXCR5, C-X-C chemokine receptor type 5; DC, dendritic cells; EBV, Epstein-Barr virus; Fcγ R, Fc γ receptor; FDCs, follicular dendritic cells; FGFR3, fibroblast growth factor receptor 3; FOXP1, forkhead box P1; GC, germinal center; GSK3β, glycogen synthase kinase 3β; IFNα, interferon α; Ig, immunoglobulin; IGF-1, insulin-like growth factor-1; IKBA, inhibitor of nuclear factor-κBα; IKBE, inhibitor of nuclear factor-κBε; IL-2, interleukin 2; IL-6, interleukin 6; LEF, lymphoid enhancing factor; LNs, lymph nodes; MALT, mucosa-associated lymphoid tissue; MAPK, mitogen-activated protein kinase; MB, memory B cell; MCL-1, myeloid cell leukemia; MMPs, matrix metalloproteinases; moAbs, monoclonal antibodies; mTOR, mechanistic (mammalian) target of rapamycin; MZ, marginal zone; NF-κB, nuclear factor-κB; NK, natural killer cell; NLCs, nurse-like cells; PAX5, paired box gene 5; pHSCs, pluripotent hematopoietic stem cells; PI3Kγ, phosphoinositide 3-kinase; PKC, protein kinase C; PU.1, purine rich sequence box; RB2, retinoblastoma-related gene 2; SOCS1, suppressor of cytokine signaling 1; SYK, spleen tyrosine kinase; TAMs, tumor-associated macrophages; TNF-α, tumor necrosis factor-α; WIF1, WNT inhibitory factor 1.
References
1. Howlader N, Noone A, Krapcho M, Garshell J, Neyman N, Altekruse S, et al. SEER Cancer Statistics Review, 1975–2012. Rockville, MD: National Cancer Institute 2015. The statistics of cancer in the United States Google Scholar (2016).
2. Hulkkonen J, Vilpo L, Hurme M, Vilpo J. Surface antigen expression in chronic lymphocytic leukemia: clustering analysis, interrelationships and effects of chromosomal abnormalities. Leukemia (2002) 16:178–85. doi: 10.1038/sj.leu.2402363
3. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. (2013) 131:959–71. doi: 10.1016/j.jaci.2013.01.046
4. Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. (2016) 48:253–64. doi: 10.1038/ng.3488
5. Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. (2015) 282:20143085. doi: 10.1098/rspb.2014.3085
6. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science (2010) 327:656–61. doi: 10.1126/science.1178331
7. Harvey BP, Gee RJ, Haberman AM, Shlomchik MJ, Mamula MJ. Antigen presentation and transfer between B cells and macrophages. Eur J Immunol. (2007) 37:1739–51. doi: 10.1002/eji.200636452
8. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. (2013) 34:592–601. doi: 10.1016/j.it.2013.07.002
9. Duhren-von Minden M, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature (2012) 489:309–12. doi: 10.1038/nature11309
10. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature (2015) 526:525–30. doi: 10.1038/nature15395
11. Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, et al. Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID). Nature (1995) 377:65–8. doi: 10.1038/377065a0
12. Russell SM, Keegan AD, Harada N, Nakamura Y, Noguchi M, Leland P, et al. Interleukin-2 receptor gamma chain: a functional component of the interleukin-4 receptor. Science (1993) 262:1880–3.
13. Schaffner C, Stilgenbauer S, Rappold GA, Dohner H, Lichter P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood (1999) 94:748–53.
14. Stankovic T, Weber P, Stewart G, Bedenham T, Murray J, Byrd PJ, et al. Inactivation of ataxia telangiectasia mutated gene in B-cell chronic lymphocytic leukaemia. Lancet (1999) 353:26–9. doi: 10.1016/S0140-673610117-4
15. Vaandrager JW, Schuuring E, Zwikstra E, de Boer CJ, Kleiverda KK, van Krieken JH, et al. Direct visualization of dispersed 11q13 chromosomal translocations in mantle cell lymphoma by multicolor DNA fiber fluorescence in situ hybridization. Blood (1996) 88:1177–82.
16. Camacho E, Hernández L, Hernández S, Tort F, Bellosillo B, Beà S, et al. ATM gene inactivation in mantle cell lymphoma mainly occurs by truncating mutations and missense mutations involving the phosphatidylinositol-3 kinase domain and is associated with increasing numbers of chromosomal imbalances. Blood (2002) 99:238–44. doi: 10.1182/blood.V99.1.238
17. Wlodarska I, Nooyen P, Maes B, Martin-Subero JI, Siebert R, Pauwels P, et al. Frequent occurrence of BCL6 rearrangements in nodular lymphocyte predominance Hodgkin lymphoma but not in classical Hodgkin lymphoma. Blood (2003) 101:706–10. doi: 10.1182/blood-2002-05-1592
18. Jungnickel B, Staratschek-Jox A, Bräuninger A, Spieker T, Wolf J, Diehl V, et al. Clonal deleterious mutations in the IκBα gene in the malignant cells in Hodgkin's lymphoma. J Exp Med. (2000) 191:395–402.
19. Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Dohner K, et al. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol. (2003) 201:413–20. doi: 10.1002/path.1454
20. Muschen M, Re D, Brauninger A, Wolf J, Hansmann ML, Diehl V, et al. Somatic mutations of the CD95 gene in Hodgkin and Reed-Sternberg cells. Cancer Res. (2000) 60:5640–3.
21. Avet-Loiseau H, Li JY, Facon T, Brigaudeau C, Morineau N, Maloisel F, et al. High incidence of translocations t(11;14)(q13;q32) and t(4;14)(p16;q32) in patients with plasma cell malignancies. Cancer Res. (1998) 58:5640–5.
22. Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. (1997) 16:260–4. doi: 10.1038/ng0797-260
23. Chesi M, Bergsagel PL, Shonukan OO, Martelli ML, Brents LA, Chen T, et al. Frequent dysregulation of the c-maf proto-oncogene at 16q23 by translocation to an Ig locus in multiple myeloma. Blood (1998) 91:4457–63.
24. Landowski TH, Qu N, Buyuksal I, Painter JS, Dalton WS. Mutations in the Fas antigen in patients with multiple myeloma. Blood (1997) 90:4266–70.
25. Iida S, Rao P, Nallasivam P, Hibshoosh H, Butler M, Louie D, et al. The t(9; 14)(p13; q32) chromosomal translocation associated with lymphoplasmacytoid lymphoma involves the PAX-5 gene. Blood (1996) 88:4110–7.
26. Melzner I, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Barth TF, et al. Biallelic mutation of SOCS-1 impairs JAK2 degradation and sustains phospho-JAK2 action in the MedB-1 mediastinal lymphoma line. Blood (2005) 105:2535–42. doi: 10.1182/blood-2004-09-3701
27. Ye BH, Rao PH, Chaganti RS, Dalla-Favera R. Cloning of bcl-6, the locus involved in chromosome translocations affecting band 3q27 in B-cell lymphoma. Cancer Res. (1993) 53:2732–5.
28. Weiss LM, Warnke RA, Sklar J, Cleary ML. Molecular analysis of the t(14;18) chromosomal translocation in malignant lymphomas. N Engl J Med. (1987) 317:1185–9. doi: 10.1056/NEJM198711053171904
29. Ladanyi M, Offit K, Jhanwar SC, Filippa DA, Chaganti RS. MYC rearrangement and translocations involving band 8q24 in diffuse large cell lymphomas. Blood (1991) 77:1057–63.
30. Gronbaek K, Straten PT, Ralfkiaer E, Ahrenkiel V, Andersen MK, Hansen NE, et al. Somatic Fas mutations in non-Hodgkin's lymphoma: association with extranodal disease and autoimmunity. Blood (1998) 92:3018–24.
31. Gronbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P. ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood (2002) 100:1430–7. doi: 10.1182/blood-2002-02-0382
32. Koduru PR, Raju K, Vadmal V, Menezes G, Shah S, Susin M, et al. Correlation between mutation in P53, p53 expression, cytogenetics, histologic type, and survival in patients With B-Cell non-Hodgkin9s lymphoma. Blood (1997) 90:4078–91.
33. Moller MB, Ino Y, Gerdes AM, Skjodt K, Louis DN, Pedersen NT. Aberrations of the p53 pathway components p53, MDM2 and CDKN2A appear independent in diffuse large B cell lymphoma. Leukemia (1999) 13:453–9.
34. Dalla-Favera R, Martinotti S, Gallo RC, Erikson J, Croce CM. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science (1983) 219:963–7.
35. Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Nat Acad Sci USA. (1982) 79:7837–41.
36. Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA. (1991) 88:5413–7.
37. Cinti C, Leoncini L, Nyongo A, Ferrari F, Lazzi S, Bellan C, et al. Genetic alterations of the retinoblastoma-related gene RB2/p130 identify different pathogenetic mechanisms in and among Burkitt's lymphoma subtypes. Am J Pathol. (2000) 156:751–60. doi: 10.1016/S0002-9440(10)64941-3
38. Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, et al. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11;18)(q21;q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood (1999) 93:3601–9.
39. Willis TG, Jadayel DM, Du M-Q, Peng H, Perry AR, Abdul-Rauf M, et al. Bcl10 is involved in t (1; 14)(p22; q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell (1999) 96:35–45.
40. Zhang Q, Siebert R, Yan M, Hinzmann B, Cui X, Xue L, et al. Inactivating mutations and overexpression of BCL10, a caspase recruitment domain-containing gene, in MALT lymphoma with t(1;14)(p22;q32). Nat Genet. (1999) 22:63–8. doi: 10.1038/8767
41. Streubel B, Lamprecht A, Dierlamm J, Cerroni L, Stolte M, Ott G, et al. T(14;18)(q32;q21) involving IGH and MALT1 is a frequent chromosomal aberration in MALT lymphoma. Blood (2003) 101:2335–9. doi: 10.1182/blood-2002-09-2963
42. Streubel B, Vinatzer U, Lamprecht A, Raderer M, Chott A. T(3;14)(p14.1;q32) involving IGH and FOXP1 is a novel recurrent chromosomal aberration in MALT lymphoma. Leukemia (2005) 19:652–8. doi: 10.1038/sj.leu.2403644
43. Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA, et al. Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood (2014) 123:1207–13. doi: 10.1182/blood-2013-07-515361
44. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Nasman-Glaser B, Young E, et al. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica (2017) 102:562–72. doi: 10.3324/haematol.2016.151100
45. Smallwood DT, Apollonio B, Willimott S, Lezina L, Alharthi A, Ambrose AR, et al. Extracellular vesicles released by CD40/IL-4-stimulated CLL cells confer altered functional properties to CD4+ T cells. Blood (2016) 128:542–52. doi: 10.1182/blood-2015-11-682377
46. Herndon TM, Chen SS, Saba NS, Valdez J, Emson C, Gatmaitan M, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia (2017) 31:1340–7. doi: 10.1038/leu.2017.11
47. Girbl T, Hinterseer E, Grossinger EM, Asslaber D, Oberascher K, Weiss L, et al. CD40-mediated activation of chronic lymphocytic leukemia cells promotes their CD44-dependent adhesion to hyaluronan and restricts CCL21-induced motility. Cancer Res. (2013) 73:561–70. doi: 10.1158/0008-5472.CAN-12-2749
48. Wu J, Xu X, Lee EJ, Shull AY, Pei L, Awan F, et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget (2016) 7:40558–70. doi: 10.18632/oncotarget.9941
49. McClanahan F, Riches JC, Miller S, Day WP, Kotsiou E, Neuberg D, et al. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the Emicro-TCL1 CLL mouse model. Blood (2015) 126:212–21. doi: 10.1182/blood-2015-02-626754
50. Kabanova A, Sanseviero F, Candi V, Gamberucci A, Gozzetti A, Campoccia G, et al. Human cytotoxic T lymphocytes form dysfunctional immune synapses with B cells characterized by non-polarized lytic granule release. Cell Rep. (2016) 15:9–18. doi: 10.1016/j.celrep.2016.02.084
51. Naik E, Dixit VM. Usp9X is required for lymphocyte activation and homeostasis through its control of ZAP70 ubiquitination and PKCbeta kinase activity. J Immunol. (2016) 196:3438–51. doi: 10.4049/jimmunol.1403165
52. Deng J, Pennati A, Cohen JB, Wu Y, Ng S, Wu JH, et al. GIFT4 fusokine converts leukemic B cells into immune helper cells. J Transl Med. (2016) 14:106. doi: 10.1186/s12967-016-0865-1
53. Vardi A, Vlachonikola E, Karypidou M, Stalika E, Bikos V, Gemenetzi K, et al. Restrictions in the T-cell repertoire of chronic lymphocytic leukemia: high-throughput immunoprofiling supports selection by shared antigenic elements. Leukemia (2017) 31:1555–61. doi: 10.1038/leu.2016.362
54. Jadidi-Niaragh F, Ghalamfarsa G, Yousefi M, Tabrizi MH, Shokri F. Regulatory T cells in chronic lymphocytic leukemia: implication for immunotherapeutic interventions. Tumor Biol. (2013) 34:2031–9. doi: 10.1007/s13277-013-0832-x
55. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+ HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124:750–60. doi: 10.1182/blood-2013-12-546416
56. Hus I, Bojarska-Junak A, Chocholska S, Tomczak W, Woś J, Dmoszynska A, et al. Th17/IL-17A might play a protective role in chronic lymphocytic leukemia immunity. PLoS ONE (2013) 8:e78091. doi: 10.1371/journal.pone.0078091
57. Idler I, Giannopoulos K, Zenz T, Bhattacharya N, Nothing M, Döhner H, et al. Lenalidomide treatment of chronic lymphocytic leukaemia patients reduces regulatory T cells and induces Th17 T helper cells. Br J Haematol. (2010) 148:948–50. doi: 10.1111/j.1365-2141.2009.08014.x
58. Lad DP, Varma S, Varma N, Sachdeva MUS, Bose P, Malhotra P. Regulatory T-cell and T-helper 17 balance in chronic lymphocytic leukemia progression and autoimmune cytopenias. Leuk Lymphoma (2015) 56:2424–8. doi: 10.3109/10428194.2014.986479
59. Matsuno K, Ueta H, Shu Z, Xue-Dong X, Sawanobori Y, Kitazawa Y, et al. The microstructure of secondary lymphoid organs that support immune cell trafficking. Arch Histol Cytol. (2010) 73:1–21. doi: 10.1679/aohc.73.1
60. Rhee I, Zhong MC, Reizis B, Cheong C, Veillette A. Control of dendritic cell migration, T cell-dependent immunity, and autoimmunity by protein tyrosine phosphatase PTPN12 expressed in dendritic cells. Mol Cell Biol. (2014) 34:888–99. doi: 10.1128/MCB.01369-13
61. Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. (2014) 262:36–55. doi: 10.1111/imr.12223
62. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41:14–20. doi: 10.1016/j.immuni.2014.06.008
63. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. (2005) 175:342–9. doi: 10.4049/jimmunol.175.1.342
64. Lahmar Q, Keirsse J, Laoui D, Movahedi K, Van Overmeire E, Van Ginderachter JA. Tissue-resident versus monocyte-derived macrophages in the tumor microenvironment. BBA Rev Cancer (2016) 1865:23–34. doi: 10.1016/j.bbcan.2015.06.009
65. Burgess M, Ellis J, Mapp S, Mollee P, Mazzieri R, Mattarollo S, et al. Transcriptomic analysis of monocytes and macrophages derived from CLL patients which display differing abilities to respond to therapeutic antibody immune complexes. Genom Data (2016) 7:4–6. doi: 10.1016/j.gdata.2015.11.010
66. Filip AA, Cisel B, Koczkodaj D, Wasik-Szczepanek E, Piersiak T, Dmoszynska A. Circulating microenvironment of CLL: are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol Dis. (2013) 50:263–70. doi: 10.1016/j.bcmd.2012.12.003
67. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177:7303–11. doi: 10.1038/srep12524
68. Tsukada N, Burger JA, Zvaifler NJ, Kipps TJ. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood (2002) 99:1030–7. doi: 10.1182/blood.V99.3.1030
69. Bürkle A, Niedermeier M, Schmitt-Gräff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood (2007) 110:3316–25. doi: 10.1182/blood-2007-05-089409
70. Nishio M, Endo T, Tsukada N, Ohata J, Kitada S, Reed JC, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood (2005) 106:1012–20. doi: 10.1182/blood-2004-03-0889
71. Deaglio S, Vaisitti T, Bergui L, Bonello L, Horenstein AL, Tamagnone L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood (2005) 105:3042–50. doi: 10.1182/blood-2004-10-3873
72. Burger JA, Quiroga MP, Hartmann E, Bürkle A, Wierda WG, Keating MJ, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood (2009) 113:3050–8. doi: 10.1182/blood-2008-07-170415
73. DiLillo DJ, Weinberg JB, Yoshizaki A, Horikawa M, Bryant JM, Iwata Y, et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia (2013) 27:170–82. doi: 10.1038/leu.2012.165
74. di Celle PF, Mariani S, Riera L, Stacchini A, Reato G, Foa R. Interleukin-8 induces the accumulation of B-cell chronic lymphocytic leukemia cells by prolonging survival in an autocrine fashion. Blood (1996) 87:4382–9.
75. Burgess M, Cheung C, Chambers L, Ravindranath K, Minhas G, Knop L, et al. CCL2 and CXCL2 enhance survival of primary chronic lymphocytic leukemia cells in vitro. Leuk Lymphoma (2012) 53:1988–98. doi: 10.3109/10428194.2012.672735
76. Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood (2005) 106:1824–30. doi: 10.1182/blood-2004-12-4918
77. Yaktapour N, Übelhart R, Schüler J, Aumann K, Dierks C, Burger M, et al. Insulin-like growth factor-1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood (2013) 122:1621–33. doi: 10.1182/blood-2013-02-484386
78. Ticchioni M, Essafi M, Jeandel P, Davi F, Cassuto J, Deckert M, et al. Homeostatic chemokines increase survival of B-chronic lymphocytic leukemia cells through inactivation of transcription factor FOXO3a. Oncogene (2007) 26:7081–91. doi: 10.1038/sj.onc.1210519
79. Blunt MD, Carter MJ, Larrayoz M, Smith LD, Aguilar-Hernandez M, Cox KL, et al. The PI3K/mTOR inhibitor PF-04691502 induces apoptosis and inhibits microenvironmental signaling in CLL and the Emicro-TCL1 mouse model. Blood (2015) 125:4032–41. doi: 10.1182/blood-2014-11-610329
80. Georgoudaki AM, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Ostling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
81. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature (2016) 539:443–7. doi: 10.1038/nature20554
82. Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, et al. PI3Kγ is a molecular switch that controls immune suppression. Nature (2016) 539:437–42. doi: 10.1038/nature19834
83. Rezvany MR, Jeddi-Tehrani M, Biberfeld P, Soderlund J, Mellstedt H, Osterborg A, et al. Dendritic cells in patients with non-progressive B-chronic lymphocytic leukaemia have a normal functional capability but abnormal cytokine pattern. Br J Haematol. (2001) 115:263–71. doi: 10.1046/j.1365-2141.2001.03117.x
84. Orsini E, Guarini A, Chiaretti S, Mauro FR, Foa R. The circulating dendritic cell compartment in patients with chronic lymphocytic leukemia is severely defective and unable to stimulate an effective T-cell response. Cancer Res. (2003) 63:4497–506.
85. Toniolo PA, Liu S, Yeh JE, Ye DQ, Barbuto JA, Frank DA. Deregulation of SOCS5 suppresses dendritic cell function in chronic lymphocytic leukemia. Oncotarget (2016) 7:46301–14. doi: 10.18632/oncotarget.10093
86. Heinig K, Gatjen M, Grau M, Stache V, Anagnostopoulos I, Gerlach K, et al. Access to follicular dendritic cells is a pivotal step in murine chronic lymphocytic leukemia B-cell activation and proliferation. Cancer Discov. (2014) 4:1448–65. doi: 10.1158/2159-8290.CD-14-0096
87. Palma M, Hansson L, Mulder TA, Adamson L, Nasman-Glaser B, Eriksson I, et al. Lenalidomide as immune adjuvant to a dendritic cell vaccine in chronic lymphocytic leukemia patients. Eur J Haematol. (2018) 101:68–77. doi: 10.1111/ejh.13065
88. Junevik K, Werlenius O, Fogelstrand L, Karlsson-Parra A, Andersson PO. High functional CD70 expression on alpha-type 1-polarized dendritic cells from patients with chronic lymphocytic leukaemia. Scand J Immunol. (2014) 79:415–22. doi: 10.1111/sji.12172
89. Ghosh AK, Kay NE. Critical signal transduction pathways in CLL. Adv Exp Med Biol. (2013) 792:215–39. doi: 10.1007/978-1-4614-8051-8_10
90. Herishanu Y, Katz B-Z, Lipsky A, Wiestner A. Biology of chronic lymphocytic leukemia in different microenvironments: clinical and therapeutic implications. Hematol Oncol Clin North Am. (2013) 27:173–206. doi: 10.1016/j.hoc.2013.01.002
91. Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood (2008) 111:846–55. doi: 10.1182/blood-2007-05-089037
92. Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. (2013) 369:32–42. doi: 10.1056/NEJMoa1215637
93. Ding W, Shanafelt TD, Lesnick CE, Erlichman C, Leis JF, Secreto C, et al. Akt inhibitor MK2206 selectively targets CLL B-cell receptor induced cytokines, mobilizes lymphocytes and synergizes with bendamustine to induce CLL apoptosis. Br J Haematol. (2014) 164:146–50. doi: 10.1111/bjh.12564
94. Cuesta-Mateos C, López-Giral S, Alfonso-Pérez M, de Soria VGG, Loscertales J, Guasch-Vidal S, et al. Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp Hematol. (2010) 38:756–64, 64.e1–4. doi: 10.1016/j.exphem.2010.05.003
95. Krysov S, Dias S, Paterson A, Mockridge CI, Potter KN, Smith KA, et al. Surface IgM stimulation induces MEK1/2-dependent MYC expression in chronic lymphocytic leukemia cells. Blood (2012) 119:170–9. doi: 10.1182/blood-2011-07-370403
96. Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene (2007) 26:3279–90. doi: 10.1038/sj.onc.1210421
97. Igawa T, Sato Y, Takata K, Fushimi S, Tamura M, Nakamura N, et al. Cyclin D2 is overexpressed in proliferation centers of chronic lymphocytic leukemia/small lymphocytic lymphoma. Cancer Sci. (2011) 102:2103–7. doi: 10.1111/j.1349-7006.2011.02046.x
98. Solvason N, Wu WW, Kabra N, Wu X, Lees E, Howard MC. Induction of cell cycle regulatory proteins in anti-immunoglobulin-stimulated mature B lymphocytes. J Exp Med. (1996) 184:407–17.
99. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. (2005) 446:475–82. doi: 10.1007/s00428-005-1264-9
100. Bhattacharya N, Reichenzeller M, Caudron-Herger M, Haebe S, Brady N, Diener S, et al. Loss of cooperativity of secreted CD40L and increased dose-response to IL4 on CLL cell viability correlates with enhanced activation of NF-kB and STAT6. Int J Cancer (2015) 136:65–73. doi: 10.1002/ijc.28974
101. Shono Y, Tuckett AZ, Liou HC, Doubrovina E, Derenzini E, Ouk S, et al. Characterization of a c-Rel Inhibitor that mediates anticancer properties in hematologic malignancies by blocking NF-kappaB-controlled oxidative stress responses. Cancer Res. (2016) 76:377–89. doi: 10.1158/0008-5472.CAN-14-2814
102. Fabbri G, Rasi S, Rossi D, Trifonov V, Khiabanian H, Ma J, et al. Analysis of the chronic lymphocytic leukemia coding genome: role of NOTCH1 mutational activation. J Exp Med. (2011) 208:1389–401. doi: 10.1084/jem.20110921
103. Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood (2009) 113:856–65. doi: 10.1182/blood-2008-02-139725
104. De Falco F, Sabatini R, Del Papa B, Falzetti F, Di Ianni M, Sportoletti P, et al. Notch signaling sustains the expression of Mcl-1 and the activity of eIF4E to promote cell survival in CLL. Oncotarget (2015) 6:16559–72. doi: 10.18632/oncotarget.4116
105. Filarsky K, Garding A, Becker N, Wolf C, Zucknick M, Claus R, et al. Kruppel-like factor 4 (KLF4) inactivation in chronic lymphocytic leukemia correlates with promoter DNA-methylation and can be reversed by inhibition of NOTCH signaling. Haematologica (2016) 101:e249–53. doi: 10.3324/haematol.2015.138172
106. Jordaan G, Liao W, Sharma S. E-cadherin gene re-expression in chronic lymphocytic leukemia cells by HDAC inhibitors. BMC Cancer (2013) 13:88. doi: 10.1186/1471-2407-13-88
107. Gutierrez A, Tschumper RC, Wu X, Shanafelt TD, Eckel-Passow J, Huddleston PM, et al. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood (2010) 116:2975–83. doi: 10.1182/blood-2010-02-269878
108. K Gandhirajan R, J Poll-Wolbeck S, Gehrke I, Kreuzer K-A. Wnt/β-catenin/LEF-1 signaling in chronic lymphocytic leukemia (CLL): a target for current and potential therapeutic options. Curr Cancer Drug Targets (2010) 10:716–27. doi: 10.2174/156800910793605794
109. Li PP, Lu K, Geng LY, Zhou XX, Li XY, Wang X. Bruton's tyrosine kinase inhibitor restrains Wnt signaling in chronic lymphocytic leukemia. Mol Med Rep. (2016) 13:4934–8. doi: 10.3892/mmr.2016.5111
110. Janovska P, Poppova L, Plevova K, Plesingerova H, Behal M, Kaucka M, et al. Autocrine signaling by Wnt-5a deregulates chemotaxis of leukemic cells and predicts clinical outcome in chronic lymphocytic leukemia. Clin Cancer Res. (2016) 22:459–69. doi: 10.1158/1078-0432.CCR-15-0154
111. Seeberger H, Starostik P, Schwarz S, Knörr C, Kalla J, Ott G, et al. Loss of Fas (CD95/APO-1) regulatory function is an important step in early MALT-type lymphoma development. Lab Invest. (2001) 81:977–86. doi: 10.1038/labinvest.3780310
112. Jäger U, Böcskör S, Le T, Mitterbauer G, Bolz I, Chott A, et al. Follicular lymphomas9 BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t (14; 18) translocation. Blood (2000) 95:3520–9.
113. Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer (2006) 6:184–92. doi: 10.1038/nrc1819
114. Bar N, Dikstein R. miR-22 forms a regulatory loop in PTEN/AKT pathway and modulates signaling kinetics. PLoS ONE (2010) 5:e10859. doi: 10.1371/journal.pone.0010859
115. Balakrishnan K, Peluso M, Fu M, Rosin NY, Burger JA, Wierda WG, et al. The phosphoinositide-3-kinase (PI3K)-delta and gamma inhibitor, IPI-145 (Duvelisib), overcomes signals from the PI3K/AKT/S6 pathway and promotes apoptosis in CLL. Leukemia (2015) 29:1811–22. doi: 10.1038/leu.2015.105
116. Richardson SJ, Matthews C, Catherwood MA, Alexander HD, Carey BS, Farrugia J, et al. ZAP-70 expression is associated with enhanced ability to respond to migratory and survival signals in B-cell chronic lymphocytic leukemia (B-CLL). Blood (2006) 107:3584–92. doi: 10.1182/blood-2005-04-1718
117. Cuni S, Perez-Aciego P, Perez-Chacon G, Vargas J, Sanchez A, Martin-Saavedra F, et al. A sustained activation of PI3K/NF-κB pathway is critical for the survival of chronic lymphocytic leukemia B cells. Leukemia (2004) 18:1391–400. doi: 10.1038/sj.leu.2403398
118. Kern D, Regl G, Hofbauer SW, Altenhofer P, Achatz G, Dlugosz A, et al. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene (2015) 34:5341–51. doi: 10.1038/onc.2014.450
119. Pike KA, Tremblay ML. TC-PTP and PTP1B: regulating JAK-STAT signaling, controlling lymphoid malignancies. Cytokine (2016) 82:52–7. doi: 10.1016/j.cyto.2015.12.025
120. Guo A, Lu P, Coffey G, Conley P, Pandey A, Wang YL. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget (2017) 8:12953–67. doi: 10.18632/oncotarget.14588
121. You Y, Myers RC, Freeberg L, Foote J, Kearney JF, Justement LB, et al. Marginal zone B cells regulate antigen capture by marginal zone macrophages. J Immunol. (2011) 186:2172–81. doi: 10.4049/jimmunol.1002106
122. Veninga H, Borg EG, Vreeman K, Taylor PR, Kalay H, Kooyk Y, et al. Antigen targeting reveals splenic CD169+ macrophages as promoters of germinal center B-cell responses. Eur J Immunol. (2015) 45:747–57. doi: 10.1002/eji.201444983
123. Batista FD, Harwood NE. The who, how and where of antigen presentation to B cells. Nature Rev Immunol. (2009) 9:15–27. doi: 10.1038/nri2454
124. Cerutti A, Cols M, Puga I. Activation of B cells by non-canonical helper signals. EMBO Rep. (2012) 13:798–810. doi: 10.1038/embor.2012.111
125. Craxton A, Magaletti D, Ryan EJ, Clark EA. Macrophage- and dendritic cell–dependent regulation of human B-cell proliferation requires the TNF family ligand BAFF. Blood (2003) 101:4464–71. doi: 10.1182/blood-2002-10-3123
126. Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science (1999) 285:260–3.
127. O'Garra A, Chang R, Go N, Hastings R, Haughton G, Howard M. Ly-1 B (B-1) cells are the main source of B cell-derived interleukin 10. Eur J Immunol. (1992) 22:711–7. doi: 10.1002/eji.1830220314
128. Thies FG, Laurindo MFL, Perez EC, e Brito RRN, Mariano M, Popi AF. Cross talk between peritoneal macrophages and B-1 cells in vitro. PLoS ONE (2013) 8:e62805. doi: 10.1371/journal.pone.0062805
129. Popi AF, Lopes JD, Mariano M. Interleukin-10 secreted by B-1 cells modulates the phagocytic activity of murine macrophages in vitro. Immunology (2004) 113:348–54. doi: 10.1111/j.1365-2567.2004.01969.x
130. Wong SC, Puaux AL, Chittezhath M, Shalova I, Kajiji TS, Wang X, et al. Macrophage polarization to a unique phenotype driven by B cells. Eur J Immunol. (2010) 40:2296–307. doi: 10.1002/eji.200940288
131. Sivina M, Hartmann E, Vasyutina E, Boucas JM, Breuer A, Keating MJ, et al. Stromal cells modulate TCL1 expression, interacting AP-1 components and TCL1-targeting micro-RNAs in chronic lymphocytic leukemia. Leukemia (2012) 26:1812–20. doi: 10.1038/leu.2012.63
132. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood (2011) 117:563–74. doi: 10.1182/blood-2010-05-284984
133. Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood (2009) 114:3367–75. doi: 10.1182/blood-2009-06-225326
134. Ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: implications for disease pathogenesis AND treatment. BBA Mol Cell Res. (2016) 1863:401–13. doi: 10.1016/j.bbamcr.2015.07.009
135. Pedersen IM, Kitada S, Leoni LM, Zapata JM, Karras JG, Tsukada N, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood (2002) 100:1795–801.
136. Ramsay AG, Clear AJ, Fatah R, Gribben JG. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: establishing a reversible immune evasion mechanism in human cancer. Blood (2012) 120:1412–21. doi: 10.1182/blood-2012-02-411678
137. Bologna C, Buonincontri R, Serra S, Vaisitti T, Audrito V, Brusa D, et al. SLAMF1/CD150 is a signaling receptor in a subset of chronic lymphocytic leukemia cells (46.9). J Immunol. (2012) 188(Suppl. 1):46.9.
138. Gordiienko I, Shlapatska L, Kholodniuk V, Sklyarenko L, Gluzman DF, Clark EA, et al. The interplay of CD150 and CD180 receptor pathways contribute to the pathobiology of chronic lymphocytic leukemia B cells by selective inhibition of Akt and MAPK signaling. PLoS ONE (2017) 12:e0185940. doi: 10.1371/journal.pone.0185940
139. Yurchenko MY, Kovalevska LM, Shlapatska LM, Berdova GG, Clark EA, Sidorenko SP. CD150 regulates JNK1/2 activation in normal and Hodgkin's lymphoma B cells. Immunol Cell Biol. (2010) 88:565–74. doi: 10.1038/icb.2010.14
140. Kutsch N, Hallek M, Eichhorst B. Emerging therapies for refractory chronic lymphocytic leukemia. Leuk Lymphoma (2015) 56:285–92. doi: 10.3109/10428194.2014.917641
141. MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue-and tumor-associated macrophages but does not inhibit inflammation. Blood (2010) 116:3955–63. doi: 10.1182/blood-2010-02-266296
142. Cannarile MA, Ries CH, Hoves S, Rüttinger D. Targeting tumor-associated macrophages in cancer therapy and understanding their complexity. Oncoimmunology (2014) 3:e955356. doi: 10.4161/21624011.2014.955356
143. Polk A, Lu Y, Wang T, Seymour E, Bailey NG, Singer JW, et al. Colony-stimulating Factor-1 receptor is required for nurse-like cell survival in chronic lymphocytic leukemia. Clin Cancer Res. (2016) 22:6118–28. doi: 10.1158/1078-0432.CCR-15-3099
144. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
145. Galletti G, Scielzo C, Barbaglio F, Rodriguez TV, Riba M, Lazarevic D, et al. Targeting macrophages sensitizes chronic lymphocytic leukemia to apoptosis and inhibits disease progression. Cell Rep. (2016) 14:1748–60. doi: 10.1016/j.celrep.2016.01.042
146. Skarzynski M, Niemann CU, Lee YS, Martyr S, Maric I, Salem D, et al. Interactions between ibrutinib and anti-CD20 antibodies: competing effects on the outcome of combination therapy. Clin Cancer Res. (2016) 22:86–95. doi: 10.1158/1078-0432.CCR-15-1304
147. Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell (2013) 23:249–62. doi: 10.1016/j.ccr.2013.01.008
148. Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs (2015) 7:303–10. doi: 10.1080/19420862.2015.1011450
149. Chanan-Khan AA, Cheson BD. Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol. (2008) 26:1544–52. doi: 10.1200/JCO.2007.14.5367
150. Schulz A, Durr C, Zenz T, Dohner H, Stilgenbauer S, Lichter P, et al. Lenalidomide reduces survival of chronic lymphocytic leukemia cells in primary cocultures by altering the myeloid microenvironment. Blood (2013) 121:2503–11. doi: 10.1182/blood-2012-08-447664
151. Karp M, Kosior K, Karczmarczyk A, Zajac M, Zaleska J, Tomczak W, et al. Cytotoxic activity of valproic Acid on primary chronic lymphocytic leukemia cells. Cytogenetics (2015) 41:55–62. doi: 10.17219/acem/29264
152. Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. (2001) 20:6969–78. doi: 10.1093/emboj/20.24.6969
153. Stamatopoulos B, Meuleman N, De Bruyn C, Mineur P, Martiat P, Bron D, et al. Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia (2009) 23:2281–9. doi: 10.1038/leu.2009.176
154. Dong LH, Cheng S, Zheng Z, Wang L, Shen Y, Shen ZX, et al. Histone deacetylase inhibitor potentiated the ability of MTOR inhibitor to induce autophagic cell death in Burkitt leukemia/lymphoma. J Hematol Oncol. (2013) 6:53. doi: 10.1186/1756-8722-6-53
155. Elknerova K, Myslivcova D, Lacinova Z, Marinov I, Uherkova L, Stöckbauer P. Epigenetic modulation of gene expression of human leukemia cell lines-induction of cell death and senescence. Neoplasma (2011) 58:35–44. doi: 10.4149/neo_2011_01_35
156. Yoshida GJ. Therapeutic strategies of drug repositioning targeting autophagy to induce cancer cell death: from pathophysiology to treatment. Journal of hematology & oncology (2017) 10:67. doi: 10.1186/s13045-017-0436-9
157. Grzywnowicz M, Karczmarczyk A, Skorka K, Zajac M, Zaleska J, Chocholska S, et al. Expression of programmed death 1 ligand in different compartments of chronic lymphocytic leukemia. Acta Haematol. (2015) 134:255–62. doi: 10.1159/000430980
Keywords: B cell, chronic lymphocytic leukemia, crosstalk, dendritic cell, leukemia therapy, macrophage, T cell
Citation: Haseeb M, Anwar MA and Choi S (2018) Molecular Interactions Between Innate and Adaptive Immune Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Front. Immunol. 9:2720. doi: 10.3389/fimmu.2018.02720
Received: 27 June 2018; Accepted: 05 November 2018;
Published: 26 November 2018.
Edited by:
Rayne Rouce, Baylor College of Medicine, United StatesReviewed by:
Antonella Sistigu, Università Cattolica del Sacro Cuore, ItalyPremal Lulla, Baylor College of Medicine, United States
Copyright © 2018 Haseeb, Anwar and Choi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangdun Choi, c2FuZ2R1bmNob2lAYWpvdS5hYy5rcg==